
Abstract
Diffuse intrinsic pontine glioma (DIPG) is the deadliest central nervous system tumor in children. The survival of affected children has remained poor despite treatment with radiation therapy (RT) with or without chemotherapy. We reviewed the medical records of all surviving patients with DIPG treated at our institution between October 1, 1992 and May 31, 2011. Blinded central radiologic review of the magnetic resonance imaging at diagnosis of all surviving patients and 15 controls with DIPG was performed. All surviving patients underwent neurocognitive assessment during follow-up. Five (2.6 %) of 191 patients treated during the study period were surviving at a median of 9.3 years from their diagnosis (range 5.3–13.2 years). Two patients were younger than 3 years, one lacked signs of pontine cranial nerve involvement, and three had longer duration of symptoms at diagnosis. One patient had a radiologically atypical tumor and one had a tumor originating in the medulla. All five patients received RT. Chemotherapy was variable among these patients. Neurocognitive assessments were obtained after a median interval of 7.1 years. Three of four patients who underwent a detailed evaluation showed cognitive function in the borderline or mental retardation range. Two patients experienced disease progression at 8.8 and 13 years after diagnosis. A minority of children with DIPG experienced long-term survival with currently available therapies. These patients remained at high risk for tumor progression even after long follow-ups. Four of our long-term survivors had clinical and radiologic characteristics at diagnosis associated with improved outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diffuse intrinsic pontine glioma (DIPG) is the deadliest central nervous system cancer in children [1]. The survival of children with DIPG has remained poor despite the use of radiation therapy (RT) with or without chemotherapy [1]. Local RT, the mainstay of therapy, provides only a temporary benefit. Chemotherapy has shown no benefit in the treatment of children with DIPG [1].
DIPG is commonly called a fatal cancer [2, 3]. The majority of clinical trials for children with DIPG reported no survivors beyond two years from diagnosis [1]. Although several studies reported exceptional surviving patients, most of them had been followed for <5 years [4–10]. Only a few studies reported children who survived for more than 5 years after the diagnosis of DIPG [11–14]. When information was available, several of these surviving patients had clinical and radiologic characteristics at diagnosis associated with a better outcome, including young age, long latency between onset of symptoms and diagnosis, and atypical magnetic resonance imaging (MRI) characteristics [10–13]. Only one of these manuscripts provided limited information about the neurocognitive status of long-term survivors of DIPG [11].
We provide detailed information about all surviving patients with DIPG treated at our institution over a period of almost 20 years. Unfortunately, these patients remained at high risk for tumor progression even after long follow-ups. Furthermore, we showed that four of our patients had clinical and radiologic characteristics at diagnosis which could predict their improved outcome.
Patients and methods
We identified all surviving patients with DIPG treated at our institution between October 1, 1992 and May 31, 2011. The beginning of the inclusion period coincided with the availability of MRI technology which provided good quality studies at our institution. Clinical characteristics, treatment, and long-term neurologic and endocrinologic sequelae were abstracted from the medical records of all surviving patients. This study was approved by the Institutional Review Board at our institution.
The brain MRIs at diagnosis and during follow-up of all surviving patients were initially reviewed by two experienced neuro-oncologists (A.B., A.G.) to ascertain that these were primary pontine tumors with poorly marginated margins which involved more than 50 % of the pons [15, 16].
As part of their clinical care, all surviving patients underwent neurocognitive assessment of full scale intellectual quotient (IQ) and academic achievement. IQ and academic achievement were measured with instruments that have been standardized and normed with large representative samples. Scores for IQ and academic measures were standardized by age with a mean of 100 and standard deviation of 15. Patients were administered the Wechsler intelligence scale for children, fourth edition (WISC-IV; n = 4) or Wechsler adult intelligence scale, fourth edition (WAIS-IV; n = 1) based on their age at the time of assessment [17, 18]. One patient was administered the WISC-IV using modified procedures to take into account his significant dysarthria. Assessment of academic achievement included reading and math subtests from the Woodcock-Johnson tests of achievement, third edition (WJ-III; n = 3), Wechsler individual achievement test, second edition (WIAT-II; n = 1), and gray diagnostic reading test (GDRT; n = 1) [19–21]. Three assessments included the behavior rating inventory of executive function (BRIEF), a commonly used rater measure that examines the real-world manifestation of various executive functions in children [22]. T-scores for the BRIEF assessment have a mean of 50 and standard deviation of 10, with higher scores reflective of greater concern on all clinical scales and indices.
A single senior neuro-radiologist (Z.P.) blindly reviewed the brain MRIs at diagnosis of all surviving patients and at least two controls (range 2–4; total number of controls: 15) per patient to confirm their diagnosis. The controls were comprised of patients with the institutional diagnosis of DIPG who were selected as appropriately as possible to match the age and year of diagnosis of surviving patients. All control patients died of disease progression after a median survival of 1 year (range 0.5–2.2 years). While still blinded, the neuro-radiologist reviewed the brain MRIs at the end of RT, and at approximately 6 months after diagnosis of the same study population. Finally, the most recent brain MRI of all surviving patients was reviewed. The purpose of this review was to identify possible imaging features that could distinguish long-term survivors from those with the short survival characteristic of DIPG. The imaging features evaluated consisted of primary site of the tumor, signal properties before and after administration of the contrast, presence of necrosis, hemorrhage, and exophytic growth.
A review of RT fields and isodoses of surviving patients was performed by a radiation oncologist (A.P.P.) to assess the relationship between these parameters and neurocognitive deficits.
The Kaplan–Meier method was used to estimate the overall survival of all patients with newly diagnosed DIPG treated at our institution during the study period.
Results
Five (2.6 %) of 191 patients with newly diagnosed DIPG treated at our institution during the study period were surviving for at least 5 years from their diagnosis. A summary of their clinical characteristics at diagnosis is provided in Table 1. Two of five patients were younger than 3 years at the time of diagnosis. Three patients had an interval longer than 6 months between onset of symptoms and diagnosis. Significant neurologic compromise, which is associated with a poor outcome in children with DIPG, was documented at diagnosis in four of the surviving patients [23]. Although patient 5 showed lower cranial nerve palsies (IX and X) and cerebellar signs at diagnosis, his neurologic deficits were mild. He lacked involvement of cranial nerves VI and VII, which is commonly seen in DIPG and associated with worst outcome [24]. None of the surviving patients had neurofibromatosis type 1.
Histologic confirmation was attempted in two cases at the time of progression. Patient 1 was diagnosed with a low-grade astrocytoma, likely a fibrillary astrocytoma. His tumor lacked BRAF duplication by fluorescence in situ hybridization. The biopsy of patient 2 was non-diagnostic.
The blinded central radiologic review confirmed the diagnosis of a typical DIPG in three patients (patients 1, 4, and 5). The tumor in patient 2 was called atypical. Although her tumor originated from the ventral pons and involved more than two-thirds of its cross-sectional area, its borders were considered well defined. It also contained a prominent exophytic component that extended towards the suprasellar cistern and temporal fossa. The tumor of patient 3 was called a medullary tumor although it involved 85 % of the pons. All tumors except one (patient 4) involved more than one segment of the brainstem. All tumors showed hyperintense signal on T2- and four were hypointense on T1-weighted MRI at diagnosis. The tumor of patient 5 had a T2-weighted isointense signal. Minimal necrosis within the tumor was present at diagnosis in patients 1 and 2, but completely disappeared over time. Only one patient (patient 1) had very subtle tumor enhancement at diagnosis. We could not find any associations between the radiologic characteristics of the tumors at diagnosis and survival.
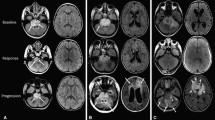
Figure 1a shows the brain MRI at diagnosis of patient 2. The brain MRIs of all surviving patients still showed abnormalities at the time of their last follow-up, some of them still quite significant. Figure 1b shows the brain MRI of patient 2 more than 12 years after her diagnosis.

a T2-weighted axial brain MRI of patient 2 at diagnosis. b T2-weighted axial brain MRI of patient 2 obtained 12.2 years after her diagnosis and before tumor progression
The 1-, 2-, and 5-year overall survival for the 191 patients treated at our institution during the study period were 46.6 % (SE 3.6 %), 11.7 % (SE 2.3 %), and 4.4 % (SE 1.5 %) respectively.
Treatment
Two surviving patients required a ventriculo-peritoneal shunt (n = 1; patient 1) or a third ventriculostomy (n = 1; patient 3) at diagnosis because of hydrocephalus.
All surviving patients underwent local RT. Table 1 provides additional information about their upfront therapy. Two of the patients received therapy in a multicentric and one in an institutional clinical trial [8, 10, 25]. Doses of RT varied between 54 and 55.8 Gy. RT fields included the tumor bed with margins of 10–20 mm to account for microscopic disease.
Patient 1 received further chemotherapy consisting of carboplatin, vincristine, and etoposide after he experienced clinical and radiologic tumor progression at 8.8 years after diagnosis. Patient 2 initially received two cycles of carboplatin and vincristine. She went on to receive local RT because of clinical and radiologic progression. She experienced further tumor progression after RT and required additional chemotherapy, including treatment in two clinical trials which used carmustine in combination with O6-benzylguanine and lonafarnib (SCH66336, Sarasar; Schering-Plough, Kenilworth, NJ) [26, 27]. Her tumor then remained stable without any therapy for 8.6 years when she experienced further tumor progression 13 years after diagnosis.
Long-term sequelae and neurocognitive outcome
Neurologic exam has remained quite intact in three patients (patients 3–5). Patient 1 continued to have significant dysarthria. Patients 1 and 2 maintained moderate cerebellar signs and cranial nerve palsies until they experienced further tumor progression 8.8 and 13 years after diagnosis, respectively. Endocrinologic deficits were diagnosed in four patients consisting of hypothyroidism (n = 2), adrenal (n = 2), and growth hormone insufficiency (n = 4). All these deficits, except for growth hormone insufficiency, were appropriately corrected with supplements.
Neurocognitive evaluations were obtained after a median interval of 7.1 years from diagnosis (range 4.4–11.4 years). The mean IQ score for the combined group was below average [standard score (SS) = 76.0 ± 15.6]. Patient 5 had an overall IQ score in the average range (SS = 94). Patient 1 underwent limited evaluation because of significant dysarthria. All three other patients had scores in the borderline range (patient 2, SS = 76; patient 3, SS = 78) or consistent with a diagnosis of mental retardation (patient 4, SS = 56). Figure 2 provides a summary of intelligence scores for all patients. Similarly, mean academic performance was well below age expectations for the combined group (word reading SS = 72.0 ± 26.0; math calculation SS = 71.0 ± 9.1). Only patient 5 displayed largely intact academic skills.

Intelligence scores of surviving patients. Of note, patient 1 only underwent testing by Wechsler intelligence scale for children, 4th edition (WISC-IV) because of significant dysarthria. FSIQ full scale intellectual quotient, VCI verbal comprehension index, PRI perceptual reasoning index, WMI working memory index, PSI processing speed index
The BRIEF inventory was completed for patients 2, 4, and 5. Significant concerns were raised in the areas of working memory (T = 74.7 ± 28.0) and planning/organization (T = 65.0 ± 15.7) for the combined group. When examining the informants’ responses for the patients individually, two patients were rated as displaying clinically significant problems with working memory (patients 2 and 4) and one patient was rated as displaying clinically significant problems with planning/organization (patient 2). All clinical BRIEF scales were within normal limits for patient 5.
The doses of RT were variable among patients. The minimum, maximum, and mean temporal lobe doses for the five patients were 7.8, 56.9, and 38.4 Gy, respectively. The minimum, maximum, and mean frontal lobe doses were 1.7, 54.3, and 15.1 Gy respectively. For example, patient 3 received between 30 and 40 Gy to 50 % of each temporal lobe and approximately 20 Gy to 50 % of her frontal lobes. Patient 4 received approximately 50 Gy to the same volumes.
Discussion
This is the largest single-institution report to date to address the outcome of long-term survivors of DIPG. Although we confirmed that a minority of children with DIPG survived for long periods of time, it was disappointing to observe that these patients remain at high risk for tumor progression even many years after their diagnosis. All surviving patients received therapies that were otherwise considered ineffective for the majority of children with DIPG [8, 10, 25, 28, 29]. Four of the surviving patients had characteristics at diagnosis associated with improved outcome consisting of young age (patients 1 and 2), long interval between the onset of symptoms and diagnosis (patient 1, 2, and 3), lack of pontine cranial nerve palsies (patient 5), and atypical radiologic imaging (patients 2 and 3) [1, 23, 24].
When considering studies which provided enough details, only 12 children with DIPG who survived for more than 5 years after diagnosis have been reported [11, 13, 14]. At least 8 of 12 long-term survivors had clinical and radiologic characteristics at diagnosis associated with a more favorable outcome [11, 13, 14]. Freeman et al. [11] reported 9 of 130 children with DIPG treated in a prospective clinical trial who survived for more than 5 years. Six of these 9 long-term survivors had either a latency between onset of symptoms and diagnosis of at least 6 months (n = 3) or atypical MRI characteristics (n = 4). Of the three remaining patients, two presented with only cranial nerve palsies or a history suspicious for a longer involvement by the tumor.
We report detailed neurocognitive assessment in long-term survivors of DIPG. It was not surprising to see that only the oldest patient at diagnosis (patient 5) had a normal intellectual function. The neurocognitive deficits observed in four patients showed a pattern of global compromise not restricted to particular domains. Although all surviving patients received only local conformal RT, we demonstrated that their temporal and frontal lobes were exposed to high doses of irradiation. Since the temporal and frontal lobes play important roles in multiple cognitive functions including attention regulation, executive functions, and memory, disruption of these brain areas has the potential to affect learning and academic function [30]. Therefore, it was reasonable to assume that the neurocognitive impairment in these children was secondary to the treatment with RT. Unfortunately we did not have a formal evaluation of neurocognitive function before RT in any of our patients. Three patients had other risk factors for cognitive impairment consisting of diagnosis at an early age (patients 1 and 2) and hydrocephalus (patients 1 and 3). Once effective therapies are available for children with DIPG, it will be critical to use more conformal RT techniques to spare as much as possible other nonaffected cerebral areas.
A few studies reported cognitive assessment in adults who experienced isolated pontine infarcts or hemorrhages [31–33]. The majority of these adults demonstrated significant, long-lasting, and often debilitating dysfunction in several domains, including executive function, attention, and memory, which were independent of motor sequelae [31–33]. This global cognitive dysfunction was attributed to the remote effects of the disruption of corticopontine pathways (diaschisis) [32, 33]. It is possible that this mechanism may also play a role in the neurocognitive impairment of long-term survivors of DIPG.
The blinded radiologic review of our patients and age-matched controls resulted in two discordant diagnoses. Although our radiologist considered the tumor of patient 3 to be arising mostly from the medulla oblongata, this patient’s tumor involved more than two-thirds of the pons. However, since she received therapy in a phase II multicentric clinical trial, her brain MRI at diagnosis had already been centrally reviewed by another expert neuro-radiologist and was considered a typical DIPG [10]. Histologic confirmation was attempted without success in patient 2, who was considered to have an atypical tumor. The criteria for defining radiologically atypical DIPGs have not been standardized and are very subjective. Our definition of atypical tumors was based on the extent of pontine involvement, the presence of well-defined margins, unusual extension outside the pons, or of a dorsal exophytic component. Unfortunately, we did not systematically obtain MR spectroscopy or other advanced techniques from our patients with DIPG to make any comments about their usefulness in predicting outcome. The lack of association between the radiologic characteristics of the tumors at diagnosis and survival in this study was not surprising based on the small number of surviving patients and because 3 of 5 long-term survivors had typical radiologic characteristics at diagnosis.
Recent molecular studies in tumors obtained at biopsy or autopsy unveiled key new molecular findings in DIPG, particularly the presence of two different histone mutations in the majority of tumors [34, 35]. One of these studies has already shown that patients with DIPG harboring the H3F3A K27M mutation have a worse outcome compared to patients with wild-type tumors [35]. Hopefully, future studies will unveil new molecular findings that may help guide the therapy of children with DIPG.
We confirmed that the majority of long-term survivors of DIPG treated with currently available therapies had clinical and radiologic characteristics at diagnosis associated with improved prognosis. Our patients remained at high risk for tumor progression even after long follow-ups. Although the radiologic criteria to define DIPG based on standard MRI were described more than 20 years ago [15], our results confirmed that the radiologic definition of a typical DIPG remains contentious.
References
Hargrave D, Bartels U, Bouffet E (2006) Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 7:241–248
Puget S, Philippe C, Bax DA et al (2012) Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One 7:e30313
Monje M, Mitra SS, Freret ME et al (2011) Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci USA 108:4453–4458
Broniscer A, Leite CC, Lanchote VL, Machado TM, Cristófani LM (2000) Radiation therapy and high-dose tamoxifen in the treatment of patients with diffuse brainstem gliomas: results of a Brazilian cooperative study. Brainstem Glioma Cooperative Group. J Clin Oncol 18:1246–1253
Greenberg ML, Fisher PG, Freeman C et al (2005) Etoposide, vincristine, and cyclosporin A with standard-dose radiation therapy in newly diagnosed diffuse intrinsic brainstem gliomas: a Pediatric Oncology Group phase I study. Pediatr Blood Cancer 45:644–648
Wagner S, Warmuth-Metz M, Emser A et al (2006) Treatment options in childhood pontine gliomas. J Neurooncol 79:281–287
Massimino M, Spreafico F, Biassoni V et al (2008) Diffuse pontine gliomas in children: changing strategies, changing results? a mono-institutional 20-year experience. J Neurooncol 87:355–361
Broniscer A, Baker JN, Tagen M et al (2010) Phase I study of vandetanib during and after radiotherapy in children with diffuse intrinsic pontine glioma. J Clin Oncol 28:4762–4768
Haas-Kogan DA, Banerjee A, Poussaint TY et al (2011) Phase II trial of tipifarnib and radiation in children with newly diagnosed diffuse intrinsic pontine gliomas. Neuro Oncol 13:298–306
Pollack IF, Stewart CF, Kocak M et al (2011) A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: a report from the pediatric brain tumor consortium. Neuro Oncol 13:290–297
Freeman CR, Bourgouin PM, Sanford RA, Cohen ME, Friedman HS, Kun LE (1996) Long term survivors of childhood brain stem gliomas treated with hyperfractionated radiotherapy. Clinical characteristics and treatment related toxicities. The Pediatric Oncology Group. Cancer 77:555–562
Allen J, Siffert J, Donahue B et al (1999) A phase I/II study of carboplatin combined with hyperfractionated radiotherapy for brainstem gliomas. Cancer 86:1064–1069
Hargrave D, Chuang N, Bouffet E (2008) Conventional MRI cannot predict survival in childhood diffuse intrinsic pontine glioma. J Neurooncol 86:313–319
Warren K, Bent R, Wolters PL et al (2012) A phase 2 study of pegylated interferon α-2b (PEG-Intron(®)) in children with diffuse intrinsic pontine glioma. Cancer 118:3607–3613
Barkovich AJ, Krischer J, Kun LE et al (1990/1991) Brain stem gliomas: a classification system based on magnetic resonance imaging. Pediatr Neurosurg 16:73–83
Fischbein NJ, Prados MD, Wara W, Russo C, Edwards MS, Barkovich AJ (1996) Radiologic classification of brain stem tumors: correlation of magnetic resonance imaging appearance with clinical outcome. Pediatr Neurosurg 24:9–23
Wechsler D (2003) Wechsler intelligence scale for children. The Psychological Corporation, San Antonio
Wechsler D (2008) Wechsler adult intelligence scale. The Psychological Corporation, San Antonio
Woodcock RW, McGrew KS, Mather N (2001) Woodcock–Johnson tests of achievement III. Riverside Publishing, Itasca
Wechsler D (2002) Wechsler individual achievement test. The Psychological Corporation, San Antonio
Bryant BR, Wiederholt L, Bryant DP (2004) Gray diagnostic reading tests. PRO-ED Inc., Austin
Gioia GA, Isquith PK, Guy SC, Kenworthy L (2000) Behavior rating inventory of executive function. Psychological Assessment Resources Inc., Odessa
Sanford RA, Freeman CR, Burger P, Cohen ME (1988) Prognostic criteria for experimental protocols in pediatric brainstem gliomas. Surg Neurol 30:276–280
Fisher PG, Breiter S, Carson BS et al (2000) A clinicopathologic reappraisal of brain stem tumor classification. Identification of pilocytic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer 89:1569–1576
Pollack IF, Jakacki RI, Blaney SM et al (2007) Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a pediatric brain tumor consortium report. Neuro Oncol 9:145–160
Adams DM, Zhou T, Berg SL, Bernstein M, Neville K, Blaney SM (2008) Children’s Oncology Group. phase 1 trial of O6-benzylguanine and BCNU in children with CNS tumors: a Children’s Oncology Group study. Pediatr Blood Cancer 50:549–553
Kieran MW, Packer RJ, Onar A et al (2007) Phase I and pharmacokinetic study of the oral farnesyltransferase inhibitor lonafarnib administered twice daily to pediatric patients with advanced central nervous system tumors using a modified continuous reassessment method: a pediatric brain tumor consortium study. J Clin Oncol 25:3137–3143
Broniscer A, Iacono L, Chintagumpala M et al (2005) Role of temozolomide after radiotherapy for newly diagnosed diffuse brainstem glioma in children: results of a multiinstitutional study (SJHG-98). Cancer 103:133–139
Cohen KJ, Heideman RL, Zhou T et al (2011) Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children’s Oncology Group. Neuro Oncol 13:410–416
Lezak MD, Howieson DB, Loring DW (2004) Neuropsychological assessment. Oxford University Press, New York
Garrard P, Bradshaw D, Jäger HR, Thompson AJ, Losseff N, Playford D (2002) Cognitive dysfunction after isolated brain stem insult. An underdiagnosed cause of long term morbidity. J Neurol Neurosurg Psychiatry 73:191–194
Hoffmann M, Watts A (1998) Cognitive dysfunction in isolated brainstem stroke: a neuropsychological and SPECT study. J Stroke Cerebrovasc Dis 7:24–31
Hoffmann M, Schmitt F (2004) Cognitive impairment in isolated subtentorial stroke. Acta Neurol Scand 109:14–24
Wu G, Broniscer A, McEachron TA et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253
Khuong-Quang DA, Buczkowicz P, Rakopoulos P et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447
Acknowledgments
This work was supported by the US National Institutes of Health Cancer Center Support (CORE) Grant P30 CA21765 and by the American Lebanese Syrian Associated Charities (ALSAC). All procedures described in this manuscript were performed in compliance with clinical research regulations in the United States.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jackson, S., Patay, Z., Howarth, R. et al. Clinico-radiologic characteristics of long-term survivors of diffuse intrinsic pontine glioma. J Neurooncol 114, 339–344 (2013). https://doi.org/10.1007/s11060-013-1189-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-013-1189-0