
Abstract
The last decade has seen significant breakthroughs for the management of metastatic melanoma. The search for molecular targets and the new understanding of tumor’s interplay with the host immune system have led to the development of therapies that have changed the management and outcome of these patients. Biological agents have become front-line options for patients with melanoma. BRAF inhibitors and immune checkpoint blockade suppression by monoclonal antibodies are new standards of care. In this review we will evaluate the diagnosis and clinical management approach to metastatic or high risk melanoma patients, emphasizing immunology and molecular analysis to define therapy.
Support from the Lake Champlain Cancer Research Organization
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Clinical Overview
Melanoma is the most aggressive of cutaneous malignancies. It accounts for less than 5 % of skin cancer cases, but for the majority of deaths from skin cancer. The incidence rates have increased in the last 30 years [1]. Before the age of 40, the incidence is higher in women, and after 40, higher in men. There were about 76,000 new cases and 9,000 deaths from melanoma in the United States in 2013. The estimated death rate is 2.6 in 100,000 [2]. In Australia and New Zealand the death rate is higher at 3.5 per 100,000, and in Western Europe, slightly lower at 1.8 per 100,000 [2, 3]. The median survival of patients affected with metastatic melanoma is about 1 year. The most important prognostic factors include the Breslow, which is the thickness of the melanoma measured in millimeters, the stage (Table 28.1), and the presence or absence of ulceration of the overlying epithelium. These factors have been included in the TNM staging system that was most recently updated in 2009 [4].
The mainstay of treatment for early melanoma is surgery, which helps staging patients and has a curative intent. Definitive surgery includes a wide excision with or without sentinel lymph node biopsy (SLNB). The role of SLNB on overall survival is unclear. The NCCN guidelines recommend a wide excision as category 1 evidence, but the SLN is only a category 2B and should be discussed and advocated for lesions thicker than 0.75 mm (Stage 1A) [5]. Sentinel lymph node biopsy is preferred over observation because it provides staging and prognostic information on the risk of stage upgrade with increasing Breslow [6]. The incidence of sentinel node micrometastases is 15–20 % in patients with intermediate thickness primary melanoma (1.2–3.5 mm). High risk features for positive sentinel lymph node are high mitotic rate, ulceration and lymph vascular invasion [5]. Patients with lymph node metastases should undergo lymphadenectomy which improves prognosis and survival rate and be offered adjuvant immunotherapy on a clinical trial or with interferon. Patients with metastatic disease need systemic therapy. In the last 5 years, there has been substantial development in the treatment of advanced melanoma. New targeted therapies and immunotherapies are benefiting a subset of patients who derive a longer survival [2]. Therapeutic options include targeted therapies, immune-based treatments, chemotherapy, or a combination thereof.
2 Molecular Signaling Pathways
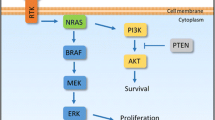
The mitogen activated protein kinase (MAPK) pathway is activated in the majority of melanomas, through the neuroblastoma RAS viral oncogene homolog (NRAS) (15–20 %) or the v-raf murine sarcoma viral oncogene homolog B1 (BRAF) (40–50 %). NRAS and BRAF are components of the MAPK pathway, also called RAS-RAF-MEK-ERK signal transduction pathway (Fig. 28.1) [7]. Under physiological conditions the MAPK pathway transmits extracellular signals to the nucleus which leads to the expression of genes that drive cell proliferation, differentiation, and survival [8, 9]. The MAPK pathway is a critical component of oncogenic RAS signaling. In normal cells, the most important downstream mediators through this pathway are BRAF found in the testes, some hematopoietic precursors, and some brain cells, and CRAF which is essential to the daily function of most other cells. Both are serine/threonine kinases. The RAF proteins are major mediators of this pathway and signal through phosphorylation and activation of downstream kinases. RAF homodimerization or heterodimerization interacts with MEK and initiates its phosphorylation that leads to the phosphorylation and activation of ERK (also called MAPK), its substrate.

MAPK pathway. TRK Tyrosine Kinase. When a ligand binds to a receptor on the cell surface it stimulates the activity of RAS. There are three isoforms of RAS: HRAS, KRAS and NRAS (RAS is the most commonly mutated oncogene in human malignancies). NRAS is commonly mutated in melanomas and can signal through MAPK and non MAPK pathways (PI3K pathways) [7, 8]
The activation of ERK leads to pro-growth signals that alter gene transcription. CRAF can have oncogenic effects through MEK independent pathways leading to nuclear factor kappa B (NF-kB) activation and inhibition of critical regulators of apoptosis (ASK-1 and MST-2). Activated BRAF has no other substrates other than MEK [8, 9].
There are more than 65 BRAF mutations reported in the literature. BRAF mutations occur most frequently in exon 15 at codon 600 (V600). The most common is BRAF V600E, comprising 90 % of all BRAF mutations. There are several substitutions that have been documented including valine by glutamic acid (V600E, 75 %), valine by lysine (V600K, 10–30 %), valine by arginine (V600R, 1–7 %) and lysine by glutamic acid (K601E, 1–4 %). Several characteristics are attributable to different BRAF mutations, as described in Tables 28.2 and 28.3 [7, 10]. Other pathway interferences by mutated BRAF include activation of NF-kB and others. For example, BRAF mutation (BRAF V600E) is also associated with activation of the mammalian target of rapamycin (mTOR) pathway. Activated ERK inhibits the tumor suppressor LKB1, a serine/threonine protein kinase mutated in autosomal dominantly inherited Peutz-Jeghers syndrome, a disease characterized by increased risk of benign and malignant tumors in multiple tissues. The LKB1 tumor suppressor negatively regulates mTOR signaling. ERBB 4 is activated in 19 % of melanomas, which leads to the activation of the PI3K pathway. This pathway involves PTEN and AKT, as described in Fig. 28.1. Normally PTEN is a tumor suppressor protein that negatively regulates the PI3K pathway, but when it is mutated it can activate the PI3k pathway by increasing expression of AKT. Selective activation of AKT (a downstream factor) is seen in 53 % of primary and 67 % of metastatic melanomas. PTEN mutation or deletion has been reported in up to 30 % of melanomas and can occur concurrently with BRAF, but not NRAS mutations [11].
3 BRAF Testing
Testing for a BRAF mutation involves the extraction of genomic DNA from the tumor sample and a real time polymerase chain reaction (PCR) assay that detects both wild type and mutant BRAF. The Food and Drug Administration (FDA) has approved two tests cobas 4800 BRAF V600 Mutation Test (Roche Molecular Systems Inc., Pleasanton, CA, U.S.A.) and THxID®-BRAF KIT [12]. The Cobas 4800 test can identify 96 % of mutations across all specimen types with 5 % mutant alleles at a DNA input of 125 ng, an amount readily obtained from one 5 μm section of formalin-fixed paraffin-embedded tissue. The test can also identify V600K and V600D mutations, although the limit of detection is lower than that for V600E. Eighteen percent mutant alleles in a specimen are required for detection [13]. Other testing methods reported in the literature but not readily available in all institutions, include immunohistochemistry, pyrosequencing and next generation sequencing [12]. In our institution, BRAF testing is a send-out test and usually takes around 14 days to be reported. We recommend that the reader familiarizes him/herself with the turnaround time at their institution or vendors.
4 Chemotherapy for Metastatic Melanoma
Dacarbazine is the standard chemotherapy option for metastatic melanoma and the only FDA approved cytotoxic drug. The response rate is about 15 % with a median overall survival of 6–8 months [13]. Complete responses are observed in 5 % of patients with a 2–6 % survival at 5 years [14].
Temozolomide is an oral prodrug of the active metabolite of dacarbazine. It has been used to treat advanced melanoma and crosses the blood brain barrier, a theoretical advantage for patients with brain metastases. In a phase III study that compared temozolomide with dacarbazine in patients with no brain metastases, the median survival time was 7.7 and 6.4 months, respectively (HR 1.18; 95 % Confidence Interval (CI), 0.92–1.52). The median PFS was longer in patients who receive temozolomide (1.9 months) compared to dacarbazine (1.5 months) (p 0.012). There was no difference in overall survival or overall response rate [15].
Current NCCN guidelines list the following agents as category 2B for systemic chemotherapy of melanoma: nab paclitaxel, dacarbazine or temozolomide, dacarbazine, cisplatin, and vinblastine (CVD) with or without interferon alpha, and carboplatin with paclitaxel [5]. Combination chemotherapy usually yields a 25 % response rate with no improvement in survival. Biochemotherapy combines interleukin and interferon with CVD. This combination failed to demonstrate a survival benefit despite higher response rates. Chemotherapy remains a good option for patients who have potentially resectable oligometastases, and who obtain a response to systemic treatment given as a neoadjuvant modality prior to surgery. Patients should be carefully selected for this multidisciplinary approach.
5 Targeted Therapies for Metastatic Melanoma
5.1 c-KIT Inhibitors
In a phase II open label trial of 28 patients with advanced unresectable melanoma bearing a c- KIT mutation, imatinib, at 400 mg twice a day, yielded an overall response rate of 16 % (95 % confidence interval, 2–30 %) with a median time to progression of 12 weeks and a median overall survival of 46 weeks. While these results demonstrate the targeted effects, better patient selection is needed to narrow the targets that imatinib affects. Further studies with c- KIT inhibitors are underway in melanoma [16].
5.2 BRAF and MEK Inhibitors
To date, the FDA has approved three BRAF inhibitors vemurafenib, dabrafenib and trametinib, along with the combination of dabrafenib with trametinib (Table 28.4).
5.2.1 Sorafenib
The first BRAF inhibitor to be tested was sorafenib, however a double-blind, randomized, placebo-controlled phase III study failed to improve overall survival when given in combination with carboplatin and paclitaxel for chemotherapy-naïve patients with metastatic melanoma. The median overall survival was 11.3 months (95 % CI, 9.8–12.2 months) for carboplatin and paclitaxel and 11.1 months (95 % CI, 10.3–12.3 months) for carboplatin, paclitaxel and sorafenib; the difference in overall survival distribution was not statistically significant. The reason for sorafenib failure could be attributed to the fact that is a non-specific inhibitor and that the trial included an unselected population [23].
5.2.2 Vemurafenib
Several clinical trials have established the clinical efficacy of vemurafenib for BRAF V600E mutated melanoma. The dose of vemurafenib is 960 mg orally twice a day. The overall response rate is 53 %, with 6 % and 47 % of complete and partial responses, respectively. The median duration of response is 6.7 months, the median progression-free survival (PFS), 6.8 months (95 % CI, 5.6–8.1), and the median overall survival, 15.9 months (95 % CI, 11.6–18.3 months) [24, 25]. The phase 3 trial, BRIM-3 Study Group, eventually led to FDA approval. The study enrolled 675 previously untreated patients with BRAF V600E mutated melanoma who were randomized between vemurafenib and dacarbazine. At 6 months, the overall survival was 84 % (95 % CI, 78–89) in the vemurafenib group and 64 % (95 % CI, 56–73) in the dacarbazine group. The study allowed a crossover from dacarbazine to vemurafenib. Vemurafenib was associated with a relative reduction of 63 % in the risk of death, compared to dacarbazine (p < 0.001). However, when the melanoma recurs, the prognosis is terrible. About 50 % of patients died of disease progression within 28 days of the last vemurafenib dose. Patients who progress after BRAF inhibitors have rapid clinical deterioration [2]. The most common adverse events included grade 1 and 2 photosensitivity, fatigue, alopecia, arthralgia, rash, serositis, keratoacanthoma and squamous cell carcinoma, and nausea and diarrhea. Squamous cell carcinoma was diagnosed in 18–26 % of patients [2, 25]. These skin cancers develop secondary to a paradoxical activation of the MAPK pathway and proliferation of HRAS Q61L transformed keratinocytes. This creates a decreased latency and accelerated growth of cutaneous squamous cell carcinomas and keratoacanthomas. Vemurafenib is not a tumor promoter but has been shown to accelerate the growth of preexisting RAS-mutant subclinical lesions [26]. All patients should have a dermatology evaluation before starting treatment with vemurafenib and a skin screening every 2 months afterwards. They should be aware of new lesions and report them to their oncologist. Before starting therapy it is recommended to perform an electrocardiogram to monitor for QT prolongation, to consult an ophthalmologist for a baseline eye exam, and to protect skin with regular sunscreen.
5.2.3 Dabrafenib
Dabrafenib is a selective inhibitor of mutant BRAF kinase. The first phase 1 study enrolled 184 patients including 156 patients with melanoma with or without asymptomatic brain metastases. The median PFS was 5.5 months in patients without brain metastasis and 4.2 months in patients with brain metastases. The dose is 150 mg orally twice a day. The phase 2 study enrolled 92 melanoma patients with histologically confirmed BRAF mutations (76 with BRAF V600E and 16 with BRAF V600K mutations). A 59 % response rate was seen in patients with BRAF V600E mutation, but only two patients with BRAF V600K mutation obtained a complete response. The median PFS was 6.3 months for BRAF V600E and 4.5 months for BRAF V600K. After a follow up of 11.9 months, the median overall survival was 13.1 and 12.9 months for BRAF V600E and BRAF V600K, respectively. The median time to response for BRAF V600E was 1.3 months [27]. The phase 3 study included 250 patients with stage IV or unresectable stage III BRAF V600E mutation positive melanoma randomly assigned to receive dabrafenib 150 mg orally twice a day or dacarbazine 1,000 mg/m2 intravenously every 3 weeks in a 3/1 ratio. The median PFS was 5.1 months for dabrafenib and 2.7 months for dacarbazine, with a hazard ratio of 0.30 (95 % CI 0.18–0.51; p < 0.0001) [17]. The most common adverse events were cutaneous squamous cell carcinoma, keratoacanthoma, fatigue, pyrexia, headache, nausea, and arthralgia. A panniculitis has also been described in patient obtaining remissions. The development of squamous cell carcinoma led to studies of the combination of BRAF inhibitors with MEK inhibitors to inhibit the squamous cell carcinoma pathway [27, 28].
5.2.4 Trametinib
Activated BRAF phosphorylates and activates MEK proteins (MEK1 and MEK2), which then activate downstream MAP kinases. Trametinib, a selective inhibitor of MEK1 and MEK2, is administered orally. The phase 3 study enrolled 322 patients with stage IIIC or IV cutaneous melanoma with a V600E (281 patients) or V600K BRAF mutations (40 patients). All patients were naïve to BRAF and/or MEK inhibition, or to ipilimumab. Patients with stable brain metastases were also allowed to enroll. Patients were randomized in a 2:1 ratio to 2 mg of trametinib once daily or chemotherapy consisting of either dacarbazine (1,000 mg/m2) or paclitaxel (175 mg/m2), every 3 weeks. The median PFS was 4.8 months in the trametinib group and 1.5 months in the chemotherapy group (HR, 0.45; 95 % CI, 0.33–0.63; P < 0.001). The 6-month overall survival rate was 81 % for trametinib and 67 % for chemotherapy. Crossover was allowed during this trial and 47 % of patients treated with chemotherapy crossed over to trametinib. The median duration of response was 5.5 months in the trametinib group. Adverse events of trametinib include rash, diarrhea, peripheral edema, fatigue, and dermatitis acneiform. There was a decrease in ejection fraction of 7 % in the trametinib group. There were no reports of cutaneous squamous cell carcinomas in patients receiving trametinib [18].
5.2.5 Cobimetinib
Cobimetinib is a potent selective MEK inhibitor, administered orally. The phase 3 study enrolled 495 patients with advanced stage IIIC or stage IV melanoma with a BRAF V600 mutation. They included patients with stable metastatic disease to the brain. Patients were randomized to vemurafenib + cobimetinib or placebo. The dose of vemurafenib was 960 mg BID and the dose of cobimetinib was 60 mg daily for 21 days and 7 days off. The median PFS was 9.9 months in the combination group and 6.2 months in the control group (HR for death or disease progression, 0.51; 95 % confidence interval [CI], 0.39–0.68; P < 0.001). The rate of CR or PR in the combination group was 68 %, and 45 % in the control group (P < 0.001). The interim analyses of overall survival showed 9-month survival rates of 81 % (95 % CI, 75–87) in the combination group and 73 % (95 % CI, 65–80) in the control group. Median duration of response was not reached in the combination group but was only 7.3 months in the vemurafenib and placebo arm. Adverse events in the combination group included central serous retinopathy, gastrointestinal events (diarrhea, nausea, or vomiting), photosensitivity, elevated aminotransferase levels, and an increased creatinine kinase level; most of them were grade 1 or 2. Most common grade 4 AE was elevation of creatinine kinase in the combination group (4 %), thought to be a class effect of MEK inhibition [19].
5.2.6 Combination of BRAF Inhibitors and MEK Inhibitors
In order to overcome resistance to BRAF inhibitors, several studies are underway to evaluate alternative combination of kinase inhibitors. Patients with BRAF V600 mutated metastatic melanoma were randomized to receive the combination of dabrafenib 150 mg orally daily and trametinib 1 or 2 mg, or dabrafenib monotherapy. The maximum tolerated doses for this combination were not reached in this study. The recommended phase 2 dose is the combination of dabrafenib 150 mg with trametinib 2 mg, which combines the recommended monotherapy dose for each agent. The median PFS of these 247 patients was 9.4 months for the combination and 5.8 months for single agent dabrafenib (HR, 0.39; 95 % CI, 0.25–0.62; p < 0.001). The overall response rate was 76 % for the combination group and 54 % for dabrafenib single agent (p = 0.03). Only 7 % of patients developed cutaneous squamous cell carcinomas when treated with the combination, but 19 % did with the monotherapy (p = 0.09). This combination was approved by the FDA in 2013.
In a preplanned interim overall survival analysis, the overall survival rate at 12 months was 72 % (95 % confidence interval [CI], 67–77) in the combination-therapy group and 65 % (95 % CI, 59–70) in the vemurafenib group (hazard ratio for death in the combination-therapy group, 0.69; 95 % CI, 0.53–0.89; P = 0.005). Median PFS was 11.4 months in the combination group and 7.3 months in the vemurafenib group (hazard ratio, 0.56; 95 % CI, 0.46–0.69; P < 0.001). The objective response rate in the combination group was 64 % and 51 % in the vemurafenib group (P < 0.001) [20, 21].
5.3 Mechanism of Resistance to BRAF Inhibitors
Tumor resistance develops in a median of 5–7 months. There are different mechanisms by which tumors develop resistance. The MAPK pathway dependent mechanism includes de novo mutations in NRAS (upstream) and MEK (downstream). Overexpression of mitogen-activated protein kinase kinase kinase 8 (MAP3K8) drives resistance to RAF inhibition in BRAF V600E cell lines. MAP3K8 activates ERK primarily through MEK-dependent mechanisms that do not require RAF signaling. Moreover, MAP3K8 expression is associated with de novo resistance in BRAF V600E cultured cell lines and acquired resistance in melanoma cells and tissue obtained from relapsing patients following treatment with MEK or RAF inhibitors [14]. Another MAPK independent pathway mechanism involves the overexpression or overactivation of PDGFR-β or IGF1R inducing oncogenic signaling through PI3K-AKT-mTOR pathway (Fig. 28.2) [30]. Resistance to the combination of dabrafenib and trametinib was tested by whole exome and whole transcriptome sequencing, on five patients with acquired resistance. Three patients had additional MAPK pathway alterations including a novel MEK2 mutation that conferred resistance to RAF/MEK inhibition in vitro [31]. Acquired resistance to these targeted therapies need to be further studied to determine alternative treatment strategies. These may include combination therapies, addition of downstream targeted therapies, and dosing adjustment, among others.

Mechanisms of resistance to BRAF inhibitors [29]. 1. NRAS mutations. 2. BRAF V600E slice variant: creates a truncated form of BRAF mRNA and this mutated BRAF protein has enhanced interaction with RAS. This leads to dimer formation between the truncated, activated BRAF kinase and wild type RAF kinases. Once dimerized, BRAF inhibitors (such as Vemurafenib) can induce transactivation and then reactivation of MAPK pathway. 3. MEK-1 mutation. 4. BRAF inhibitors lead to decreased activation of ERK. There is decreased level of negative regulators which then leads to decreased suppression of RAS. RAS reactivates and then dimerizes and activates BRAF. 5. IGFR activation leads to non-MAPK pathway activation. 6. PDGFRβ activation leads to non-MAPK pathway activation
A phase I/II trial evaluated the combination of dabrafenib and trametinib after disease progression with a BRAF inhibitor. The ORR was 14 % (95 % CI, 7–24 %), and an additional 46 % of patients had stable disease 8 weeks; median PFS was 3.6 months. This regimen may be a therapeutic strategy in patients who had previously been exposed to single agent BRAF inhibitor for >6 months. In patients with rapid development of resistance, less than 6 months, derived no benefit on further combination therapy and had rapid progression [32].
6 Immunotherapy for Metastatic Melanoma
Melanoma is associated with immune-related phenomena, including spontaneous remission in the absence of active therapy or vitiligo. Rare patients who developed infections and fever have been found to have tumor regression [33]. About 16 % of patients with advanced melanoma respond to high-dose interleukin-2 (IL-2), a non-specific type of immunotherapy that activates T cells [34]. Cytotoxic T lymphocyte (CTL) activation requires antigen-specific recognition. Co-stimulatory and co-inhibitory signals are also required to orchestrate this process [35] (Fig. 28.3 and Table 28.8). Immunomodulation of co-inhibitory signals, including CTLA-4 and PD-1, have become pivotal targets for the treatment of melanoma. In the last 5 years, such new targeted immunotherapy drugs have revolutionized the treatment of advanced melanoma. Gradual understanding of immune-specialized cell interplay will lead to newer therapeutic approaches.

T cell activation and mechanism of action of ipilimumab. When an antigen (Ag) is presented in the context of the major histocompatibility complex (MHC) to the T cell receptor (TCR), binding of B7 with CD28 occurs which activates the T cell. Slightly later, the activated T cell stimulates CTLA4 which also binds to B7 to down-regulate the T cell. Ipilimumab inactivates the binding of CTLA4 with B7, allowing the T cell to remain activated [36]
6.1 Evaluation of Response after Immunotherapy for Melanoma
Response Evaluation Criteria in Solid Tumors (RECIST) or WHO criteria are conventionally employed to evaluate the response to chemotherapy in solid tumors. Tumor response to immunotherapy has a different pattern. Tumor shrinkage induced by immunotherapy may be preceded by inflammatory changes, initially causing tumor swelling. New immune-related response criteria (irRC) have been proposed [49]. The irRC approach attempts to not separate index lesions from new lesions. Instead, irRC considers index lesions and new measurable lesions together to measure total tumor burden and defines immune-related complete response (irCR), immune-related partial response (irPR), and immune-related stable disease (irSD). As long as the total tumor burden is decreased to more than 50 %, progression of some lesions or the appearance of new lesions is acceptable to adjudicate partial response. In most clinical trials of immunotherapy in advanced melanoma, irRC are used with RECIST and/or WHO criteria in parallel or in tandem (Table 28.5).
6.2 Immunotherapy Drugs
6.2.1 Interferon Alpha
Single treatment with interferon alpha was primarily tested for adjuvant therapy in high-risk melanoma. The Eastern Cooperative Oncology Group trial 1684 compared high dose adjuvant interferon versus observation and showed a prolonged median survival of 3.8 years compared to 2.8 years for observation [50]. Interferon was therefore approved by the FDA in 1996. There is a debate whether interferon alpha improves overall survival or not, as some of the earlier trials and pooled meta-analyses did not reveal statistical significant HR for overall survival [51]. The most recent meta-analysis included 10,499 patients enrolled on 18 eligible randomized clinical trials [52]. This pooled analysis demonstrated a HR of 0.83 in favor of adjuvant interferon for PFS, and 0.91 for overall survival, both statistically significant (Table 28.6).
6.2.2 Interleukin-2
Interleukine-2 (IL-2) is an immune-modulatory cytokine that enhances cellular immune responses through inducing lymphocyte proliferation and promoting lymphokine production [54]. High-dose bolus of recombinant IL-2 (600,000–720,000 international units per kg administrated intravenously as a 15 min bolus every 8 h over five consecutive days up to a maximum of 28 doses per course) was given to patients with advanced melanoma [54]. Eight clinical trials using high dose IL-2 with or without lymphokine-activated killer (LAK) cells were conducted from 1985–1993, recruiting 270 patients with advanced melanoma. The pooled analysis of these trials confirms that the use of high-dose IL-2 in advanced melanoma results in a low but durable response rate [34]. The overall response rate is 16 % (95 % confidence interval, 12–21 %; complete response, 6 %; partial response, 10 %). Of the responding 43 patients, 20 (47 %) patients were alive at a median follow up of 62 months and 15 (35 %) survived more than 10 years [34]. High-dose IL-2 was approved by the FDA for treatment of metastatic melanoma in 1998. Until recently, IL-2 has been the mainstay of treatment, either alone, or as part of biochemotherapy. IL-2 is difficult to administer because of side effects. Treatment with high-dose IL-2 requires expertise and intensive care access. IL-2 administration is limited to patients with excellent performance status of 0–1, age less than 65 years old, and with excellent organ function. Treatment related mortality is 1–2 %. Common IL-2 toxicities include hypotension, cardiac arrhythmias, metabolic acidosis, nausea and vomiting, diarrhea, fevers and chills, dyspnea, peripheral edema, elevated creatinine, elevated transaminases, neurotoxicity, skin rash, and pruritus [34]. Although patients are carefully selected to receive high-dose IL-2, it is not possible to predict who will respond. However, NRAS mutation status might correlate with response to IL-2 [55]. Among patients with NRAS mutation, 47 % responded to high dose IL-2, while 23 % with proved BRAF mutation responded to IL-2. Among patients without NRAS or BRAF mutation, the response rate was only 12 % [55]. Gene expression profiling and other newer technologies will provide more answers, but this is not yet applicable to clinical practice.
6.2.3 Anti CTLA-4 Therapy
Cytotoxic T-lymphocyte antigen-4 (CTLA-4, CD152) is an antigen that is expressed on CTLs (Fig. 28.3). It competes with the co-stimulatory molecule CD28 for its shared ligand family B7 on the surface of antigen presenting cells (APCs) [56]. CTLA-4 is up-regulated and becomes functional only after T-cell activation. This physiological delay in CTLA-4 up-regulation allows for initial T-cell activation by CD28, followed by a regulatory feedback inhibition by CTLA-4 [36]. Therefore, CTLA-4 functions as a negative regulator of activated T cells, and is commonly called an immunocheckpoint protein. To generate more effective immune responses to tumors, one approach is to block the co-inhibitory effect of CTLA-4 so that CTL activity is persistent.
Ipilimumab is a fully human monoclonal antibody against anti-CTLA-4. The first randomized phase III trial, compared ipilimumab to the GP100 peptide vaccine in patients with recurrent metastatic melanoma. Ipilimumab was administrated at 3 mg/kg every 3 weeks for four doses. Recurring patients, who had achieved a response or stabilized their disease after completion of the initial treatment, could be reinduced at the same dose. The median duration of survival increased from 6 to 10 months [37]. In addition, the 1-year survival was 45.6 % versus 25.3 % for the vaccine (Table 28.9) [37], suggesting the possibility of a durable response. Among 177 patients who were treated with ipilimumab, 15 patients achieved a complete response. The longest response lasted over 99 months [61]. Based on these positive results, ipilimumab was approved in 2011 by the FDA. Of patients who were previously treated with high-dose IL-2, 48 (23 %) received ipilimumab and had a median overall survival of 12 months, suggesting that these patients exposed to IL-2 also benefit from ipilimumab [62]. After 11 years of clinical studies, a pooled analysis of 12 prospective and retrospective trials, including the expanded access program was performed [63]. Two different doses were tried, 3 mg/kg in 965 patients and 10 mg/kg in 706 patients. The median overall survival was 11.4 months (95 % CI: 10.7–12.1) and the 3 year overall survival, 22 %.
Tremelimumab appears to have similar activity to ipilimumab in phase I and II trials [64], but there was no trial designed to compare this two drugs head to head. In a randomized, open-label phase III trial, tremelimumab failed to demonstrate a survival benefit compared to standard-of-care chemotherapy (temozolomide or dacarbazine). Of note, ipilimumab became available to patients while this trial was ongoing, and at least 16 % of patients in the control arm received ipilimumab, which might explain the negative result [38].
The toxicity profile associated with ipilimumab and tremelimumab is the result of activation of auto-immunity due to the blockage of CTLA-4. Common immune-related adverse effects (irAEs) include skin reaction such as rash, pruritus, vitiligo; gastrointestinal reaction, diarrhea, colitis; endocrine effect, hypothyroidism, thyroiditis, adrenal insufficiency, hypophysitis; hepatitis; ophthalmological inflammation, uveitis and conjunctivitis. Cutaneous and gastrointestinal side effects are very common while other organ systems are usually less frequently affected (Table 28.7) [65].
6.2.4 Anti PD-1 Therapy
PD-1, also called programmed cell death 1 protein or CD 279, is a member of the extended CD28/CTLA-4 family of T cell regulators. It is another co-inhibitory checkpoint protein, negatively immune-modulating T cell activity (Table 28.8). PD-1 has 2 ligands: PD-L1 and PD-L2. PD-L1 is expressed on tumor cells; PD-L2 expression is more restricted and mainly identified on dendritic cells, macrophages, as well as mast cells. Within the tumor and its microenvironment, the interaction of PD-1 with PD-L1 down-regulates T cell activity, which helps tumors escape immune recognition. CTLA-4 is expressed on various antigen presenting cells including tumor cells, while PD-L1 expression is commonly restricted to tumor cells.
Nivolumab, also known as MDX-1106, BMS-936558, or Ono-4538, was the first drug in this class to be tested. It is a fully human monoclonal anti-PD-1 antibody, has a high affinity to PD-1 (KD ~3 nM), and competitively blocks both PD-L1 and PD-L2. In a phase I/II trial, nivolumab was administrated to 296 patients with metastatic pre-treated solid tumors, including 107 patients with melanoma. Although patients in the melanoma cohort had received various prior systemic therapies, responses were seen throughout the range of doses given every 2 weeks (0.1–10 mg/kg), with an overall response rate of 31 % [41]. The best response was seen in patients treated at 3 mg/kg with an overall response rate of 41 %. Pretreated tumors from 42 patients in this trial were tested for PD-L1 expression. Among 25 patients who were positive for PD-L1 expression, the objective response rate to nivolumab was 36 %. In contrast, among 17 patients who were negative for PD-L1 expression, none of them responded to nivolumab [41], suggesting a correlation between PD-L1 expression and overall response. The toxicity profile is similar to anti-CTLA-4, but less pronounced. There are 11–14 % of grade 3 and 4 irAEs, fewer than with ipilimumab (18.4 %). Nivolumab has less severe gastrointestinal effects (3.4 % of grade 3 and 4) [41], in contrast to ipilimumab (10 %) with colitis and bowel perforation being potentially life threatening. The main gastrointestinal side effect of nivolumab is diarrhea, while colitis is seen in less than 1 % of cases. Cutaneous grade 3 and 4 reactions are seen in 0.3 % after nivolumab and in 2.6 % after ipilimumab. Pneumonitis occurs in 3 % of patients and seems unique to Nivolumab [41], but it was not reported in the subsequent phase III trial [42]. Pneumonitis is rarely seen in ipilimumab, with one case described [66]. Nivolumab has been approved by the FDA in December 2014 to treat patients who have progressed on Ipilimumab.
Pembrolizumab (formally called Lambrolizumab) is another anti-PD-1 antibody tested in a phase I study for patients with advanced melanoma [39]. Overall response rate across all dose level cohorts was 38 %. The highest response rate (52 %) was observed in the cohort that received Lambrolizumab at 10 mg/kg every 2 weeks. Of 135 patients, 48 (36 %) had received prior treatment with ipilimumab. The response rate did not differ significantly between patients who had received ipilimumab and those who did not (28 %), confirming that PD-1/PD-L1 and CTLA-4/B7 may have non-redundant functions. The overall incidence of grade 3 and 4 side effects appears to be lower compared to the one observed with CTLA-4 blockade antibodies. Pembrolizumab (2 mg/kg administered as an intravenous infusion over 30 min every 3 weeks) has been approved by the FDA in September 2014 to treat patients who have progressed on Ipilimumab.
Many novel anti-PD-1 drugs are currently undergoing clinical testing. Their class, targets, and status of clinical development (through February 2015) are summarized in Table 28.8.
6.2.5 Anti PD-L1 Therapy
PD-L1, also called B7-H1 or CD274, is a ligand that is expressed on tumor cells (occasionally tumor infiltrating macrophages) within the tumor microenvironment. PD-L1 is expressed on many solid tumors including melanoma, and increasingly identified in hematological malignancies. With evidence from preclinical and translational studies that PD-L1 expression is one of the mechanisms for tumors to evade immune recognition, blockade with anti-PD-L1 provides a novel strategy to enhance T cell activity.
BMS-936559, a fully human anti-PD-L1 antibody, was tested in a phase I/II trial on 56 patients with metastatic melanoma [43]. All had received prior immunotherapy and 9 % had received prior BRAF inhibitor therapy. The overall objective response rate was 17 % and ranged from 6 % to 29 % across dose levels (0.3–10 mg/kg). The highest response rate was observed at 3 mg/kg dosage instead of 10 mg/kg. Of nine patients who responded, response lasted for over a year in five patients. Twenty-seven percent had stable disease for longer than 6 months. Toxicity was generally mild. Of 207 patients, 39 % had an immune adverse event of any grade, and only 5 % reported a toxicity of grade 3 or higher. No case of pneumonitis was reported. There was no significant difference in toxicities across dose levels, except that infusion reactions were more common in those who received the highest dose, 10 mg/kg.
MPDL3280A is another anti-PD-L1 human monoclonal antibody that was tested in 45 patients with locally advanced or metastatic melanomas and yielded an overall response rate of 26 % [44].
6.2.6 Blockage of Other Co-inhibitory Molecules
Lymphocyte activation gene 3 (LAG-3), also known as CD223, is another checkpoint protein. Its ligands are MHC class II molecules which are upregulated in some cancers and tumor-infiltrated macrophages. Blockage by the anti-LAG-3 antibody, IMP 321, is currently being tested in clinical trials (Table 28.8). B7-H3 is a newly identified B7 family member, thought to inhibit T cell activation. Its overexpression is seen in some tumor cells and correlates with disease severity, therefore, it might help tumor evade immune recognition [67]. One Fc-enhanced anti-B7-H3 monoclonal antibody, MGA 271, is being tested in phase I.
6.2.7 Upregulation of Other Co-stimulatory Molecules
Glucocorticoid-induced TNFR (GITR) was initially identified as a new family member of the Tumor Necrosis Factor (TNF) receptor superfamily. It is upregulated by T cell activation and functions as one of the co-stimulatory factors. Agonist anti-GITR antibody, TRX518, just started to be tested in phase I trial. Similarly, one drug that activates CD40 is also being tested in pilot studies.
6.2.8 Cancer Vaccines
Cancer vaccines include tumor cell-based vaccines, dendritic cell-based vaccines, or DNA vaccines. Vaccines against cancer have been tested for the last 50 years. However, the knowledge of the microenvironmental immunity of tumors was lacking and led to a lack of understanding of vaccine function. Thus, randomized controlled trials failed to prove a benefit of cancer vaccines for the treatment of advanced or metastatic melanoma [68]. Recently, new vaccine concepts have emerged, using DNA addition to modify gene translation. The cancer-killing virus talimogene laherparepvec (T-VEC) constitutes the first oncolytic immunotherapy to demonstrate therapeutic benefit against melanoma in a phase III trial. T-VEC was engineered by introducing genetic mutations to knock out the infectious genes of herpes simplex virus type-1 and at the same time introducing the gene encoding the granulocyte-macrophage colony-stimulating factor (GM-CSF). The study compared T-VEC to GM-CSF in patients with metastatic disease. Response rates were 26.4 % for T-VEC compared to 5.7 % for GM-CSF [69]. A durable response defined as a complete or partial response that lasted 6 months or more and was mainly observed in patients who had non-visceral disease and in those who received T-VEC as first-line therapy. This vaccine is injected in the largest tumor. The OPTiM study randomized 436 patients with unresectable stage IIIB, IIIC, or IV melanoma in a 2:1 ratio to receive intratumoral T-VEC or subcutaneous GM-CSF [69]. The median time to treatment failure was 8.2 months with T-VEC compared to 2.9 months with GM-CSF (hazard ratio 0.42, 95 % CI [0.32, 0.54]; p < 0.0001). The study continues to monitor patients for survival. The most common grade 3 and 4 adverse event after T-VEC was cellulitis in 2.1 % of patients. Other common symptoms included fatigue (50.3 %), chills (48.6 %), fever (42.8 %), and nausea (35.6 %).
6.2.9 Combination of Dual Immunotherapy
CTLA-4 and PD-1 are not redundant. The CTLA-4/B7 axis plays an important role in attenuating the early activation of naïve and memory T cells, while the PD-1/PD-L1 interaction is observed within the peripheral tumor microenvironments. The combination of ipilimumab and nivolumab (together or sequentially) yields an overall response rate of 40 % [59]. At the maximum dose, the response rate is 53 %. However, the incidence of grade 3 or 4 side effects is also higher.
6.2.10 Immunotherapy Combined with Systemic Chemotherapy
One hallmark study compared ipilimumab (10 mg/kg every 3 weeks for four doses) plus dacarbazine versus dacarbazine plus placebo. The median overall survival was 11.2 months among patients receiving ipilimumab plus dacarbazine compared to 9.1 months among patients receiving dacarbazine alone. Durable survival rates for the combination compared to dacarbazine were at 1 year 47.3 % versus 36.3 %; at 2 years, 28.5 % versus 17.9 %; and at 3 years 20.8 % versus 12.2 %. Of note, the incidence of grade 3 and 4 events was significantly increased for the combination (56.3 % versus 27.5 % for dacarbazine) [57].
The combination of ipilimumab (10 mg/kg every 3 weeks for four doses) and GM-CSF had a similar overall response than ipilimumab single agent, but the overall survival seemed improved with 17.5 months for the combination versus 12.7 months for ipilimumab alone (Table 28.9).
7 Management of Melanoma in the Twenty-First Century
BRAF inhibitors induce rapid responses but the median time to progression is less than 7 months. When exposed to targeted inhibitors (such as the BRAF inhibitor, MEK inhibitor, NRAS inhibitor, c-KIT inhibitor), the tumor itself dies quickly, potentially increasing endogenous antigenicity. Combined with immunotherapy such as high-dose IL-2, anti-CTLA-4 or anti-PD-1, immune effect may be enhanced. Therefore, concurrent or sequential combinations of immunotherapy and targeted therapy have a strong rationale and potentially a huge impact in the management of advanced or metastatic melanoma. Currently, there is no randomized trial to provide insight on the appropriate sequencing of all the available choices. A recent single institution retrospective analysis included 34 BRAF mutation positive patients. Six patients received ipilimumab and then BRAF inhibitor and 28 patients were treated with BRAF inhibitor before receiving ipilimumab. Among the 28 patients that received BRAF inhibitor first, the median time to disease progression was 3.6 months and 12 out of 28 patients had rapid disease progression that resulted in death. These 12 patients were unable to complete induction doses with ipilimumab and their overall survival was 5.7 months. In the 16 patients that were able to complete induction therapy with ipilimumab, the medial overall survival was 18.6 months (95 % CI: 3.2–41.3; p < 0.0001). The median overall survival for all patients in this group was 14.3 months [70]. The six patients that received ipilimumab followed by BRAF inhibitors, were alive at 11.2 months. The median time to progression with ipilimumab was 3.4 months. The authors suggested to consider starting therapy with ipilimumab first and then follow with BRAF inhibition [70].
We propose a management algorithm for patients with advanced melanoma (Fig. 28.4). Currently there are no published guidelines that have established which drug to use front line or how to combine with immunotherapies. On-going clinical trials are elucidating this question. Our recommendation is to offer patients with melanoma participation in judicious clinical trials.

Proposed algorithm for treatment of advanced melanoma
References
SEER (2014) SEER stat fact sheets: melanoma of the skin. (cited 18 Jan 2014). Available from: http://seer.cancer.gov/statfacts/html/melan.html
Chapman PB et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516
Ferlay J et al (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127(12):2893–2917
Gershenwald JE et al (2008) Staging and prognostic factors for stage IV melanoma: initial results of an American Joint Committee on Cancer (AJCC) international evidence-based assessment of 4,895 melanoma patients. ASCO meeting abstracts. 26(15_suppl):9035
Coit DG et al (2013) Melanoma, version 2.2013: featured updates to the NCCN guidelines. J Natl Compr Canc Netw 11(4):395–407
Morton DL et al (2006) Sentinel-node biopsy or nodal observation in melanoma. N Engl J Med 355(13):1307–1317
Mandala M, Voit C (2013) Targeting BRAF in melanoma: biological and clinical challenges. Crit Rev Oncol Hematol 87(3):239–255
Sullivan RJ, Flaherty K (2013) MAP kinase signaling and inhibition in melanoma. Oncogene 32(19):2373–2379
Rahman MA, Salajegheh A, Smith RA, Lam AK-Y (2014) BRAF inhibitors: from the laboratory to clinical trials. Crit Rev Oncol Hematol 90(3):220–232
Menzies AM, Long GV (2013) Recent advances in melanoma systemic therapy. BRAF inhibitors, CTLA4 antibodies and beyond. Eur J Cancer 49(15):3229–3241
Johnson DB, Sosman JA (2013) Update on the targeted therapy of melanoma. Curr Treat Options Oncol 14(2):280–292
Spagnolo F et al (2015) BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther 8:157–168
Gonzalez D et al (2013) BRAF mutation testing algorithm for vemurafenib treatment in melanoma: recommendations from an expert panel. Br J Dermatol 168(4):700–707
Johannessen CM et al (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468(7326):968–972
Middleton MR et al (2000) Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 18(1):158–166
Carvajal RD et al (2011) KIT as a therapeutic target in metastatic melanoma. JAMA 305(22):2327–2334
Hauschild A et al (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380(9839):358–365
Flaherty KT et al (2012) Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367(2):107–114
Larkin J et al (2014) Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371(20):1867–1876
Flaherty KT et al (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367(18):1694–1703
Robert C et al (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372(1):30–39
Young K, Minchom A, Larkin J (2012) BRIM-1, -2 and -3 trials: improved survival with vemurafenib in metastatic melanoma patients with a BRAF(V600E) mutation. Future Oncol 8(5):499–507
Flaherty KT et al (2013) Phase III trial of carboplatin and paclitaxel with or without sorafenib in metastatic melanoma. J Clin Oncol 31(3):373–379
Flaherty KT et al (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363(9):809–819
Sosman JA et al (2012) Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366(8):707–714
Su F et al (2012) RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366(3):207–215
Ascierto PA et al (2013) Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 31(26):3205–3211
Falchook GS et al (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379(9829):1893–1901
Chapman PB (2013) Mechanisms of resistance to RAF inhibition in melanomas harboring a BRAF mutation. Am Soc Clin Oncol Educ Book 80–82
Jang S, Atkins MB (2013) Which drug, and when, for patients with BRAF-mutant melanoma? Lancet Oncol 14(2):e60–e69
Wagle N et al (2014) MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov 4(1):61–68
Johnson DB et al (2014) Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol 32(33):3697–3704
Kleef R et al (2001) Fever, cancer incidence and spontaneous remissions. Neuroimmunomodulation 9(2):55–64
Atkins MB et al (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17(7):2105–2116
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12(4):252–264
Verschraegen C (2012) The monoclonal antibody to cytotoxic T lymphocyte antigen 4, ipilimumab, in the treatment of melanoma. Cancer Manag Res 4:1–8
Hodi FS et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723
Ribas A et al (2013) Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol 31(5):616–622
Hamid O et al (2013) Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 369(2):134–144
Robert C et al (2014) Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 384(9948):1109–1117
Topalian SL et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366(26):2443–2454
Robert C et al (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372(4):320–330
Brahmer JR et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366(26):2455–2465
Hamid O et al (2013) Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). ASCO meeting abstracts. 31(15_suppl):9010
Brignone C et al (2009) A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res 15(19):6225–6231
Sznol M, Hodi F, Margolin K et al (2008) Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA). J Clin Oncol 26(No 15S (May 20 Supplement)):3007
Li SY, Liu Y (2013) Immunotherapy of melanoma with the immune costimulatory monoclonal antibodies targeting CD137. Clin Pharmacol 5(Suppl 1):47–53
Vonderheide RH et al (2007) Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol 25(7):876–883
Wolchok JD et al (2009) Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 15(23):7412–7420
Kirkwood JM et al (1996) Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14(1):7–17
Wheatley K et al (2003) Does adjuvant interferon-alpha for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treat Rev 29(4):241–252
Mocellin S et al (2013) Interferon alpha for the adjuvant treatment of cutaneous melanoma. Cochrane Database Syst Rev 6:Cd008955
Mocellin S et al (2010) Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. J Natl Cancer Inst 102(7):493–501
Chang AE, Rosenberg SA (1989) Overview of interleukin-2 as an immunotherapeutic agent. Semin Surg Oncol 5(6):385–390
Joseph RW et al (2012) Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother 35(1):66–72
Chen L et al (1992) Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell 71(7):1093–1102
Robert C et al (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364(26):2517–2526
Atkins MB, Mario Sznol RRK, McDermott DF, Lotem M, Schachter J, Wolchok JD, Urba WJ, Kuzel T, Schuchter LM, Slingluff CL, Ernstoff MS, Fay JW, Friedlander PA, Gajewski T, Zarour HM, Rotem-Yehudar R, Sosman JA (2014) Phase 2, multicenter, safety and efficacy study of pidilizumab in patients with metastatic melanoma. 2014 ASCO annual meeting. J Clin Oncol
Wolchok JD et al (2013) Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369(2):122–133
Hodi FS et al (2014) Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA 312(17):1744–1753
Prieto PA et al (2012) CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res 18(7):2039–2047
Joseph RW et al (2012) Characterizing the clinical benefit of ipilimumab in patients who progressed on high-dose IL-2. J Immunother 35(9):711–715
Schadendorf D, Hodi S, Robert C et al (2013) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. European Cancer Congress 2013, late breaking abstract 24
Kirkwood JM et al (2010) Phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma. Clin Cancer Res 16(3):1042–1048
Ibrahim RA et al (2011) Ipilimumab safety profile: summary of findings from completed trials in advanced melanoma. ASCO meeting abstracts. 29(15_suppl):8583
Mis L, Clarke JM (2013) Ipilimumab-induced pneumonitis: a case report. J Pharm Technol 29(2):94–98
Hofmeyer KA, Ray A, Zang X (2008) The contrasting role of B7-H3. Proc Natl Acad Sci U S A 105(30):10277–10278
Eggermont AM (2009) Immunotherapy: vaccine trials in melanoma – time for reflection. Nat Rev Clin Oncol 6(5):256–258
Ingemar Andtbacka RH, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, Spitler LE, Puzanov I, Doleman S, Ye Y, Vanderwalde AM, Coffin R, Kaufman H (2013) OPTiM: a randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol 31(suppl; abstr LBA9008)
Ascierto PA et al (2012) Sequencing of BRAF inhibitors and ipilimumab in patients with metastatic melanoma: a possible algorithm for clinical use. J Transl Med 10:107
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Monterroso, J., Ji, Y., Emmons, S., Verschraegen, C. (2015). Clinical Approach to Advanced Melanoma for Today and Tomorrow. In: de Mello, R., Tavares, Á., Mountzios, G. (eds) International Manual of Oncology Practice. Springer, Cham. https://doi.org/10.1007/978-3-319-21683-6_28
Download citation
DOI: https://doi.org/10.1007/978-3-319-21683-6_28
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-21682-9
Online ISBN: 978-3-319-21683-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)