
Abstract
In recent years, melanoma treatment has radically changed with the emergence of targeted therapies and immunotherapies. Both have led to improved survival for patients with advanced or unresectable melanoma. Targeted therapies with BRAF inhibitors in the lead use the presence of activating driver mutations to inhibit tumour growth. Forty to 60% of melanomas harbour BRAF mutations, which makes them susceptible to treatment with BRAF and/or MEK inhibitors. In parallel, the development of immunotherapeutic agents has also expanded. These agents stimulate the endogenous immune system of the patient to eradicate cancer cells. Immune checkpoint inhibitors targeting cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) and programmed death 1 (PD-1) resulted in durable responses in a subset of patients. An important issue with immunotherapy lies in the identification of patients who will benefit from treatment. In this review, we will discuss these recent developments in melanoma therapy and highlight the role of the pathologist in both types of treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Melanoma has an increasing incidence worldwide [1, 2]. In early stages, complete surgical resection is often curative, but once metastases have occurred, the prognosis looks grim [1, 3]. Melanoma is remarkably resistant to various types of chemotherapy with few responders and very few durable responses [2]. Historically, the median survival for patients with stage IV disease was ≤1 year [2]. The limited effectiveness of chemotherapy has prompted the search for new therapeutic agents.
In recent years, two therapeutic strategies have changed the landscape of melanoma treatment and have led to improved survival for patients with advanced or unresectable melanoma. First, the targeted therapies have emerged as the result of the ever-advancing knowledge of underlying molecular pathways and the identification of activating driver mutations. Unfortunately, these therapies are nearly exclusively suitable for patients with certain genetic aberrations in their melanoma and resistance to targeted therapy develops in the majority of patients. Second, immunotherapy in melanoma enhances the patients’ own immune system in order to eradicate the cancer cells. Theoretically, this treatment is applicable to all patients with metastatic or unresectable melanoma, irrespective of mutation status.
In this review, we will discuss these recent developments in melanoma therapy and we will highlight the role of the pathologist in both types of treatment.
Targeted therapies
Unravelling the molecular genetics of melanoma
Primary melanoma of the skin is characterised by an enormous number of mutations, partly due to the carcinogenic effects of ultraviolet (UV) light [4–6]. Although most of these mutations are ‘passenger’ mutations, some of them are ‘driver’ mutations affecting critical genes involved in the cell cycle. These driver mutations constitute unique targets for treatment.
The mitogen-activated protein kinase (MAPK) pathway has been the main target of research into the genetic mechanisms underlying melanoma development. Important members of this pathway are RAS, BRAF, MEK and ERK [7]. Activation of the MAPK pathway leads to increased cell proliferation and survival. When RAS is activated, it drives BRAF dimerization and activation. Activated BRAF phosphorylates and hence activates MEK, which subsequently phosphorylates and activates ERK (Fig. 1). Within the MAPK pathway, we encounter two proteins whose genes frequently harbour mutations in melanoma, namely RAS and BRAF.

The mitogen-activated protein kinase (MAPK) pathway and the phosphatidylinositol 3′ kinase (PI3K) pathway. Activated receptor tyrosine kinases can transmit signals to NRAS. This signal can be transduced via the MAPK pathway consisting of BRAF, MEK and ERK, leading to proliferation and survival as well as via the PI3K pathway consisting of PI3K and AKT, leading to survival (RTK receptor tyrosine kinase)
Mutations in BRAF, a serine/threonine kinase, occur in approximately 40–60% of cutaneous melanomas [7–11]. Hereby, BRAF is activated independently of upstream stimulation and of RAS activity, resulting in an increased kinase activity and a constitutively active cascade [10, 12]. The mutations in BRAF are clustered in two regions of the protein: the P-loop and the activation segment [10, 12]. Interactions between these two regions regulate the active and inactive conformation of BRAF. Oncogenic mutations destabilise the inactive conformation of BRAF, as a result of which the active state of BRAF is greatly promoted [12]. The most frequent BRAF mutation, V600E, is a valine to glutamic acid substitution within the activation segment [7, 10, 11]. The V600E mutation accounts for approximately 80% of the BRAF mutations encountered in melanoma [9–11]. The V600K mutation (a valine to lysine substitution) is the second most common [9, 13, 14]. In comparison to V600E mutations, V600K mutations are more common in older patients and occur more frequently in skin with chronic sun-induced damage (CSD) [14].
BRAF mutations in general occur more often in melanomas originating in skin, intermittently exposed to UV. They are relatively rare in skin with CSD and are more frequent in younger patients [4, 9, 15–17]. The effect of BRAF mutations on prognosis and survival of patients is biased by the use of BRAF inhibitors (cf. infra). Long et al. found no significant impact on the disease-free interval from diagnosis of the primary melanoma to first distant metastasis, but there was a trend towards poorer outcome of BRAF mutant (not treated with a BRAF inhibitor) versus BRAF wild-type metastatic melanoma [9]. Houben et al. reported similar data [11]. The presence of a BRAF/NRAS mutation was associated with a poorer prognosis in metastatic, but not in primary, lesions. However, not all series confirm these findings [9]. BRAF mutant melanomas have a tendency to metastasise to regional lymph nodes, while BRAF wild-type melanomas more often show in transit or systemic metastases [17].
Melanomas harbouring BRAF mutations show some distinct morphological features [16–18]. They show more pagetoid spread and nest formation of intraepidermal melanocytes. The involved epidermis is thickened, and the demarcation with the surrounding skin is sharper. The cells are usually larger, more epithelioid and more pigmented (Fig. 2).

Morphological features of a BRAF-mutated melanoma. Superficial spreading melanoma harbouring a BRAF V600E mutation. Presence of pagetoid spread and nest formation of intraepidermal melanocytes, thickened epidermis and sharp demarcation with the surrounding skin. The cells are large, epithelioid and pigmented (original magnification ×100)
RAS is another protein of the MAPK pathway, in which frequent oncogenic mutations occur. In melanomas, almost all mutations in RAS concern NRAS [5, 15]. They occur in roughly 20% of melanomas and are almost exclusively present in melanomas without BRAF mutation [11, 15, 19, 20]. The oncogenic mutations in NRAS lead to constitutive activation of the NRAS protein due to decreased GTP-ase activity and accumulation of the active, GTP-bound protein [7]. NRAS activates both the MAPK pathway and the phosphatidylinositol 3′ kinase (PI3K) pathway (Fig. 1) [7, 15]. In contrast to BRAF mutant melanomas, melanomas with NRAS mutations do not seem to possess distinct morphological features [16]. However, Broekaert et al. found an association between low or absent scatter of intraepidermal melanocytes and better circumscription on the one hand and NRAS mutant status on the other hand [17]. NRAS mutant tumours are also associated with thicker primary tumours and increased mitotic rate [20]. Most of the NRAS mutations in melanoma have been linked to a poorer overall survival (OS) although a mutation in NRAS, protecting melanoma patients from metastasis, has also been described [20, 21].
Multiple other genetic aberrations are present in melanomas. Mutations and amplification of KIT occur in a relatively low proportion of melanomas but are more frequent in mucosal and acral melanomas (15–40%) and in melanomas arising in skin with CSD [4, 22, 23]. They are associated with worse prognosis [24]. KIT is a receptor tyrosine kinase with a number of different effector pathways, including the MAPK and the PI3K pathway [22–24]. In melanoma, mutations in KIT are widely distributed over the coding region [23]. This makes it harder to separate driver from passenger mutations.
Inactivating mutations of NF1 are associated with a high mutational burden and occur in older patients [5]. They arise in 14% of melanomas and more often in desmoplastic/neurotropic melanomas and in melanomas arising in skin with CSD [4, 5, 25]. NF1 normally downregulates RAS activity, hence its inactivation leads to increased RAS signalling [5].
PTEN is an inhibitory protein of the PI3K pathway. Loss of functional PTEN leads to upregulation of the PI3K pathway and increased survival of the melanoma cell (Fig. 1) [15]. This occurs more frequently in BRAF-mutated melanomas [5].
Uveal melanomas have a different genetic background [4, 26, 27]. In comparison with cutaneous melanomas, uveal melanomas display a different pattern of mutated driver genes, among which GNAQ and GNA11 [27]. Mutations in GNAQ and GNA11 arise early in the development of uveal melanoma. These can be found in 83% of uveal melanomas in a mutually exclusive manner [26]. The mutations in both genes cause an upregulation of the MAPK pathway and have the same effect as the V600E mutation in BRAF [26, 27].
The insights into the underlying molecular mechanisms of melanoma have inspired some researchers to propose another classification scheme of melanoma. Nowadays, we are used to the classification scheme of Clark on which the current WHO classification is based [4, 28]. On morphological grounds, one can distinguish four main subtypes: superficial spreading melanoma (SSM), lentigo maligna melanoma (LMM), nodular melanoma (NM) and acral lentiginous melanoma (ALM). These subtypes have very little impact on clinical decision making on optimal treatment [15, 16]. The OS or response to treatment does not differ significantly between these morphological groups when tumours of equivalent microstaging (encompassing thickness, presence or absence of ulceration and mitotic count) were compared [16].
An alternative type of classification subdivides the melanomas on non-glabrous skin in lesions occurring in skin without chronic sun-induced damage (non-CSD) and lesions in skin with CSD. The first group has frequent BRAF mutations and no KIT mutations. The second group has less frequent BRAF mutations but more NF1 and KIT mutations. Melanomas on glabrous skin and the nail apparatus are termed acral melanomas, those on mucosal membranes, mucosal melanomas. Finally, uveal melanomas and intradermal melanocytic proliferations (blue nevus spectrum) are discerned. They show frequent mutations in the same genes (GNAQ and GNA11) [4, 17].
Yet another option is to create a genomic classification with subtypes based on the presence or absence of mutated genes. The Cancer Genome Atlas Network proposes four genomic subtypes: mutant BRAF, mutant RAS, mutant NF1 and Triple-WT (wild type) [5]. The Triple-WT subtype is enriched for KIT mutations and amplification. Although there is no significant correlation between outcome and genomic group, this classification might be of help in guiding treatment choices, since it is based on the type of driver mutation [5].
Therapeutic agents
Not all oncogenic mutations in melanoma are targetable. At the moment, only two BRAF inhibitors (vemurafenib and dabrafenib) and two MEK inhibitors (trametinib and cobimetinib) are European Medicines Agency (EMA)- and FDA-approved to be used in metastatic or unresectable melanoma with BRAF V600 mutations [29, 30]. Many more agents are under evaluation in clinical trials. For example, encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) are currently assessed in a phase 3 trial with encouraging results in line with those of other combinations (NCT01909453) [31].
Vemurafenib and dabrafenib are potent and highly specific inhibitors of the BRAF V600 mutant protein [8]. They produce objective responses (ORs) in roughly half of the patients with metastatic melanoma harbouring a BRAF V600 mutation, and they have showed survival benefit in these patients [32, 33]. In spite of this success, most patients develop resistance to these small molecule inhibitors and disease progression after 6 to 7 months [7, 8, 34].
The mechanisms of resistance are diverse. Consistent with their temporal occurrence, they can be divided into intrinsic and acquired resistance. In intrinsic resistance, the tumour virtually does not respond to therapy. Upon administration of a BRAF inhibitor, there are rapid re-adjustments of signalling pathways, which render the drug ineffective [7, 8]. This is also the case in many other malignancies with BRAF mutations such as colorectal adenocarcinoma and papillary thyroid cancer [8]. In most cases of acquired resistance, the tumour cell manages to reactivate the MAPK pathway in the presence of the BRAF inhibitor. The most frequent mechanisms are NRAS or KRAS mutations, BRAF splice variants, BRAF V600E/K amplification and MEK mutations [7, 8, 35]. Another mechanism of resistance is the upregulation of the PI3K pathway, for instance by loss of PTEN function [8, 36]. Table 1 gives an overview of different mechanisms of resistance to BRAF inhibitors in melanoma [7, 8, 35, 36]. Much research is directed towards finding solutions to overcome these different patterns of resistance [1, 7, 8].
Frequent adverse events of BRAF inhibitors are arthralgia, pyrexia, rash, fatigue, keratoacanthoma or squamous cell carcinoma, photosensitivity, nausea, diarrhoea and liver function abnormalities [32, 33]. Rarely, these drugs can also cause QT prolongations, pericarditis and potentially pneumonitis. Specific attention should be paid to the emergence of cutaneous malignancies, which is related to the paradoxical activation of the MAPK pathway in cells without the BRAF V600 mutation (via RAF dimerization) [7, 8, 37]. Work is done to create ‘paradox breaking’ BRAF inhibitors that do not cause this paradoxical MAPK activation in non-mutant cells [1, 7, 8].
MEK inhibitors use a downstream target of BRAF [7]. As a single agent, their activity is rather modest [7, 38]. However, the combination of MEK and BRAF inhibition has the potential to achieve a more robust inhibition of the MAPK pathway than BRAF inhibition alone. Due to the good rationale for combining BRAF and MEK inhibitors, this combination was tested in clinical trials. Besides a higher efficacy, these studies also aimed at overcoming or delaying resistance to BRAF inhibition and blocking paradoxical activation of the MAPK pathway [7, 39]. Two MEK inhibitors are approved by the EMA and FDA: trametinib in combination with dabrafenib and cobimetinib in combination with vemurafenib. These combinations yielded a significantly improved response rate, a significantly better progression-free survival and OS compared to BRAF inhibition alone [39–41]. Patients treated with the combination regime developed fewer cutaneous malignancies, a finding consistent with suppression of paradoxical MAPK pathway activation [8, 39, 41]. This is achieved, however, at the expense of adding MEK inhibitor-specific adverse events such as ophthalmological complications, decreased left ventricular ejection fraction and fluid retention [38, 41]. MEK inhibition has shown very modest activity in uveal melanomas, where more active drugs or combinations are necessary [1, 20].
AKT, PI3K and ERK inhibitors are under development [1, 7, 8]. Direct pharmacological inhibition of NRAS has proven to be difficult [7, 20]. A part of the efforts is targeting post-translational modifications (farnesylation, prenylation) of NRAS to prevent its attachment to the cell membrane, a step essential for NRAS activation [7]. NRAS mutant tumours are resistant to BRAF inhibition [19, 20, 37]. Trials are ongoing to test the efficacy of MEK inhibition and combination of MEK and CDK4/6 inhibition (a downstream target of ERK) in patients with NRAS-mutated melanomas [20, 37]. The results of a phase 3 trial comparing binimetinib to chemotherapy (dacarbazine) in unresectable or metastatic NRAS mutant melanoma indicate a small but statistically significant improvement in progression-free survival with no significant difference in OS [42]. Currently, there are no approved therapeutic agents specifically for NRAS mutant melanoma [20].
Inhibitors of KIT, such as imatinib, dasatinib, nilotinib and sunitinib, have shown activity in patients whose melanomas harbour mutations or amplification of KIT [22–24]. Although trials in unselected patients with advanced melanoma were negative, studies focussing on selected patients with melanomas harbouring KIT alterations could identify responders, albeit in a relatively small number of patients compared to MAPK pathway inhibition in BRAF-mutated melanoma [23, 24]. Activating mutations in KIT were therapeutically relevant, i.e. associated with an objective response to KIT inhibitors, whereas an increased copy number of wild-type KIT seems to be associated with a lower clinical activity [23, 24]. In most studies, clinical benefit was largely transient [22]. Targeting downstream components of the MAPK and PI3K pathways may be an attractive, alternative approach [22].
Techniques to discover the presence of molecular targets
Next to giving the appropriate diagnosis, the pathologist has an important role in the detection of oncogenic mutations. The pathologist is frequently regarded as ‘the guardian’ of the patient’s tissue. He or she is obliged to the rational use of tissue, especially when the available amount is limited. To investigate the mutation status of a molecular target, tissue of the most recent metastasis is preferred, because metastases can acquire additional mutations; subclones can be selected or generated. Heterogeneity between the primary tumour and the metastases has been reported in literature, although at a relatively low frequency [11, 18, 43]. Colombino et al. found that the distribution of BRAF/NRAS mutations was highly consistent between primary melanomas and lymph node or visceral metastases, whereas rates of consistency between primary tumour and brain or skin metastases were significantly lower [43]. This heterogeneity poses a challenge for targeted therapies since it can influence treatment choices. Retesting patients with initially BRAF wild-type melanoma can be considered when new metastases appear, since there is a small chance that a BRAF mutation becomes detectable making patients eligible for MAPK pathway inhibition [44, 45]. Apart from tissue, tumour-derived circulating cell-free DNA (cfDNA) from patient plasma is a potential alternative source to assess mutation status. This is of special interest in patients with lesions not easily accessible for repeated biopsy like patients with brain metastases [46].
Several methods exist to detect BRAF, NRAS and/or KIT mutations: High-resolution-melting PCR, real-time (allele specific amplification) PCR including competitive amplification of differentially melting amplicons, Sanger sequencing, pyrosequencing, mismatch ligation assay, ligase detection reaction, denaturating high-performance liquid chromatography, SNAPshot and mass spectrometry [13, 18, 47–49]. In recent years, next-generation sequencing (NGS) has become more widely available. All these assays show differences in sensitivity, specificity, costs and time to result. When tumour cell fraction is low, some tests require macro-/microdissection to improve sensitivity [48, 49]. It is important to bear in mind that not all assays detect all possible mutations of a certain gene. For example in BRAF, some tests do not detect all possible mutations in V600 or other non-V600 mutations. Table 2 gives a summary of sensitivities, advantages and disadvantages of various techniques, including immunohistochemistry (IHC) [48–51].
For certain molecular targets, IHC can be an alternative to molecular testing. This is certainly the case for the detection of the BRAFV600E-mutated protein. VE1 is a BRAFV600E mutant-specific monoclonal antibody (Fig. 3). In a meta-analysis conducted by Anwar et al., pooled sensitivity of IHC for detection of the BRAFV600E-mutated protein was 96%, and pooled specificity was 100% [47]. The VE1 antibody is very specific for the V600E mutation but occasional cross reactivity with other V600 mutations, for example V600K, has been variably reported [13, 49, 52]. Staining with the VE1 antibody is in most cases strong, diffuse and cytoplasmic [18, 37, 53]. However, several authors have reported difficulty in interpreting weak staining in a subset of samples [13, 37, 53]. Different ways to report IHC results are used. Some use a dichotomous positive versus negative system [18, 37, 49, 52]. Others use a four-tier system to report staining results from 0 (no staining) to 3+ (strong staining) [13, 53].

Immunohistochemical demonstration of mutated BRAF protein in scattered melanoma cells (Immunoperoxidase, VE1 antibody, counterstained with haematoxylin, original magnification ×400)
Most of the molecular assays are costly and take several days to produce results, especially when the tissue has to be transferred to another laboratory [47, 52]. In comparison, IHC is cheaper and more widely available in most pathology laboratories [37, 49, 52, 53]. Results can be obtained the next day. IHC is particularly suitable for very small samples or samples with a very low tumour cell content [47, 52]. For example, in a lymph node with single or small clusters of tumour cells, IHC shows single cell-resolution images [52]. In samples with low-quality or non-amplifiable DNA, proteins may be better preserved [47, 53].
IHC can be used as a first-line method to yield rapid results and direct patients to appropriate treatment, for instance in the case of symptomatic brain metastasis. This has to be followed by molecular confirmation and detection of other V600 mutations [37, 47, 49, 52, 53]. Another option is to perform molecular testing only on samples with negative or equivocal IHC results. To produce clinically applicable results, stringent antibody optimisation and quality control is required [47].
Concerning NRAS, antibodies are available for some of the most prevalent mutations (NRAS Q61R and NRAS Q61L) [37]. The anti-NRASQ61R IHC has been reported to be highly sensitive and specific [19, 37].
KIT expression or KIT copy number does not correlate with the presence of activating KIT mutations, and KIT expression does not always predict response to KIT inhibitors in melanoma. Therefore, IHC has little use in molecular profiling for KIT aberrations [54].
Immunotherapy
How does it work?
Melanoma is an immunogenic cancer with a high mutational burden that triggers the adaptive immune system. This can even result in spontaneous partial or complete regression [4, 6, 36, 55]. The presence of tumour-infiltrating lymphocytes (TILs) in cutaneous melanomas with a vertical growth phase is a significant and independent prognostic factor [56]. Also, immune infiltration of metastatic melanoma has been correlated with a more favourable prognosis [5]. Unfortunately, due to immunomodulation and immunoediting, this immune response is often abrogated during tumour progression [55]. Immunotherapy tries to overcome this abrogation.
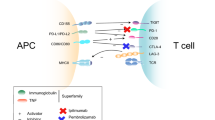
A central role in cancer immunotherapy is reserved for the T cell (Fig. 4). Activation of the T cell requires stimulation via the T cell receptor and a co-stimulatory interaction between CD28 and its ligand B7-1 or B7-2 [36, 55, 57]. To avoid overactivation of the T cell with the risk of developing autoimmune reactions and to promote self-tolerance, also activation of co-inhibitory molecules comes into play [58, 59]. These are known as immune checkpoints and include cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) and programmed death 1 (PD-1) [36, 58, 59]. CTLA-4 binds to B7-1 or B7-2 with greater affinity than CD28 and downregulates T cell activation. Also, binding of PD-1 and its ligands programmed death ligand 1 (PD-L1) and programmed death ligand 2 (PD-L2) limits T cell activation [58–60]. Interferon gamma (IFNɣ), produced by activated T cells and natural killer cells, is an important stimulator of PD-L1 expression [61]. Since PD-L1, among others, may be expressed on the tumour cell membrane, these inhibitory signals are obstacles to T cell antitumour activity and make it possible for tumour cells to evade immune reaction [36, 58]. Hence, antibodies, referred to as immune checkpoint inhibitors, which block these co-inhibitory signals, are expected to augment T cell antitumour activity and may enhance pre-existing immune responses to tumour antigens [36, 58].

Mechanism of action of immune checkpoints. For activation of the immune response, T cells require stimulation by the major histocompatibility complex (MHC) presenting antigens. Interaction between CD28 and B7 provides a co-stimulatory signal, predominantly during interactions with the antigen-presenting cells (APCs) (the afferent arm of the T cell response). To avoid uncontrolled stimulation, immune checkpoints, including cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) and programmed death 1 (PD-1), provide co-inhibitory interactions. CTLA-4 acts mainly in the afferent arm of the T cell response, while PD-1 operates primarily during the effector phase of the T cell response. These interactions lead to a decreased activation of the T cell and consequently a decreased immune response (TCR T cell receptor, PD-L1 programmed death ligand 1)
Other treatments deploying the immunogenicity of cutaneous melanoma include vaccination strategies, adoptive transfer of autologous T cells directed against melanoma antigens, T cell engineering therapy and oncolytic virotherapy [36]. The latter is approved by the EMA and FDA [29, 30]. Further discussion on these therapeutic principles is beyond the scope of this review.
Therapeutic agents
One of the first immunotherapeutic drugs was high-dose interleukin-2 (IL-2). It induces responses in 15–20% of patients with durable responses in 5–8% of patients. Toxicities are severe and often life threatening. However, it established a role for immunotherapy in the treatment of advanced melanoma [1, 3, 20, 36]. Also, interferon alpha (IFNα) is an immunotherapeutic agent that can be used in the treatment of melanoma. IFNα adjuvant therapy, in patients with high-risk cutaneous melanoma, showed improvement in both disease-free survival and OS, although the gain in OS was relatively small and most of the studies failed to establish a significant impact of adjuvant cytokine treatment on OS [62].
With the immune checkpoint inhibitors, a new type of immunotherapeutic agent has emerged. To date, three such antibodies are approved by the EMA and FDA: ipilimumab, nivolumab and pembrolizumab [29, 30]. Many more agents are under evaluation in clinical trials.
Ipilimumab is a monoclonal antibody that blocks CTLA-4. Next to a decreased inhibition of T cell activation, it also promotes cytotoxic T lymphocyte trafficking to the tumour and a decline in the number of (inhibitory) regulatory T cells in the tumour microenvironment (TME) [1, 63]. In two phase 3 trials, ipilimumab significantly improved OS in previously treated and untreated patients with metastatic or unresectable melanoma [64, 65]. ORs are achieved in 11% of patients with a disease control rate (the proportion of patients with a partial or complete response or stable disease) of 29% [64]. Treatment with ipilimumab has shown durable tumour regression despite relatively brief therapy durations. An analysis of long-term survival in melanoma patients treated with ipilimumab showed a 3-year survival rate of 22%. Interestingly, at this time, the survival curve reaches a plateau which extended up to 10 years in some patients [66].
Ipilimumab toxicities are frequent and include immune-related adverse events. These consist of rash, pruritus, fatigue, diarrhoea, endocrinopathy, colitis, hepatitis, pancreatitis and other rare autoimmune phenomena [64, 65]. These adverse events require patient education, a close follow-up and prompt medical intervention [64, 65].
Nivolumab and pembrolizumab are monoclonal antibodies that block PD-1. They have shown improved OS in patients with metastatic or unresectable melanoma in comparison to chemotherapy or ipilimumab. They have shown OR in 30–40% of patients with many appearing durable [67–69].
Immune-related adverse events occurred less commonly than with ipilimumab [20, 69]. The most common adverse events include fatigue, pruritus, nausea and diarrhoea [67–69]. PD-1 antibodies have gained broad approvals, including use as first-line therapy regardless of BRAF mutation status [1].
Responses to immunotherapy show sometimes an unconventional pattern. A portion of patients on immunotherapy achieves an objective response after initial progression or after a period with stable disease [58, 66, 68, 70]. Conventional response criteria like RECIST 1.1 may therefore be less adequate to accurately assess the activity of immunotherapeutic agents.
Immunotherapies can be associated with intrinsic resistance and the emergence of acquired resistance too. Sharma et al. wrote an excellent review on this topic [71]. Low tumour immunogenicity and an immunosuppressive TME are associated with intrinsic resistance. Mutations affecting the sensitivity of tumour cells to T cell-derived interferons and mutations which limit tumour-cell antigen presentation have been reported [72, 73]. For example, loss of function mutations in Janus kinase 1 (JAK1) or Janus kinase 2 (JAK2) lead to a lack of response to IFNɣ and have been associated with both intrinsic and acquired resistance to PD-1 blockade [74, 75]. Another route to resistance consists of the upregulation of alternative immune checkpoints or vascular endothelial growth factor (VEGF) [73, 76]. Table 3 gives an overview of different mechanisms of resistance to immune checkpoint inhibitors [61, 71–76].
Ipilimumab and nivolumab or pembrolizumab act via different signalling pathways, suggesting that they could have a synergistic effect [60]. Clinical trials using the combination of ipilimumab and nivolumab have shown a higher activity than either agent alone. In a recently published phase 3 trial, the combination of ipilimumab and nivolumab yielded an objective response in 58% of patients [77]. However, there was also an increase in the number and severity of adverse events seen in patients treated with the combination regime [77].
Many other antibodies are currently under evaluation in clinical trials. Besides anti-CTLA-4 and anti-PD-1 antibodies, these include also anti-PD-L1 and anti-PD-L2 antibodies. Anti-PD-L1 has gained much attention in other diseases and is currently evaluated in melanoma in several ongoing trials [57, 63, 68]. Next to these and other co-inhibitory receptors and ligands, also co-stimulatory targets are the subject of active research [60]. Different combinations of immunotherapeutic agents are evaluated with the success of the ipilimumab-nivolumab combination in mind [60, 63].
Predictive biomarkers?
Theoretically, every patient with metastatic melanoma could benefit from immunotherapy. There is no need for the presence of a certain genetic aberration to respond to this type of therapy, unlike, for example, therapy with a BRAF inhibitor. This offers a treatment opportunity even for patients not eligible for targeted therapy due to absence of a targetable genetic aberration. Even patients who developed resistance to targeted therapies can benefit from immunotherapy. Unfortunately, only a subset of patients does respond to immunotherapy. It would therefore be useful to identify those patients who are most likely to benefit from immunotherapy. Such an approach avoids treatment-related adverse events in non-responders and would also be more cost-effective.
Finding predictive markers has proven difficult. It is a hot research area. Studies have shown that a high mutational load and a high neo-antigen load are associated with a better response to immune checkpoint inhibitors [55, 78, 79]. This association could be an explanation for the lower responses to immunotherapies in uveal melanomas as compared to cutaneous melanomas, since they have a lower mutational burden [27]. However, mutational burden and neo-antigen load are not sufficient to predict clinical benefit [55, 78, 79].
There is increasing evidence supporting the hypothesis that an immune-active TME correlates with clinical benefit from immune checkpoint inhibitors. An immune gene expression signature, reflecting a cellular and humoral immune response, for example expression of cytolytic markers, is associated with clinical benefit from CTLA-4 or PD-1 inhibition [76, 78, 80–82]. Also an inflammatory TME, (CD8+) TILs, CD8+ T cells at the invasive tumour margin, PD-1+ cells, expression of forkhead box P3 (FOXP3) (a marker of regulatory T cells) and indoleamine 2,3-dioxygenase (IDO) (associated with an immunosuppressive TME) have been claimed to predict clinical benefit from immune checkpoint inhibitors [76, 80–85]. However, none of these is perfectly predictive and biomarker profiles between patients with or without clinical benefit are often overlapping [76].
In a study of Chen et al., adaptive immune signatures in early on-treatment tumour biopsies were predictive of response to immune checkpoint blockade [76]. Similar to this, Hamid et al. found that an increase in TILs between baseline and 3 weeks after start of treatment with ipilimumab was associated with clinical efficacy [81]. Tumeh et al. reported a proliferation of CD8+ TILs in regressing tumours under PD-1 blockade [83]. These findings are illustrated in Fig. 5 showing melanoma metastases before and during successful treatment with an anti-PD-1 antibody (Fig. 5).

Cutaneous melanoma metastasis before (a, c, e, g) and after two doses of Pembrolizumab 2 mg/kg (b, d, f, h), stained for H&E (a, b), CD8 (c, d), PD-L1 (e, f) and PD-1 (g, h). During treatment, there is a sharp increase in the number of T cells (b) and expression of CD8 (d) and PD-1 (h) with a concomitant increased expression of PD-L1 on the melanoma cells (f)
For treatment with anti-PD-1 or anti-PD-L1 antibodies, multiple studies examined the association of PD-L1 expression by the TME (assessed with an immunohistochemical assay) and objective response rate (ORR). These studies are difficult to compare since they use different methodologies such as different antibodies and thresholds to determine PD-L1 positivity [1]. PD-L1 can be expressed on tumour cells and on the immune infiltrate [84, 85]. Also, the dynamic nature and possible heterogeneity of PD-L1 expression within a patient adds complexity to its use as a predictive marker [63, 69]. Several studies have shown an association between PD-L1 expression and ORR to monotherapy with PD-1 pathway blockade in patients with metastatic melanoma [3, 63, 84]. But, ORs were not limited to the PD-L1-positive tumours and also patients with PD-L1-negative tumours benefited from treatment [67, 68]. In a phase 3 trial, nivolumab-treated patients had improved OS in comparison with dacarbazine-treated patients, regardless of PD-L1 status [67]. The ORR in nivolumab-treated patients was 52.7% in the subgroup with positive PD-L1 status versus 33.1% in the subgroup with negative or undetermined PD-L1 status [67]. Hence, PD-L1 status alone does not seem suitable for selection of patients for treatment with an anti-PD-1 antibody in monotherapy. Larkin et al. suggested that patients with PD-L1-negative tumours may benefit more from combination therapies [63, 77]. More clinical data are needed to fully evaluate the potential of PD-L1 status in making treatment decisions. Currently, PD-L1 status should not be used as guidance for directing treatment choices in patients with metastatic or unresectable melanoma.
It is possible that in the near future, pathologists will be asked to make a certain assessment of the immune TME in combination with PD-L1 expression or evaluate on-treatment tumour biopsies in order to predict response to immunotherapy. However, the ideal predictive biomarker has yet to be found.
Conclusion
Targeted therapy and immunotherapy have both become first-line treatments in patients with metastatic or unresectable melanoma. For patients without targetable genetic aberrations, it is mainly immunotherapy that can be applied. Combinations of both strategies are under evaluation. Most likely, they generate a synergistic effect [60, 63]. Although these therapies have greatly improved the prognosis of patients with metastatic or unresectable melanoma, challenges still remain. The costs, the increasing pressure on healthcare systems and the adverse events necessitate the identification of better predictive biomarkers. A tool allowing distinguishing responding from non-responding patients as well as patients at risk for developing early treatment resistance is of utmost importance. Strategies to increase the proportion of responders and to overcome resistance would be of great value. Treatment options are evolving rapidly and clinicians have to select the most appropriate therapy for individual patients. In this selection process, the pathologist will play a crucial role. He or she is undoubtedly a part of the collaborated effort to further advance and improve precision medicine.
Reference
Margolin K (2016) The promise of molecularly targeted and immunotherapy for advanced melanoma. Curr Treat Options in Oncol 17:48. doi:10.1007/s11864-016-0421-5
Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM (2011) Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist 16:5–24. doi:10.1634/theoncologist.2010-0190
Teixido C, Gonzalez-Cao M, Karachaliou N, Rosell R (2015) Predictive factors for immunotherapy in melanoma. Ann Transl Med 3:208. doi:10.3978/j.issn.2305-5839.2015.05.07
Bastian BC (2014) The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol Mech Dis 9:239–271. doi:10.1146/annurev-pathol-012513-104658
Cancer Genome Atlas Network (2015) Genomic classification of cutaneous melanoma. Cell 161:1681–1696. doi:10.1016/j.cell.2015.05.044
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA Jr, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinsk M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Andrew Futreal P, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. doi:10.1038/nature12477
Samatar AA, Poulikakos PI (2014) Targeting RAS–ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13:928–942. doi:10.1038/nrd4281
Holderfield M, Deuker MM, McCormick F, McMahon M (2014) Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14:455–467. doi:10.1038/nrc3760
Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, Hughes TM, Thompson JF, Scolyer RA, Kefford RF (2011) Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol 29:1239–1246. doi:10.1200/JCO.2010.32.4327
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, JWC H, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. doi:10.1038/nature00766
Houben R, Becker JC, Kappel A, Terheyden P, Bröcker E-B, Goetz R, Rapp UR (2004) Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog 3:6. doi:10.1186/1477-3163-3-6
Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-duvaz D, Good VM, Project CG, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867
Harlé A, Salleron J, Franczak C, Dubois C, Filhine-Tressarieu P, Leroux A, Merlin J-L (2016) Detection of BRAF mutations using a fully automated platform and comparison with high resolution melting, real-time allele specific amplification, immunohistochemistry and next generation sequencing assays, for patients with metastatic melanoma. PLoS One 11:e0153576. doi:10.1371/journal.pone.0153576
Menzies AM, Haydu LE, Visintin L, Carlino MS, Howle JR, Thompson JF, Kefford RF, Scolyer RA, Long GV (2012) Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res 18:3242–3249. doi:10.1158/1078-0432.CCR-12-0052
Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho K-H, Aiba S, Bröcker E-B, LeBoit PE, Pinkel D, Bastian BC (2005) Distinct sets of genetic alterations in melanoma. N Engl J Med 353:2135–2147. doi:10.1056/NEJMoa050092
Viros A, Fridlyand J, Bauer J, Lasithiotakis K, Garbe C, Pinkel D, Bastian BC (2008) Improving melanoma classification by integrating genetic and morphologic features. PLoS Med 5:e120. doi:10.1371/journal.pmed.0050120
Broekaert SMC, Roy R, Okamoto I, van den Oord J, Bauer J, Garbe C, Barnhill RL, Busam KJ, Cochran AJ, Cook MG, Elder DE, McCarthy SW, Mihm MC, Schadendorf D, Scolyer RA, Spatz A, Bastian BC (2010) Genetic and morphologic features for melanoma classification. Pigment Cell Melanoma Res 23:763–770. doi:10.1111/j.1755-148X.2010.00778.x
Verlinden I, van den Hurk K, Clarijs R, Willig AP, Stallinga CMHA, Roemen GMJM, van den Oord JJ, zur Hausen A, Speel E-JM, Winnepenninckx VJL (2014) BRAFV600E immunopositive melanomas show low frequency of heterogeneity and association with epithelioid tumor cells. Medicine (Baltimore) 93:e285. doi:10.1097/MD.0000000000000285
Ilie M, Long-Mira E, Funck-Brentano E, Lassalle S, Butori C, Lespinet-Fabre V, Bordone O, Gay A, Zahaf K, Poissonnet G, Lacour J-P, Bahadoran P, Ballotti R, Gros A, Dutriaux C, Saiag P, Merlio J-P, Vergier B, Emile JF, Hofman V, Hofman P (2015) Immunohistochemistry as a potential tool for routine detection of the NRAS Q61R mutation in patients with metastatic melanoma. J Am Acad Dermatol 72:786–793. doi:10.1016/j.jaad.2015.01.012
Johnson DB, Puzanov I (2015) Treatment of NRAS-mutant melanoma. Curr Treat Options in Oncol 16:15. doi:10.1007/s11864-015-0330-z
Demunter A, Ahmadian MR, Libbrecht L, Stas M, Baens M, Scheffzek K, Degreef H, De Wolf-Peeters C, Van den Oord JJ (2001) A novel N-ras mutation in malignant melanoma is associated with excellent prognosis. Cancer Res 61:4916–4922
Carlino MS, Todd JR, Rizos H (2014) Resistance to c-kit inhibitors in melanoma: insights for future therapies. Oncoscience 1:423. doi:10.18632/oncoscience.51
Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, Pavlick AC, Fusco A, Cane L, Takebe N, Vemula S, Bouvier N, Bastian BC, Schwartz GK (2011) KIT as a therapeutic target in metastatic melanoma. JAMA 305:2327. doi:10.1001/jama.2011.746
Bastian BC, Esteve-Puig R (2013) Targeting activated KIT signaling for melanoma therapy. J Clin Oncol 31:3288–3290. doi:10.1200/JCO.2013.50.3227
Wiesner T, Kiuru M, Scott SN, Arcila M, Halpern AC, Hollmann T, Berger MF, Busam KJ (2015) NF1 mutations are common in desmoplastic melanoma. Am J Surg Pathol 39:1357–1362. doi:10.1097/PAS.0000000000000451
Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, Heguy A, Dolgalev I, Khanin R, Busam K, Speicher MR, O’Brien J, Bastian BC (2010) Mutations in GNA11 in uveal melanoma. N Engl J Med 363:2191–2199. doi:10.1056/NEJMoa1000584
Helgadottir H, Hoiom V (2016) The genetics of uveal melanoma: current insights. Appl Clin Genet Volume 9:147–155. doi:10.2147/TACG.S69210
Clark WH, From L, Bernardino EA, Mihm MC (1969) The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res 29:705–727
Caroline Robert, Boguslawa Karaszewska, Jacob Schachter, Piotr Rutkowski, Andrzej Mackiewicz, Daniil Stroiakovski, Michael Lichinitser, Reinhard Dummer, Florent Grange, Laurent Mortier, Vanna Chiarion-Sileni, Kamil Drucis, Ivana Krajsova, Axel Hauschild, Paul Lorigan, Pascal Wolter, Georgina V. Long, Keith Flaherty, Paul Nathan, Antoni Ribas, Anne-Marie Martin, Peng Sun, Wendy Crist, Jeff Legos, Stephen D. Rubin, Shonda M. Little, Dirk Schadendorf, Caroline Robert, Boguslawa Karaszewska, Jacob Schachter, Piotr Rutkowski, Andrzej Mackiewicz, Daniil Stroiakovski, Michael Lichinitser, Reinhard Dummer, Florent Grange, Laurent Mortier, Vanna Chiarion-Sileni, Kamil Drucis, Ivana Krajsova, Axel Hauschild, Paul Lorigan, Pascal Wolter, Georgina V. Long, Keith Flaherty, Paul Nathan, Antoni Ribas, Anne-Marie Martin, Peng Sun, Wendy Crist, Jeff Legos, Stephen D. Rubin, Shonda M. Little, Dirk Schadendorf, (2015) Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. New England Journal of Medicine 372 (1):30–39
James Larkin, Paolo A. Ascierto, Brigitte Dréno, Victoria Atkinson, Gabriella Liszkay, Michele Maio, Mario Mandalà, Lev Demidov, Daniil Stroyakovskiy, Luc Thomas, Luis de la Cruz-Merino, Caroline Dutriaux, Claus Garbe, Mika A. Sovak, Ilsung Chang, Nicholas Choong, Stephen P. Hack, Grant A. McArthur, Antoni Ribas, James Larkin, Paolo A. Ascierto, Brigitte Dréno, Victoria Atkinson, Gabriella Liszkay, Michele Maio, Mario Mandalà, Lev Demidov, Daniil Stroyakovskiy, Luc Thomas, Luis de la Cruz-Merino, Caroline Dutriaux, Claus Garbe, Mika A. Sovak, Ilsung Chang, Nicholas Choong, Stephen P. Hack, Grant A. McArthur, Antoni Ribas, (2014) Combined Vemurafenib and Cobimetinib in-Mutated Melanoma. New England Journal of Medicine 371 (20):1867–1876
Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, Sileni VC, Dutriaux C, De Groot JWB, Yamazaki N, Loquai C, Parseval LAM, Pickard MD, Sandor V, Robert C, Flaherty KT (2016) Results of COLUMBUS part 1: a phase 3 trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in BRAF-mutant melanoma. Society for Melanoma Research (SMR) Annual Congress. http://www.arraybiopharma.com/files/6314/7865/9329/COLUMBUSprimary_SMR2016OralFINAL_110916.pdf. Accessed 5 Mar 2017
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AMM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. doi:10.1056/NEJMoa1103782
Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH, Kaempgen E, Martín-Algarra S, Karaszewska B, Mauch C, Chiarion-Sileni V, Martin A-M, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, Chapman PB (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380:358–365. doi:10.1016/S0140-6736(12)60868-X
Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, Puzanov I, Lewis KD, Amaravadi RK, Chmielowski B, Lawrence HJ, Shyr Y, Ye F, Li J, Nolop KB, Lee RJ, Joe AK, Ribas A (2012) Survival in BRAF V600–mutant advanced melanoma treated with vemurafenib. N Engl J Med 366:707–714. doi:10.1056/NEJMoa1112302
Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, Gogas H, Kefford RF, Thompson JF, Becker JC, Berking C, Egberts F, Loquai C, Goldinger SM, Pupo GM, Hugo W, Kong X, Garraway LA, Sosman JA, Ribas A, Lo RS, Long GV, Schadendorf D (2015) Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer 51:2792–2799. doi:10.1016/j.ejca.2015.08.022
Zhu Z, Liu W, Gotlieb V (2016) The rapidly evolving therapies for advanced melanoma—towards immunotherapy, molecular targeted therapy, and beyond. Crit Rev Oncol Hematol 99:91–99. doi:10.1016/j.critrevonc.2015.12.002
Kakavand H, Walker E, Lum T, Wilmott JS, Selinger CI, Smith E, Saw RPM, Yu B, Cooper WA, Long GV, O’Toole SA, Scolyer RA (2016) BRAF V600E and NRAS Q61L/Q61R mutation analysis in metastatic melanoma using immunohistochemistry: a study of 754 cases highlighting potential pitfalls and guidelines for interpretation and reporting. Histopathology 69:680–686. doi:10.1111/his.12992
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JMG, Utikal J, Dreno B, Nyakas M, Middleton MR, Becker JC, Casey M, Sherman LJ, Wu FS, Ouellet D, Martin A-M, Patel K, Schadendorf D (2012) Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367:107–114. doi:10.1056/NEJMoa1203421
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandalà M, Millward M, Arance A, Bondarenko I, Haanen JBAG, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Casey M, Ouellet D, Martin A-M, Le N, Patel K, Flaherty K (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med 371:1877–1888. doi:10.1056/NEJMoa1406037
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA, Falchook G, Algazi A, Lewis K, Long GV, Puzanov I, Lebowitz P, Singh A, Little S, Sun P, Allred A, Ouellet D, Kim KB, Patel K, Weber J (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367:1694–1703. doi:10.1056/NEJMoa1210093
Ascierto PA, McArthur GA, Dréno B, Atkinson V, Liszkay G, Di Giacomo AM, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, Yan Y, Wongchenko M, Chang I, Hsu JJ, Koralek DO, Rooney I, Ribas A, Larkin J (2016) Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 17:1248–1260. doi:10.1016/S1470-2045(16)30122-X
Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, Rutkowski P, Del Vecchio M, Gutzmer R, Mandala M, Thomas L, Demidov L, Garbe C, Hogg D, Liszkay G, Queirolo P, Wasserman E, Ford J, Weill M, Sirulnik LA, Jehl V, Bozón V, Long GV, Flaherty K (2017) Binimetinib versus dacarbazine in patients with advanced NRAS -mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2045:1–11. doi:10.1016/S1470-2045(17)30180-8
Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, Massi D, Fonsatti E, Staibano S, Nappi O, Pagani E, Casula M, Manca A, Sini M, Franco R, Botti G, Caracò C, Mozzillo N, Ascierto PA, Palmieri G (2012) BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol 30:2522–2529. doi:10.1200/JCO.2011.41.2452
Heinzerling L, Baiter M, Kühnapfel S, Schuler G, Keikavoussi P, Agaimy A, Kiesewetter F, Hartmann A, Schneider-Stock R (2013) Mutation landscape in melanoma patients clinical implications of heterogeneity of BRAF mutations. Br J Cancer 109:2833–2841. doi:10.1038/bjc.2013.622
Bradish JR, Richey JD, Post KM, Meehan K, Sen JD, Malek AJ, Katona TM, Warren S, Logan TF, Fecher LA, Cheng L (2015) Discordancy in BRAF mutations among primary and metastatic melanoma lesions: clinical implications for targeted therapy. Mod Pathol 28:480–486. doi:10.1038/modpathol.2014.136
Santiago-Walker A, Gagnon R, Mazumdar J, Casey M, Long GV, Schadendorf D, Flaherty K, Kefford R, Hauschild A, Hwu P, Haney P, O’Hagan A, Carver J, Goodman V, Legos J, Martin A-M (2016) Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin Cancer Res 22:567–574. doi:10.1158/1078-0432.CCR-15-0321
Anwar MAF, Murad F, Dawson E, Abd Elmageed ZY, Tsumagari K, Kandil E (2016) Immunohistochemistry as a reliable method for detection of BRAF-V600E mutation in melanoma: a systematic review and meta-analysis of current published literature. J Surg Res 203:407–415. doi:10.1016/j.jss.2016.04.029
Lade-Keller J, Rømer KM, Guldberg P, Riber-Hansen R, Hansen LL, Steiniche T, Hager H, Kristensen LS (2013) Evaluation of BRAF mutation testing methodologies in formalin-fixed, paraffin-embedded cutaneous melanomas. J Mol Diagnostics 15:70–80. doi:10.1016/j.jmoldx.2012.08.003
Colomba E, Hélias-Rodzewicz Z, Von Deimling A, Marin C, Terrones N, Pechaud D, Surel S, Côté J-F, Peschaud F, Capper D, Blons H, Zimmermann U, Clerici T, Saiag P, Emile J-F (2013) Detection of BRAF p.V600E mutations in melanomas. J Mol Diagnostics 15:94–100. doi:10.1016/j.jmoldx.2012.09.001
Bruno W, Martinuzzi C, Andreotti V, Pastorino L, Spagnolo F, Dalmasso B, Cabiddu F, Gualco M, Ballestrero A, Bianchi-Scarrà G, Queirolo P, Grillo F, Mastracci L, Ghiorzo P, Melanoma Intergroup (IMI) on behalf of the I (2017) Heterogeneity and frequency of BRAF mutations in primary melanoma: comparison between molecular methods and immunohistochemistry. Oncotarget 8:8069–8082. doi:10.18632/oncotarget.14094
Franczak C, Salleron J, Dubois C, Filhine-Trésarrieu P, Leroux A, Merlin J-L, Harlé A (2017) Comparison of five different assays for the detection of BRAF mutations in formalin-fixed paraffin embedded tissues of patients with metastatic melanoma. Mol Diagn Ther 1–8. doi:10.1007/s40291-017-0258-z
Long GV, Wilmott JS, Capper D, Preusser M, Zhang YE, Thompson JF, Kefford RF, von Deimling A, Scolyer RA (2013) Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am J Surg Pathol 37:61–65. doi:10.1097/PAS.0b013e31826485c0
Long E, Ilie M, Lassalle S, Butori C, Poissonnet G, Washetine K, Mouroux J, Lespinet V, Lacour JP, Taly V, Laurent-Puig P, Bahadoran P, Hofman V, Hofman P (2015) Why and how immunohistochemistry should now be used to screen for the BRAFV600E status in metastatic melanoma? The experience of a single institution (LCEP, nice, France). J Eur Acad Dermatology Venereol 29:2436–2443. doi:10.1111/jdv.13332
Kong Y, Kumar SM, Xu X (2010) Molecular pathogenesis of sporadic melanoma and melanoma-initiating cells. Arch Pathol Lab Med 134:1740–1749. doi:10.1043/2009-0418-RAR.1
Munhoz RR, Postow MA (2016) Recent advances in understanding antitumor immunity. F1000Research 5:2545. doi:10.12688/f1000research.9356.1
Clemente CG, Mihm MC, Bufalino R, Zurrida S, Collini P, Cascinelli N (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77:1303–1310. doi:10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5
Cree IA, Booton R, Cane P, Gosney J, Ibrahim M, Kerr K, Lal R, Lewanski C, Navani N, Nicholson AG, Nicolson M, Summers Y (2016) PD-L1 testing for lung cancer in the UK: recognizing the challenges for implementation. Histopathology 69:177–186. doi:10.1111/his.12996
Gogas H, Polyzos A, Kirkwood J (2013) Immunotherapy for advanced melanoma: fulfilling the promise. Cancer Treat Rev 39:879–885. doi:10.1016/j.ctrv.2013.04.006
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T (2000) Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192:1027–1034. doi:10.1084/jem.192.7.1027
Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, Haanen J (2015) Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 15:457–472. doi:10.1038/nrc3973
Zhao X, Subramanian S (2017) Intrinsic resistance of solid tumors to immune checkpoint blockade therapy. Cancer Res 77:817–822. doi:10.1158/0008-5472.CAN-16-2379
Mocellin S, Pasquali S, Rossi CR, Nitti D (2010) Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. JNCI J Natl Cancer Inst 102:493–501. doi:10.1093/jnci/djq009
Mahoney KM, Freeman GJ, McDermott DF (2015) The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther 37:764–782. doi:10.1016/j.clinthera.2015.02.018
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723. doi:10.1056/NEJMoa1003466
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain J-F, Testori A, Grob J-J, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen T-T, Humphrey R, Hoos A, Wolchok JD (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364:2517–2526. doi:10.1056/NEJMoa1104621
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen T-T, Berman DM, Wolchok JD (2015) Pooled analysis of long-term survival data from phase II and phase III trials of Ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33:1889–1894. doi:10.1200/JCO.2014.56.2736
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbé C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V, Ascierto PA (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372:320–330. doi:10.1056/NEJMoa1412082
Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob J-J, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16:375–384. doi:10.1016/S1470-2045(15)70076-8
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A (2015) Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 372:2521–2532. doi:10.1056/NEJMoa1503093
Robert C, Schadendorf D, Messina M, Hodi FS, O’Day S (2013) Efficacy and safety of retreatment with ipilimumab in patients with pretreated advanced melanoma who progressed after initially achieving disease control. Clin Cancer Res 19:2232–2239. doi:10.1158/1078-0432.CCR-12-3080
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168:707–723. doi:10.1016/j.cell.2017.01.017
O’Donnell JS, Smyth MJ, Teng MWL (2016) Acquired resistance to anti-PD1 therapy: checkmate to checkpoint blockade? Genome Med 8:111. doi:10.1186/s13073-016-0365-1
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong K-K, Hammerman PS (2016) Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7:10501. doi:10.1038/ncomms10501
Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, Palaskas N, Rodriguez GA, Parisi G, Azhdam A, Chmielowski B, Cherry G, Seja E, Berent-Maoz B, Shintaku IP, Le DT, Pardoll DM, Diaz LA, Tumeh PC, Graeber TG, Lo RS, Comin-Anduix B, Ribas A (2017) Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 7:188–201. doi:10.1158/2159-8290.CD-16-1223
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TNM, Lo RS, Ribas A (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375:819–829. doi:10.1056/NEJMoa1604958
Chen P-L, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, De Macedo MP, Austin-Breneman JL, Jiang H, Chang Q, Reddy SM, Chen W-S, Tetzlaff MT, Broaddus RJ, Davies MA, Gershenwald JE, Haydu L, Lazar AJ, Patel SP, Hwu P, Hwu W-J, Diab A, Glitza IC, Woodman SE, Vence LM, Wistuba II, Amaria RN, Kwong LN, Prieto V, Davis RE, Ma W, Overwijk WW, Sharpe AH, Hu J, Futreal PA, Blando J, Sharma P, Allison JP, Chin L, Wargo JA (2016) Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov 6:827–837. doi:10.1158/2159-8290.CD-15-1545
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373:23–34. doi:10.1056/NEJMoa1504030
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA (2015) Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350:207–211 doi:10.1126/science.aad0095
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371:2189–2199. doi:10.1056/NEJMoa1406498
Seremet T, Koch A, Jansen Y, Schreuer M, Wilgenhof S, Del Marmol V, Liènard D, Thielemans K, Schats K, Kockx M, Van Criekinge W, Coulie PG, De Meyer T, van Baren N, Neyns B (2016) Molecular and epigenetic features of melanomas and tumor immune microenvironment linked to durable remission to ipilimumab-based immunotherapy in metastatic patients. J Transl Med 14:232. doi:10.1186/s12967-016-0990-x
Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, Guida M, Hyams DM, Gómez H, Bastholt L, Chasalow SD, Berman D (2011) A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med 9:204. doi:10.1186/1479-5876-9-204
Ji R-R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, Jackson JR, Shahabi V (2012) An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 61:1019–1031. doi:10.1007/s00262-011-1172-6
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571. doi:10.1038/nature13954
Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 20:5064–5074. doi:10.1158/1078-0432.CCR-13-3271
Herbst RS, Soria J-C, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, HEK K, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515:563–567. doi:10.1038/nature14011
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C. Melis, J.J. van den Oord and O. Bechter declare that they have no conflict of interest. A. Rogiers received travel support from BMS and Novartis.
Rights and permissions
About this article

Cite this article
Melis, C., Rogiers, A., Bechter, O. et al. Molecular genetic and immunotherapeutic targets in metastatic melanoma. Virchows Arch 471, 281–293 (2017). https://doi.org/10.1007/s00428-017-2113-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00428-017-2113-3