
Opinion statement
Melanoma is the most aggressive of the cutaneous malignancies, causing more than 9,000 deaths in the past year in the United States. Historically, systemic therapies have been largely ineffective, because melanoma is usually resistant to cytotoxic chemotherapy. However, during the past few years, several targeted therapies have proved effective in this challenging disease. These recent advances have been facilitated by an improved understanding of the driving genetic aberrations of melanoma, particularly mutations in the mitogen-activated protein kinase (MAPK) pathway. Vemurafenib, a BRAF inhibitor, demonstrated an overall survival advantage in phase III trials and is an appropriate option for first-line therapy in metastatic BRAF mutant melanoma. Dabrafenib, another BRAF inhibitor, and trametinib, a MEK inhibitor, also have been shown to be effective in phase III trials for BRAF mutant melanoma and may be additional treatment options as monotherapy or in combination pending regulatory approval. Additionally, imatinib is a promising targeted therapy for patients whose tumors harbor a KIT mutation in exons 11 and 13. Although these targeted agents cause objective responses and clinical benefit in patients with metastatic melanoma, resistance invariably develops. New targets and strategies to overcome acquired resistance are urgently needed. Furthermore, no effective targeted therapy has been developed for NRAS mutant tumors or in melanomas with as yet unknown driver mutations. In this review, we discuss current molecular targeted treatment options and promising ongoing research to develop new strategies to treat melanoma.
Similar content being viewed by others



Avoid common mistakes on your manuscript.
Introduction
Melanoma is a common and aggressive cutaneous malignancy, with more than 76,000 new cases and 9,000 deaths in the United States in the past year [1]. In contrast to many other types of cancer, the incidence and mortality of melanoma has increased during the past 10 years [2•]. Metastatic melanoma portends a poor prognosis, historically with a median survival of well under 1 year. However, with the advent of effective genotype directed therapy, as well as more effective immunotherapy, outcomes have improved for patients with metastatic disease. Although surgical resection remains the mainstay of therapy for localized disease, molecular targeted therapy has an expanding role in the recurrent and metastatic setting.
Historically, melanoma has been classified based on clinical characteristics, such as primary tumor thickness and ulceration. More recently, it has become clear that particular driver mutations have important therapeutic implications, allowing for additional melanoma classification based on tumor genetics. The MAP kinase pathway is constitutively activated in >90 % of melanomas, driving unregulated cellular proliferation and unrestrained tumor growth. These aberrations consist of activating mutations in the G-protein NRAS (15-20 %), the serine-threonine kinase BRAF (40-50 %), or the receptor tyrosine kinase KIT (2-3 %) [3–6]. Additionally, other melanomas lacking these mutations have been found to rarely carry other MAP kinase pathway mutations (i.e., NF1, H-RAS, MAP2K1, MAP2K2) or demonstrate ERK phosphorylation without a known driver mutation [7–9]. Targeted treatments now exist for BRAF and KIT mutant melanoma and are discussed below. These mutations also appear to associate with the clinical presentation of melanoma, as well as prognosis. BRAF mutant tumors are especially common on skin without chronic sun damage, whereas NRAS mutant melanoma is equally common on skin with chronic or intermittent sun damage [6]. KIT mutations occur more frequently in two uncommon subtypes: acral and mucosal melanoma [5]. GNAQ and GNA11 mutations, both rarely occurring, are nearly exclusively seen in uveal melanoma [10]. Additionally, both BRAF and NRAS mutations appear to confer a worse prognosis in the metastatic setting compared with tumors without known driver mutations (WT) [3, 11].
Treatment
-
Surgical resection is the principle therapy for localized disease. For unresectable and metastatic melanoma, the primary two treatment modalities are molecular targeted therapy and immune based therapies. Cytotoxic chemotherapy has a limited role in the treatment of advanced or metastatic disease. For the purpose of this review, we will focus on targeted therapy.
Pharmacologic Treatment
BRAF Inhibitors
-
Somatic mutations in BRAF, a serine-threonine kinase, are present in 40-50 % of metastatic melanoma [6, 12]. The vast majority of mutations involve a substitution for valine at the 600th amino acid position, to glutamine in 80 % of cases (V600E). BRAF V600K mutations make up the majority of the remainder, with rare V600R, V600D, V600M, V600E’, L597, and K601E mutations [4, 13, 14].
Vemurafenib
-
Vemurafenib (PLX4032 [Zelboraf], Roche) was developed as a specific BRAF inhibitor. Preclinical data showed specificity for the BRAF kinase with the V600E mutation and potent inhibition of cellular proliferation in vitro [15].
-
Phase I and II trials demonstrated that vemurafenib has significant antitumor activity, with objective response rates of 53 % (confirmed) to 81 % (including unconfirmed) [16, 17]. Tumor responses often were rapid and dramatic, and the large majority of patients had at least stabilization of their disease. Complete responses were seen in less than 10 % of patients. Resistance to therapy and tumor progression developed within 2 years for nearly all patients, although 5-10 % of responders remain in remission at 24 months.
-
A phase III trial (BRIM-3) showed an objective response rate of 59 % with vemurafenib in BRAF V600E mutant melanoma. Overall survival (OS) at 6 months was improved compared to dacarbazine (84 % vs. 64 %), with a median overall survival of 9.7 months for chemotherapy and 13.6 months for vemurafenib (hazard ratio (HR) = 0.7). Median progression-free survival (PFS) also was improved (5.9 vs. 1.6 months) [18••].
-
Vemurafenib has similar activity in patients with V600K mutations, with preclinical data suggesting efficacy in other V600 and possibly rare L597 mutations as well [14, 17, 19].
-
Although vemurafenib is well tolerated, adverse effects are not trivial, and include fatigue, arthralgia, edema, nausea, and prolonged QT interval. Secondary cutaneous squamous cell carcinomas and keratoacanthomas, often with RAS mutations, occur in approximately 20 % of patients, usually in the first 2–3 months of therapy [18••, 20•].
-
In nonmelanoma cells without mutant BRAF, the MAP kinase pathway may be paradoxically activated by BRAF inhibitors through a CRAF dependent mechanism [21]. This is likely the cause for secondary cutaneous squamous cell neoplasms. Additionally, there is concern that heightened MAP kinase signaling may lead to unmasking of other secondary malignancies. Notably, new primary melanomas without BRAF mutations appear to occur at a higher rate while on BRAF inhibitor therapy [22]. More recently, reports of colonic adenomatous polyps and chronic myelomonocytic leukemia have been seen following initiation of BRAF inhibitor therapy [23, 24]. In all of these cases, the BRAF inhibitor does not cause the malignancies but acts to accelerate its clinical presence.
-
Recommended starting dose for vemurafenib is 960 mg twice daily, which can be reduced to 720 mg twice daily or even to 480 mg twice daily if intolerable side effects develop. Recently, preclinical work seems to imply alternate scheduling of drug may delay the development of resistance.
Dabrafenib
-
Dabrafenib (GSK2118436, GlaxoSmithKline) is another specific BRAF V600 inhibitor found to have preclinical activity against melanoma in cell lines and xenografts.
-
Early phase trials also showed that dabrafanib is a highly active drug in BRAF mutant melanoma [25]. Objective responses were seen in 53-69 % of patients with V600E or V600K mutations. Timing of responses and development of resistance were very similar to vemurafenib.
-
A phase III trial (BREAK-3) compared dabrafenib with dacarbazine in BRAF V600E mutant metastatic melanoma. Response rates (53 % vs. 19 %) and PFS (5.3 vs. 2.7 months) were improved, although the trial allowed crossover and was not powered to detect an overall survival benefit [26•]. Patients with either BRAF V600E or V600K mutations and patients with brain metastases were included in the trial and early responses were observed in the brain parenchyma. This was confirmed in a recently published article (BREAK-MB) where 29 of 74 (39.2 %, 95 % confidence interval (CI) 28.0–51.2) newly diagnosed and untreated brain metastases from V600E BRAF-mutant melanoma achieved an overall intracranial response as did 20 of 65 (30.8 %, 19.9–43.4) in progressive brain metastases following failure of local treatment [27•].
-
Side-effect profile differs in several ways from vemurafenib. The incidence of cutaneous squamous cell carcinomas or keratoacanthomas (6-19 %) appeared to be slightly lower than seen with vemurafenib treatment. This might be explained by a higher affinity to the mutant BRAF and less cross-reactivity to CRAF leading to enhanced MAP kinase inhibition. However, fevers and chills appeared to be more common (25 %).
-
The initial starting dose for dabrafenib is 150 mg twice daily. Regulatory approval for dabrafenib is pending and is expected soon.
MEK Inhibitors
-
MEK inhibition blocks signaling in the MAP kinase pathway downstream from BRAF and mechanistically could have activity in both melanoma with BRAF mutations and NRAS mutations. However, although MEK inhibitors may have some activity in NRAS mutant disease, preclinical data and clinical evidence is strongest that these agents effectively inhibit cellular proliferation and tumor growth in BRAF mutant melanoma [28••]. Combination therapy with BRAF inhibitors will be discussed in the combination section.
Trametinib
-
Trametinib (GSK1120212, GlaxoSmithKline) is a highly specific inhibitor of MEK1 and 2 and is the best evaluated MEK inhibitor in the clinic. Early phase trials showed effective MAP kinase signaling inhibition and tolerable side effects [29].
-
In a phase III trial (METRIC), trametinib was compared with dacarbazine in patients with BRAF mutations [30•]. Overall survival was improved at 6 months (81 % vs. 67 %) despite a high crossover rate to trametinib at time of progression (47 %) in the dacarbazine arm. Progression-free survival also was increased (4.8 vs. 1.5 months), with an objective response rate of 22 %.
-
Treatment was relatively well tolerated, although a drop in ejection fraction was seen in 7 % of patients, causing treatment discontinuation in two patients. An acneiform rash, peripheral edema, diarrhea, and blurred vision were other side effects. Central serous retinopathy (CSR) is an uncommon but well-known cause of blurred vision in patients on MEK inhibitors, although was not seen in this trial [29]. All patients with visual symptoms should be evaluated for both CSR and retinal vein thrombosis. Notably, no squamous cell skin cancers were seen.
-
The role of trametinib in BRAF mutant melanoma requires further clarification. It is unlikely to match the efficacy of the selective BRAF inhibitors as a single agent. Furthermore, single-agent trametinib is not effective when patients progress on a BRAF inhibitor (Kim K, et. al, in press, J Clin Oncol). Combination therapy with dabrafenib is likely the most appropriate setting to use trametinib (see “Combination Therapy”).
-
The phase III dose of trametinib was 2-mg daily. Regulatory approval is pending and is expected soon.
Other MEK Inhibitors
-
MEK162 (Novartis) is another selective MEK1 and MEK2 inhibitor in early-phase trials, which appears to have modest activity in both BRAF and NRAS mutant melanoma [31]. Eight of 35 (23 %) patients with BRAF mutations had objective responses. In patients with NRAS mutations, 6 of 28 (21 %) had objective responses. This is the first molecularly targeted therapy showing activity in NRAS mutant melanoma.
-
Selumetinib (AZD6244, AstraZeneca) also inhibits MEK1/2 and has modest activity in melanoma. However, in unselected patients with metastatic disease, it was not superior to temozolomide and in those retrospectively found to have a BRAF V600E containing melanoma, responses were observed in only 5 of 45 patients (11 %) [32]. Several trials are ongoing to evaluate whether combination therapy with other targeted agents is efficacious.
Resistance to BRAF Inhibition
-
The vast majority of patients with BRAF mutant melanoma will have objective responses or temporary disease stabilization (by RECIST 1.1) when treated with a BRAF inhibitor, although approximately 15 % appear to have progressed by the time of the first tumor reevaluation (6–8 weeks) [18••]. However, all patients inevitably relapse, frequently in the first 1 to 2 years of therapy.
-
Most other tumors that develop resistance to targeted agents acquire a secondary mutation in the site of binding to the inhibiting agent. For example, in chronic myelogenous leukemia, resistance to imatinib is derived from secondary BCR-ABL mutations, and 50 % of non-small cell lung cancers develop a T790M secondary mutation in EGFR in response to EGFR inhibitors. In contrast, no secondary BRAF mutations have been described in melanoma. Resistance is derived from a variety of mechanisms, including the following:
-
Acquired alterations in the MAP kinase pathway may induce resistance in the following ways (Figure 1):
Figure 1 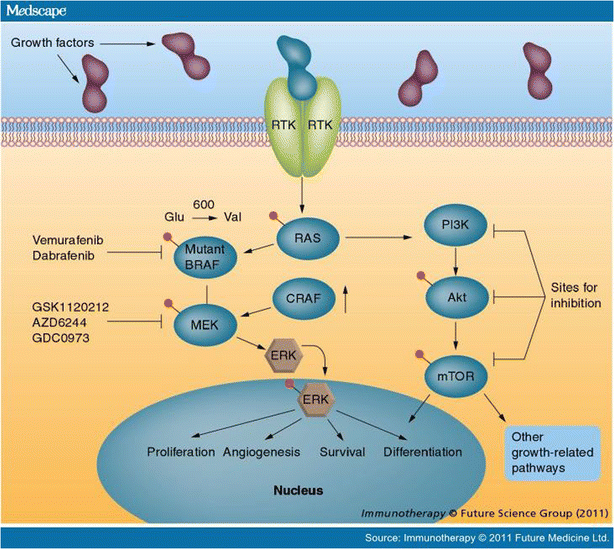
Reproduced with permission from Future Medicine Ltd. from Immunotherapy, December 2011;3(12):1461–1469.
-
Secondary NRAS mutations may arise and bypass the inactivated mutant BRAF protein, likely utilizing CRAF to activate MAPK signaling [33].
-
MEK1 mutations may constitutively activate the pathway downstream of the inhibited mutant BRAF [7].
-
Elevated levels of CRAF have been described, allowing for heterodimerization with BRAF and renewed cell signaling [34].
-
Enhanced expression and activation of COT, a serine/threonine kinase, can reinduce MAP kinase signaling by activating MEK in a BRAF independent manner [35].
-
Alternate splicing of the BRAF V600E kinase with enhanced dimerization can restore MAP kinase pathway activity independent of upstream RAS signaling [36].
-
Additionally, BRAF resistance can develop though alternate cell signaling pathways independent of the MAP kinase pathway, including the following:
-
PDGFRβ and other tyrosine kinase overexpression (EGFR) may lead to renewed proliferation and tumor growth [33, 37].
-
Upregulation of insulin growth factor receptor 1 (IGFR1) and activation of the PI3K-AKT pathway leads to cellular proliferation [38].
-
Hepatocyte growth factor (HGF) induces resistance by activating the HGF receptor MET, which restores cell signaling and tumor growth through the PI3K-AKT and MAP kinase pathways [39, 40•]. This mechanism also may cause primary resistance to BRAF inhibitors.
-
-
-
-
Overcoming resistance is a major problem that requires innovative solutions given the diversity of mechanisms. Multiple strategies, usually involving combination therapies, are being evaluated in clinical and preclinical studies (see Combination Therapy and Ongoing Research below).

Combination Therapy
Dabrafenib/Trametinib
-
Preclinical studies showed that combination therapy could potentially prevent or delay the development of acquired resistance to BRAF inhibitors by complete abrogation of MAP kinase signaling [41]. Additionally, cutaneous toxicities appeared to be diminished with combination therapy, likely due to inhibition of paradoxical MAP kinase activation caused by BRAF inhibitors [20•].
-
A phase I/II open label trial evaluated in a 1:1:1 ratio combination therapy at two different doses and dabrafenib monotherapy [42••]. The median PFS for combination therapy (at the 150 mg/2 mg arm) was 9.4 months compared with 5.8 months with monotherapy. Objective response rate was 76 % for combination therapy and 54 % in the monotherapy group. Median overall survival had not been reached in either group at the time of publication.
-
Therapy was relatively well tolerated. Squamous cell carcinomas occurred less frequently in the combination group (7 % and 2 % at 2 and 1 mg/kg of trametinib, respectively) compared with the monotherapy group (19 %). Other toxicities included those expected with MEK inhibitors, such as acneiform dermatitis, pyrexia, peripheral edema, ocular events, and rarely decreased cardiac ejection fraction.
-
Currently, a phase III trial is ongoing comparing combination therapy with vemurafenib or dabrafenib monotherapy.
-
Combination therapy appears to delay the development of resistance to targeted therapy but still invariably develops. Mechanisms of resistance to dual therapy are being elucidated.
KIT Inhibitors
-
Somatic mutations in KIT occur infrequently, with an incidence of 2-3 % of unselected cases of metastatic melanoma [5, 43]. However, they more often are present in primary tumors found on acral surfaces (soles of the feet, nailbeds) or mucosal melanoma (10-20 %) [44, 45]. Most commonly, KIT mutations occur in the juxtamembrane domain (L576P, V559A, W557R), although they also may occur in the kinase domain (K642E, D816H).
Imatinib
-
Efforts to target KIT with tyrosine kinase inhibitors have been successful in gastrointestinal stromal tumor (GIST), generating interest in exploiting this target in melanoma. Preclinical data in melanoma and studies in GIST patients predict that mutations in the juxtamembrane domain will be sensitive to imatinib, whereas some mutations in the kinase domain may be resistant (D816H).
-
Two phase II trials have been completed using imatinib in KIT mutant melanoma. In a trial from Memorial Sloan Kettering, 28 patients with KIT mutations or amplifications were treated with imatinib, with a durable response rate of 16 % and a median progression-free survival of 12 weeks [46•]. The majority of patients (72 %) had at least temporary disease stabilization.
-
In the second trial, 43 patients with KIT aberrations were treated with imatinib, with an objective response rate of 23 %, temporary stabilization of disease in an additional 30 %, and a median PFS of 3.5 months [47•].
-
In both trials, mutations in the juxtamembrane domain (L596, V559), as well as K642E, appear to display enhanced sensitivity to imatinib therapy. KIT amplified tumors without mutations also were sensitive in some cases. Notably, dose escalation to 800 mg did not restore disease control in responding patients. Therefore, the recommended dose of imatinib is 400 mg daily.
Other KIT Inhibitors
-
Dasatinib: Objective responses to dasatinib in KIT mutant patients have been noted in case reports and as part of a phase II trial [48, 49].
-
Sunitinib: This agent also appears to have activity, producing objective responses in three of four patients with KIT mutations (although only in one of six patients with KIT amplifications) [50].
-
Sorafenib: Cases of temporary responses to sorafenib also have been reported [51, 52].
“Targeted” Immunotherapy
-
Immunotherapy is the other major class of therapy for metastatic melanoma, and includes interleukin-2, anti-CTLA-4 (ipilimumab), and promising new agents including anti-PD-1/PD-L1. Currently these agents are being applied to melanoma patients regardless of tumor mutational status.
-
Significant interest exists in developing a biomarker to predict response to immunotherapy to enhance patient selection and develop “targeted” immunotherapy. Candidates include host gene polymorphisms, tumor gene expression profiles, and tumor genotype, among others.
-
PD-L1 (programmed death ligand 1) expression is a promising candidate for predicting response to anti-PD-1 therapy. Forty-two patients treated with anti-PD-1 (18 with melanoma) had tumor specimens assayed for PD-L1 expression. Of 17 patients without ligand expression, no patients had objective responses, compared with 9 of 25 in patients with expression [53••]. This biomarker is currently being assessed further in ongoing trials.
-
Additionally, one trial that evaluated nearly 100 patients treated with IL-2 suggests that NRAS mutant melanoma may respond better to IL-2 than melanoma without known driver mutations [54].
Emerging Therapies and Ongoing Areas of Investigation
BRAF Mutant
-
In patients with BRAF mutant tumors, it is unclear whether first-line BRAF inhibition or immunotherapy is preferable. In our opinion, when a rapid response is needed due to symptomatic metastases, BRAF inhibitor therapy is the preferred first option. However, if patients are relatively asymptomatic, immunotherapy should be considered due to the possibility of durable benefit.
-
Combining BRAF inhibitors and immunotherapy is an intriguing possibility. This approach is supported by preclinical rationale: BRAF inhibitors increase tumor infiltrating lymphocytes and may enhance the tumor-specific immune response [55]. Additionally, resistance to BRAF inhibitors are associated with increased PD-L1 expression [56]. Trials are planned to assess vemurafenib or the combination of dabrafenib/trametinib and ipilimumab or anti-PD1/PD-L1 based combination therapy.
-
Acquired resistance to BRAF inhibition remains a major problem, with intensive ongoing efforts to overcome this resistance. Preclinical rationale exists for combinations with BRAF inhibitors, including inhibitors of MET, and heat shock protein [39, 57]. These agents appear to prevent development of acquired resistance in cell lines. Trials of these agents are in various early stages of planning or accrual.
NRAS Mutant
-
NRAS mutant melanoma currently has no effective targeted therapy. Effective agents for this population (15-20 % of metastatic melanoma) are urgently needed. Targeting RAS has proved very difficult in melanoma and other tumor types. BRAF and MEK inhibitors are thought to be largely ineffective due to reliance on CRAF signaling (bypassing BRAF) and RAS-activated PI3K-AKT pathway signaling (bypassing MEK) [58]. Trials evaluating combination therapy with MEK inhibitors and PI3 kinase or AKT inhibitors are ongoing.
-
In NRAS mutant melanoma, MEK inhibitor monotherapy appears to activate apoptosis, but does not achieve cell cycle arrest. Intriguing preclinical data shows that combination therapy with MEK inhibitors and cyclin dependent kinase (CDK4) inhibitors achieves apoptosis and cell cycle arrest, providing significant therapeutic synergy similar to genetic knockdown of NRAS [59].
Other Areas of Investigation
-
Anti-angiogenic agents do not have a clear role in melanoma. Axitinib monotherapy was associated with a 19 % objective response rate and median PFS of 2.9 months; trials are ongoing to assess whether this is an effective therapy in combination with other agents [60]. Additionally, bevacizumab has been combined with carboplatin and paclitaxel in a phase II trial (BEAM), which showed a trend to improved survival compared with chemotherapy alone (12.3 vs. 9.3 months; p = 0.19) [61]. Phase III trials are being considered.
-
Inhibitors of ERK have generated interest. These agents are in very early-stage development.
-
Currently, there are no direct targeted agents for GNAQ or GNA11 mutant tumors, primarily found in uveal melanoma. These tumors often are resistant to targeted therapies and immunotherapy. Efforts are ongoing to develop targeted agents for these patients. These trials have primarily evaluated downstream targets, such as MEK or epigenetic changes secondary to the loss of the BAP1 function [62, 63].
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
American Cancer Society. Cancer Facts and Figures 2012. Atlanta: American Cancer Society; 2012.
Jemal A, Saraiya M, Patel P, et al. Recent trends in cutaneous melanoma incidence and death rates in the United States, 1992–2006. J Am Acad Dermatol. 2011;65:S17–25. This study highlights the epidemiology and mortality trends in melanoma.
Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239–46.
Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype-driven therapeutic trials. PLoS One. 2012;7:e35309.
Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14:6821–8.
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47.
Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96.
Maertens O, Johnson B, Hollstein P, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2012. doi:10.1158/2159-8290.CD-12-0313.
Nikolaev SI, Rimoldi D, Iseli C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–9.
Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–9.
Jakob JA, Bassett Jr RL, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–23.
Maldonado JL, Fridlyand J, Patel H, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–90.
Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med. 2010;8:67.
Dahlman KB, Xia J, Hutchinson K, et al. BRAF L597 mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2012;2:791–7.
Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–6.
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19.
Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14.
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. This phase III trial was the first time a mortality benefit was demonstrated for targeted therapy in metastatic melanoma.
Yang H, Higgins B, Kolinsky K, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010;70:5518–27.
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–15. This study demonstrates the molecular basis for secondary squamous cell carcinomas, a unique side effect of BRAF inhibitor therapy.
Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5.
Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375–83.
Chapman PB, Metz D, Sepulveda A, et al. Development of colonic adenomas and gastric polyps in BRAF mutant melanoma patients treated with vemurafenib. Pigment Cell Melanoma Res. 2012;25:847.
Callahan MK, Rampal R, Harding JJ, et al. Progression of RAS-Mutant Leukemia during RAF Inhibitor Treatment. N Engl J Med. 2012;367:2316–21.
Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–901.
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. Phase III trial demonstrating a mortality benefit in metastatic melanoma for dabrafenib.
Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087–95. Phase II trial demonstrating the activity of BRAF inhibitor therapy in patients with brain metastases.
Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2012. doi:10.1038/onc.2012.345.A review article detailing therapy targeting the MAP kinase signaling pathway.
Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–81.
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. Phase III trial demonstrating mortality benefit in metastatic melanoma for trametinib.
Ascierto PA, Berking C, Agarwala SS. Efficacy and safety of oral MEK162 in patients with locally advanced and unresectable or metastatic cutaneous melanoma harboring BRAFV600 or NRAS mutations. J Clin Oncol. 2012;30:8511.
Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67.
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7.
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61.
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72.
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480:387–90.
Lito P, Pratilas CA, Joseph EW, et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell. 2012;22:668–82.
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95.
Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9.
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. Translational work detailing the newest discovered mechanism of BRAF inhibitor resistance.
Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–30.
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N Engl J Med. 2012;367:1694–703. Phase I/II trial demonstrating improved outcomes and fewer cutaneous toxicities from combination BRAF and MEK inhibition compared with monotherapy.
Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res. 2010;23:210–5.
Satzger I, Schaefer T, Kuettler U, et al. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. Br J Cancer. 2008;99:2065–9.
Torres-Cabala CA, Wang WL, Trent J, et al. Correlation between KIT expression and KIT mutation in melanoma: a study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod Pathol. 2009;22:1446–56.
Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–34.
Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904–9. Reference 46 and 47 are two phase II trials demonstrating activity of imatinib in KIT mutant melanoma.
Woodman SE, Trent JC, Stemke-Hale K, et al. Activity of dasatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009;8:2079–85.
Kluger HM, Dudek AZ, McCann C, et al. A phase 2 trial of dasatinib in advanced melanoma. Cancer. 2011;117:2202–8.
Minor DR, Kashani-Sabet M, Garrido M, et al. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18:1457–63.
Handolias D, Hamilton AL, Salemi R, et al. Clinical responses observed with imatinib or sorafenib in melanoma patients expressing mutations in KIT. Br J Cancer. 2010;102:1219–23.
Quintas-Cardama A, Lazar AJ, Woodman SE, et al. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol. 2008;5:737–40.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. Phase I/II trial demonstrating significant activity of anti-PD-1 therapy as well as biomarker data for PD-L1 expression.
Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother. 2012;35:66–72.
Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–94.
Jiang X, Zhou J, Giobbie-Hurder A, et al. The Paradoxical Activation of MAPK in Melanoma Cells Resistant to BRAF Inhibition Promotes PD-L1 Expression that is Reversible by MEK and PI3K inhibition. Clin Cancer Res. 2012;19:598–609.
Paraiso KH, Haarberg HE, Wood E, et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res. 2012;18:2502–14.
Jaiswal BS, Janakiraman V, Kljavin NM, et al. Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS One. 2009;4:e5717.
Kwong LN, Costello JC, Liu H, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–10.
Fruehauf J, Lutzky J, McDermott D, et al. Multicenter, phase II study of axitinib, a selective second-generation inhibitor of vascular endothelial growth factor receptors 1, 2, and 3, in patients with metastatic melanoma. Clin Cancer Res. 2011;17:7462–9.
Kim KB, Sosman JA, Fruehauf JP, et al. BEAM: a randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. J Clin Oncol. 2012;30:34–41.
Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–9.
Aoude LG, Vajdic CM, Kricker A, et al. Prevalence of germline BAP1 mutation in a population-based sample of uveal melanoma cases. Pigment Cell Melanoma Res. 2012. doi:10.1111/pcmr.12046.
Conflicts of Interest
Douglas B. Johnson declares that he has no conflicts of interest.
Jeffrey A. Sosman has consulted for Roche/Genentech and Millennium, has grants/grants pending with BMS, GSK, and Roche, and has received honoraria from Roche.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johnson, D.B., Sosman, J.A. Update on the Targeted Therapy of Melanoma. Curr. Treat. Options in Oncol. 14, 280–292 (2013). https://doi.org/10.1007/s11864-013-0226-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-013-0226-8