
Abstract
Chronic myeloid leukaemia (CML) was the first leukaemia associated with a unique genetic abnormality, the Philadelphia chromosome. This results from a reciprocal translocation between chromosomes 9 and 22, which generates the BCR-ABL1 fusion gene encoding a constitutively active tyrosine kinase. The complex intracellular signalling initiated by BCR-ABL1 is responsible for disease development, and targeted tyrosine kinase inhibitors have been the most successful therapeutic advance in CML. In this chapter, we review the implications of BCR-ABL1 signalling in CML, how this knowledge revolutionized CML treatment, and discuss approaches to further improving therapeutic response by the targeting of leukaemic stem cells.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Chronic myeloid leukaemia (CML) was probably the first form of leukaemia to be independently recognized in 1845 by John H. Bennett, in Scotland, and Rudolf Virchow, in Germany [1]. The discovery of the Philadelphia (Ph) chromosome, in 1960 [2] was the first consistent chromosomal abnormality associated with a specific type of leukaemia, and was not only a breakthrough in cancer biology but also the first important clue to CML pathogenesis. In 1973, Rowley identified the Ph chromosome as a shortened chromosome 22 (22q-), result of a reciprocal t(9;22)(q34.1;q11.2) translocation [3]. In the next decade, the Ph chromosome was shown to carry a unique fusion gene, BCR-ABL1 [4], the deregulated BCR-ABL1 tyrosine kinase activity was defined as the pathogenetic principle of CML [5], and the first animal models were developed [6]. Ultimately, this knowledge provided the basis for the design of a targeted therapy for CML with the development of ABL1 specific tyrosine kinase inhibitors (TKIs), which selectively inhibit the growth of BCR-ABL1 positive cells in vitro and in vivo [7–9].
2 CML Characteristics and Disease Phases
CML is a clonal myeloproliferative disease originating in a single haematopoietic stem cell (HSC). It represents 15–20 % of the leukaemias in adults and has a relatively low incidence (1–1.5 new cases per 100,000 people per year). However, its prevalence is on the rise due to the significant improvement in its treatment over the past 15 years [10]. In the Western countries, the median age of patients at diagnosis is 55–65 years old, whereas it is significantly lower, averaging 38–41 years, in Asia, Africa, Southern/Eastern Europe and Latin America [11]. The disease affects both sexes, with a slight male preponderance (male:female ratio of 1.3:1).
The only known predisposing factor to CML is high-dose ionizing radiation, as best demonstrated by studies of survivors of the Hiroshima and Nagasaki atomic bomb explosions [12]. Apart from a borderline increased risk of CML in first-degree relatives of patients with myeloproliferative disorders [13], there is no evidence of an inherited disposition or association with chemical exposure.
In its natural history, CML is a tri-phasic disease, predominantly presenting in a chronic phase (CP) averaging around 3–7 years. In most cases of CP CML, the neoplastic expansion involves a leukaemic clone that differentiates into mature granulocytes which function normally, despite being derived from malignant progenitors. The ‘indolent’ phenotype of CP means that some patients are asymptomatic, and the diagnosis is frequently an incidental finding; however, the majority typically present with mild symptoms of fatigue, weight loss and sweats [14, 15]. CP progresses to either the transitional accelerated phase (AP) or transforms directly into blast crisis (BC). When present, the AP precedes BC by 2–15 months [16]. Transformation to BC is characterized by the presence, in the peripheral blood or bone marrow (BM), of 20 % or more blasts, which can be of myeloid (approximately 70 % of cases) or lymphoid (30 %) origin [17]. BC is clinically indistinguishable from acute leukaemia and can present leukocytosis, cytopenia, hepatosplenomegaly, enlarged lymph nodes, and marked refractoriness to treatment which results in a dismal clinical outcome, with a historical median survival of no more than 3–6 months [18]. Even with the advent of TKIs, response of BC to this type of therapy is minimal, and median survival is still only 9 months [19, 20].
Until the emergency of TKIs, the only curative treatment for CML was HSC transplantation, but this was restricted to a minority of patients, due to age restrictions and the need for a histocompatible donor. The prognosis has now substantially improved for most CML patients who respond well to TKIs, a proportion of whom are able to survive indefinitely without evidence of disease.
3 Molecular Pathogenesis
3.1 The BCR-ABL1 Gene
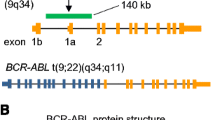
The t(9;22)(q34.1;q11.2) reciprocal translocation gives rise to two pathognomonic fusion genes, BCR-ABL1 on the der(22) (Ph) chromosome, and ABL1-BCR, on the der(9) (Fig. 7.1). Although the latter is transcribed, there is no evidence that it has functional relevance to the disease [21]. Thus, it is the translation of the BCR-ABL1 gene into an abnormal fusion protein that is responsible for the leukaemic process.

Schematic representation of the t(9;22)(q34.1;q11.2) chromosomal translocation, the cytogenetic hallmark of chronic myeloid leukaemia. Breaks within the ABL1 and BCR genes on chromosomes 9 and 22, respectively, followed by recombination of the broken DNA ends, give origin to two derivative chromosomes, the der(22) or Philadelphia (Ph) and the der(9). The BCR-ABL1 gene, which is the pathogenetic product of this translocation, is housed in the Ph chromosome
The breakpoints within ABL1 at 9q34.1 can occur anywhere over a large (>300 kb) area at its 5′ end, either upstream of exon 1b, downstream of exon 1a or, more frequently, between the two [22]. Regardless of the exact location of the breakpoint, splicing of the transcript yields an mRNA molecule where BCR is fused to ABL1 exon a2 (Fig. 7.2).

Schematic representation of the ABL1 and the BCR genes disrupted in the t(9;22)(q34.1;q11.2). Exons are represented by boxes and introns by connecting horizontal lines. Breakpoint regions in ABL1 are illustrated as vertical arrows, and in BCR by the three double-headed horizontal arrows. The lower half of the figure shows the structure of the various BCR-ABL1 mRNA transcripts which are formed in accordance with the position of the breakpoint in BCR. Breaks in m-bcr give origin to BCR-ABL1 mRNA molecules with an e1a2 junction. The breaks in M-bcr occur either between exons e13 and e14 or between e14 and e15, generating fusion transcripts with a e13a2 or a e14a2 junction, respectively. Breakpoints in μ-bcr, the most 3′ cluster region, result in BCR-ABL1 transcripts with an e19a2 junction
In contrast to ABL1, breakpoints within BCR localize to one of three breakpoint cluster regions (bcr). In most CML cases and in about one third of Ph + acute lymphoblastic leukaemias (ALL), the break occurs within BCR exons 12–16 (previously exons b1–b5), defined as the major bcr (M-bcr) [22]. Due to alternative splicing, the mRNA usually contains the BCR-ABL1 junctions e13a2 or e14a2 (originally b2a2 or b3a2) and is translated into a 210 kDa protein (P210BCR-ABL). The majority of Ph + ALL and very rare cases of CML, characterized by prominent monocytosis [23, 24], have breakpoints further upstream between exons e2′ and e2, termed the minor bcr (m-bcr). The resulting e1a2 mRNA is translated into a 190 kDa protein (P190BCR-ABL). A third bcr (μ-bcr) is located downstream of exon 19, giving rise to a 230 kDa fusion protein (P230BCR-ABL), which is sometimes associated with an uncommon neutrophilic variant of CML [25, 26]. Although all three major BCR-ABL1 fusion proteins induce a CML-like disease in mice, they differ in their ability to induce lymphoid leukaemia [27].
The mechanism by which the Ph chromosome is first formed and the time required for overt disease to appear are unknown. BCR-ABL1 fusion transcripts can be induced in haematopoietic cells by exposure to ionizing radiation in vitro [28]; such induced translocations may not be random events but may depend on the cellular background and the particular genes involved. Translocations between BCR and ABL1 may be favoured by their relative proximity during the interphase of cycling haematopoietic cells [29]. Furthermore, a 76 kb ‘duplicon’ near ABL1 and BCR has been implicated in the translocation, but this mechanism is purely speculative [30].
The BCR-ABL1 gene is expressed in all CML patients, but the reciprocal ABL1-BCR gene on the der(9) occurs in only 70 % of cases [21]. Approximately 20 % of CML patients have deletions on the der(9) and have significantly shorter survival than those lacking the deletions [31, 32]. Notably, absence of the ABL1-BCR gene, which is always included in the deleted region, does not by itself have the same ominous prognostic implication [33]. Similarly, no prognostic relevance of the der(9) deletions was observed on patients treated with TKIs [34, 35].
The idea that CML may result from a multi-step process was first broached over 30 years ago [36] but there is little evidence of additional abnormalities that precede the t(9;22) translocation. Even so, the presence of BCR-ABL1 in any haematopoietic cell is not in itself sufficient to cause leukaemia, since BCR-ABL1 is detectable at low frequency in the blood of many normal individuals [37, 38]. Thus the generation of a correct BCR-ABL1 in a multipotent HSC, possibly under reduced immunological surveillance, is necessary to initiate the clonal expansion that leads to CML. This hypothesis is supported by the production of a CML-like disease in mice transplanted with BCR-ABL1-positive stem cells [6, 39, 40]. However, once established, the ‘tempo’ or aggressiveness of the CP disease varies in different patients and must be influenced by other factors.
3.2 The BCR-ABL1 Protein
The BCR-ABL1 oncoprotein includes several important domains of its parental BCR and ABL1 normal counterparts, which endow it of specific biological properties (Fig. 7.3).

Schematic representation of the normal ABL1 (p145), the normal BCR (p160) and the leukaemia-associated BCR-ABL1 fusion proteins. Note that the variation between the three forms of BCR-ABL1 proteins is due to the different contributions of BCR rather than of ABL1 sequences to the hybrid product. The arrows in BCR indicate the sites of protein fusion arising from m-bcr (p190BCR-ABL), M-bcr (p210BCR-ABL) and μ-bcr (p230BCR-ABL) breakpoints. Some special features and regions of these proteins are shown: In the ABL1 protein these are the myristoylation (MYR) site present in the human type 1b protein, the regulatory src-homology (SH) regions SH3 and SH2, the SH1 (kinase domain) with its principal site of autophosphorylation (Y412), the nuclear localisation signal (NLS), the DNA- and the actin-binding domains. In the BCR protein these are the dimerization domain (DD), the phospho-serine/threonine (P-S/T)-rich SH2-binding domain, the dbl-like and the GAPrac domains
In ABL1, they include the SRC-homology SH1, SH2 and SH3, a nuclear localisation signal, DNA and actin-binding domains, and in BCR a coiled-coil motif contained in amino acids 1–63 [41], the tyrosine at position 177 [42] and phosphoserine/threonine rich sequences between amino acids 192–242 and 298–413 [43]. The most important feature for its leukaemogenic potential resides in the fact that the tyrosine kinase of the ABL1 protein is constitutively activated by the juxtaposition of BCR. The BCR dimerization domains connect two BCR-ABL1 molecules which then phosphorylate their respective partners on tyrosine residues in the kinase activation loops [41]. The consequent increase of phosphotyrosine residues on BCR-ABL1 itself creates binding sites for the SH2 domains of other proteins. A host of substrates can be tyrosine phosphorylated by BCR-ABL1, the net result of which is deregulated cellular proliferation, decreased adherence of leukaemia cells to the BM stroma, reduced response to apoptotic stimuli, increased genomic instability and increased capacity for self-renewal [44, 45].
Tyrosine phosphatases counterbalance and regulate the effects of tyrosine kinases under physiological conditions. Two tyrosine phosphatases, SYP and PTPN1, have been shown to form complexes with BCR-ABL1, and both appear to dephosphorylate BCR-ABL1 [46, 47]. On the other hand, BCR-ABL1 protects itself from the protein tyrosine phosphatase 1 (PTPN6/SHP1), which can dephosphorylate BCR-ABL1 and induce its proteasomal degradation, by inhibiting the PTPN6/SHP1 activator PP2A [48].
3.3 Signalling and Disease
3.3.1 Proliferation and Survival
BCR-ABL1 shifts the balance towards inhibition of apoptosis while simultaneously providing a proliferative stimulus through multiple signals. These are frequently difficult to separate but mostly involve PI3K/AKT1, JAK/STAT, RAS/RAF/MEK/ERK and MYC pathways (Fig. 7.4).

Some of the major signalling pathways directly or indirectly regulated by BCR-ABL1 (see text for detailed descriptions)
Once the adapter molecule GRB2 binds to P-Tyr177 on BCR-ABL1, it recruits SOS and constitutively activates RAS, which, in turn, activates MAPK3/ERK1 and MAPK1/ERK2 [42, 49]. Two other adapter molecules, SHC1 and CRKL, can also activate RAS after binding to BCR-ABL1 [50, 51]. Ultimately, activated MAPKs indirectly induce gene transcription and cell proliferation [49, 52].
Signalling from RAS can be relayed via RAC GTPases [53] to activate MAPK8/JNK, which is required for BCR-ABL1 malignant transformation [54]. Accordingly, downregulation of the JNK pathway negative regulator JUNB, by promoter hypermethylation, has been described in CML primary cells [55]. RAC GTPases themselves play an important role in BCR-ABL1 leukaemogenesis, activating STAT5, PI3K and MAPKs pathways [56]. Moreover, concomitant loss of Rac1 and Rac2 impaired the development of a myeloproliferative disease and increased survival of mice transplanted with BCR-ABL1-expressing cells [57].
Constitutive phosphorylation of STAT1 and STAT5 has been reported in several BCR-ABL1 positive cell lines [58] and primary CML cells [59], and seems to be independent of JAK. STAT5 can be directly activated by BCR-ABL1 [60] or indirectly through GRB2/RAS/RAC or HCK [56] to then up-regulate target genes, such as CCND1 (leading to cell cycle progression) and the anti-apoptotic BCL2L1/BCL-XL [61, 62]. Although one study found that BCR-ABL1 induced a CML like disease in Stat5a/b−/− mice [63], another reported that complete deletion of Stat5a/b locus turned mice resistant to BCR-ABL1 transformation [64]. In addition, knock-down of STAT5 in primary CML cells blocks Ph + colony formation [62], and cells expressing a mutant BCR-ABL1 unable to activate STAT5 or wild type BCR-ABL1 with a dominant negative STAT5 are more apoptotic than wild type cells [65]. Altogether, these results support a role for STAT5 in BCR-ABL1 transformation.
BCR-ABL1 forms complexes with PI3K, CBL and the adapters CRK and CRKL [66], in which PI3K, and the downstream AKT1 and mTOR, are constitutively activated [67]. In addition, activation of RAS and the adapter GAB2 by GRB2 cause constitutive activation of PI3K [62]. PI3K exerts its oncogenic effects mainly by activation of mTOR, which forms the mTORC1 and mTORC2 complexes that play important roles in the proliferation and survival of BCR-ABL1-positive cells [49, 68]. PI3K activity is required for BCR-ABL1-mediated leukaemogenesis, since its inhibition impairs BCR-ABL1 transformation of HSCs [49, 67]. PI3K also hyperphosphorylates the transcription factor (TF) IRF8/ICSBP, preventing its DNA binding and reverting its transcriptional repression of the antiapoptotic BCL2 gene [69].
AKT1 itself is an oncogene, and is essential for the resistance to apoptosis of BCR-ABL1-positive cells. It phosphorylates BAD, which promotes its sequestration by 14-3-3, and blocks its binding to BCL2 family members, consequently inhibiting apoptosis [70]. AKT1 also blocks apoptosis through phosphorylation of caspases [67], and downregulation of antiapoptotic BCL2L11/BIM [49].
Activation of MYC by BCR-ABL1 is dependent on the SH2 domain [71]. In addition, RAS/MAPK and PI3K/AKT1 pathways contribute to inducing MYC transcription or promoting MYC stability [67, 72]. Depending on the cellular context, MYC may transduce proliferative or apoptotic signals [67]; however, considering BCR-ABL1-mediated antiapoptotic mechanisms, the apoptotic arm of MYC is most likely inhibited in CML. Proliferation, on the other hand, may be induced by MYC’s activation of cyclin and CDK transcription, repression of CIP/KIP family cyclin/CDK inhibitors’ expression, and indirect induction of mTORC1 transcription [67].
3.3.2 Progression to Blast Crisis
CML progression is characterized by the occurrence of non-random chromosomal abnormalities. The most frequent are trisomy 8 (33 %), an additional Ph (30 %), isochromosome 17 (20 %), trisomy 19 (12 %), loss of the Y chromosome (8 % of males), trisomy 21 (7 %) and monosomy 7 (5 %) [73]. Although these changes are used as markers of disease progression, they may not necessarily be causal agents of transformation. Two important mechanisms and phenotypes related to the emergence of BC are addressed below.
3.3.2.1 Block in Differentiation
With progression of CML, the leukaemic clone undergoes differentiation arrest, resulting in a major increase of immature blasts at the expense of the terminally differentiated leucocytes. This differentiation arrest implies pathological interference with differentiation programmes involving the targeted activation/inactivation of tissue-specific genes by TF [74].
Abnormal CTNNB1/β-catenin signalling leads granulocyte-macrophage progenitors to acquire the stem cell-like capacity of unrestricted self-renewal [75]. In addition, interaction between CTNNB1 and BCR-ABL1 increases β-catenin transcriptional activity influencing leukaemic stem cell (LSC) lineage commitment as early as in CP, and loss of CTNNB1impairs the self-renewal of CML stem-cells [76, 77].
Another mechanism of differentiation arrest is the down-modulation of the TF CEBPA by BCR-ABL1, in BC but not in CP, through regulation of pre- and post-transcriptional mechanisms [78, 79]. CEBPA activates transcription of the CSFR3/GCSFR and ID1 genes in myeloid cells, and its ectopic expression restores differentiation in BCR-ABL1-transformed cell lines or BC CML primary cells [80–83].
Additional causes of the block in differentiation in BC CML include mutations, translocations or deletions in genes that regulate differentiation and self-renewal of haematopoietic stem and progenitor cells, such as GATA2 [84, 85], RUNX1 [86–88], ASXL1 [79, 87, 89], IKZF1 [87, 90, 91] and PAX5 [90, 92].
3.3.2.2 Genomic Instability
BCR-ABL1-transformed cell lines and CD34+ primary CML cells produce 2–6 times more ROS than the normal controls [93]. ROS can damage the DNA generating oxidized bases and double strand brakes (DSB). Accordingly, CD34+ CML cells accumulate three to eight times more oxidized bases and DSBs than normal cells [93]. At the same time, they display defective mismatch repair; stimulate DSBs repair but with low fidelity, through homologous recombination repair (HRR), nonhomologous end-joining (NHEJ), and single strand annealing (SSA) repair mechanisms; and induce mutagenic nucleotide excision repair (NER), all of which exacerbate genomic instability and contribute to disease progression (Fig. 7.5). The mechanisms of altered DNA repair in CML are addressed below.

BCR-ABL1 enhances DNA damage and deregulates DNA repair, the two main components of genomic instability. BCR-ABL1 positive cells accumulate more DNA lesions induced by endogenous and exogenous DNA genotoxic agents and, in parallel, activates cellular pathways which favour unfaithful DNA repair mechanisms. The overarching consequence of the two processes is the generation of improperly repaired DNA molecules containing point-mutations, insertions or deletions in genes which, once inappropriately expressed and/or activated, lead to the transformation into blast crisis (Figure modified from [94])
ATR is a DNA damage ‘sensor’ that controls cell cycle check points. BCR-ABL1 was reported to translocate to the nucleus, following exposure to genotoxic agents, where it bound and inhibited ATR and CHEK1, allowing inappropriate DNA replication [95, 96]. In a contradictory study, however, ATR signalling was stimulated in BCR-ABL1-positive cells in response to genotoxic agents [97]. This result was further corroborated by recent findings that BCR-ABL1 inhibition reduces CHEK1 activation and cell cycle arrest in G2/M phase, and induces apoptosis in cells exposed to genotoxic agents [98]. Therefore, ATR signalling might contribute to chemotherapeutics resistance in CML.
The tumour suppressor BRCA1 is another ‘sensor’ that detects DNA damage and mediates cell cycle check points and HRR [99]. BRCA1 is virtually undetectable in CML cells and BCR-ABL1-transformed cell lines [100] and this absence contributes to the genomic instability observed in BCR-ABL1 cells [101]. To overcome BRCA1 deficiency, HRR occurs through the alternative RAD52-RAD51 pathway [102, 103].
Both HRR and NHEJ promote less faithful ROS-induced DSB repair in BCR-ABL1-transformed cells [104]. Downregulation of PRKDC/DNA-PKcs, LIG4/DNA ligase IV and DCLRE1C/Artemis, and upregulation of LIG3/DNA ligase IIIα, WRN nuclease and RBBP8/CtIP in BCR-ABL1-positive cells may be responsible for the alternative error-prone NHEJ pathway observed in CML [105–108]. HRR, in turn, is abnormally stimulated to the detriment of its fidelity in CML due to BCR-ABL1-mediated overexpression and activation of RAD51, which promotes erroneous HRR when overstimulated [109–111]. Incorrect DNA repair can be prevented by mismatch repair, but BCR-ABL1 inhibits this process by abrogating heterodimerization of the mismatch repair proteins MLH1 and PMS2 [112]. SSA is a rare and unfaithful mechanism of DSB repair and BCR-ABL1 stimulates SSA activity in a dose-dependent manner and through up-regulation of RBBP8/CtIP [108, 113].
NER activity status in CML is controversial. In initial reports, BCR-ABL1 was found to interfere with NER proteins reducing NER activity [114, 115]. It was later suggested that P210BCR-ABL induced NER in myeloid but repressed it in lymphoid cell lines [116]. However, more recent findings reported no difference in NER activity between lymphoid and myeloid CML cell lines, and a BCR-ABL1 kinase-dependent increase in NER activity in CML cell lines [117].
Expression of BCR-ABL1 is also associated with upregulation of DNA polymerase β [118, 119], an enzyme involved in HRR, NER and base excision repair (BER) [120–122]. Due to its low-fidelity DNA repair, it might be expected that DNA polymerase β overexpression contributes to CML genomic instability. Accumulation of point mutations in CML might also result from BCR-ABL1 inhibition of UNG, the most active glycosylase during BER, in both CML primary and BCR-ABL1-transformed cells [123].
4 Targeted Therapy
The knowledge on BCR-ABL1 structure and function that accumulated over the past 30 years set up the scene for the design of ‘molecularly targeted’ therapy for CML. Since the tyrosine kinase activity of BCR-ABL1 is essential for disease development, it was the most attractive target for designer therapy, although not the only one approached [124–132]. Undoubtedly, the advent of TKIs, which block or prevent BCR-ABL1 oncogenic signalling, has been so far the most exciting and successful therapeutic advance in CML.
4.1 First Generation TKI: Imatinib
Imatinib mesilate (IM) is a small chemical compound which competes with ATP for binding to its pocket in the BCR-ABL1 kinase domain (KD), thus blocking the BCR-ABL1 oncogenic signal [45]. IM inhibits the kinase activity of all ABL1- and ARG-containing proteins, the PDGFR family and the KIT receptor [133–135]. Such inhibition results in transcriptional modulation of various genes involved in the control of cell cycle, cell adhesion and cytoskeletal organization, leading the Ph + cell to an apoptotic death [44]. In addition, IM inhibits growth of CML primary cells and cell lines in vitro and in vivo [7, 8, 136].
In a phase I trial, IM showed little toxicity but proved to be highly effective [137]. The 8-year follow-up of the phase III IRIS trial reported an overall free survival rate (excluding discontinuation of therapy) of 85 % for CP CML patients under IM as first-line therapy, with 86 % of major molecular responses (MMR) [138]. In contrast, most of the responses of patients in BC are short-lived with very low (12–17 %) cytogenetic responses and median survival of 6.5–10 months [20].
4.1.1 Resistance to IM
While the efficacy of IM is unquestioned, resistance to TKIs became a pressing challenge in CML treatment. The persistence of minimal residual disease and, more worryingly, the development of refractoriness to single drug therapy, have dampened the initial enthusiasm. At the 8-year follow-up on the IRIS study, only 55 % of patients remained on IM therapy, and in 16 % of those who discontinued this was due to unsatisfactory therapeutic outcome [138]. Other studies have reported even higher resistance rates, varying from 12 to 50 % [19].
The definition of resistance can be based on its time of onset as primary resistance, i.e., failure to achieve a significant cytogenetic response, and secondary or acquired resistance, i.e., progressive reappearance of the leukaemic clone after an initial response to the drug. In addition, resistance can also be classified as BCR-ABL1-dependent and -independent. The first group encompasses the emergence of leukaemic clones with mutations in the BCR-ABL1 KD [139], overexpression of the BCR-ABL1 protein [140, 141] and amplification of the BCR-ABL1 oncogene [142, 143]. The mechanisms of BCR-ABL1-independent resistance include mostly defects in drug transport in and out of the leukaemic cells, and activation of oncogenic pathways downstream of BCR-ABL1 [144].
The most common mechanism for acquired IM resistance is through the development of point mutations in the ABL1 KD of BCR-ABL1 [144]. These mutations are not induced by the drug but, rather, confer resistance to rare populations of progenitors which are selected due to their capacity to survive and expand in the presence of the drug.
Mutations can be broadly categorized into four groups: (i) those which directly impair IM binding; (ii) those within the ATP binding site; (iii) those within the activation loop; and (iv) those within the catalytic domain (Fig. 7.6).

Incidence of reported mutations within the kinase domain by percentage of total. The seven most frequent mutations are depicted in red and the following eight in blue; mutations shown in green have been reported in less than 2 % of clinical resistance cases. Specific regions of the kinase domain are indicated as P-loop or ATP binding site (P), imatinib binding site (B), catalytic domain (C) and activation loop (A). Also shown as SH2 and SH3 are the contact regions with SH2 and SH3 domain-containing proteins (Data based on [145, 146])
The substitution of isoleucine for threonine at position 315 of ABL1, or T315I, reduces the affinity for the drug by preventing the formation of a hydrogen bond between T315 and the secondary amino group of IM, and by sterically preventing the binding of IM [143]. Another amino acid that makes contact with IM is phenylalanine 317, and its mutation to leucine (F317L) also leads to resistance.
The ATP-binding loop (or P-loop) domain spans amino acids 248–256 [147]. Mutations in this domain are the most common and modify the flexibility of the P-loop destabilizing the conformation required for IM binding [148]. The most frequent of such mutations are substitutions at G250, Q252, Y253 and E255. An additional feature of clinical relevance is that IM-treated patients who harbour P-loop mutations have a worse prognosis than those with non-P-loop mutations [149–152].
The activation loop of the ABL1 kinase begins at amino acid 381 and can adopt a closed (inactive) or an open (active) conformation. IM forces ABL1 into the inactive conformation and is incapable of binding to the active one [153]. Mutations in the activation loop may disturb the energetic balance required to stabilize the closed conformation of the loop and, thus, favour the open conformation resulting in IM resistance [148].
Finally, some substitutions cluster in the catalytic domain (amino acids 350–363), a region that has a close topologic relation to the base of the activation loop. Therefore, mutations in this region can also influence IM binding [148].
The degree of IM resistance varies between mutations and is predicted to affect prognosis and response to treatment. Thus far, more than 100 different point mutations leading to a substitution of approximately 50 amino acids in the ABL1 KD have been identified in patients resistant to IM and this number is likely to increase with more sensitive methods of detection [154].
4.2 Second and Third Generation TKIs
4.2.1 Dasatinib
Dasatinib is a dual SRC/ABL1 kinase inhibitor that also binds to the ATP-binding site, but extends in the opposite direction from IM. It binds the inactive and active conformation of the ABL1 KD, has a greater affinity to this domain, and is more potent than IM [155]. In clinical trials, dasatinib showed significantly higher MMR and overall survival rates than IM for CP CML patients [156, 157]. CML patients in advanced phase also showed improved complete cytogenetic response (CCyR) rates under dasatinib; however, those are still low, at 32 % [158].
Dasatinib requires fewer contact points with ABL1 residues; therefore, it is active against several IM-associated mutations. The T315I and F317L mutations, however, lead to the least favourable responses [159–162]. Due to a direct interaction between F317 and dasatinib, several amino acid substitutions in this position result in dasatinib-resistant mutants, such as F317L, F317V, F317I, and F317S [150, 163]. In a phase III study of dasatinib in CP CML patients, development of mutations T315I, F317L, V299L, and, rarely, E255K correlated with loss of response [19].
4.2.2 Nilotinib
Nilotinib was designed as a chemical modification of IM and is 10–50 times more potent [164]. It also inhibits the activity of ARG, KIT, and PDGFRA and PDGFRB, but not SRC kinase. CP patients treated with nilotinib showed higher CCyR, MMR and overall survival rates, and lower transformation events than those under IM [19, 165, 166]. Moreover, in trials for patients with advanced CML, nilotinib treatment also resulted in higher CCyR rates than IM [19].
Similar to dasatinib, nilotinib inhibits the in vitro proliferation of most of the clinically relevant BCR-ABL1 mutants, except for the T315I [167–170]. Likewise, the degree of sensitivity/resistance to nilotinib also varies for individual mutants. Accordingly, the mutations T315I, E255K/V, F359C/V, and Y253H have shown association with lack of CCyR to nilotinib, followed by disease progression [170].
4.2.3 Bosutinib
Bosutinib is a potent second generation TKI that, like dasatinib, also has SRC inhibitory activity. In a phase III trial it showed higher MMR rates, and lower disease progression than IM [171]. Bosutinib also induces CCyR, albeit at a low rate (23 %), in patients resistant to IM or to either nilotinib or dasatinib [172]. At present, bosutinib is registered in many countries as a second- or third-line therapeutic agent.
4.2.4 Ponatinib
Ponatinib is a third generation TKI rationally designed to inhibit the T315I mutation, whilst still keeping activity against the unmutated and the majority of other BCR-ABL1 mutants. It also inhibits VEGFA, FGF, KIT and SRC kinases [173]. In a clinical trial of patients resistant or intolerant to nilotinib or dasatinib, or with the T315I mutation, ponatinib treatment caused CCyR and MMR in 46 % and 34 % of CP patients, respectively [174]. Moreover, 24 % of AP patients achieved CCyR and 16 % MMR, while only 18 % of BC patients experienced CCyR. Ponatinib’s toxicity profile, however, can be a major drawback, since 5 % of patients suffered pancreatitis, and there was a significant association between ponatinib treatment and cardiovascular, cerebrovascular, and peripheral vascular events [174]. As a consequence, its indication is currently restricted to patients with a T315I mutation or for whom no other TKI is indicated.
4.2.5 Rebastinib
Rebastinib (or DCC-2036) is a switch pocket TKI rationally designed to induce an inactive conformation on BCR-ABL1. It retains full activity against the majority of BCR-ABL1 mutations, including T315I, but five P-loop mutants, G250E, Q252H, Y253H and E255K/V, in addition to F359I, have shown resistance to it [175, 176]. Preliminary results from a phase I trial (NCT00827138; www.clinicaltrials.gov) suggest it has anti-leukaemic activity in patients intolerant/refractory to other TKIs or positive for T315I [177], but a Phase II trial is not presently planned.
5 LSC as a Therapeutic Target in CML
Despite the success of TKI treatment, the persistence of minimal residual disease or the recurrence of disease upon cessation of therapy in most patients with undetectable BCR-ABL1, indicate that LSC persist even when response to treatment is optimal [178, 179].
Although primitive CML cells were shown to stop proliferating and enter a reversible cell cycle arrest upon IM treatment, they are resistant to TKI-induced apoptosis both in vitro and in vivo, even when BCR-ABL1 signalling is effectively inhibited [180–188]. These results suggest that the LSCs are capable of surviving independently of BCR-ABL1.
It has been suggested that the LSC quiescent state was responsible for their resistance to IM. In fact, stimulating quiescent LSCs to enter the cell cycle with CSF3/G-CSF reduces the overall non-cycling cell population in vitro [189, 190]; however, in clinical practice, this does not impact on disease outcome [191]. Accordingly, even the cycling primitive CML cells resist apoptosis due to BCR-ABL1 inhibition [187]. Therefore, LSCs capacity to survive BCR-ABL1 inhibition may be mediated by their ability to escape apoptosis and/or to self-renew, or by interactions with the BM stroma [192]. Indeed, the resistance of primitive LSC is not confined to apoptosis induced by TKI but apparently extends to multiple pro-apoptotic agents, such as cytosine arabinoside and arsenic trioxide [183].
From the self-renewal aspect the Wnt/CTNNB1 and Hedgehog (Hh) pathways are altered in CML and are potential targets [193, 194]. For instance, knockout or pharmacological inhibition of either CTNNB1 or SMO in combination with TKI efficiently reduces LSC numbers in vivo and delays disease relapse [195, 196]. These data support the hypothesis that targeting self-renewal is effective to eradicate LSCs and is the basis of ongoing clinical trials with inhibitors of these pathways (NCT01606579, NCT01357655, NCT01218477, NCT01456676; www.clinicaltrials.gov).
It has been proposed that sequestration of LSCs in the BM niche induces the phenotype of environment-mediated drug resistance (EMDR) [197]. The mechanisms so far identified for EMDR include interaction of β1 integrins and CD44 with fibronectin on BM stromal cells, degradation of BCL2L11/BIM due to β1 integrin-mediated cell adhesion, activation of AKT1 through integrin-linked kinase, activation of JAK/STAT and HIF1A pathways, increase in STAT3 phosphorylation and subsequent expression of anti-apoptotic proteins, and interactions of CXCR4 in CML cells with extra cellular-matrix components and BM stromal cells [197, 198]. Special focus on CXCR4 as a possible drug target in CML has produced contradictory results with two studies showing that combination of CXCR4 antagonists with TKIs reduced leukaemia burden on CML mouse models [199, 200], while a third showed that combination of plerixafor with dasatinib had no advantage over dasatinib alone [201].
On a different approach, a farnesyltransferase inhibitor, BMS-214662, was found to selectively kill quiescent and dividing CML stem/progenitor cells in vitro, and its effect was enhanced when combined with either TKIs or a MEK inhibitor, PD184352, making it a promising agent for clinical development [202, 203].
Recent reports have also focused on manipulating the PP2A tumour suppressor activity to target LSCs. PP2A reactivation had been shown to effectively kill CML lines and primary cells from BC and both TKI-sensitive and -resistant patients [48, 204, 205]. Recently, the same group demonstrated that reactivating PP2A can erradicate quiescent LSCs, but not normal HSCs, through inhibition of the BCR-ABL1-JAK2-CTNNB1 signalling axis [206].
Other potential molecular targets in the CML LSC are listed on Table 7.1.
6 Conclusion
The knowledge about the biology of CML increased exponentially since the Ph chromosome was first described. Even though this knowledge has led to the development of TKIs, which revolutionized CML treatment, there are still challenges to be overcome. Progression to BC, due to either primary failure to respond to a TKI or ‘acquired’ resistance, is still a major problem, since this aggressive disease stage is refractory to all types of available therapy. In addition, persistence of minimal residual disease in the majority of patients means they will have to continue under TKI therapy indefinitely. This raises two main problems, i.e., the risk that these patients develop resistance, which can then cause progression to BC, and the financial burden to families and/or Government’s medical systems, which will have to provide lifelong expensive treatment for those patients. Therefore, there is still much to be investigated and learned about this apparently benign leukaemia before we can achieve the final goal of a cure for the great majority of patients.
References
Piller G (2001) Leukaemia – a brief historical review from ancient times to 1950. Br J Haematol 112:282–292
Nowell PC, Hungerford DA (1960) A minute chromosome in human chronic granulocytic leukemia. Science 132:1497
Rowley JD (1973) Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 243:290–293
Shtivelman E, Lifshitz B, Gale RP, Canaani E (1985) Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 315:550–554
Lugo TG, Pendergast AM, Muller AJ, Witte ON (1990) Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 247:1079–1082
Daley GQ, Van Etten RA, Baltimore D (1990) Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247:824–830
Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM et al (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2:561–566
Deininger MWN, Goldman JM, Lydon NB, Melo JV (1997) The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL positive cells. Blood 90:3691–3698
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E et al (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 344:1031–1037
Hoglund M, Sandin F, Hellstrom K, Bjoreman M, Bjorkholm M et al (2013) Tyrosine kinase inhibitor usage, treatment outcome, and prognostic scores in CML: report from the population-based Swedish CML registry. Blood 122:1284–1292
Mendizabal AM, Garcia-Gonzalez P, Levine PH (2013) Regional variations in age at diagnosis and overall survival among patients with chronic myeloid leukemia from low and middle income countries. Cancer Epidemiol 37:247–254
Bizzozero OJ Jr, Johnson KG, Ciocco A (1966) Radiation-related leukemia in Hiroshima and Nagasaki, 1946–1964. I. Distribution, incidence and appearance time. N Engl J Med 274:1095–1101
Landgren O, Goldin LR, Kristinsson SY, Helgadottir EA, Samuelsson J et al (2008) Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood 112:2199–2204
Hehlmann R, Heimpel H, Hasford J, Kolb HJ, Pralle H et al (1993) Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. The German CML Study Group. Blood 82:398–407
Savage DG, Szydlo RM, Goldman JM (1997) Clinical features at diagnosis in 430 patients with chronic myeloid leukaemia seen at a referral centre over a 16-year period. Br J Haematol 96:111–116
Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB et al (2002) Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood 99:3530–3539
Vardiman JW, Melo JV, Baccarani M, Thiele J (2008) Chronic myelogenous leukaemia, BCR-ABL1 positive. In: Swerdlow SH, Campo E, Harris NL (eds) WHO classification of tumours of heaematopoietic and lymphoid tissues. International Agency for Research in Cancer (IARC), Lyon, pp 32–37
Wadhwa J, Szydlo RM, Apperley JF, Chase A, Bua M et al (2002) Factors affecting duration of survival after onset of blastic transformation of chronic myeloid leukemia. Blood 99:2304–2309
Radich JP (2012) Measuring response to BCR-ABL inhibitors in chronic myeloid leukemia. Cancer 118:300–311
Hehlmann R (2012) How I treat CML blast crisis. Blood 120:737–747
Melo JV, Gordon DE, Cross NC, Goldman JM (1993) The ABL-BCR fusion gene is expressed in chronic myeloid leukemia. Blood 81:158–165
Melo JV (1996) The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype [editorial]. Blood 88:2375–2384
Melo JV, Myint H, Galton DA, Goldman JM (1994) P190BCR-ABL chronic myeloid leukaemia: the missing link with chronic myelomonocytic leukaemia? Leukemia 8:208–211
Ravandi F, Cortes J, AlBitar M, Arlinghaus R, Qiang GJ et al (1999) Chronic myelogenous leukaemia with p185(BCR/ABL) expression: characteristics and clinical significance. Br J Haematol 107:581–586
Pane F, Frigeri F, Sindona M, Luciano L, Ferrara F et al (1996) Neutrophilic-chronic myeloid leukemia (CML-N): a distinct disease with a specific molecular marker (BCR/ABL with C3/A2 junction). Blood 88:2410–2414
Gotlib J, Maxson JE, George TI, Tyner JW (2013) The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood 122:1707–1711
Li S, Ilaria RL Jr, Million RP, Daley GQ, Van Etten RA (1999) The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med 189:1399–1412
Deininger MW, Bose S, Gora TJ, Yan XH, Goldman JM et al (1998) Selective induction of leukemia-associated fusion genes by high-dose ionizing radiation. Cancer Res 58:421–425
Neves H, Ramos C, da Silva MG, Parreira A, Parreira L (1999) The nuclear topography of ABL, BCR, PML, and RARalpha genes: evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation [see comments]. Blood 93:1197–1207
Saglio G, Storlazzi CT, Giugliano E, Surace C, Anelli L et al (2002) A 76-kb duplicon maps close to the BCR gene on chromosome 22 and the ABL gene on chromosome 9: possible involvement in the genesis of the Philadelphia chromosome translocation. Proc Natl Acad Sci U S A 99:9882–9887
Sinclair PB, Nacheva EP, Leversha M, Telford N, Chang J et al (2000) Large deletions at the t(9;22) breakpoint are common and may identify a poor-prognosis subgroup of patients with chronic myeloid leukemia. Blood 95:738–743
Huntly BJ, Reid AG, Bench AJ, Campbell LJ, Telford N et al (2001) Deletions of the derivative chromosome 9 occur at the time of the Philadelphia translocation and provide a powerful and independent prognostic indicator in chronic myeloid leukemia. Blood 98:1732–1738
de la Fuente J, Merx K, Steer EJ, Muller M, Szydlo RM et al (2001) ABL-BCR expression does not correlate with deletions on the derivative chromosome 9 or survival in chronic myeloid leukemia. Blood 98:2879–2880
Quintas-Cardama A, Kantarjian H, Talpaz M, O’Brien S, Garcia-Manero G et al (2005) Imatinib mesylate therapy may overcome the poor prognostic significance of deletions of derivative chromosome 9 in patients with chronic myelogenous leukemia. Blood 105:2281–2286
Castagnetti F, Testoni N, Luatti S, Marzocchi G, Mancini M et al (2010) Deletions of the derivative chromosome 9 do not influence the response and the outcome of chronic myeloid leukemia in early chronic phase treated with imatinib mesylate: GIMEMA CML Working Party analysis. J Clin Oncol 28:2748–2754
Fialkow PJ, Martin PJ, Najfeld V, Penfold GK, Jacobson RJ et al (1981) Evidence for a multistep pathogenesis of chronic myelogenous leukemia. Blood 58:158–163
Biernaux C, Sels A, Huez G, Stryckmans P (1996) Very low level of major BCR-ABL expression in blood of some healthy individuals. Bone Marrow Transplant 17(Suppl 3):S45–S47
Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV (1998) The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood 92:3362–3367
Pear WS, Miller JP, Xu L, Pui JC, Soffer B et al (1998) Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 92:3780–3792
Zhang X, Ren R (1998) Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood 92:3829–3840
McWhirter JR, Galasso DL, Wang JY (1993) A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol 13:7587–7595
Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z et al (1993) BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 75:175–185
Pendergast AM, Muller AJ, Havlik MH, Maru Y, Witte ON (1991) BCR sequences essential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non- phosphotyrosine-dependent manner. Cell 66:161–171
Deininger MW, Vieira S, Mendiola R, Schultheis B, Goldman JM et al (2000) BCR-ABL tyrosine kinase activity regulates the expression of multiple genes implicated in the pathogenesis of chronic myeloid leukemia. Cancer Res 60:2049–2055
Druker BJ (2008) Translation of the Philadelphia chromosome into therapy for CML. Blood 112:4808–4817
Tauchi T, Feng GS, Shen R, Song HY, Donner D et al (1994) SH2-containing phosphotyrosine phosphatase Syp is a target of p210bcr- abl tyrosine kinase. J Biol Chem 269:15381–15387
LaMontagne KR Jr, Flint AJ, Franza BR Jr, Pandergast AM, Tonks NK (1998) Protein tyrosine phosphatase 1B antagonizes signalling by oncoprotein tyrosine kinase p210 bcr-abl in vivo. Mol Cell Biol 18:2965–2975
Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW et al (2005) The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 8:355–368
Juan WC, Ong ST (2012) The role of protein phosphorylation in therapy resistance and disease progression in chronic myelogenous leukemia. Prog Mol Biol Transl Sci 106:107–142
Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD et al (1994) Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J Biol Chem 269:22925–22928
Pelicci G, Lanfrancone L, Salcini AE, Romano A, Mele S et al (1995) Constitutive phosphorylation of Shc proteins in human tumors. Oncogene 11:899–907
Cahill MA, Janknecht R, Nordheim A (1996) Signalling pathways: jack of all cascades. Curr Biol 6:16–19
Skorski T, Wlodarski P, Daheron L, Salomoni P, Nieborowska-Skorska M et al (1998) BCR/ABL-mediated leukemogenesis requires the activity of the small GTP- binding protein Rac. Proc Natl Acad Sci U S A 95:11858–11862
Raitano AB, Halpern JR, Hambuch TM, Sawyers CL (1995) The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci U S A 92:11746–11750
Yang MY, Liu TC, Chang JG, Lin PM, Lin SF (2003) JunB gene expression is inactivated by methylation in chronic myeloid leukemia. Blood 101:3205–3211
Thomas EK, Cancelas JA, Zheng Y, Williams DA (2008) Rac GTPases as key regulators of p210-BCR-ABL-dependent leukemogenesis. Leukemia 22:898–904
Thomas EK, Cancelas JA, Chae HD, Cox AD, Keller PJ et al (2007) Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell 12:467–478
Ilaria RL Jr, Van Etten RA (1996) P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem 271:31704–31710
Chai SK, Nichols GL, Rothman P (1997) Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol 159:4720–4728
Nieborowska-Skorska M, Wasik MA, Slupianek A, Salomoni P, Kitamura T et al (1999) Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J Exp Med 189:1229–1242
Horita M, Andreu EJ, Benito A, Arbona C, Sanz C et al (2000) Blockade of the bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of bcl-x(L). J Exp Med 191:977–984
Quintas-Cardama A, Cortes J (2009) Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 113:1619–1630
Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP et al (2000) Stat5a/b contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcr/abl-induced transformation are independent of stat5. Blood 96:2277–2283
Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W et al (2006) Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 107:4898–4906
Kantarjian HM, Giles F, Quintas-Cardama A, Cortes J (2007) Important therapeutic targets in chronic myelogenous leukemia. Clin Cancer Res 13:1089–1097
Sattler M, Salgia R, Okuda K, Uemura N, Durstin MA et al (1996) The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3′ kinase pathway. Oncogene 12:839–846
Shortt J, Johnstone RW (2012) Oncogenes in cell survival and cell death. Cold Spring Harb Perspect Biol 4:a009829
Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen SC et al (1995) Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood 86:726–736
Middleton MK, Zukas AM, Rubinstein T, Jacob M, Zhu P et al (2006) Identification of 12/15-lipoxygenase as a suppressor of myeloproliferative disease. J Exp Med 203:2529–2540
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278:687–689
Sawyers CL, Callahan W, Witte ON (1992) Dominant negative MYC blocks transformation by ABL oncogenes. Cell 70:901–910
Zou X, Rudchenko S, Wong K, Calame K (1997) Induction of c-myc transcription by the v-Abl tyrosine kinase requires Ras, Raf1, and cyclin-dependent kinases. Genes Dev 11:654–662
Johansson B, Fioretos T, Mitelman F (2002) Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol 107:76–94
Melo JV, Barnes DJ (2007) Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 7:441–453
Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C et al (2004) Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351:657–667
Zhao C, Blum J, Chen A, Kwon HY, Jung SH et al (2007) Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 12:528–541
Chen R, Hu T, Mahon GM, Tala I, Pannucci NL et al (2013) Ubiquitin-mediated interaction of p210 BCR/ABL with beta-catenin supports disease progression in a murine model for chronic myelogenous leukemia. Blood 122:2114–2124
Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G et al (2002) BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet 30:48–58
Neviani P (2014) Genetic events other than BCR-ABL1. Curr Hematol Malig Rep 9:24–32
Smith LT, Hohaus S, Gonzalez DA, Dziennis SE, Tenen DG (1996) PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood 88:1234–1247
Ferrari-Amorotti G, Keeshan K, Zattoni M, Guerzoni C, Iotti G et al (2006) Leukemogenesis induced by wild-type and STI571-resistant BCR/ABL is potently suppressed by C/EBPalpha. Blood 108:1353–1362
Guerzoni C, Bardini M, Mariani SA, Ferrari-Amorotti G, Neviani P et al (2006) Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood 107:4080–4089
Wagner K, Zhang P, Rosenbauer F, Drescher B, Kobayashi S et al (2006) Absence of the transcription factor CCAAT enhancer binding protein alpha results in loss of myeloid identity in bcr/abl-induced malignancy. Proc Natl Acad Sci U S A 103:6338–6343
Zhang SJ, Ma LY, Huang QH, Li G, Gu BW et al (2008) Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci U S A 105:2076–2081
Zhang SJ, Shi JY, Li JY (2009) GATA-2 L359 V mutation is exclusively associated with CML progression but not other hematological malignancies and GATA-2 P250A is a novel single nucleotide polymorphism. Leuk Res 33:1141–1143
Mitani K, Ogawa S, Tanaka T, Miyoshi H, Kurokawa M et al (1994) Generation of the AML1-EVI-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J 13:504–510
Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C et al (2011) A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9 % of cases. Leukemia 25:557–560
Zhao LJ, Wang YY, Li G, Ma LY, Xiong SM et al (2012) Functional features of RUNX1 mutants in acute transformation of chronic myeloid leukemia and their contribution to inducing murine full-blown leukemia. Blood 119:2873–2882
Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J et al (2002) Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 99:121–129
Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J et al (2008) BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453:110–114
Suzuki K, Ono R, Ohishi K, Masuya M, Kataoka I et al (2012) IKAROS isoform 6 enhances BCR-ABL1-mediated proliferation of human CD34+ hematopoietic cells on stromal cells. Int J Oncol 40:53–62
Casolari DA, Makri M, Yoshida C, Muto A, Igarashi K et al (2013) Transcriptional suppression of BACH2 by the Bcr-Abl oncoprotein is mediated by PAX5. Leukemia 27:409–415
Skorski T (2012) Genetic mechanisms of chronic myeloid leukemia blastic transformation. Curr Hematol Malig Rep 7:87–93
Perrotti D, Jamieson C, Goldman J, Skorski T (2010) Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest 120:2254–2264
Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M et al (2003) An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11:203–213
Dierov J, Dierova R, Carroll M (2004) BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell 5:275–285
Nieborowska-Skorska M, Stoklosa T, Datta M, Czechowska A, Rink L et al (2006) ATR-Chk1 axis protects BCR/ABL leukemia cells from the lethal effect of DNA double-strand breaks. Cell Cycle 5:994–1000
Kurosu T, Nagao T, Wu N, Oshikawa G, Miura O (2013) Inhibition of the PI3K/Akt/GSK3 pathway downstream of BCR/ABL, Jak2-V617F, or FLT3-ITD downregulates DNA damage-induced Chk1 activation as well as G2/M arrest and prominently enhances induction of apoptosis. PLoS One 8:e79478
Foulkes WD, Shuen AY (2013) In brief: BRCA1 and BRCA2. J Pathol 230:347–349
Deutsch E, Jarrousse S, Buet D, Dugray A, Bonnet ML et al (2003) Down-regulation of BRCA1 in BCR-ABL-expressing hematopoietic cells. Blood 101:4583–4588
Valeri A, Alonso-Ferrero ME, Rio P, Pujol MR, Casado JA et al (2010) Bcr/Abl interferes with the Fanconi anemia/BRCA pathway: implications in the chromosomal instability of chronic myeloid leukemia cells. PLoS ONE 5:e15525
Feng Z, Scott SP, Bussen W, Sharma GG, Guo G et al (2011) Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci U S A 108:686–691
Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA et al (2013) Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 122:1293–1304
Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T et al (2004) BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 104:3746–3753
Deutsch E, Dugray A, AbdulKarim B, Marangoni E, Maggiorella L et al (2001) BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 97:2084–2090
Brady N, Gaymes TJ, Cheung M, Mufti GJ, Rassool FV (2003) Increased error-prone NHEJ activity in myeloid leukemias is associated with DNA damage at sites that recruit key nonhomologous end-joining proteins. Cancer Res 63:1798–1805
Sallmyr A, Tomkinson AE, Rassool FV (2008) Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood 112:1413–1423
Salles D, Mencalha AL, Ireno IC, Wiesmuller L, Abdelhay E (2011) BCR-ABL stimulates mutagenic homologous DNA double-strand break repair via the DNA-end-processing factor CtIP. Carcinogenesis 32:27–34
Dubrez L, Eymin B, Sordet O, Droin N, Turhan AG et al (1998) BCR-ABL delays apoptosis upstream of procaspase-3 activation. Blood 91:2415–2422
Slupianek A, Schmutte C, Tombline G, Nieborowska-Skorska M, Hoser G et al (2001) BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell 8:795–806
Slupianek A, Dasgupta Y, Ren SY, Gurdek E, Donlin M et al (2011) Targeting RAD51 phosphotyrosine-315 to prevent unfaithful recombination repair in BCR-ABL1 leukemia. Blood 118:1062–1068
Stoklosa T, Poplawski T, Koptyra M, Nieborowska-Skorska M, Basak G et al (2008) BCR/ABL inhibits mismatch repair to protect from apoptosis and induce point mutations. Cancer Res 68:2576–2580
Cramer K, Nieborowska-Skorska M, Koptyra M, Slupianek A, Penserga ET et al (2008) BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res 68:6884–6888
Takeda N, Shibuya M, Maru Y (1999) The BCR-ABL oncoprotein potentially interacts with the xeroderma pigmentosum group B protein. Proc Natl Acad Sci U S A 96:203–207
Maru Y, Bergmann E, Coin F, Egly JM, Shibuya M (2001) TFIIH functions are altered by the P210BCR-ABL oncoprotein produced on the Philadelphia chromosome. Mutat Res 483:83–88
Canitrot Y, Falinski R, Louat T, Laurent G, Cazaux C et al (2003) p210 BCR/ABL kinase regulates nucleotide excision repair (NER) and resistance to UV radiation. Blood 102:2632–2637
Sliwinski T, Czechowska A, Szemraj J, Morawiec Z, Skorski T et al (2008) STI571 reduces NER activity in BCR/ABL-expressing cells. Mutat Res 654:162–167
Canitrot Y, Lautier D, Laurent G, Frechet M, Ahmed A et al (1999) Mutator phenotype of BCR–ABL transfected Ba/F3 cell lines and its association with enhanced expression of DNA polymerase beta. Oncogene 18:2676–2680
Canitrot Y, Laurent G, Astarie-Dequeker C, Bordier C, Cazaux C et al (2006) Enhanced expression and activity of DNA polymerase beta in chronic myelogenous leukemia. Anticancer Res 26:523–525
Canitrot Y, Hoffmann JS, Calsou P, Hayakawa H, Salles B et al (2000) Nucleotide excision repair DNA synthesis by excess DNA polymerase beta: a potential source of genetic instability in cancer cells. FASEB J 14:1765–1774
Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366–374
Canitrot Y, Capp JP, Puget N, Bieth A, Lopez B et al (2004) DNA polymerase beta overexpression stimulates the Rad51-dependent homologous recombination in mammalian cells. Nucleic Acids Res 32:5104–5112
Slupianek A, Falinski R, Znojek P, Stoklosa T, Flis S et al (2013) BCR-ABL1 kinase inhibits uracil DNA glycosylase UNG2 to enhance oxidative DNA damage and stimulate genomic instability. Leukemia 27:629–634
Quintas-Cardama A (2008) Experimental non-ATP-competitive therapies for chronic myelogenous leukemia. Leukemia 22:932–940
Rohon P (2012) Biological therapy and the immune system in patients with chronic myeloid leukemia. Int J Hematol 96:1–9
Kaymaz BT, Selvi N, Gokbulut AA, Aktan C, Gunduz C et al (2013) Suppression of STAT5A and STAT5B chronic myeloid leukemia cells via siRNA and antisense-oligonucleotide applications with the induction of apoptosis. Am J Blood Res 3:58–70
Kaymaz BT, Selvi N, Gunduz C, Aktan C, Dalmizrak A et al (2013) Repression of STAT3, STAT5A, and STAT5B expressions in chronic myelogenous leukemia cell line K-562 with unmodified or chemically modified siRNAs and induction of apoptosis. Ann Hematol 92:151–162
Koldehoff M, Zakrzewski JL, Beelen DW, Elmaagacli AH (2013) Additive antileukemia effects by GFI1B- and BCR-ABL-specific siRNA in advanced phase chronic myeloid leukemic cells. Cancer Gene Ther 20:421–427
Liu Y, Song Y, Ma W, Zheng W, Yin H (2013) Decreased microRNA-30a levels are associated with enhanced ABL1 and BCR-ABL1 expression in chronic myeloid leukemia. Leuk Res 37:349–356
Li Y, Wang H, Tao K, Xiao Q, Huang Z et al (2013) miR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Exp Cell Res 319:1094–1101
Schurch CM, Riether C, Ochsenbein AF (2013) Dendritic cell-based immunotherapy for myeloid leukemias. Front Immunol 4:496
Valencia-Serna J, Gul-Uludag H, Mahdipoor P, Jiang X, Uludag H (2013) Investigating siRNA delivery to chronic myeloid leukemia K562 cells with lipophilic polymers for therapeutic BCR-ABL down-regulation. J Control Release 172:495–503
Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M et al (1996) Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res 56:100–104
Okuda K, Weisberg E, Gilliland DG, Griffin JD (2001) ARG tyrosine kinase activity is inhibited by STI571. Blood 97:2440–2448
Savage DG, Antman KH (2002) Imatinib mesylate – a new oral targeted therapy. N Engl J Med 346:683–693
le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E et al (1999) In vivo eradication of human BCR/ABL positive leukemia cells with an ABL kinase inhibitor. J Natl Cancer Inst 91:163–168
Druker BJ, Talpaz M, Resta D, Peng B, Buchdunger E et al (1999) Clinical efficacy and safety of an Abl specific tyrosine kinase inhibitor as targeted therapy for chronic myelogenous leukemia [abstract]. Blood 94:368a
Deininger M, O’Brien SG, Guilhot F, Goldman JM, Hochhaus A et al (2009) International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib [abstract]. Blood (ASH Annual Meeting Abstracts) 114:1126
Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R et al (2003) Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol 4:75–85
Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J et al (2000) Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood 96:1070–1079
Weisberg E, Griffin JD (2000) Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines. Blood 95:3498–3505
le Coutre P, Tassi E, Varella-Garcia M, Barni R, Mologni L et al (2000) Induction of resistance to the Abelson inhibitor STI571 in human leukemic cells through gene amplification. Blood 95:1758–1766
Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R et al (2001) Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293:876–880
Diamond JM, Melo JV (2011) Mechanisms of resistance to BCR-ABL kinase inhibitors. Leuk Lymphoma 52(Suppl 1):12–22
Apperley JF (2007) Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol 8:1018–1029
Soverini S, Branford S, Nicolini FE, Talpaz M, Deininger MW et al (2014) Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leuk Res 38:10–20
Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC et al (2002) Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 16:2190–2196
Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL et al (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2:117–125
Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A et al (2003) Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 102:276–283
Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F et al (2006) Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res 12:7374–7379
Nicolini FE, Corm S, Le QH, Sorel N, Hayette S et al (2006) Mutation status and clinical outcome of 89 imatinib mesylate-resistant chronic myelogenous leukemia patients: a retrospective analysis from the French intergroup of CML (Fi(phi)-LMC GROUP). Leukemia 20:1061–1066
Khorashad JS, Milojkovic D, Mehta P, Anand M, Ghorashian S et al (2008) In vivo kinetics of kinase domain mutations in CML patients treated with dasatinib after failing imatinib. Blood 111:2378–2381
Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR et al (2002) Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res 62:4236–4243
Jabbour E, Soverini S (2009) Understanding the role of mutations in therapeutic decision making for chronic myeloid leukemia. Semin Hematol 46:S22–S26
Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC et al (2004) Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem 47:6658–6661
Cortes JE, Jones D, O’Brien S, Jabbour E, Ravandi F et al (2010) Results of dasatinib therapy in patients with early chronic-phase chronic myeloid leukemia. J Clin Oncol 28:398–404
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S et al (2010) Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 362:2260–2270
Kantarjian H, Cortes J, Kim DW, Dorlhiac-Llacer P, Pasquini R et al (2009) Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood 113:6322–6329
Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J et al (2007) Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood 110:4005–4011
Jabbour E, Kantarjian HM, Jones D, Reddy N, O’Brien S et al (2008) Characteristics and outcome of chronic myeloid leukemia patients with F317L BCR-ABL kinase domain mutation after therapy with tyrosine kinase inhibitors. Blood 112:4839–4842
Jabbour E, Kantarjian H, Jones D, Breeden M, Garcia-Manero G et al (2008) Characteristics and outcomes of patients with chronic myeloid leukemia and T315I mutation following failure of imatinib mesylate therapy. Blood 112:53–55
Muller MC, Cortes JE, Kim DW, Druker BJ, Erben P et al (2009) Dasatinib treatment of chronic-phase chronic myeloid leukemia: analysis of responses according to preexisting BCR-ABL mutations. Blood 114:4944–4953
Tokarski JS, Newitt JA, Chang CY, Cheng JD, Wittekind M et al (2006) The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res 66:5790–5797
Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW et al (2005) Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7:129–141
Cortes JE, Jones D, O’Brien S, Jabbour E, Konopleva M et al (2010) Nilotinib as front-line treatment for patients with chronic myeloid leukemia in early chronic phase. J Clin Oncol 28:392–397
Larson RA, Hochhaus A, Hughes TP, Clark RE, Etienne G et al (2012) Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia 26:2197–2203
Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S et al (2006) Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 354:2542–2551
Kantarjian H, O’Brien S, Talpaz M, Borthakur G, Ravandi F et al (2007) Outcome of patients with Philadelphia chromosome-positive chronic myelogenous leukemia post-imatinib mesylate failure. Cancer 109:1556–1560
le Coutre P, Ottmann OG, Giles F, Kim DW, Cortes J et al (2008) Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood 111:1834–1839
Hughes T, Saglio G, Branford S, Soverini S, Kim DW et al (2009) Impact of baseline BCR-ABL mutations on response to nilotinib in patients with chronic myeloid leukemia in chronic phase. J Clin Oncol 27:4204–4210
Cortes JE, Kim DW, Kantarjian HM, Brummendorf TH, Dyagil I et al (2012) Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol 30:3486–3492
Cortes JE, Kantarjian HM, Brummendorf TH, Kim DW, Turkina AG et al (2011) Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 118:4567–4576
O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM et al (2009) AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16:401–412
Cortes JE, Kim DW, Pinilla-Ibarz J, Le Coutre PD, Paquette R et al (2013) A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med 369:1783–1796
Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL et al (2011) Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell 19:556–568
Eide CA, Adrian LT, Tyner JW, Mac PM, Anderson DJ et al (2011) The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res 71:3189–3195
Cortes JE, Talpaz M, Kantarjian HM, Smith H, Bixby D et al (2011) A phase 1 study o DCC-2036, a novel oral inhibitor of BCR-ABL kinase, in patients with Philadelphia chromosome positive (Ph+) leukemias including patients with T315I mutation. [abstract]. Blood (ASH Annual Meeting Abstracts) 118:601
Melo JV, Ross DM (2011) Minimal residual disease and discontinuation of therapy in chronic myeloid leukemia: can we aim at a cure? Hematology. Am Soc Hematol Educ Program 2011:136–142
Kinstrie R, Copland M (2013) Targeting chronic myeloid leukemia stem cells. Curr Hematol Malig Rep 8:14–21
Holyoake T, Jiang X, Eaves C, Eaves A (1999) Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood 94:2056–2064
Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ et al (2002) Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 99:319–325
Holtz MS, Slovak ML, Zhang F, Sawyers CL, Forman SJ et al (2002) Imatinib mesylate (STI571) inhibits growth of primitive malignant progenitors in chronic myelogenous leukemia through reversal of abnormally increased proliferation. Blood 99:3792–3800
Holtz MS, Forman SJ, Nonproliferating BR, CML (2005) CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia 19:1034–1041
Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK et al (2006) Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood 107:4532–4539
Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL (2007) Nilotinib exerts equipotent anti-proliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 109:4016–4019
Konig H, Holtz M, Modi H, Manley P, Holyoake TL et al (2008) Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia 22:748–755
Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW et al (2011) Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest 121:396–409
Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S et al (2012) Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 119:1501–1510
Jorgensen HG, Copland M, Allan EK, Jiang X, Eaves A et al (2006) Intermittent exposure of primitive quiescent chronic myeloid leukemia cells to granulocyte-colony stimulating factor in vitro promotes their elimination by imatinib mesylate. Clin Cancer Res 12:626–633
Holtz M, Forman SJ, Bhatia R (2007) Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer Res 67:1113–1120
Drummond MW, Heaney N, Kaeda J, Nicolini FE, Clark RE et al (2009) A pilot study of continuous imatinib vs pulsed imatinib with or without G-CSF in CML patients who have achieved a complete cytogenetic response. Leukemia 23:1199–1201
Barnes DJ, Melo JV (2006) Primitive, quiescent and difficult to kill: the role of non-proliferating stem cells in chronic myeloid leukemia. Cell Cycle 5:2862–2866
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A et al (2009) Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458:776–779
Crews LA, Jamieson CH (2013) Selective elimination of leukemia stem cells: hitting a moving target. Cancer Lett 338:15–22
Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR et al (2008) Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 14:238–249
Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA et al (2012) Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 10:412–424
Kuroda J, Shimura Y, Yamamoto-Sugitani M, Sasaki N, Taniwaki M (2013) Multifaceted mechanisms for cell survival and drug targeting in chronic myelogenous leukemia. Curr Cancer Drug Targets 13:69–79
Li X, Miao H, Zhang Y, Li W, Li Z et al (2015) Bone marrow microenvironment confers imatinib resistance to chronic myelogenous leukemia and oroxylin A reverses the resistance by suppressing Stat3 pathway. Arch Toxicol 89:121–136
Weisberg E, Azab AK, Manley PW, Kung AL, Christie AL et al (2012) Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 26:985–990
Beider K, rash-Yahana M, Blaier O, Koren-Michowitz M, Abraham M et al (2014) Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Mol Cancer Ther 13:1155–1169
Agarwal A, Fleischman AG, Petersen CL, MacKenzie R, Luty S et al (2012) Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood 120:2658–2668
Copland M, Pellicano F, Richmond L, Allan EK, Hamilton A et al (2008) BMS-214662 potently induces apoptosis of chronic myeloid leukemia stem and progenitor cells and synergizes with tyrosine kinase inhibitors. Blood 111:2843–2853
Pellicano F, Simara P, Sinclair A, Helgason GV, Copland M et al (2011) The MEK inhibitor PD184352 enhances BMS-214662-induced apoptosis in CD34+ CML stem/progenitor cells. Leukemia 25:1159–1167
Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M et al (2007) FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest 117:2408–2421
Agarwal A, Mackenzie RJ, Pippa R, Eide CA, Oddo J et al (2014) Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res 20:2092–2103
Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ et al (2013) PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest 123:4144–4157
Chen Y, Hu Y, Zhang H, Peng C, Li S (2009) Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet 41:783–792
Chen Y, Hu Y, Michaels S, Segal D, Brown D et al (2009) Inhibitory effects of omacetaxine on leukemic stem cells and BCR-ABL-induced chronic myeloid leukemia and acute lymphoblastic leukemia in mice. Leukemia 23:1446–1454
Robert F, Carrier M, Rawe S, Chen S, Lowe S et al (2009) Altering chemosensitivity by modulating translation elongation. PLoS ONE 4:e5428
Goff DJ, Court R, Sadarangani A, Chun HJ, Barrett CL et al (2013) A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 12:316–328
Hurtz C, Hatzi K, Cerchietti L, Braig M, Park E et al (2011) BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med 208:2163–2174
Zhang H, Peng C, Hu Y, Li H, Sheng Z et al (2012) The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat Genet 44:861–871
Schurch C, Riether C, Matter MS, Tzankov A, Ochsenbein AF (2012) CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J Clin Invest 122:624–638
Krause DS, Lazarides K, von Andrian UH, Van Etten RA (2006) Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med 12:1175–1180
Goessling W, North TE, Loewer S, Lord AM, Lee S et al (2009) Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 136:1136–1147
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T et al (2010) TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463:676–680
Yamamoto-Sugitani M, Kuroda J, Ashihara E, Nagoshi H, Kobayashi T et al (2011) Galectin-3 (Gal-3) induced by leukemia microenvironment promotes drug resistance and bone marrow lodgment in chronic myelogenous leukemia. Proc Natl Acad Sci U S A 108:17468–17473
Reddiconto G, Toto C, Palama I, De Leo S, de Luca E et al (2012) Targeting of GSK3beta promotes imatinib-mediated apoptosis in quiescent CD34+ chronic myeloid leukemia progenitors, preserving normal stem cells. Blood 119:2335–2345
Wang Y, Liu Y, Malek SN, Zheng P, Liu Y (2011) Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 8:399–411
Zhang H, Li H, Xi HS, Li S (2012) HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 119:2595–2607
Peng C, Brain J, Hu Y, Goodrich A, Kong L et al (2007) Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood 110:678–685
Chen M, Gallipoli P, DeGeer D, Sloma I, Forrest DL et al (2013) Targeting primitive chronic myeloid leukemia cells by effective inhibition of a new AHI-1-BCR-ABL-JAK2 complex. J Natl Cancer Inst 105:405–423
Lim S, Saw TY, Zhang M, Janes MR, Nacro K et al (2013) Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci U S A 110:E2298–E2307
Chen Y, Sullivan C, Peng C, Shan Y, Hu Y et al (2011) A tumor suppressor function of the Msr1 gene in leukemia stem cells of chronic myeloid leukemia. Blood 118:390–400
Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G et al (2008) PML targeting eradicates quiescent leukaemia-initiating cells. Nature 453:1072–1078
Peng C, Chen Y, Yang Z, Zhang H, Osterby L et al (2010) PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood 115:626–635
Zhang H, Li H, Ho N, Li D, Li S (2012) Scd1 plays a tumor-suppressive role in survival of leukemia stem cells and the development of chronic myeloid leukemia. Mol Cell Biol 32:1776–1787
Pelletier SD, Hong DS, Hu Y, Liu Y, Li S (2004) Lack of the adhesion molecules P-selectin and intercellular adhesion molecule-1 accelerate the development of BCR/ABL-induced chronic myeloid leukemia-like myeloproliferative disease in mice. Blood 104:2163–2171
Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R et al (2005) Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A 102:9194–9199
Sullivan C, Chen Y, Shan Y, Hu Y, Peng C et al (2011) Functional ramifications for the loss of P-selectin expression on hematopoietic and leukemic stem cells. PLoS ONE 6:e26246
Oakley K, Han Y, Vishwakarma BA, Chu S, Bhatia R et al (2012) Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood 119:6099–6108
Yuan H, Wang Z, Li L, Zhang H, Modi H et al (2012) Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 119:1904–1914
Li L, Wang L, Li L, Wang Z, Ho Y et al (2012) Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 21:266–281
Zhang B, Irvine D, Ho YW, Buonamici M, Manley P et al (2010) Inhibition of chronic myeloid leukemia stem cells by the combination of the Hedgehog pathway inhibitor LDE225 with nilotinib [abstract]. Blood (ASH Annual Meeting Abstracts) 116:514
Jamieson C, Cortes JE, Oehler V, Baccaranni M, Kantarjian HM et al (2011) Phase 1 dose-escalation study of PF-04449913, an oral hedgehog (Hh) inhibitor, in patients with select hematologic malignancies [abstract]. Blood (ASH Annual Meeting Abstracts) 118:424
Katagiri S, Tauchi T, Okabe S, Minami Y, Kimura S et al (2013) Combination of ponatinib with Hedgehog antagonist vismodegib for therapy-resistant BCR-ABL1-positive leukemia. Clin Cancer Res 19:1422–1432
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Casolari, D.A., Melo, J.V. (2015). Chronic Myeloid Leukaemia. In: Rowley, J., Le Beau, M., Rabbitts, T. (eds) Chromosomal Translocations and Genome Rearrangements in Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-19983-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-19983-2_7
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-19982-5
Online ISBN: 978-3-319-19983-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)