
Abstract
Chronic myeloid leukaemia (CML) is a myeloproliferative disorder of the pluripotent stem cell. Understanding the molecular biology of CML began with the discovery of the Philadelphia (Ph) chromosome. This genomic abnormality is the result of a t(9;22) translocation, leading to the fusion oncogene, BCR-ABL1. The resulting protein is capable of hijacking a vast repertoire of cellular functions that drive myeloid hyperplasia, characteristic of the initial, chronic phase of the disease. However, the mechanisms of resistance and disease progression are less well defined. In this chapter, we explore the various pathways involved in the pathogenesis of CML and also the biological events underpinning progression to the more advanced stages of the disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
2.1 The Molecular Biology of CML
2.1.1 The t(9;22) Translocation and the BCR-ABL1 Gene
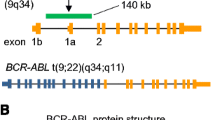
The Philadelphia (Ph) chromosome is formed by a reciprocal t(9;22)(q34;q11) translocation between the long arms of chromosomes 9 and 22, causing the juxtaposition of the BCR (breakpoint cluster region) and ABL1 (Abelson) genes. The BCR-ABL1 fusion gene consists of the 5′ end of the BCR gene and the 3′ end of the ABL1 gene (Fig. 2.1a). The location of the BCR and ABL1 genomic breakpoints is highly variable, but the recombination usually involves fusion of intron 13 or 14 of BCR with a 140-kilobase (kb) region of ABL1 surrounding exons 1b and 2 (Fig. 2.1a) [1, 2]. Regardless of the breakpoint location on the ABL1 gene, mRNA splicing gives rise to major BCR-ABL1 transcripts with e13a2 (BCR exon 13 and ABL1 exon 2) or e14a2 junctions, originally referred to as b2a2 and b3a2, respectively. Both transcripts result in the expression of a 210 kDa BCR-ABL1 protein with a 75-amino acid difference. In <2% of chronic phase (CP)-CML, ‘atypical’ transcripts can form when the breakpoint occurs between exons 1 and 2 (e1a2 transcript) or exons 19 and 20 (e19a2) of BCR. Alternative atypical transcripts have also been described although even less frequently [3, 4].

The gene and protein structure of BCR-ABL1. (a) The BCR-ABL1 fusion gene consists of the 5′ end of the BCR gene and the 3’end of ABL1. The location of the translocation usually involves fusion of intron 13 or 14 of BCR with a 140-kilobase (kb) region of ABL1 surrounding exons 1b and 2. Examples of the two BCR-ABL1 major mRNA isoforms are shown to highlight the BCR breakpoint variants. Depending on the breakpoint on the ABL1 gene, exons 1a and/or 1b may be included in the primary transcript but are always excluded from the mRNA because they lack a splice acceptor sequence. (b) The BCR-ABL1 protein contains the dimerisation or coiled-coil (C-C), the Ser-Thr kinase and the Rho/GEF domains of BCR, as well as the SH-domains, Proline-rich (PxxP) nuclear localisation signal (NLS), DNA-binding nuclear export signal (NES) and Actin-binding domains from ABL. The ATP-binding site in the SH1 domain is indicated, highlighting the site of traditional tyrosine kinase inhibitor binding. The tyrosine residues in the Ser/Thr and SH1 kinase domains have been highlighted with a Y. The diagrams in A and B are not to scale
There has been much debate regarding the consequence of a patient expressing either the e13a2 or e14a2 transcripts [2]. Before the tyrosine kinase inhibitor (TKI) era, most reports on large series refuted the importance of the BCR breakpoint [5,6,7,8]. However, a recent revival of this debate has found consistent evidence that patients with either the e14a2 transcript or both the e14a2 and e13a2 transcripts exhibit a higher platelet count, approximating 1.5 times higher than that in the e13a2 group [3, 9, 10]. Several laboratories have also found that patients with e14a2 transcripts achieve optimal ELN-defined responses more rapidly, including the deep molecular response which is mandated for consideration of a treatment-free remission attempt [3, 11,12,13]. Patients expressing both transcripts tend to track with the e14a2 group [3, 10]. More recently, the transcript type has been demonstrated to influence long-term treatment-free remission outcomes with e14a2 expression correlating with higher treatment-free remission success [13, 14]. Furthermore, despite the rarity, atypical BCR-ABL1 transcripts are generally associated with inferior outcomes [15, 16].
BCR-ABL1 has also been detected in healthy individuals with neither clinical nor laboratory evidence of CML [17,18,19,20,21]. With limited follow-up, these patients do not develop CML, likely due to these events being detected in terminally differentiated leukocytes as opposed to the leukaemic stem cell [19]. The absence of BCR-ABL1 in the pluripotent stem cell explains the lack of proliferative potential, corroborating that this genetic event must develop in the leukaemic stem cell for CML to develop.
2.1.2 Protein Structure
The 210 kDa BCR-ABL1 protein observed in CML contains more than ten protein domains (Fig. 2.1b). The SH1 tyrosine kinase region is the most studied due to its inherent role in CML pathogenesis and, consequently, the target for TKIs [22]. However other important regions include the SH2, SH3 and the N-terminal cap [23]. Myristoyl modification of the N-terminal cap permits the regulation of the kinase domain by SH2 and SH3 [24]. The fusion of BCR to ABL1 eventuates in loss of the N-terminal cap which results in constitutive activation of the SH1 kinase domain, inducing uncontrolled signal transduction and abnormal cellular proliferation [22]. TKIs, such as imatinib, compete with ATP for binding at the catalytic domain, inhibiting the phosphorylation of the tyrosine residues on substrates and impeding the downstream signalling effects of the oncogenic protein [22, 24]. In contrast, asciminib mimics the actions of the myristoyl site of the N-terminal cap, leading to the allosteric inhibition of BCR-ABL1 [25].
2.1.3 The Consequence of BCR-ABL1
The BCR-ABL1 protein gives rise to aberrant activation of cell signalling pathways and a shift to a micro-environment that is optimal for the development of leukaemia. For example, CML cells exhibit changes in growth-factor dependence, apoptosis, proliferation and cell adhesion [24]. These changes result in excessive proliferation of granulocytes, leading to the clinical features observed in CP-CML [26, 27]. The importance of BCR-ABL1 signalling (particularly via the tyrosine kinase domain) is ultimately corroborated by the efficacy of TKI therapy.
BCR-ABL1 is a multi-faceted fusion gene with a marked effect on downstream signalling pathways, all of which promote the leukaemic phenotype observed in CML. Early work involving the transplantation of murine bone marrow transfected with BCR-ABL1 induced a CML-like disease in transplanted mice [26, 28, 29]. Additional experiments confirmed the oncogenic potential of BCR-ABL1 through gradual disruption of cellular differentiation, dysregulated proliferation, growth factor independence and interference of apoptosis through downstream signalling pathways [30,31,32]. Moreover, studies targeting BCR-ABL1 by antisense oligonucleotides [33,34,35,36] demonstrated that BCR-ABL1 was crucial for maintenance of the leukaemic process. These early observations underpinned the function of BCR-ABL1 and provided affirmation that this fusion oncogene is the sole driver of CP-CML. However evolution to the more aggressive stages of CML is likely dependent on the cooperation of BCR-ABL1 with other genetic events implicated in malignancy [37].
2.2 Important Pathways Affected by BCR-ABL1 Activity
2.2.1 JAK/STAT
The JAK/STAT signalling pathway has been heavily implicated in leukaemogenesis, including the pathogenesis of CML [38]. BCR-ABL1 augments activation of JAK2 through enhanced efficiency of JAK2 phosphorylation, promoting cell growth/survival while numerous STAT proteins are activated by the JAK receptor [39, 40]. Furthermore, murine models have illustrated the pivotal role of STAT5 signalling in the development and maintenance of CML. One experiment utilizing retroviral transduction of BCR-ABL1 in STAT5-knockout bone marrow failed to induce CML in recipient mice after both primary and secondary transplantation [41]. In a second model, STAT5 deletion resulted in marked depletion of BCR-ABL1-expressing leukaemic cells, demonstrating the importance of STAT5 in the maintenance of CML [42]. Furthermore, enhanced STAT5 expression reduced imatinib-mediated cytotoxicity in BCR-ABL1-positive cells, potentially linked to marked anti-apoptotic activity mediated by increased STAT5 downstream signalling [43]. Increased STAT3 levels have also correlated with imatinib resistance [44]. Regardless of the mechanism, JAK inhibitors have exhibited efficacy against BCR-ABL1-positive cells, overcoming TKI resistance [45]. Furthermore, the combination of the JAK-inhibitor, ruxolitinib, and nilotinib has been demonstrated to induce undetectable BCR-ABL1 levels in patients with low-level disease [46].
2.2.2 PI3K/AKT and Autophagy
PI3K proteins communicate extra-cellular signals to modulate transcription factor activation and programming that favour cell growth/survival and inhibition of cell death. AKT is a downstream effector of PI3K and plays a major role in its signalling [47]. BCR-ABL1 can stimulate PI3K signalling through the adapter proteins Grb2/Gab2 [48] and CBL [49] but also through loss of function of the tumour suppressor gene PTEN, which is frequently silenced in malignancy [50]. Several reports indicate that the PI3K/AKT pathway is critical for BCR-ABL1-induced leukaemogenesis and for CML maintenance [51] and that its interruption can circumvent BCR-ABL1 oncogenesis [52, 53]. Another consequence of PI3K activation is stimulation of the mTOR pathway [54], which is responsible for controlling protein synthesis, cell growth/size and autophagy.
Autophagy can occur following cell stress (i.e. loss of BCR-ABL1 signalling) to promote cell hibernation as opposed to apoptosis, and can be reversed with restoration of the optimal environment. Recent studies have observed that whilst BCR-ABL1 inhibits autophagy, TKI treatment restores this pathway and may contribute to molecular relapse in failed treatment-free remission attempts, despite undetectable BCR-ABL1 levels prior to TKI discontinuation [55]. Moreover, BCR-ABL1-positive stem cells with knockdown of genes vital for the autophagy process failed to proliferate in an optimized environment [56]. Therefore, autophagy may be an appropriate mechanism to target for the future.
2.2.3 Ras/MEK Pathway
Activation of Ras GTPases/MEK kinases stimulates cell growth via a membrane receptor-binding cascade to activate transcription of a number of growth factor genes and is a key pathway deregulated in cancer [57]. BCR-ABL1 activates Ras via Grb2/Gab2 phosphorylation to promote cell growth [58, 59], and persistence of Ras activity has been demonstrated in TKI-resistant CML cells [60]. Disruption of Ras signalling impairs development of BCR-ABL1-induced CML-like disease in mice [49, 61]. In addition, MEK inhibitors can induce apoptosis in blast crisis (BC)- and drug-resistant CML cells with targeting of CML progenitors [60, 62]. Further work is required to investigate the true potential of inhibition of this pathway in CML.
2.2.4 Src Kinases
The Src-family kinases (SFKs) are another group of widely studied downstream targets of BCR-ABL1. Their role is to coordinate cell growth, differentiation and motility in response to extracellular signals [63]. Initial CML cell line models showed that BCR-ABL1 expression significantly activated the Hck and Lyn SFKs [64]. Subsequent studies demonstrated that Hck, Lyn and Fyn were required for BCR-ABL1 cell line transformation as well as functionally phosphorylating several BCR-ABL1 tyrosines [65, 66]. One mechanism by which SFKs contribute to disease is in assisting BCR-ABL1 in its activation of STAT5 and AKT [67, 68]. In addition, knockdown of Lyn exhibited impressive killing of BC cells, and its upregulation in BC-CML suggested a potential role for promoting disease progression [69, 70]. However, the importance of SFKs in CML remains unclear because mouse models show that SFKs are not required for initiation of CML but, rather, support the generation of acute lymphoid leukaemia [71, 72]. The second generation TKIs, dasatinib and bosutinib, are dual Src/Abl1 inhibitors, so defining the role of SFKs in CML could have an impact on both understanding its biology and treatment [73].
2.2.5 Crkl
The adaptor protein Crkl is constitutively activated by BCR-ABL1 [74]. Protein networks involving BCR-ABL1 and Crkl include Cbl, STAT, PI3K, paxillin and Ras [75]. Indeed, loss of the interaction between Ckrl and BCR-ABL1 impaired BCR-ABL1-induced transformation in mice [76]. The potent phosphorylation of Crkl by BCR-ABL1 allows the measurement of the percentage of phospho-Crkl as a surrogate to BCR-ABL1 phosphorylation levels (which are more difficult to measure) in order to experimentally examine the patient’s response to TKI therapy and to predict outcome [77].
2.2.6 Long Non-coding (lnc) RNA
LncRNAs are heavily involved in normal haematopoiesis and have increasingly been implicated in haematological malignancies [78]. In CML cell line models, lncRNA-BGL3 sensitizes BCR-ABL1-positive cells to imatinib-induced apoptosis [79]. It also acts as a decoy for several microRNAs that target the tumour suppressor gene PTEN, leading to its stabilisation and associated inhibition of leukaemogenesis [79]. In contrast, lncRNA-H19 facilitates leukaemogenesis in CML through upregulation of MYC, and its knockdown perturbs the pathogenicity of BCR-ABL1 in CML cell lines [80]. Further work is required to understand the full mechanisms and impact of these lncRNAs in CML.
2.2.7 Apoptosis Deregulation
In addition to promoting cell proliferation, BCR-ABL1 can disrupt cell death. An example of this involves a BCR-ABL1, Bad, BCL2 and BCL-XL circuit (Fig. 2.2). Expression of BCR-ABL1 can inhibit apoptosis by increasing expression of the anti-apoptotic proteins BCL2 and BCL-XL [81]. Both STAT5 and PI3K signalling are important mediators of BCR-ABL1’s anti-apoptotic function. STAT5 activation by BCR-ABL1 causes increased BCL-XL expression [82, 83]. Furthermore, phosphorylation of the pro-apoptotic protein Bad by PI3K/Akt facilitates the interaction between the chaperone protein 14–3-3 and Bad, which restricts Bad to the cytoplasm [84]. This prevents Bad opposing BCL2 and BCL-XL inhibition of apoptosis in the mitochondrion.

An example of apoptotic circuitry controlled by BCR-ABL1. BCR-ABL1 promotes the expression of anti-apoptotic genes BCL2 and BCL-XL and inhibits the function of pro-apoptotic protein Bad via phosphorylation (grey circle) and cytoplasmic sequestration
2.3 CML Stem Cells
2.3.1 Leukaemic Stem Cells (LSCs) Are Refractory to TKIs
A seminal paper from the Holyoake laboratory showed that BCR-ABL1 inhibition reduced LSC proliferation but failed to deplete quiescent LSCs [85]. Furthermore, LSCs have also been shown to be insensitive to more potent second-generation TKIs, despite complete silencing of BCR-ABL1 activity [86, 87]. These studies raised the possibility of early relapse despite TKI therapy, but long-term TKI usage has rebuffed this theory [88]. Subsequent studies have strengthened the notion that survival of the LSC is independent of BCR-ABL1 activity [89, 90]. It has also been reported that therapy-refractory LSCs exhibit a bias for low BCR-ABL1 expression [91, 92]. Persistence of the LSCs have been postulated to be the primary causes of molecular relapse following a treatment-free remission attempt despite long-term BCR-ABL1 negativity [88]. Several pathways have been shown to play key roles in stem cell biology (Fig. 2.3), and targeting them could lead to a promising strategy to eliminate the LSC in CML.

LSC circuitry of genes discussed in this chapter. STAT5, JAK2 and PI3K all feature to control LSC-effector genes. However, quiescent stem cells have intrinsic counters to prevent potent BCR-ABL1 signalling depleting the LSC population, such as MSI2/TGF-β, PTEN, FOXO transcription factors and Fbw7. In the context of BC-CML, the reliance on countering BCR-ABL1 is not as important due to the incapacity of leukaemic progenitor cells to differentiate. This may explain how enhanced pathway activation (JAK2 / β-catenin) is compatible with expansion of the stem/progenitor compartment in BC. Hes1 activity enhances that of PI3K while the Hh pathway via the transmembrane receptors of PTCH and Smo regulate Gli signalling, also important for LSC maintenance
2.3.2 Wnt/β-Catenin Pathway
The Wnt signalling pathway has been demonstrated to be crucial for LSC self-renewal [93], and β-catenin is one of its components [94, 95]. Binding of Wnt to its receptor, Frizzled, causes disruption of ubiquitin-mediated degradation of β-catenin, freeing the molecule for nuclear translocation to activate the transcription of target genes such as the cyclin D1 and MYC oncogenes [96]. BCR-ABL1 induces aberrant PI3K/AKT signalling, resulting in upregulated β-catenin activity [53], which has also been implicated in risk of progression to BC [97]. Enhanced β-catenin signalling in BC-CML confers stem-cell-like characteristics to progenitor cells leading to cellular expansion [98]. Future strategies may be to target both β-catenin and BCR-ABL1, as murine models have demonstrated that this approach is synergistic, delaying disease progression while depleting CML-LSCs [99] (Fig. 2.4).

Complex control of β-catenin in CML. BCR-ABL1 stabilises β-catenin signalling via PI3K, JAK2 and inhibition of IRF8. Canonical stability of β-catenin is controlled by protein ubiquitination (grey circles). Thus, in CML, this pathway is activated to promote a stem-cell like environment. However, inhibition of, e.g. PP2A activation, can reverse pathogenic β-catenin signalling and synergise with BCR-ABL1 inhibition to enhance treatment efficacy
2.3.3 Hedgehog (Hh) Pathway
Signalling in the Hh pathway is critical for LSC self-renewal and contributes to tissue homeostasis, regeneration and healing [100]. In BCR-ABL1-positive progenitor cells, increased Hh signalling is observed with marked upregulation in BC-CML [101, 102]. It also induces malignant expansions of LSCs in murine models [103]. Upregulation of Smoothened (Smo), a membrane receptor for the hedgehog ligand, has been found to augment LSCs and to drive disease progression [104]. Activation of Smo, in turn, activates Gli transcription factors, which drive expression of their downstream transcriptional targets [105]. Studies on primary CML cells found that Smo/Gli2 promoted LSC dormancy via cell cycle arrest, and an enhanced hedgehog pathway signature is observed in BC patients. Inhibition of Gli2 was able to restore LSC cycling and sensitise LSCs to TKI eradication [106]. Dual targeting with Smo inhibitors and TKIs may be a future therapeutic strategy to target both stem and progenitor cells as in vitro data suggest a reduced rate of leukaemic progression [100].
2.3.4 Notch Pathway
The Notch pathway has been demonstrated to be vital for cellular signalling and is dysregulated in multiple malignancies, including haematological cancers [107]. A member of the Notch family, Hairy enhancer of split 1 (Hes1), cooperates with BCR-ABL1 to induce BC-CML in murine models [108]. Furthermore, over-expression of Hes1 has been shown in BC but not CP, while dominant-negative Hes1 deterred growth of Hes1-expressing cell lines [108]. Interfering with the cross-talk between Notch signalling and BCR-ABL1 may be achievable with combined targeting of both pathways and may be a treatment option for future exploration [109].
2.3.5 FoxO Family
BCR-ABL1 promotes deregulation of several transcription factors, including forkhead box class O (FoxO), through activation of the PI3K/Akt pathway [94]. Members of the FoxO family, in particular FoxO3a, are vital to the maintenance of LSCs [110]. BCR-ABL1 promotes nuclear export and deactivation of these transcription factors via PI3K/Akt [111]. In mature cells, Akt signalling is strong and assists propagation of BCR-ABL1’s proliferative advantage. However, in LSCs, Akt signalling is inhibited by PTEN [112] and TGF-β [113]. This reverses BCR-ABL1 inactivation of FoxO3a and allows for BCL6 transcription, which favours quiescence and self-renewal [112]. Targeting this mechanism with BCL6 or TGF-β inhibitors together with TKIs perturbed CML development and induced cell death/turnover of primitive CML cells [112, 113].
2.3.6 BCL2 Family
The proteins in the BCL2 family are key regulators of apoptosis and crucial for LSC survival [114]. BCL2 anti-apoptotic protein expression is increased in CML and is further increased in CML-BC. BCR-ABL1 signalling also promotes CML cell survival by upregulation of anti-apoptotic BCL2 proteins, including BCL-XL [115]. Furthermore, BCL2 acts synergistically with BCR-ABL1 to induce BC-CML [116]. Another member of the BCL2 family, the BH3-only pro-apoptotic protein (BIM), is also downregulated in CML, supporting LSC survival [116]. TKI therapy leads to upregulation of pro-apoptotic proteins, including BIM [117]. The presence of a common synonymous variant in the BH3 functional domain of BIM has been associated with imatinib resistance and inferior molecular target achievement [118]. Selective inhibition of BCL-2 through combined therapy with venetoclax (a novel BCL2 inhibitor primarily utilized in chronic lymphocytic leukaemia) and TKI has been demonstrated to target the LSC in a BCR-ABL1 transgenic mouse model, potentially offering a long-term cure in CML [114].
2.3.7 PP2A-JAK2-SET
BCR-ABL1 was reported to circumvent the requirement for JAK2 in its activation of STAT5 [119], but there are data demonstrating a role for JAK2 within the LSC compartment. A network involving PP2A/JAK2/Set/GSK-3β was shown to play a critical role in LSC survival [120]. Central to this pathway is PP2A, a tyrosine phosphatase whose activity is impaired in CML. ‘Active’ PP2A has the ability to silence key pathways which are activated by BCR-ABL1, including BCR-ABL1 itself [121]. In CML-LSCs, BCR-ABL1/JAK2 signalling overcomes PP2A activity by enhancing the activity of SET, a PP2A-inhibitor. Blocking the PP2A inhibitory role of SET restores PP2A function and impairs the self-renewal and survival of CML-LSCs but not normal haematopoietic stem cells (HSCs) [120]. A major mechanism by which PP2A activation affects LSC maintenance is thought to be the loss of β-catenin signalling via GSK-3β mediated ubiquitination. This is coupled with PP2A silencing of BCR-ABL1 to allow for LSC turnover and reduced leukaemic potential.
2.3.8 Bone Marrow Microenvironment
HSCs reside in the bone marrow, which provides an environment that controls haemopoiesis by coordinating HSC renewal and differentiation into functional blood cells. The bone marrow supportive environment comprises the osteoblast and vascular niches [122, 123]. The former promotes self-renewal and quiescence, while the vascular niche is permissive of differentiation into progenitor and subsequent functional cells. In CML, it is thought that the osteoblast niche nurtures LSCs, which may explain why LSCs do not require BCR-ABL1 kinase activity to survive TKI exposure [124, 125]. This may also contribute to BC. Since progenitor cells attain stem cell like properties, a progenitor-contingent may retreat towards the osteoblast niche for protection against TKIs, whilst retaining cycling properties that allow for faster accumulation of mutations (compared to LSCs) required for transformation.
2.4 Biology of Blast Crisis
The mechanism of disease evolution to BC-CML is still incompletely understood. This stage of the disease is characterised by the expansion of haemopoietic progenitors that fail to differentiate and interfere with normal haematopoiesis. These progenitor cells gain self-renewal capacity, differentiation arrest and survival properties that lead to uncontrolled proliferation, [98] exhibiting more stem-cell like characteristics compared to CP-progenitors. This is partially attributed to increased β-catenin activity [98] but also marked genomic and genetic instability [126, 127]. Extra chromosomal abnormalities are observed in approximately 80% of BC patients (e.g. Ph duplication, trisomy 8 or 19, loss of 17p) [128]. Pathogenic mutations in tumour suppressor and oncogenes have also been detected in BC-CML [129], and it is hypothesised that these additional hits contribute to the transition into BC [127, 129]. The rapid recent technological advances in next-generation sequencing has not only enabled attempts at unmasking the genomic landscape involved in BC-CML but has further highlighted the vast gaps of knowledge which yet remain.
2.4.1 BCR-ABL1 and CML-BC
Inhibition of BCR-ABL1 kinase activity effectively delays the onset of BC but does not eliminate the primitive population that establishes advanced disease. One interpretation is that BCR-ABL1 signalling is required for transition to BC, especially since progression is rare in TKI responsive patients. A number of studies have found increased expression of BCR-ABL1 in BC compared to CP. This was observed when comparing matched CP and BC samples (from the same patient) at both the mRNA [131,132,133,133] and protein levels [121, 130, 134]. Additionally, it has been shown that cells expressing higher amounts of BCR-ABL1 have an increase in genomic instability as well as perturbed differentiation, which are intrinsic properties of BC-CML [127, 135]. These findings imply more than a passenger role for BCR-ABL1 in BC-transformation.
2.4.2 DNA Damage/Repair
BCR-ABL1 has been shown to facilitate genomic instability via disrupting DNA repair pathways, generating reactive oxygen species and inhibiting DNA-damage-induced apoptosis, all of which may lead to retention of genomic mutations [137,138,139,140,140]. These events are in part tied to the level of BCR-ABL1 expression [141]. CML CD34+ cells express high levels of BCR-ABL1 as compared to mature cells [132], and they are highly susceptible to genomic instability compared to their healthy counterparts [89]. Although not formally shown, it is reasonable to suggest that BCR-ABL1 provides progenitor cells with the genomic plasticity required for malignant transformation [127, 142, 143].
2.4.3 C/EBPα and hnRNP-E2
Required for myeloid differentiation [144], C/EBPα expression is reduced in cell lines expressing BCR-ABL1 [145]. These lines responded poorly to growth-factor-induced differentiation [135], but ectopic expression of C/EBPα and BCR-ABL1 kinase inhibition were able to reverse this differentiation block [145]. Further experiments revealed that BCR-ABL1 negatively regulates the expression of C/EBPα via upregulation of hnRNP-E2, an RNA-binding protein which inhibits C/EBPα expression [135]. Interestingly, analysis of CML-patient cells found that loss of C/EBPα and expression of hnRNP-E2 was restricted to BC [135]. In addition, hnRNP-E2 upregulation and C/EBPα downregulation were directly proportional to increasing levels of BCR-ABL1 [135]. To add extra complexity to this pathway, it was recently shown that the microRNA miR-328 acts in a non-canonical way to block hnRNP-E2 regulation of C/EBPα and promotes myeloid differentiation [146]. The expression of miR-328 negatively correlates with BCR-ABL1 expression levels and is thus downregulated in BC [146]. These experiments provide evidence of a sophisticated circuit by which enhanced BCR-ABL1 expression can facilitate a switch to BC by disrupting myeloid differentiation.
2.4.4 Important Pathways Involved in BC-CML
2.4.4.1 MYC
The MYC proto-oncogene was one of the first genes implicated in CML disease progression. MYC is a transcription factor which governs the expression of genes enabling cell growth and proliferation and, thus, commonly activated in cancer [147]. It was originally observed that patients with BC exhibited higher levels of MYC compared to CP patients [148]. This was followed by reports that ABL1 expression enhances MYC expression and that MYC is required for BCR-ABL1-induced transformation [149, 150]. Although excess MYC can induce apoptosis [151], early cell line models show that BCR-ABL1 activation of BCL2 can inhibit MYC apoptotic activity whilst retaining its proliferative advantage [152]. This is one of many examples by which BCR-ABL1 creates ‘a perfect storm’ to promote leukaemogenesis.
BCR-ABL1 can control MYC expression via PI3K, JAK2 and the transcription factor E2F1 [51, 154,155,155], while maintaining protein stability via MEK and hnRNP-K [156]. A CML mouse model demonstrated that MYC expression is required for CML maintenance and progression. It also showed that high levels of MYC are harmful for LSCs, and ubiquitination (degradation) of MYC by ubiquitin ligase Fbw7 keeps MYC levels in check in LSCs [157]. This provides a rationale for the constrained BCR-ABL1 kinase activity observed in quiescent LSCs [120] and selection of low BCR-ABL1 expression in TKI-refractive LSCs [91, 92] (suggesting that enhanced BCR-ABL1 signalling is toxic for quiescent cells). These findings, coupled with MYC’s established role in myeloid differentiation [158], present MYC deregulation as a strong candidate for driving BC-transformation in CML.
2.4.4.2 p53
The normal function of p53 is to respond to cell stress events, where it becomes activated and drives transcription of genes that decide cell fate (apoptosis, DNA repair, cell cycle arrest or senescence) [159]. Early genetic studies observed inactivating mutations of p53 in approximately 20% of CML patients who progressed to BC [160, 161]. Regulation of p53 by BCR-ABL1 is complex and unclear, with both p53 activation [162, 163] and inactivation [164, 165] being reported. However, loss or inhibition of p53 promotes BC-like disease in mice [166,167,167], and stabilisation of p53 in BC cells induces apoptosis [167, 168]. It has also been shown that MYC over-expression is only toxic to LSCs if p53 is present [157].
2.4.4.3 XPO1
The nuclear export protein, XPO1, is another novel candidate for the regulation of BC. Its expression is enhanced in BC patients, and pharmacological blockade of its function was shown as sufficient to kill both CP and BC-primary CD34+ cells [169]. Inhibition of XPO1 in BCR-ABL1-positive cell lines demonstrated that impaired nuclear transport could explain XPO1-inhibition lethality. For example, both SET and p53 were abnormally enriched in the nucleus leading to their inactivation [169]. Additional experiments revealed that long-term XPO1 inhibition caused BCR-ABL1 degradation (via loss of SET control of PP2A activity) whereas short-term inhibition shutdown STAT5, AKT and MEK signalling prior to affecting BCR-ABL1 activity [169]. This suggests that both BCR-ABL1-dependent and -independent cell death results through XPO1 inhibition. Remarkably, an XPO1 inhibitor reversed CML symptoms (WBC count/splenomegaly) in a patient with disease progression and who was resistant to TKI therapy, highlighting a potential strategy to treat advanced disease [169].
2.4.4.4 SIRT1
Expression of SIRT1 is enhanced in CML and is, in part, regulated by BCR-ABL1/STAT5 [170]. This protein-deacetylase has been linked to CML BC due to its disruption of LSC turnover and DNA repair. SIRT1 suppression of p53/FoxO-controlled LSC maintenance is believed to prolong the survival of CML LSCs [170, 171]. In contrast, knockout or inhibition of SIRT1 impairs CML development and disease progression in mice by reducing proliferative and self-renewal capacity of LSCs [170, 171]. SIRT1 regulation of the DNA repair protein Ku70 in CML cell lines causes enhancement of less faithful non-homologous end joining DNA repair, which enhances mutations [172]. The knowledge that SIRT1 provides a route for LSC survival and genomic instability—the key drivers of BC-CML—offers strong evidence that SIRT1 has a major role in BC development.
2.4.4.5 ADAR1
ADAR1 is an RNA editor whose enzymatic activity converts adenosine to inosine in RNA, resulting in these nucleotides being interpreted as guanine in the ribosome, thus altering RNA behaviour and protein amino acid composition. Analysis of ADAR1 expression in CML patients showed a marked increase in expression from CP to BC, and was correlated with BCR-ABL1 levels [173]. The BC samples also had enhanced A to I editing and altered expression of RNA-edited genes, providing evidence that the increased expression of ADAR1 in BC had a functional effect on its downstream targets [173]. Two mouse models have been developed which successfully demonstrate the important role that ADAR1 plays in CML stem cells. Following disruption of ADAR1 expression in CML mouse models, leukaemia development, maintenance and BC onset were all impaired due to the loss of primitive leukaemic cells [174]. In contrast, ADAR1 over-expression caused myeloid progenitor expansion [173]. Moreover, specific deletion of the ADAR1’s RNA-editing moiety demonstrated that RNA editing is vital for CML progenitor self-renewal [174]. It is known that the RNA-editing activity of ADAR1 is required for HSC survival [175], so it is speculated that the enhanced activity of ADAR1 in BC locks the LSCs in a primitive state.
2.4.4.6 Polycomb Repressive Complexes (PRCs) and Epigenetic Regulation
Dysregulation of PRCs have been implicated in a number of haematological malignancies, including CML [176]. Early data indicated that overexpression of BMI1, a member of PRC1, correlated with inferior survival and higher risk of BC transformation [177]. Enhanced EZH2 activity, a catalytic subunit of PRC2, has also been demonstrated as necessary for the propagation of CML [178]. More recent exploration of the BC genome indicates substantial enrichment for mutations affecting the PRCs: transcriptomic interrogation of BC progenitors demonstrated both upregulation and depletion of PRC1- and PRC2-related gene sets, respectively [179].
The impact of epigenetic reprogramming is still an emerging area of research in CML. The PRCs are heavily involved with epigenetic reprogramming in BC-CML with PRC2-driven DNA hypermethylation being responsible for arrested myeloid differentiation and loss of tumour suppressor function [179]. However, DNA methylation inhibitors have failed to produce durable responses in BC-CML [179]. Gene expression analysis of BC-cells treated with hypomethylating agents revealed failure to normalize the majority of the gene expression changes associated with DNA-methylation, indicating additional layers of unidentified epigenetic regulation [179]. However, in vitro combinatorial therapy with directed inhibition of BMI1 and hypomethylating agents reduced colony formation in BC-CML cell lines by approximately 90% [179].
2.4.4.7 Mutational Landscape
While BCR-ABL1 alone is sufficient to induce CP-CML, it is unlikely to be the sole event in more advanced stages of the disease. BCR-ABL1 has been associated with substantial genetic instability [180], assisting in the acquisition of additional mutational events that could trigger progression to BC. In order to identify putative BC driver genes, Giotopoulos et al. utilised an impressive mouse model. The experiment centred on a transposable cassette array in the presence or absence of BCR-ABL1 [181]. Transposition of the cassettes can either activate or deactivate the genes in proximity to the genomic insertion site. Gene activation is achieved by a transposition event within the 5′ region of the gene due to enhancer/promoter sequences in the cassette [181]. Conversely, intragenic transposition can disrupt genes causing loss-of-function. Mice with a BCR-ABL1 only genetic background succumbed to a CML-CP phenotype, whilst 85% of the BCR-ABL1/transposon mice exhibited CML-BC, 5% CP and 10% AP-like disease [181]. Microarray gene expression analysis of the mice showed clustering within disease type and inter-type separation, identifying several genes known to be involved in in the development of BC. Transposition events within the BC sample cohort included STAT5, XPO1, PTEN, MYC-target genes and JAK1 [181].
The current era has been characterized by dramatic technological advances in next-generation sequencing which have enabled the identification of somatic mutational profiles that characterize various haematological malignancies, influencing diagnosis, treatment and prognosis [183,184,184]. In BC-CML, most patients have been identified to harbour additional mutational events in known cancer genes [185, 186] seen in up to 95% of patients in one study [185]. Mutations in RUNX1, ASXL1 and IKZF1 exon deletions are the most frequently observed events [187] while single nucleotides, insertions, deletions, fusions and aberrant splicing in multiple different cancer-related genes have all been described in BC-CML. Aberrant RAG-mediated recombination has also been demonstrated to contribute to structural rearrangements in lymphoid BC [188]. A novel class of variant, termed ‘Ph-associated rearrangements’, involving gene rearrangements and novel fusions on the chromosome arms involved with the inciting Ph-translocation, has also been observed in poor outcome patients at the time of diagnosis, including those progressing to BC-CML [185]. Moreover, the Ph-associated rearrangements were more frequently identified in patients progressing to lymphoid BC [185]. While there are minimal data regarding this novel group of mutations, their presence may highlight a cohort of patients with increased genetic instability and, therefore, increased propensity for adverse outcomes. Kinase domain mutations can be identified in approximately 50% of patients in BC-CML [185], more frequently in lymphoid BC [185]. However, these are rarely the sole event [185], frequently co-occurring with IKZF1 variants. Additionally, cancer-gene variants often pre-date the development of kinase domain mutations in approximately 60% of patients, emphasizing the genomic instability associated with the acquisition of cancer-gene mutations [185].
2.5 Concluding Remarks
The biology of CML is centred on BCR-ABL1’s constitutive kinase activity, which is sufficient to cause the clinical features of CP. The ability to readily model CML in both cell lines and mice has allowed for a large accumulation of knowledge regarding the molecular network of CML. These studies have shown that BCR-ABL1 is implicated in altering almost every process within the cell to drive CML pathogenesis. This extends to dampening its own excessive signalling in LSCs, which would be otherwise unfavourable. Current literature has shown that STAT5 stands out as a vital component of BCR-ABL1’s induction of CML as demonstrated by two conditional knockout models [41, 189]. The investigation of primitive CML-cell biology has benefitted from the utilisation of new and powerful techniques to identify a number of important genes within this compartment. The best studied are p53, MYC and β-catenin, which have prominent roles in both stem cell biology and BC transformation.
The link between LSCs and BC and treatment response has put the LSC and progenitor populations at the forefront of CML biology. Of particular interest is the finding that LSCs do not rely on BCR-ABL1 kinase activity for survival. It is unknown if another protein domain of BCR-ABL1 confers LSC survival properties. Another possibility is that BCR-ABL1 can program LSCs in such a way that its kinase activity is no longer required. It is unknown whether the HSC or progenitor compartment gives rise to the clones responsible for BC-CML. Pinpointing the latter is important because each of these compartments has discrete biological properties and, thus, requires alternative therapeutic strategies.
Next-generation sequencing and powerful experimental modelling tools will no doubt provide a flood of information regarding CML biology as well as highlight the potential drivers of disease progression. These advances are likely to generate evidence of recurrent mutations and epigenetic marks that favour or hinder CML pathogenesis or response to treatment.
In the proteomics field, improved methods to study proteins and more powerful mass spectrometers have the potential to uncover post-translational modifications and protein interactomes. The study of proteome networks is relatively untapped in CML (although elegant examples do exist [190, 191]), making this an attractive area of interest to improve the knowledge of CML biology. The same can be said of non-coding RNA (ncRNA). It is known that deregulation of these molecules occurs in CML, for example in CP versus BC, and in primitive cells versus granulocytes [192, 193]. However, most functional work is limited to a single microRNA and target. Further work is required to understand the global ncRNA circuitry in key areas within this disease. These fields of interest are bolstered with the emerging accessibility to high-powered fluorescence microscopy, which can monitor the spatiotemporal behaviour of proteins and RNA.
Finally, availability of pathway inhibitors and genome editing (including crispR) systems [194] are powerful options to functionally validate pathways identified by genomic and proteomic studies in both cell lines and mouse models. These technologies will make for an exciting time to uncover novel mechanisms behind CML pathogenesis and the potential for translation to other diseases.
References
Score J, Calasanz MJ, Ottman O, Pane F, Yeh RF, Sobrinho-Simoes MA, et al. Analysis of genomic breakpoints in p190 and p210 BCR-ABL indicate distinct mechanisms of formation. Leukemia. 2010;24(10):1742–50. https://doi.org/10.1038/leu.2010.174.
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88(7):2375–84.
Jain P, Kantarjian H, Patel KP, Gonzalez GN, Luthra R, Shamanna RK, et al. Impact of BCR-ABL transcript type on outcome in patients with chronic-phase CML treated with tyrosine kinase inhibitors. Blood. 2016;127(10):1269–75. https://doi.org/10.1182/blood-2015-10-674242.
Baccarani M, Castagnetti F, Gugliotta G, Rosti G, Soverini S, Albeer A, et al. The proportion of different BCR-ABL1 transcript types in chronic myeloid leukemia. An international overview. Leukemia. 2019;33(5):1173–83. https://doi.org/10.1038/s41375-018-0341-4.
Dowding C, Guo AP, Maisin D, Gordon MY, Goldman JM. The effects of interferon-alpha on the proliferation of CML progenitor cells in vitro are not related to the precise position of the M-BCR breakpoint. Br J Haematol. 1991;77(2):165–71.
Fioretos T, Nilsson PG, Aman P, Heim S, Kristoffersson U, Malm C, et al. Clinical impact of breakpoint position within M-bcr in chronic myeloid leukemia. Leukemia. 1993;7(8):1225–31.
Rozman C, Urbano-Ispizua A, Cervantes F, Rozman M, Colomer D, Feliz P, et al. Analysis of the clinical relevance of the breakpoint location within M-BCR and the type of chimeric mRNA in chronic myelogenous leukemia. Leukemia. 1995;9(6):1104–7.
Shepherd P, Suffolk R, Halsey J, Allan N. Analysis of molecular breakpoint and m-RNA transcripts in a prospective randomized trial of interferon in chronic myeloid leukaemia: no correlation with clinical features, cytogenetic response, duration of chronic phase, or survival. Br J Haematol. 1995;89(3):546–54.
Balatzenko G, Vundinti BR, Margarita G. Correlation between the type of bcr-abl transcripts and blood cell counts in chronic myeloid leukemia - a possible influence of mdr1 gene expression. Hematol Rep. 2011;3(1):e3. https://doi.org/10.4081/hr.2011.e3. hr.2011.e3 [pii]
Hanfstein B, Lauseker M, Hehlmann R, Saussele S, Erben P, Dietz C, et al. Distinct characteristics of e13a2 versus e14a2 BCR-ABL1 driven chronic myeloid leukemia under first-line therapy with imatinib. Haematologica. 2014;99(9):1441–7. https://doi.org/10.3324/haematol.2013.096537.
Castagnetti F, Gugliotta G, Breccia M, Iurlo A, Levato L, Albano F, et al. The BCR-ABL1 transcript type influences response and outcome in Philadelphia chromosome-positive chronic myeloid leukemia patients treated frontline with imatinib. Am J Hematol. 2017;92(8):797–805. https://doi.org/10.1002/ajh.24774.
Genthon A, Nicolini FE, Huguet F, Colin-Gil C, Berger M, Saugues S, et al. Influence of major BCR-ABL1 transcript subtype on outcome in patients with chronic myeloid leukemia in chronic phase treated frontline with nilotinib. Oncotarget. 2020;11:26.
D'Adda M, Farina M, Schieppati F, Borlenghi E, Bottelli C, Cerqui E, et al. The e13a2 BCR-ABL transcript negatively affects sustained deep molecular response and the achievement of treatment-free remission in patients with chronic myeloid leukemia who receive tyrosine kinase inhibitors. Cancer. 2019;125(10):1674–82. https://doi.org/10.1002/cncr.31977.
Claudiani S, Apperley JF, Gale RP, Clark R, Szydlo R, Deplano S, et al. e14a2 BCR-ABL1 transcript is associated with a higher rate of treatment-free remission in individuals with chronic myeloid leukemia after stopping tyrosine kinase inhibitor therapy. Haematologica. 2017;102(8):e297–e9. https://doi.org/10.3324/haematol.2017.168740.
Gong Z, Medeiros LJ, Cortes JE, Zheng L, Khoury JD, Wang W, et al. Clinical and prognostic significance of e1a2 BCR-ABL1 transcript subtype in chronic myeloid leukemia. Blood Cancer J. 2017;7(7):e583-e. https://doi.org/10.1038/bcj.2017.62.
Qin Y-Z, Jiang Q, Jiang H, Lai Y-Y, Shi H-X, Chen W-M, et al. Prevalence and outcomes of uncommon BCR-ABL1 fusion transcripts in patients with chronic myeloid leukaemia: data from a single centre. Br J Haematol. 2018;182(5):693–700. https://doi.org/10.1111/bjh.15453.
Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood. 1995;86(8):3118–22.
Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood. 1998;92(9):3362–7.
Basecke J, Griesinger F, Trumper L, Brittinger G. Leukemia- and lymphoma-associated genetic aberrations in healthy individuals. Ann Hematol. 2002;81(2):64–75. https://doi.org/10.1007/s00277-002-0427-x.
Boquett JA, Alves JR, de Oliveira CE. Analysis of BCR/ABL transcripts in healthy individuals. Genet Mol Res. 2013;12(4):4967–71. https://doi.org/10.4238/2013.October.24.8.
Ismail SI, Naffa RG, Yousef AM, Ghanim MT. Incidence of bcr-abl fusion transcripts in healthy individuals. Mol Med Rep. 2014;9(4):1271–6. https://doi.org/10.3892/mmr.2014.1951.
Goldman JM, Melo JV. Chronic myeloid Leukemia—advances in biology and new approaches to treatment. N Engl J Med. 2003;349(15):1451–64. https://doi.org/10.1056/NEJMra020777.
Hantschel O. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer. 2012;3(5–6):436–46. https://doi.org/10.1177/1947601912458584.
Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood. 2009;113(8):1619–30. https://doi.org/10.1182/blood-2008-03-144790.
Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature. 2017;543:733. https://doi.org/10.1038/nature21702. https://www.nature.com/articles/nature21702#supplementary-information
Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science (New York, NY). 1990;247(4944):824–30.
Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96(10):3343–56.
Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344(6263):251–3. https://doi.org/10.1038/344251a0.
Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci U S A. 1990;87(17):6649–53.
Bedi A, Zehnbauer BA, Barber JP, Sharkis SJ, Jones RJ. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood. 1994;83(8):2038–44.
Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci U S A. 1988;85(23):9312–6.
Hariharan IK, Adams JM, Cory S. bcr-abl oncogene renders myeloid cell line factor independent: potential autocrine mechanism in chronic myeloid leukemia. Oncogene Res. 1988;3(4):387–99.
Ratajczak MZ, Kant JA, Luger SM, Hijiya N, Zhang J, Zon G, et al. In vivo treatment of human leukemia in a scid mouse model with c-myb antisense oligodeoxynucleotides. Proc Natl Acad Sci U S A. 1992;89(24):11823–7.
Skorski T, Szczylik C, Malaguarnera L, Calabretta B. Gene-targeted specific inhibition of chronic myeloid leukemia cell growth by BCR-ABL antisense oligodeoxynucleotides. Folia Histochem Cytobiol. 1991;29(3):85–9.
Szczylik C, Skorski T, Nicolaides NC, Manzella L, Malaguarnera L, Venturelli D, et al. Selective inhibition of leukemia cell proliferation by BCR-ABL antisense oligodeoxynucleotides. Science (New York, NY). 1991;253(5019):562–5.
Engelman A, Rosenberg N. Temperature-sensitive mutants of Abelson murine leukemia virus deficient in protein tyrosine kinase activity. J Virol. 1990;64(9):4242–51.
Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103(11):4010–22. https://doi.org/10.1182/blood-2003-12-4111.
Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. 2000;19(21):2496–504. https://doi.org/10.1038/sj.onc.1203486.
Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol. 1997;159(10):4720–8.
Warsch W, Walz C, Sexl V. JAK of all trades: JAK2-STAT5 as novel therapeutic targets in BCR-ABL1+ chronic myeloid leukemia. Blood. 2013;122(13):2167–75. https://doi.org/10.1182/blood-2013-02-485573.
Walz C, Ahmed W, Lazarides K, Betancur M, Patel N, Hennighausen L, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012;119(15):3550–60. https://doi.org/10.1182/blood-2011-12-397554.
Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2(3):98–110. https://doi.org/10.1002/emmm.201000062.
Warsch W, Kollmann K, Eckelhart E, Fajmann S, Cerny-Reiterer S, Hölbl A, et al. High STAT5 levels mediate imatinib resistance and indicate disease progression in chronic myeloid leukemia. Blood. 2011;117(12):3409–20. https://doi.org/10.1182/blood-2009-10-248211.
Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther. 2008;7(10):3169–75. https://doi.org/10.1158/1535-7163.MCT-08-0314.
Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, et al. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene. 2009;28(14):1669–81. https://doi.org/10.1038/onc.2009.7.
Sweet K, Hazlehurst L, Sahakian E, Powers J, Nodzon L, Kayali F, et al. A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk Res. 2018;74:89–96. https://doi.org/10.1016/j.leukres.2018.10.002.
Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikami A, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103(44):16296–300. https://doi.org/10.1073/pnas.0607899103.
Sattler M, Salgia R, Okuda K, Uemura N, Durstin MA, Pisick E, et al. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3′ kinase pathway. Oncogene. 1996;12(4):839–46.
Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf NA, Podar K, et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell. 2002;1(5):479–92. S1535610802000740 [pii]
Morotti A, Panuzzo C, Crivellaro S, Pergolizzi B, Familiari U, Berger AH, et al. BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia. 2014;28(6):1326–33. https://doi.org/10.1038/leu.2013.370.
Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16(20):6151–61. https://doi.org/10.1093/emboj/16.20.6151.
Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene. 2002;21(38):5868–76. https://doi.org/10.1038/sj.onc.1205724.
Hu J, Feng M, Liu Z-L, Liu Y, Huang Z-L, Li H, et al. Potential role of Wnt/β-catenin signaling in blastic transformation of chronic myeloid leukemia: cross talk between β-catenin and BCR-ABL. Tumor Biol. 2016;37(12):15859–72. https://doi.org/10.1007/s13277-016-5413-3.
Mayerhofer M, Valent P, Sperr WR, Griffin JD, Sillaber C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood. 2002;100(10):3767–75. https://doi.org/10.1182/blood-2002-01-0109. 2002-01-0109 [pii]
Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118(10):2840–8. https://doi.org/10.1182/blood-2010-12-322537.
Ianniciello A, Dumas P-Y, Drullion C, Guitart A, Villacreces A, Peytour Y, et al. Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget. 2017;8(57):96984–92. https://doi.org/10.18632/oncotarget.18904.
Steelman LS, Franklin RA, Abrams SL, Chappell W, Kempf CR, Basecke J, et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia. 2011;25(7):1080–94. https://doi.org/10.1038/leu.2011.66.
Chu S, Li L, Singh H, Bhatia R. BCR-tyrosine 177 plays an essential role in Ras and Akt activation and in human hematopoietic progenitor transformation in chronic myelogenous leukemia. Cancer Res. 2007;67(14):7045–53. https://doi.org/10.1158/0008-5472.CAN-06-4312.
Puil L, Liu J, Gish G, Mbamalu G, Bowtell D, Pelicci PG, et al. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994;13(4):764–73.
Packer LM, Rana S, Hayward R, O'Hare T, Eide CA, Rebocho A, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20(6):715–27. https://doi.org/10.1016/j.ccr.2011.11.004.
Baum KJ, Ren R. Effect of Ras inhibition in hematopoiesis and BCR/ABL leukemogenesis. J Hematol Oncol. 2008;1:5. https://doi.org/10.1186/1756-8722-1-5.
Pellicano F, Simara P, Sinclair A, Helgason GV, Copland M, Grant S, et al. The MEK inhibitor PD184352 enhances BMS-214662-induced apoptosis in CD34+ CML stem/progenitor cells. Leukemia. 2011;25(7):1159–67. https://doi.org/10.1038/leu.2011.67.
Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6(10):587–95. https://doi.org/10.1038/nrclinonc.2009.129.
Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996;56(15):3589–96.
Lionberger JM, Wilson MB, Smithgall TE. Transformation of myeloid leukemia cells to cytokine independence by Bcr-Abl is suppressed by kinase-defective Hck. J Biol Chem. 2000;275(24):18581–5. https://doi.org/10.1074/jbc.C000126200. C000126200 [pii]
Wilson MB, Schreiner SJ, Choi HJ, Kamens J, Smithgall TE. Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene. 2002;21(53):8075–88. https://doi.org/10.1038/sj.onc.1206008.
Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson M, Smithgall TE, et al. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002;21(21):5766–74.
Warmuth M, Simon N, Mitina O, Mathes R, Fabbro D, Manley PW, et al. Dual-specific Src and Abl kinase inhibitors, PP1 and CGP76030, inhibit growth and survival of cells expressing imatinib mesylate-resistant Bcr-Abl kinases. Blood. 2003;101(2):664–72. https://doi.org/10.1182/blood-2002-01-0288. 2002-01-0288 [pii]
Ban K, Gao Y, Amin HM, Howard A, Miller C, Lin Q, et al. BCR-ABL1 mediates up-regulation of Fyn in chronic myelogenous leukemia. Blood. 2008;111(5):2904–8. https://doi.org/10.1182/blood-2007-05-091769.
Ptasznik A, Nakata Y, Kalota A, Emerson SG, Gewirtz AM. Short interfering RNA (siRNA) targeting the Lyn kinase induces apoptosis in primary, and drug-resistant, BCR-ABL1(+) leukemia cells. Nat Med. 2004;10(11):1187–9. https://doi.org/10.1038/nm1127.
Engelman A, Rosenberg N. bcr/abl and src but not myc and ras replace v-abl in lymphoid transformation. Mol Cell Biol. 1990;10(8):4365–9.
Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36(5):453–61. https://doi.org/10.1038/ng1343. ng1343 [pii]
Rusconi F, Piazza R, Vagge E, Gambacorti-Passerini C. Bosutinib : a review of preclinical and clinical studies in chronic myelogenous leukemia. Expert Opin Pharmacother. 2014;15(5):701–10. https://doi.org/10.1517/14656566.2014.882898.
ten Hoeve J, Arlinghaus RB, Guo JQ, Heisterkamp N, Groffen J. Tyrosine phosphorylation of CRKL in Philadelphia+ leukemia. Blood. 1994;84(6):1731–6.
Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. https://doi.org/10.1186/1478-811X-7-13.
Seo J-H, Wood LJ, Agarwal A, O'Hare T, Elsea CR, Griswold IJ, et al. A specific need for CRKL in p210BCR-ABL-induced transformation of mouse hematopoietic progenitors. Cancer Res. 2010;70(18):7325–35. https://doi.org/10.1158/0008-5472.can-10-0607.
White D, Saunders V, Lyons AB, Branford S, Grigg A, To LB, et al. In vitro sensitivity to imatinib-induced inhibition of ABL kinase activity is predictive of molecular response in patients with de novo CML. Blood. 2005;106(7):2520–6. https://doi.org/10.1182/blood-2005-03-1103.
Li W, Ren Y, Si Y, Wang F, Yu J. Long non-coding RNAs in hematopoietic regulation. Cell Regen. 2018;7(2):27–32. https://doi.org/10.1016/j.cr.2018.08.001.
Guo G, Kang Q, Zhu X, Chen Q, Wang X, Chen Y, et al. A long noncoding RNA critically regulates Bcr-Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene. 2015;34(14):1768–79. https://doi.org/10.1038/onc.2014.131.
Guo G, Kang Q, Chen Q, Chen Z, Wang J, Tan L, et al. High expression of long non-coding RNA H19 is required for efficient tumorigenesis induced by Bcr-Abl oncogene. FEBS Lett. 2014;588(9):1780–6. https://doi.org/10.1016/j.febslet.2014.03.038.
Salomoni P, Condorelli F, Sweeney SM, Calabretta B. Versatility of BCR/ABL-expressing leukemic cells in circumventing proapoptotic BAD effects. Blood. 2000;96(2):676–84.
de Groot RP, Raaijmakers JA, Lammers JW, Koenderman L. STAT5-dependent CyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol Cell Biol Res Commun. 2000;3(5):299–305. https://doi.org/10.1006/mcbr.2000.0231. S1522472400902319 [pii]
Horita M, Andreu EJ, Benito A, Arbona C, Sanz C, Benet I, et al. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5-dependent expression of Bcl-xL. J Exp Med. 2000;191(6):977–84.
Neshat MS, Raitano AB, Wang HG, Reed JC, Sawyers CL. The survival function of the Bcr-Abl oncogene is mediated by bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 2000;20(4):1179–86.
Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–25.
Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–9. https://doi.org/10.1182/blood-2005-07-2947.
Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109(9):4016–9. https://doi.org/10.1182/blood-2006-11-057521.
Vetrie D, Helgason GV, Copland M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer. 2020;20(3):158–73. https://doi.org/10.1038/s41568-019-0230-9.
Chakraborty S, Stark JM, Sun CL, Modi H, Chen W, O'Connor TR, et al. Chronic myelogenous leukemia stem and progenitor cells demonstrate chromosomal instability related to repeated breakage-fusion-bridge cycles mediated by increased nonhomologous end joining. Blood. 2012;119(26):6187–97. https://doi.org/10.1182/blood-2011-05-352252.
Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. https://doi.org/10.1172/JCI35721.
Chomel JC, Sorel N, Guilhot J, Guilhot F, Turhan AG. BCR-ABL expression in leukemic progenitors and primitive stem cells of patients with chronic myeloid leukemia. Blood. 2012;119(12):2964–5.; author reply 5-6. https://doi.org/10.1182/blood-2011-12-396226.
Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood. 2012;119(2):530–9. https://doi.org/10.1182/blood-2010-08-303495.
Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–41. https://doi.org/10.1016/j.ccr.2007.11.003.
Arrigoni E, Del Re M, Galimberti S, Restante G, Rofi E, Crucitta S, et al. Concise review: chronic myeloid Leukemia: stem cell niche and response to pharmacologic treatment. Stem Cells Transl Med. 2018;7(3):305–14. https://doi.org/10.1002/sctm.17-0175.
Grassi S, Palumbo S, Mariotti V, Liberati D, Guerrini F, Ciabatti E, et al. The WNT pathway is relevant for the BCR-ABL1-independent resistance in chronic myeloid leukemia. Front Oncol. 2019;9:532. https://doi.org/10.3389/fonc.2019.00532.
Moon RT, Kohn AD, Ferrari GVD, Kaykas A. WNT and [beta]-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5(9):691–701.
Coluccia AM, Vacca A, Dunach M, Mologni L, Redaelli S, Bustos VH, et al. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 2007;26(5):1456–66. https://doi.org/10.1038/sj.emboj.7601485.
Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–67. https://doi.org/10.1056/NEJMoa040258. 351/7/657 [pii]
Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, et al. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10(4):412–24. https://doi.org/10.1016/j.stem.2012.02.017.
Irvine DA, Zhang B, Kinstrie R, Tarafdar A, Morrison H, Campbell VL, et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci Rep. 2016;6(1):25476. https://doi.org/10.1038/srep25476.
Sengupta A, Banerjee D, Chandra S, Banerji SK, Ghosh R, Roy R, et al. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and notch signaling in chronic myeloid leukemia progression. Leukemia. 2007;21(5):949–55. https://doi.org/10.1038/sj.leu.2404657.
Long B, Zhu H, Zhu C, Liu T, Meng W. Activation of the Hedgehog pathway in chronic myelogeneous leukemia patients. J Exp Clin Cancer Res. 2011;30(1):8. https://doi.org/10.1186/1756-9966-30-8.
Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411(6835):349–54. https://doi.org/10.1038/35077219.
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–9. https://doi.org/10.1038/nature07737.
Briscoe J, Therond PP. The mechanisms of hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14(7):416–29. https://doi.org/10.1038/nrm3598.
Sadarangani A, Pineda G, Lennon KM, Chun HJ, Shih A, Schairer AE, et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J Transl Med. 2015;13:98. https://doi.org/10.1186/s12967-015-0453-9.
Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17(11):722–35. https://doi.org/10.1038/nrm.2016.94.
Nakahara F, Sakata-Yanagimoto M, Komeno Y, Kato N, Uchida T, Haraguchi K, et al. Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood. 2010;115(14):2872–81. https://doi.org/10.1182/blood-2009-05-222836.
Aljedai A, Buckle A-M, Hiwarkar P, Syed F. Potential role of notch signalling in CD34+ chronic myeloid leukaemia cells: cross-talk between notch and BCR-ABL. PLoS One. 2015;10(4):e0123016-e. https://doi.org/10.1371/journal.pone.0123016.
Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–12. https://doi.org/10.1016/j.stem.2007.02.001.
Atfi A, Abecassis L, Bourgeade MF. Bcr-Abl activates the AKT/fox O3 signalling pathway to restrict transforming growth factor-beta-mediated cytostatic signals. EMBO Rep. 2005;6(10):985–91. https://doi.org/10.1038/sj.embor.7400501.
Hurtz C, Hatzi K, Cerchietti L, Braig M, Park E, Kim YM, et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med. 2011;208(11):2163–74. https://doi.org/10.1084/jem.20110304.
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–80. https://doi.org/10.1038/nature08734.
Carter BZ, Mak PY, Mu H, Zhou H, Mak DH, Schober W, et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med. 2016;8(355):355ra117. https://doi.org/10.1126/scitranslmed.aag1180.
Horita M, Andreu EJ, Benito A, Arbona C, Sanz C, Benet I, et al. Blockade of the Bcr-Abl kinase activity induces apoptosis of chronic myelogenous leukemia cells by suppressing signal transducer and activator of transcription 5–dependent expression of Bcl-XL. J Exp Med. 2000;191(6):977–84. https://doi.org/10.1084/jem.191.6.977.
Tzifi F, Economopoulou C, Gourgiotis D, Ardavanis A, Papageorgiou S, Scorilas A. The role of BCL2 family of apoptosis regulator proteins in acute and chronic leukemias. Adv Hematol. 2012;2012:524308. https://doi.org/10.1155/2012/524308.
Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DCS, et al. Bim and bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci. 2006;103(40):14907–12. https://doi.org/10.1073/pnas.0606176103.
Marum JE, Yeung DT, Purins L, Reynolds J, Parker WT, Stangl D, et al. ASXL1 and BIM germ line variants predict response and identify CML patients with the greatest risk of imatinib failure. Blood Adv. 2017;1(18):1369–81. https://doi.org/10.1182/bloodadvances.2017006825.
Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner KU, et al. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat Chem Biol. 2012;8(3):285–93. https://doi.org/10.1038/nchembio.775.
Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest. 2013;123(10):4144–57. https://doi.org/10.1172/JCI68951.
Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8(5):355–68. https://doi.org/10.1016/j.ccr.2005.10.015.
Ellis SL, Nilsson SK. The location and cellular composition of the hemopoietic stem cell niche. Cytotherapy. 2012;14(2):135–43. https://doi.org/10.3109/14653249.2011.630729.
Ema H, Suda T. Two anatomically distinct niches regulate stem cell activity. Blood. 2012;120(11):2174–81. https://doi.org/10.1182/blood-2012-04-424507.
Hazlehurst LA, Argilagos RF, Dalton WS. Beta1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Br J Haematol. 2007;136(2):269–75. https://doi.org/10.1111/j.1365-2141.2006.06435.x.
Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood. 2013;121(10):1824–38. https://doi.org/10.1182/blood-2012-02-412890.
Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–64. https://doi.org/10.1172/JCI41246.
Skorski T. Genetic mechanisms of chronic myeloid leukemia blastic transformation. Curr Hematol Malig Rep. 2012;7(2):87–93. https://doi.org/10.1007/s11899-012-0114-5.
Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76–94. https://doi.org/10.1159/000046636.
Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7(6):441–53. https://doi.org/10.1038/nrc2147.
Barnes DJ, Palaiologou D, Panousopoulou E, Schultheis B, Yong AS, Wong A, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65(19):8912–9. https://doi.org/10.1158/0008-5472.CAN-05-0076.
Gaiger A, Henn T, Horth E, Geissler K, Mitterbauer G, Maier-Dobersberger T, et al. Increase of bcr-abl chimeric mRNA expression in tumor cells of patients with chronic myeloid leukemia precedes disease progression. Blood. 1995;86(6):2371–8.
Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21(5):926–35. https://doi.org/10.1038/sj.leu.2404609.
Marega M, Piazza RG, Pirola A, Redaelli S, Mogavero A, Iacobucci I, et al. BCR and BCR-ABL regulation during myeloid differentiation in healthy donors and in chronic phase/blast crisis CML patients. Leukemia. 2010;24(8):1445–9. https://doi.org/10.1038/leu.2010.101.
Andrews DF 3rd, Collins SJ. Heterogeneity in expression of the bcr-abl fusion transcript in CML blast crisis. Leukemia. 1987;1(10):718–24.
Chang JS, Santhanam R, Trotta R, Neviani P, Eiring AM, Briercheck E, et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2 dependent suppression of C/EBPalpha-driven myeloid differentiation. Blood. 2007;110(3):994–1003. https://doi.org/10.1182/blood-2007-03-078303.
Amos TA, Lewis JL, Grand FH, Gooding RP, Goldman JM, Gordon MY. Apoptosis in chronic myeloid leukaemia: normal responses by progenitor cells to growth factor deprivation, X-irradiation and glucocorticoids. Br J Haematol. 1995;91(2):387–93.
Bedi A, Barber JP, Bedi GC, el-Deiry WS, Sidransky D, Vala MS, et al. BCR-ABL-mediated inhibition of apoptosis with delay of G2/M transition after DNA damage: a mechanism of resistance to multiple anticancer agents. Blood. 1995;86(3):1148–58.
Dierov J, Sanchez PV, Burke BA, Padilla-Nash H, Putt ME, Ried T, et al. BCR/ABL induces chromosomal instability after genotoxic stress and alters the cell death threshold. Leukemia. 2009;23(2):279–86. https://doi.org/10.1038/leu.2008.308.
Koptyra M, Cramer K, Slupianek A, Richardson C, Skorski T. BCR/ABL promotes accumulation of chromosomal aberrations induced by oxidative and genotoxic stress. Leukemia. 2008;22(10):1969–72. https://doi.org/10.1038/leu.2008.78.
Slupianek A, Falinski R, Znojek P, Stoklosa T, Flis S, Doneddu V, et al. BCR-ABL1 kinase inhibits uracil DNA glycosylase UNG2 to enhance oxidative DNA damage and stimulate genomic instability. Leukemia. 2013;27(3):629–34. https://doi.org/10.1038/leu.2012.294.
Deutsch E, Dugray A, AbdulKarim B, Marangoni E, Maggiorella L, Vaganay S, et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood. 2001;97(7):2084–90.
Skorski T. Genomic instability: the cause and effect of BCR/ABL tyrosine kinase. Curr Hematol Malig Rep. 2007;2(2):69–74. https://doi.org/10.1007/s11899-007-0010-6.
Skorski T. BCR/ABL, DNA damage and DNA repair: implications for new treatment concepts. Leuk Lymphoma. 2008;49(4):610–4. https://doi.org/10.1080/03093640701859089.
Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21(6):853–63. https://doi.org/10.1016/j.immuni.2004.11.006.
Guerzoni C, Bardini M, Mariani SA, Ferrari-Amorotti G, Neviani P, Panno ML, et al. Inducible activation of CEBPB, a gene negatively regulated by BCR/ABL, inhibits proliferation and promotes differentiation of BCR/ABL-expressing cells. Blood. 2006;107(10):4080–9. https://doi.org/10.1182/blood-2005-08-3181.
Eiring AM, Harb JG, Neviani P, Garton C, Oaks JJ, Spizzo R, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140(5):652–65. https://doi.org/10.1016/j.cell.2010.01.007.
Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. https://doi.org/10.1016/j.cell.2012.03.003.
Preisler HD, Sato H, Yang PM, Wilson M, Kaufman C, Watt R. Assessment of c-myc expression in individual leukemic cells. Leuk Res. 1988;12(6):507–16.
Cleveland JL, Dean M, Rosenberg N, Wang JY, Rapp UR. Tyrosine kinase oncogenes abrogate interleukin-3 dependence of murine myeloid cells through signaling pathways involving c-myc: conditional regulation of c-myc transcription by temperature-sensitive v-abl. Mol Cell Biol. 1989;9(12):5685–95.
Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell. 1992;70(6):901–10. doi:0092-8674(92)90241-4 [pii]
Bissonnette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992;359(6395):552–4. https://doi.org/10.1038/359552a0.
Sanchez-Garcia I, Grutz G. Tumorigenic activity of the BCR-ABL oncogenes is mediated by BCL2. Proc Natl Acad Sci U S A. 1995;92(12):5287–91.
Birchenall-Roberts MC, Yoo YD, Bertolette DC 3rd, Lee KH, Turley JM, Bang OS, et al. The p120-v-Abl protein interacts with E2F-1 and regulates E2F-1 transcriptional activity. J Biol Chem. 1997;272(14):8905–11.
Stewart MJ, Litz-Jackson S, Burgess GS, Williamson EA, Leibowitz DS, Boswell HS. Role for E2F1 in p210 BCR-ABL downstream regulation of c-myc transcription initiation. Studies in murine myeloid cells. Leukemia. 1995;9(9):1499–507.
Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene. 2002;21(47):7137–46. https://doi.org/10.1038/sj.onc.1205942.
Notari M, Neviani P, Santhanam R, Blaser BW, Chang JS, Galietta A, et al. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood. 2006;107(6):2507–16. https://doi.org/10.1182/blood-2005-09-3732.
Reavie L, Buckley SM, Loizou E, Takeishi S, Aranda-Orgilles B, Ndiaye-Lobry D, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23(3):362–75. https://doi.org/10.1016/j.ccr.2013.01.025.
Delgado MD, Leon J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010;1(6):605–16. https://doi.org/10.1177/1947601910377495.
Pant V, Quintas-Cardama A, Lozano G. The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood. 2012;120(26):5118–27. https://doi.org/10.1182/blood-2012-05-356014.
Guinn BA, Mills KI. p53 mutations, methylation and genomic instability in the progression of chronic myeloid leukaemia. Leuk Lymphoma. 1997;26(3–4):211–26. https://doi.org/10.3109/10428199709051771.
Stuppia L, Calabrese G, Peila R, Guanciali-Franchi P, Morizio E, Spadano A, et al. p53 loss and point mutations are associated with suppression of apoptosis and progression of CML into myeloid blastic crisis. Cancer Genet Cytogenet. 1997;98(1):28–35. doi:S016546089600413X [pii]
Sionov RV, Moallem E, Berger M, Kazaz A, Gerlitz O, Ben-Neriah Y, et al. C-Abl neutralizes the inhibitory effect of Mdm2 on p53. J Biol Chem. 1999;274(13):8371–4.
Stoklosa T, Slupianek A, Datta M, Nieborowska-Skorska M, Nowicki MO, Koptyra M, et al. BCR/ABL recruits p53 tumor suppressor protein to induce drug resistance. Cell Cycle. 2004;3(11):1463–72. doi:1229 [pii]
Trotta R, Vignudelli T, Candini O, Intine RV, Pecorari L, Guerzoni C, et al. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell. 2003;3(2):145–60. doi:S1535610803000205 [pii]
Wendel HG, de Stanchina E, Cepero E, Ray S, Emig M, Fridman JS, et al. Loss of p53 impedes the antileukemic response to BCR-ABL inhibition. Proc Natl Acad Sci U S A. 2006;103(19):7444–9. https://doi.org/10.1073/pnas.0602402103.
Honda H, Ushijima T, Wakazono K, Oda H, Tanaka Y, Aizawa S, et al. Acquired loss of p53 induces blastic transformation in p210(bcr/abl)-expressing hematopoietic cells: a transgenic study for blast crisis of human CML. Blood. 2000;95(4):1144–50.
Velasco-Hernandez T, Vicente-Duenas C, Sanchez-Garcia I, Martin-Zanca D. p53 restoration kills primitive leukemia cells in vivo and increases survival of leukemic mice. Cell Cycle. 2013;12(1):122–32. https://doi.org/10.4161/cc.23031.
Peterson LF, Mitrikeska E, Giannola D, Lui Y, Sun H, Bixby D, et al. p53 stabilization induces apoptosis in chronic myeloid leukemia blast crisis cells. Leukemia. 2011;25(5):761–9. https://doi.org/10.1038/leu.2011.7.
Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122(17):3034–44. https://doi.org/10.1182/blood-2013-04-495374.
Yuan H, Wang Z, Li L, Zhang H, Modi H, Horne D, et al. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood. 2012;119(8):1904–14. https://doi.org/10.1182/blood-2011-06-361691.
Li L, Wang L, Wang Z, Ho Y, McDonald T, Holyoake TL, et al. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell. 2012;21(2):266–81. https://doi.org/10.1016/j.ccr.2011.12.020.
Wang Z, Yuan H, Roth M, Stark JM, Bhatia R, Chen WY. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene. 2013;32(5):589–98. https://doi.org/10.1038/onc.2012.83.
Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2013;110(3):1041–6. https://doi.org/10.1073/pnas.1213021110.
Steinman RA, Yang Q, Gasparetto M, Robinson LJ, Liu X, Lenzner DE, et al. Deletion of the RNA-editing enzyme ADAR1 causes regression of established chronic myelogenous leukemia in mice. Int J Cancer. 2013;132(8):1741–50. https://doi.org/10.1002/ijc.27851.
XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, et al. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci U S A. 2009;106(42):17763–8. https://doi.org/10.1073/pnas.0903324106.
Iwama A. Polycomb repressive complexes in hematological malignancies. Blood. 2017;130(1):23–9. https://doi.org/10.1182/blood-2017-02-739490.
Mohty M, Yong ASM, Szydlo RM, Apperley JF, Melo JV. The polycomb group BMI1 gene is a molecular marker for predicting prognosis of chronic myeloid leukemia. Blood. 2007;110(1):380–3. https://doi.org/10.1182/blood-2006-12-065599.
Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, et al. Chronic myelogenous leukemia- initiating cells require Polycomb group protein EZH2. Cancer Discov. 2016;6(11):1237–47. https://doi.org/10.1158/2159-8290.CD-15-1439.
Ko TK, Javed A, Lee KL, Pathiraja TN, Liu X, Malik S, et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood. 2020;135(26):2337–53. https://doi.org/10.1182/blood.2020004834.
Skorski T. Oncogenic tyrosine kinases and the dna-damage response. Nat Rev Cancer. 2002;2(5):351–60. https://doi.org/10.1038/nrc799.
Giotopoulos G, van der Weyden L, Osaki H, Rust AG, Gallipoli P, Meduri E, et al. A novel mouse model identifies cooperating mutations and therapeutic targets critical for chronic myeloid leukemia progression. J Exp Med. 2015;212(10):1551–69. https://doi.org/10.1084/jem.20141661.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid Leukemia. N Engl J Med. 2016;374(23):2209–21. https://doi.org/10.1056/NEJMoa1516192.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y-L, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15. https://doi.org/10.1056/NEJMoa1403088.
Guglielmelli P, Lasho TL, Rotunno G, Mudireddy M, Mannarelli C, Nicolosi M, et al. MIPSS70: mutation-enhanced international prognostic Score system for transplantation-age patients with primary myelofibrosis. J Clin Oncol. 2018;36(4):310–8. https://doi.org/10.1200/jco.2017.76.4886.
Branford S, Wang P, Yeung DT, Thomson D, Purins A, Wadham C, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132(9):948–61. https://doi.org/10.1182/blood-2018-02-832253.
Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25(3):557–60. http://www.nature.com/leu/journal/v25/n3/suppinfo/leu2010298s1.html.
Branford S, Kim DDH, Apperley JF, Eide CA, Mustjoki S, Ong ST, et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia. 2019;33(8):1835–50.
Thomson DW, Shahrin NH, Wang PPS, Wadham C, Shanmuganathan N, Scott HS, et al. Aberrant RAG-mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia. 2020;34:2051–63. https://doi.org/10.1038/s41375-020-0751-y.
Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. 2010;2(3):98–110. https://doi.org/10.1002/emmm.201000062.
Halbach S, Rigbolt KT, Wohrle FU, Diedrich B, Gretzmeier C, Brummer T, et al. Alterations of Gab2 signalling complexes in imatinib and dasatinib treated chronic myeloid leukaemia cells. Cell Commun Signal. 2013;11(1):30. https://doi.org/10.1186/1478-811X-11-30.
Winter GE, Rix U, Carlson SM, Gleixner KV, Grebien F, Gridling M, et al. Systems-pharmacology dissection of a drug synergy in imatinib-resistant CML. Nat Chem Biol. 2012;8(11):905–12. https://doi.org/10.1038/nchembio.1085.
Agirre X, Jimenez-Velasco A, San Jose-Eneriz E, Garate L, Bandres E, Cordeu L, et al. Down-regulation of hsa-miR-10a in chronic myeloid leukemia CD34+ cells increases USF2-mediated cell growth. Mol Cancer Res. 2008;6(12):1830–40. https://doi.org/10.1158/1541-7786.MCR-08-0167.
Machova Polakova K, Lopotova T, Klamova H, Burda P, Trneny M, Stopka T, et al. Expression patterns of microRNAs associated with CML phases and their disease related targets. Mol Cancer. 2011;10:41. https://doi.org/10.1186/1476-4598-10-41.
Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405. https://doi.org/10.1016/j.tibtech.2013.04.004.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Ethics declarations
The authors declare that they have no conflict of interest for the writing of this chapter.
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Shanmuganathan, N., Chereda, B., Melo, J.V. (2021). The Biology and Pathogenesis of Chronic Myeloid Leukaemia. In: Hehlmann, R. (eds) Chronic Myeloid Leukemia. Hematologic Malignancies. Springer, Cham. https://doi.org/10.1007/978-3-030-71913-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-71913-5_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-71912-8
Online ISBN: 978-3-030-71913-5
eBook Packages: MedicineMedicine (R0)