
Abstract
The pathophysiology of myocardial ischemia is complex and multifactorial, since, beyond epicardial coronary obstruction, other mechanisms may be involved, including vasospasm, microvascular dysfunction, and metabolic inefficiency. Although various drugs have been designed to target such pathways, anti-ischemic/antianginal therapy seems to have an overall neutral effect on prognosis, apart from beta-blockers in patients with heart failure of ischemic etiology. Therefore, according to the current guidelines, symptomatic relief and improvement of the quality of life remain the main indications and goals of anti-ischemic medications. It is however conceivable that tailoring therapeutic approaches either in monotherapy or in combination on individual pathophysiological mechanisms, as well as on patient’s specific cardiac and noncardiac comorbidities, may provide further benefits. Novel molecules (e.g., vasodilators, metabolic modulators, angiogenic factors) are emerging from preclinical and preliminary clinical studies and may provide novel opportunities to be tested in the context of adequately powered randomized controlled trials in the near future, either to treat refractory angina or to improve hard outcomes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Anti-ischemic
- Antianginal
- Nitrates
- Calcium channel blockers
- Beta-blockers
- Nicorandil
- Trimetazidine
- Ranolazine
Few things are more distressing to a physician than to stand beside a suffering patient who is anxiously looking to him for that relief from pain which he feels himself utterly unable to afford. His sympathy for the sufferer, and the regret he feels for the impotence of his art, engrave the picture indelibly on his mind, and serve as a constant and urgent stimulus in his search after the causes of the pain, and the means by which it may be alleviated.
—T. Lauder Brunton, July 27, 1867.
1 Introduction
Chronic myocardial ischemia may be a consequence of obstructive coronary artery disease (CAD), secondary to luminal stenosis and reduced coronary flow reserve, and/or of other conditions, such as vasospasm, microvascular dysfunction, and energetic mismatch [1,2,3]. According to the latest European guidelines, whenever a macro- or microvascular coronary disorder is documented, the clinical condition could be denoted as chronic coronary syndrome (CCS) [1].
Of note, myocardial ischemia is often but not always accompanied by chest pain or angina. Indeed, angina is only the final clinical manifestation of a series of pathophysiological changes induced by myocardial energetic unbalance and named the “ischemic cascade,” including diminished left ventricular compliance, decreased contractility, increased left ventricular end-diastolic pressure, and electrocardiographic changes [4]. The threshold of ischemia associated with symptoms may vary among patients and within the same patient, or may also be absent in conditions of neuropathic functional denervation (i.e., diabetes) [5, 6]: therefore, episodes of silent ischemia may occur.
Although observational studies suggested that silent myocardial ischemia could compromise contractile function and electrical stability, with negative hemodynamic consequences [7], and life-threatening arrhythmias [8,9,10,11], there is currently no evidence showing a prognostic benefit of anti-ischemic therapies in this context. Therefore, current guidelines discourage functional testing in asymptomatic individuals [1] and highlight that the main aim of medical therapy in CCS is to target angina rather than ischemia [1].
As for symptomatic patients, while meta-analyses show that all antianginal drugs are similarly efficacious in alleviating angina and increasing exercise tolerance, evidence for improvement in event-free survival is generally missing, apart from beta-blockers (BBs) in patients with heart failure and reduced ejection fraction, and nicorandil for angina-related hospitalization [12, 13].
Nonetheless, treating ischemia may prove value in specific subsets (e.g., in the presence of an extensive ischemic burden and/or of left ventricular systolic dysfunction) [1], and this topic still remains a matter of debate [14, 15].
2 Pathophysiological Mechanisms of Ischemia and Potential Targets
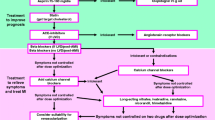
As detailed in the chapter “Pathophysiology of Ischemic Syndromes in Coronary Artery Disease”, in the last century, a plethora of elegant physiological and pharmacological studies have outlined the heterogenous pathophysiological determinants of myocardial ischemia [16]. Whereas an impaired oxygen/nutrients’ supply due to either coronary (e.g., epicardial artery stenosis, vasospasm, microvascular dysfunction) or noncoronary causes (e.g., anemia, hypoxia, toxic and metabolic disorders) and an unbalanced increase in energetic demand (secondary to increased myocardial contractility, wall stress, or heart rate) are key determinants of myocardial ischemia, more subtle abnormalities in cardiomyocyte metabolism have been observed as well [16,17,18,19]. Importantly, these mechanisms are not exclusive but could be variously intertwined and declined in the single patient, fostering the research for a tailored and integrated therapeutic approach (Fig. 1) [20, 21].

Pathophysiology-driven pharmacological management of myocardial ischemia. ACE-i angiotensin-converting enzyme inhibitors; CCBs calcium channel blockers; DHP dihydropyridines
Beyond its conduit function, coronary circulation is responsible for modulating myocardial blood flow to match energetic demand across a wide spectrum of physiological conditions, through the mechanisms of autoregulation and autonomic control [22, 23]. Accordingly, in conditions of increased myocardial requests (e.g., physical exercise, emotional stress), coronary flow increases proportionally [24]. On the other hand, in the presence of a significant luminal obstruction in an epicardial artery, the downstream flow is usually maintained at rest at the price of exhausting the vasodilatory reserve, so that myocardial ischemia may emerge when a further increase in energetic demand is not adequately counterbalanced [25]. Nevertheless, a certain degree of vasodilation may be obtained through some drugs (e.g., nitric oxide (NO) donors, nicorandil, and calcium channel blockers (CCBs)), which are therefore effective anti-ischemic agents in this setting [26,27,28].
Although such a hydraulic mechanism has long been considered the fundament of chronic myocardial ischemia and angina, it is nowadays established that this may occur also in the absence of obstructive CAD and persist also after successful revascularization [29,30,31]. In this regard, vasospasm has been identified as a potential contributor. Although the so-called resting vasospastic or Prinzmetal angina, as originally described [32], represents a rare condition, macro- and/or microvascular spasm may be frequently observed independently of the concomitant atherosclerotic burden [33, 34]. A paradoxical vasoconstrictive response to acetylcholine, which is normally associated with a NO-mediated vasodilation, characterizes coronary vasospasm, implying a pivotal role of endothelial dysfunction [35, 36]. In this context, CCBs (both dihydropyridines—DHP and non-DHP) are a well-established first-line therapy [37, 38], while other vasodilators such as nitrates and nicorandil represent possible alternatives or add-on therapies in refractory cases [39, 40]. On the contrary, BBs are usually not recommended since vasospasm could be exacerbated by the blockage of the “vasodilative” β2-adrenergic receptors and a paradoxical overstimulation of the “vasoconstrictive” α1-adrenergic receptors on coronaries’ walls [41].
Microvascular disease may underlie myocardial ischemia and angina, also in the absence of detectable epicardial coronary stenosis and vasospasm or other cardiac conditions, due to endothelial and autonomic dysfunction, exaggerated vasoconstrictive and nociceptive responses, and pro-inflammatory signals [3, 42, 43]. Although NO-mediated pathways and Ca2+ inflow modulate microvascular tone, too [44], both nitrates and CCBs are poorly effective on microvascular angina [45, 46], particularly when no vasospasm could be detected [47]. Conversely, more promising findings have been obtained for angiotensin-converting enzyme inhibitors, since angiotensin II is a direct modulator of microvascular tone [48] and for xanthines, which may favor flow redistribution toward ischemic areas (by inhibiting the arteriolar vasodilator effects of adenosine) and antagonize adenosine-mediated pain afferents, relieving angina [49].
As anticipated, also the reduction of myocardial energetic demand is an effective strategy to alleviate ischemia and angina and may be achieved by lowering blood pressure and, most importantly, heart rate [50]. Beyond reducing oxygen consumption, a lower heart rate prolongs coronary diastolic perfusion, so that the net effect of negative chronotropic drugs may be an improved contractility of ischemic regions, despite their possible negative inotropic action [51]. Therefore, BBs and non-DHP CCBs play a central role among antianginal therapies [52], while their anti-ischemic efficacy in asymptomatic patients is still controversial [50, 53]. Alternatively, a lower heart rate may be achieved by inhibiting the If current with ivabradine, considered a second-line antianginal drug, with no negative inotropic or lusitropic effect [52, 54].
Finally, further targets for anti-ischemic therapies have been identified by shifting the focus on the cardiomyocyte. Indeed, whereas its energetic metabolism is primarily based on mitochondrial oxidation of fatty acids and other substrates (e.g., glucose, ketones) are less utilized in physiological condition [55], in the presence of ischemia, such pathways may be corrupted and anaerobic glycolysis favored, resulting in acidosis, Na+ and Ca2+ overload, and decreased cardiac function [19, 55]. Promoting the shift toward a more efficient energetic asset has therefore emerged as an intriguing strategy and may be achieved by favoring glucose instead of fatty acid utilization. As detailed below in this chapter, two anti-ischemic drugs, i.e., trimetazidine and ranolazine, act in modulating these pathways [56, 57].
3 Medical Therapy of Ischemic Heart Disease
Historically, the first class of drugs that have been used as antianginal were nitrates, followed almost a century after by BBs, then CCBs, trimetazidine, nicorandil, ivabradine, and, finally, ranolazine [58,59,60,61,62,63,64] (Fig. 2).

Antianginal medications through the decades
Considering the number of antianginal drugs now available for clinical use, it may be difficult to identify the optimal treatment. Ideally, the best option should control symptoms, improve quality of life, maximize patient’s adherence, and minimize drug-related side effects. Furthermore, as suggested by the current guidelines, the therapeutic choice should also be adapted to the patient’s characteristics, such as cardiac and noncardiac comorbidities, to improve (soft) outcomes and avoid undesirable side effects [1, 18, 19]. Furthermore, targeting the pathophysiological substrate of myocardial ischemia may further improve therapeutic efficacy [65].
Antianginal drugs are classified as being first-line (BBs, CCBs, and short-acting nitrates on request) or second-line (long-acting nitrates, nicorandil, ivabradine, trimetazidine, and ranolazine) [1]. Second-line medications are usually destinated to patients who have contraindications, do not tolerate, or remain symptomatic despite first-line agents. However, no randomized clinical trial (RCT) has shown superiority of first-line over second-line treatments [1, 21]. A recent systematic review and meta-analysis has also showed that no one antianginal drug is superior to another and that equivalence has only been demonstrated for the use of BBs (atenolol), DHP CCBs (amlodipine, nifedipine), and If current inhibitors (ivabradine) [21].
Another meta-analysis supports the combination of DHP CCBs with BBs over monotherapy and to ranolazine added to either BBs or CCBs [66]. According to the same meta-analysis, adding long-acting nitrates and trimetazidine may be effective as well, although the evidence seems more scattered [66]. Similarly, ivabradine was shown to increase exercise time, angina attacks, and use on nitrates when added to BBs in the ASSOCIATE [67] and ADDITIONS [68] trials. There are no significant data for nicorandil as far as combination therapy is concerned [69].
Some authors have also highlighted that each drug/combination may have beneficial or detrimental effects on patients’ specific characteristics, and thus a “diamond” approach similar to that employed in hypertension (i.e., leaving physician free to choose the most appropriate drug/combination according to patient-specific needs) has been proposed [18]. Considering the mechanisms of action, association of BBs or ivabradine with non-DHP CCBs is not recommended, whereas other combinations (i.e., nitrates/nicorandil with CCBs or ranolazine with trimetazidine) might be partially redundant, unless specific pathophysiology is considered (e.g., vasospastic angina) [18].
3.1 Vasodilators
3.1.1 Nitrates
Short- and long-acting nitrates represent an established class of antianginal drugs, whose effects depend on the release of NO through an enzymatic process (i.e., denitrification) taking place in the vessel walls [70]. By stimulating the soluble guanylyl cyclase of smooth muscle cells, NO promotes the production of cyclic guanosine monophosphate, leading to membrane hyperpolarization and reduction of Ca2+ inflow, with consequent vasodilation [70]. Whereas at low doses nitrates act mostly on the venous system (hence reducing pre-load), arterial vasodilation occurs at higher doses, favoring epicardial coronaries and collateral blood flow perfusion (even in the presence of luminal obstruction) and reducing post-load [71]. Although the potential reduction of myocardial oxygen consumption secondary to reduced pre- and post-load may be partially counterbalanced by an autonomic mediated increase in heart rate, the concomitant use of negative chronotropic drugs (e.g., BBs) may result in synergetic anti-ischemic effect [72]. Furthermore, thanks to their NO-dependent action, nitrates are also effective in relieving vasospastic [73] but not microvascular angina, probably because of the lower sensitivity of resistance arterioles to such signals at clinically used dosages [74].
As recommended by the current guidelines, short-acting nitrates are the first-line therapy to relieve effort angina (class of recommendation (CoR) I, level of evidence (LoE) B), while long-acting nitrates are second-line choices in the long term compared to BBs and non-DHP CCBs (CoR IIa, LoE B) [52]. Indeed, several RCTs have examined the efficacy of nitrates, and in a meta-analysis of 51 studies including a total of 3595 patients with stable angina, their long-term administration was found to be beneficial in preventing angina and improving exercise tolerance but not the overall quality of life [75]. On the other hand, only a few studies have evaluated the survival benefits of chronic nitrate administration in different subsets, yielding substantially neutral results [76,77,78].
Finally, because of their intense systemic vasodilator action, the use of nitrates may exacerbate various adverse effects, including headache, flushing, and hypotension, while they are not indicated in patients with intraventricular obstruction, severe aortic or mitral stenosis, and constrictive pericarditis, and they should be used with caution in concomitance with other vasodilators [71]. Another limitation for the use of nitrates is the risk of tolerance, with a reduction in their anti-ischemic efficacy [79], so that nitrate-free or low-nitrate intervals are suggested in patients on chronic therapy (CoR IIa, LoE B) [52]. Although the underlying mechanisms are still to be completely clarified, oxidative stress may contribute [79], while the use of alternative molecules may overcome such problem [80].
3.1.2 Nicorandil
Nicorandil is a nicotinamide-nitrate ester holding anti-ischemic properties related to its NO-donor capacity and to a direct stimulation of adenosine triphosphate-sensitive K+ channels on arterial walls, together leading to vasodilatation, but also to possible metabolic effects [81,82,83]. Moreover, nicorandil may be effective in alleviating vasospasm [84], and growing evidence sustains a possible role also in the context of microvascular dysfunction, even though further research seems necessary to confirm such assumption and to clarify the biological mechanisms involved [85, 86].
The use of nicorandil in patients with stable angina has been evaluated in various RCTs, demonstrating good efficacy [20, 52]. Most notably, among 5126 patients with stable angina, nicorandil, compared to placebo, significantly reduced a composite endpoint of cardiovascular events, but not cardiac death or nonfatal myocardial infarction [87]. Therefore, it is considered a second-line treatment to reduce angina frequency and improve exercise tolerance (CoR IIa, LoE B) [52].
Despite the similar mechanisms of action, the use of nicorandil is associated with a lower risk of tolerance than nitrates, whereas nausea, vomiting, mucosal ulcerations, and, most importantly, headache are potential adverse effects, which could affect therapeutic adherence [52, 87].
3.1.3 Dihydropyridine Calcium Channel Blockers
CCBs are a heterogenous class of drugs, characterized by the inhibition of high-voltage-activated L-type Ca2+ channels on vascular smooth muscle cells and cardiomyocytes [88]. DHP CCBs (e.g., amlodipine, nicardipine, nifedipine) act more specifically on vascular channels, causing an intense coronary and systemic vasodilation, while they do not act on cardiomyocytes [89].
Beyond vasodilatation, DHP CCBs reduce myocardial oxygen demand by lowering systemic blood pressure (i.e., cardiac post-load) [88, 90] and are effective also in the case of vasospastic [91] and microvascular angina [92, 93], whereas the reflex increase in heart rate could be blunted by the use of BBs, further improving their anti-ischemic efficacy (CoR IIa, LoE B) [52, 94, 95]. In patients with stable angina, the use of nifedipine was associated with a reduced need for coronary angiography and intervention, despite no difference in cardiac death or myocardial infarction [96], while the use of amlodipine reduced the risk of adverse cardiovascular events and of atherosclerosis progression [97].
Although headache, ankle swelling, and hypotension represent possible side effects [89], DHP CCBs are usually well tolerated and represent first-line antianginal therapies (CoR I, LoE A) [52].
3.2 Drugs Reducing Myocardial Oxygen Consumption
3.2.1 Non-dihydropyridine Calcium Channel Blockers
Differently from DHP CCB, diltiazem and verapamil (i.e., non-DHP CCB) show a higher selectivity for myocardial than for vascular Ca2+ channels, and their anti-ischemic efficacy mostly relies upon the reduction of myocardial oxygen demand secondary to a negative inotropic and heart rate-dependent chronotropic effects [88, 89, 98]. The use of these drugs is therefore recommended to control heart rate and symptoms in patients with stable effort angina (CoR I, LoE A) [1], while they are routinely used also in patients with vasospastic angina [99] and may be effective in the case of microvascular dysfunction [100], where ongoing studies (e.g., NCT04777045) are expected to confirm such findings.
Although generally safe, RCTs failed to show any survival benefit with the use of diltiazem [101], while verapamil was shown to reduce adverse events only in patients after myocardial infarction and without heart failure [102]. Moreover, they share similar side effects with DHP CCBs, and they should be used with caution in patients at risk of sinus bradycardia or atrioventricular blocks and in those with systolic dysfunction [52, 88, 89].
3.2.2 Beta-Blockers
BBs are very effective antianginal therapies, as demonstrated by the high rate of patients free from anginal events after optimization of medical therapy in both the COURAGE (87% receiving BBs) [103] and the ORBITA (77% receiving BBs) [104] trials, and thus represent a first-line treatment to control heart rate and symptoms in patients with stable effort angina (CoR I, LoE A) [1].
Similarly to non-DHP CCBs, BBs’ antianginal action mainly relies on the reduction of myocardial oxygen demand [19]. Their primary action is to decrease heart rate and thus to increase diastolic duration and coronary perfusion, in particular blood flow per heartbeat [105]. Although BBs have negative inotropic effects (less than non-DHP CCBs), by decreasing oxygen consumption in the healthy myocardium, they may increase perfusion to the post-stenotic myocardium and also its regional contractility [106, 107], but only if heart rate reduction is achieved [51]. However, they may also favor coronary vasoconstriction by blocking β2-adrenergic receptors, so β1-selective compounds, or BBs with vasodilatation capability, such as carvedilol—an α-β-blocker [108] [109]—or nebivolol, through NO release [110], are usually preferred in the treatment of CCS, unless a vasospastic component is hypothesized. In that case, BBs should be used with caution (i.e., low dose or adding a vasodilator) or avoided, similarly to other conditions such as asthma, baseline bradycardia, or evidence of atrioventricular conduction abnormalities.
Although several studies have investigated the prognostic effects of BBs, according to the main RCTs and meta-analyses [111], this seems limited to patients receiving BBs early after myocardial infarction [112] or with left ventricular systolic dysfunction [113].
3.2.3 Ivabradine
As non-DHP CCBs and BBs, also ivabradine’s antianginal capacity derives from a reduction of heart rate. This is obtained through a selective inhibition of the If (or “funny,” inward Na+-K+) current of the sinoatrial node [114, 115], which has a key role in the generation of spontaneous depolarization of pacemaker cells and in mediating the autonomic control of heart rate [116]. By inhibiting If current, ivabradine causes a decrease in the slope of depolarization, lowering heart rate [105] and promoting a proportionate improvement in ischemic regional blood flow and contractile function [117].
The risk of bradycardia with ivabradine is low, since its effect is heart rate dependent by acting on open channels [118]. Furthermore, ivabradine does not affect myocardial work or vascular tone, favoring its use when such effects would be undesirable (i.e., patients with hypotension) [119].
Despite those premises, the increased risk of cardiovascular death and nonfatal myocardial infarction observed in patients with CCS treated with ivabradine in the SIGNIFY trial [120] raised some concerns, which could have been at least partially explained by the concomitant use of non-DHP CCBs (which may inhibit the ivabradine-metabolizing cytochrome p450, i.e., CYP3A4) causing bradycardia in a relevant proportion of patients. Hence, this association should be avoided. On the contrary, no safety concerns were observed when administering ivabradine with BBs, in the BEAUTIFUL trial, in which ivabradine was however shown not to improve outcome in patients with CCS and left ventricular systolic dysfunction [121], apart from decreasing the risk of hospitalization for myocardial infarction or coronary revascularization in patients with a heart rate ≥70 bpm.
Of note, ivabradine may also be useful in improving symptoms in patients with microvascular dysfunction [122], even though future studies should confirm such findings. Finally, outside the CCS scenario, ivabradine was found to decrease the combined outcome of cardiovascular mortality and hospitalization (mainly driven by reduced hospitalizations for worsening heart failure) in patients with heart failure and reduced ejection fraction (89% on BBs) [123].
3.3 Myocyte Metabolism Modulators
3.3.1 Trimetazidine
Trimetazidine increases cellular tolerance to ischemia by decreasing fatty acid metabolism through the inhibition of 3-ketoacyl CoA thiolase, shifting myocardial metabolism toward pyruvate oxidation [56, 61]. Trimetazidine also stimulates glucose metabolism and insulin sensitivity [124].
The antianginal/anti-ischemic effects of trimetazidine are similar to those obtained with BBs or CCBs [125]. Of note, the absence of relevant hemodynamic consequences [126] prompts the use of this molecule as a second-line treatment in patients that do not tolerate, have contraindications to, or whose symptoms are not adequately controlled by BBs, CCBs, and long-acting nitrates (CoR IIa, LoE B) [1]. When used in combination with metoprolol, trimetazidine was shown to decrease angina and increase exercise duration and time to ST-segment depression compared to metoprolol alone in 426 patients with stable, effort-induced angina and documented CAD (TRIMPOL II trial) [127]. Similar findings were obtained adding trimetazidine to atenolol in the VASCO trial [128] or to diltiazem [129]. The overall beneficial effect of trimetazidine on anginal attacks, daily use of nitrates, exercise duration, and time to ST-segment depression has been confirmed also in three meta-analyses [130,131,132]. Trimetazidine prolonged exercise time and time to ST depression also in patients with microvascular angina in a small placebo-controlled RCT [133]. On the contrary, ranolazine seems ineffective on major cardiovascular adverse events or angina recurrence in patients who have undergone successful percutaneous coronary intervention from the ATPCI trial (n = 6007) [134]. Trimetazidine remains contraindicated in Parkinson’s disease and motion disorders, such as tremor (shaking), muscle rigidity, walking disorders, and restless leg syndrome [1].
3.3.2 Ranolazine
Ranolazine, similarly to trimetazidine, is a metabolic antianginal agent, which inhibits fatty acid oxidation in the mitochondria and favors glucose metabolism [135]. Its main mechanism of action is however to increase myocardial relaxation by reducing Ca2+ overload caused by inhibition of late Na+ currents [136]. Like trimetazidine, also ranolazine does not affect heart rate or blood pressure and therefore may be used in patients with hypotension or bradycardia [137, 138].
The antianginal properties of ranolazine have been evaluated in several RCTs. In patients with CCS, the use of ranolazine was associated with fewer angina episodes and longer exercise duration compared to placebo [138], in both patients without other antianginal therapies or already on standard treatment [139,140,141]. Ranolazine was shown to improve angina and use of nitrate in patients with diabetes compared to placebo [142], but did not reduce angina, need for repeated revascularization, or angina-related hospitalizations in patients with incomplete revascularization: a high nonadherence to the drug may partly explain such findings [143]. Likewise, ranolazine seems ineffective in patients with microvascular disease [144]. An exception seems to be represented by women with microvascular angina, in whom ranolazine was shown to improve angina and myocardial ischemia, albeit only in those with reduced coronary flow reserve [145]. Ranolazine seems to be also not beneficial in patients with acute coronary syndrome as shown in the MERLIN-TIMI trial [146], even though a possible antiarrhythmic effect has been observed in this scenario [147].
In 2017, a Cochrane systematic review and meta-analysis on the use of ranolazine in patients with CCS has been published, highlighting the positive effect of ranolazine on angina (moderate quality of data), some evidence of increased risk of nonserious side effects (low quality of data), and an uncertain effect on both overall and cardiovascular mortality (low quality of data) [148].
Side effects of ranolazine, such as dizziness, nausea, and constipation, are dose dependent [149]. The inhibition of late sodium currents, together with its effect on delayed rectifier potassium currents, also causes prolongation of QT interval [149], and thus ranolazine should be avoided in patients with long QT interval or already taking QT-prolonging drugs. However, no significant increase in life-threatening arrythmias has been noticed in multiple safety studies [150].
4 Novel Perspectives from Animal Models and Human Studies
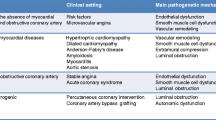
Therapeutic efficacy, safety profile, and cost-effectiveness are essential factors to be considered when designing a novel drug [151]. Standing this premise, several anti-ischemic compounds are on the pipeline. Novel vasodilators, metabolic modulators, as well as angiogenetic factors and cell therapies represent possible opportunities, especially for patients with refractory angina (Fig. 3).

Novel antianginal targets and drugs. CPT1i inhibitor of carnitine palmitoyltransferase I; FGF fibroblast growth factor; MCDi inhibitor of malonyl-CoA decarboxylase; PDE3i inhibitor of phosphodiesterase 3; RANKL-i inhibitor of the receptor activator of nuclear factor kappa-Β ligand; RCTs randomized controlled trials; SGLT2i inhibitor of sodium glucose transporter 2; VEGF vascular endothelial growth factor
4.1 Novel Compounds with Vasodilatory Effects
The small guanosine triphosphatase RhoA and its downstream effector Rho-kinase are involved in the regulation of vascular contractility, leading through inhibition myosin light-chain phosphatase to Ca2+ sensitization in response to vasoconstrictor stimuli [152]. Fasudil, a Rho-kinase inhibitor approved in Japan for the prevention of cerebral artery vasospasm in the setting of subarachnoid hemorrhage [153], has been tested in animal studies and in small trials in patients with microvascular spasm [154] and in patients with stable angina [155]. While fasudil intracoronary infusion was shown to prevent Ach-mediated vasoconstriction [153], fasudil oral administration only increased time to ST depression and had no effect on symptoms in humans [155]. To date, no Rho-kinase inhibitor has been approved for the treatment of vasospastic angina, and more clinical evidence is needed.
A selective phosphodiesterase-3-inhibitor, cilostazol, has also been shown to be efficacious in vasospastic angina in small clinical trials [156, 157], although its mechanism of action remains to be elucidated. In 49 patients with vasospastic angina, cilostazol decreased weakly angina episodes, proportion of angina-free period of angina severity compared to placebo, at the cost of increased rate of headache [157]. Still, its efficacy, dosage, and safety should be confirmed in larger RCTs.
4.2 Novel Modulators of Myocardial Metabolism
Since alterations in myocardial substrate preference contribute to energetic inefficiency, contractile dysfunction, and severity of ischemia, novel drugs inhibiting fatty acid oxidation or increasing the coupling of glycolysis to glucose oxidation represent promising approaches in CCS [158].
Decreasing myocardial fatty acid uptake may be obtained by acting on CD-36 (a sarcolemmal transporter responsible for up to 50% of cardiac fatty acid uptake) [159], and sulfo-N-succinimidyl-oleate was shown to inhibit fatty acid uptake in vitro in various cell lines including cardiomyocytes [160]. Interestingly, its infusion increased the glycolytic rate by 46% and pyruvate-dehydrogenase activity by 53%, while it decreased lactate efflux rate by 56% in the hearts of diabetic rats during hypoxia, compared with untreated rats, preventing cardiac dysfunction in hypoxic conditions. Although promising, whether this compound might be beneficial in CCS is still to be demonstrated.
The rate of cardiac fatty acid oxidation is regulated by the activity of carnitine palmitoyl-transferase-I. While the use of direct inhibitors (e.g., etomoxir, perhexiline, oxfenicine, teglicar) may be burdened by hepatotoxic and cardiotoxic effects due to unspecific mitochondrial effects [161], an indirect inhibition of this pathway by malonyl-CoA may be a promising approach. CBM-301106 inhibits malonyl-CoA decarboxylase, which catalyzes degradation of malonyl-CoA converting it to acetyl-CoA and thus decreases long-chain fatty acid metabolism [161]. This molecule reduced fatty acid oxidation and lactate production during demand-induced ischemia in various rat and pig models of ischemic heart disease [162,163,164], but it has to be tested in humans.
The role of ketones in cardiac energetics may be important in the condition of limited energy supply, as in the case of the failing heart [165]. Whether ketone metabolism may be a “super-fuel,” increasing cardiac efficiency or changing fatty acid oxidation or glucose metabolism, is still debated [166, 167]. In this respect, the positive effects of Na+-glucose-cotransporter-2-inhibitors (SGLT2-i) on cardiovascular outcomes in diabetic patients and in those with heart failure could be partially ascribed to an increased cardiac consumption of ketone bodies [168, 169]. However, the increase in ketone bodies following administration of SGLT2-i is usually mild and higher during fasting (i.e., at night) [169]. Therefore, it is currently unknown whether this may be sufficient to change myocardial metabolism (especially during daily activity), so as to have favorable effects on patients with CCS. Of note, empagliflozin has been recently shown to decrease contractile dysfunction and arrhythmias following ischemia in Langendorff-perfused rabbit heart [170], but the effects on ketone bodies were not assessed. This topic should be then addressed by dedicated studies.
4.3 Angiogenetic Factors
Vascular endothelial growth factors (VEGF) and fibroblast growth factors (FGF) have been tested in a few studies mainly in the setting of refractory angina [171], starting from the pioneering works in rabbit with hindlimb ischemia by Takeshita [172] and in humans by the group of Isner [173]. However, in the setting of RCTs, the percutaneous intracoronary administration or epicardial injection of VEGF (during bypass surgery) via naked plasmid or adenoviral vectors failed to deliver significant clinical effects, although no significant long-term side effect was observed [174].
On the other hand, intracoronary adenoviral mediated FGF-4 delivery improved exercise time in postmenopausal women as shown in a pooled analysis of the AGENT-3 and AGENT-4 trials [171]. The results of two similar trials, the Russian ASPIRE trial (NCT01550614) [174] based on intracoronary administration of Ad5FGF-4 (open-label design, no placebo, completed in 2016) and the AWARE trial [174] based on intracoronary administration of AdFGF-4 only in women with stable angina, have never been published.
Finally, intramyocardial adenoviral delivery of VEGF-D showed promising results in the KAT301 (phase I–IIa, n = 60) trial [175], where VEGF-D administration was associated with a significant improvement of myocardial perfusion reserve, reduction of angina, and improvement of quality of life, differently from placebo, especially in patients with high lipoprotein (a) levels. A larger phase IIb multicentric trial on VEFG-D is currently ongoing [171].
4.4 Cell Therapy
Similarly to angiogenetic factors, cell therapy has also been tested in refractory angina [171], but also in myocardial infarction and heart failure [176]. Although bone marrow-derived progenitors do not transform into myocytes, they may exert paracrine effects. Different pro-angiogenic cells were administered in an autologous setting, including unfractionated bone marrow-derived mononuclear cells, selected endothelial progenitors (i.e., CD34+ and CD133+ cells), or mesenchymal stem [177].
In the ACT34-CMI placebo-controlled trial (n = 167), patients with refractory angina receiving intramyocardial injection of CD34+ stem cells showed improved exercise tolerance (p = 0.01) and angina frequency (p = 0.02), also after a 2-year follow-up, where a trend of reduction in major events was observed as well [178]. Conversely, the RENEW trial was prematurely terminated by the sponsor for strategic consideration after enrolling only 112 of the 444 patients originally planned, showing a borderline reduction (p = 0.05) in angina frequency and only a trend toward an increase in exercise time at 3 months (p = 0.06), lost at 6 and 12 months. In a meta-analysis [179] including 3 phase II trials and 269 patients, intramyocardial therapy with CD34+ stem cells was superior to placebo in improving angina frequency, increasing exercise time, and decreasing mortality, without significant adverse events, thus supporting future larger trials in refractory angina patients.
5 Conclusions
Various anti-ischemic medications are currently available and extensively used in the routine clinical practice. Although their prognostic benefits are poor or scarcely investigated, they seem to be equally effective in relieving angina and improving quality of life. However, considering the multifactorial pathophysiology of myocardial ischemia and the heterogeneity of patients with CCS, a more rational patient-tailored use of anti-ischemic drugs may yield further benefits. Finally, various promising molecules are emerging from exploratory animal and preliminary clinical studies and may prove their value in the next future.
References
Knuuti J, Wijns W, Saraste A, Capodanno D, Barbato E, Funck-Brentano C, et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: the Task Force for the Diagnosis and Management of Chronic Coronary Syndromes of the European Society of Cardiology (ESC). Eur Heart J. 2020;41(3):407–77.
Kaski J-C, Crea F, Gersh BJ, Camici PG. Reappraisal of ischemic heart disease. Circulation. 2018;138(14):1463–80.
Crea F, Camici PG, Bairey Merz CN. Coronary microvascular dysfunction: an update. Eur Heart J. 2014;35(17):1101–11.
Nesto RW, Kowalchuk GJ. The ischemic cascade: temporal sequence of hemodynamic, electrocardiographic and symptomatic expressions of ischemia. Am J Cardiol. 1987;59(7):C23–30.
O’Sullivan JJ, Conroy RM, MacDonald K, McKenna TJ, Maurer BJ. Silent ischaemia in diabetic men with autonomic neuropathy. Br Heart J. 1991;66(4):313–5.
Marchant B, Umachandran V, Stevenson R, Kopelman PG, Timmis AD. Silent myocardial ischemia: role of subclinical neuropathy in patients with and without diabetes. J Am Coll Cardiol. 1993;22(5):1433–7.
Deedwania PC, Carbajal EV. Silent myocardial ischemia: a clinical perspective. Arch Intern Med. 1991;151(12):2373–82.
Group MRFITR. Exercise electrocardiogram and coronary heart disease mortality in the Multiple Risk Factor Intervention Trial. Am J Cardiol. 1985 Jan;1(55):16–24.
Yeung AC, Barry J, Orav J, Bonassin E, Raby KE, Selwyn AP. Effects of asymptomatic ischemia on long-term prognosis in chronic stable coronary disease. Circulation. 1991;83(5):1598–604.
Laukkanen JA, Kurl S, Lakka TA, Tuomainen T-P, Rauramaa R, Salonen R, et al. Exercise-induced silent myocardial ischemia and coronary morbidity and mortality in middle-aged men. J Am Coll Cardiol. 2001;38:72–9.
Sajadieh A, Nielsen OW, Rasmussen V, Hein HO, Hansen JF. Prevalence and prognostic significance of daily-life silent myocardial ischaemia in middle-aged and elderly subjects with no apparent heart disease. Eur Heart J. 2005;26(14):1402–9.
Ferrari R, Camici PG, Crea F, Danchin N, Fox K, Maggioni AP, et al. A “diamond” approach to personalized treatment of angina. Nat Rev Cardiol. 2018;15:120–32.
Group TIS. Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial. Lancet. 2002;359(9314):1269–75.
Newby DE, Williams MC. Dweck MR. Forget ischemia: it’s all about the plaque: Circulation; 2021. p. 1039–41.
Ryan M, Morgan H, Chiribiri A, Nagel E, Cleland J, Perera D. Myocardial viability testing: all STICHed up, or about to be REVIVED? Eur Heart J. 2021:ehab729.
Crossman DC. The pathophysiology of myocardial ischaemia. Heart. 2004;90(5):576.
Marzilli M, Merz CNB, Boden WE, Bonow RO, Capozza PG, Chilian WM, et al. Obstructive coronary atherosclerosis and ischemic heart disease: an elusive link! J Am Coll Cardiol. 2012;60(11):951–6.
Ferrari R, Camici PG, Crea F, Danchin N, Fox K, Maggioni AP, et al. Expert consensus document: a “diamond” approach to personalized treatment of angina. Nat Rev Cardiol. 2018;15(2):120–32.
Bertero E, Heusch G, Münzel T, Maack C. A pathophysiological compass to personalize antianginal drug treatment. Nat Rev Cardiol. 2021;18(12):838–52.
Husted SE, Ohman EM. Pharmacological and emerging therapies in the treatment of chronic angina. Lancet (London, England). 2015;386(9994):691–701.
Ferrari R, Pavasini R, Camici PG, Crea F, Danchin N, Pinto F, et al. Anti-anginal drugs–beliefs and evidence: systematic review covering 50 years of medical treatment. Eur Heart J. 2019;40(2):190–4.
Mosher P, Ross J, Mcfate P, Shaw R. Control of coronary blood flow by an autoregulatory mechanism. Circ Res. 1964;14:250–9.
Heusch G. The paradox of α-adrenergic coronary vasoconstriction revisited. J Mol Cell Cardiol. 2011;51(1):16–23.
Deussen A, Ohanyan V, Jannasch A, Yin L, Chilian W. Mechanisms of metabolic coronary flow regulation. J Mol Cell Cardiol. 2012;52(4):794–801.
Gould KL, Lipscomb K. Effects of coronary stenoses on coronary flow reserve and resistance. Am J Cardiol. 1974;34(1):48–55.
Aversano T, Becker LC. Persistence of coronary vasodilator reserve despite functionally significant flow reduction. Am J Phys 1985;248(3 Pt 2).
Canty JM, Klocke FJ. Reduced regional myocardial perfusion in the presence of pharmacologic vasodilator reserve. Circulation. 1985;71(2):370–7.
Heusch G, Guth BD, Seitelberger R, Ross J. Attenuation of exercise-induced myocardial ischemia in dogs with recruitment of coronary vasodilator reserve by nifedipine. Circulation. 1987;75(2):482–90.
Patel MR, Peterson ED, Dai D, Brennan JM, Redberg RF, Anderson HV, et al. Low diagnostic yield of elective coronary angiography. N Engl J Med. 2010;362(10):886–95.
Douglas PS, Patel MR, Bailey SR, Dai D, Kaltenbach L, Brindis RG, et al. Hospital variability in the rate of finding obstructive coronary artery disease at elective, diagnostic coronary angiography. J Am Coll Cardiol. 2011;58(8):801–9.
Arnold JR, Karamitsos TD, Van Gaal WJ, Testa L, Francis JM, Bhamra-Ariza P, et al. Residual ischemia after revascularization in multivessel coronary artery disease: insights from measurement of absolute myocardial blood flow using magnetic resonance imaging compared with angiographic assessment. Circ Cardiovasc Interv. 2013;6(3):237–45.
Prinzmetal M, Kennamer R, Merliss R, Wada T, Bor N. Angina pectoris I. A variant form of angina pectoris: preliminary report. Am J Med. 1959;27(3):375–88.
Sun H, Mohri M, Shimokawa H, Usui M, Urakami L, Takeshita A. Coronary microvascular spasm causes myocardial ischemia in patients with vasospastic angina. J Am Coll Cardiol. 2002;39(5):847–51.
Ong P, Athanasiadis A, Borgulya G, Mahrholdt H, Kaski JC, Sechtem U. High prevalence of a pathological response to acetylcholine testing in patients with stable angina pectoris and unobstructed coronary arteries: the ACOVA study (Abnormal COronary VAsomotion in patients with stable angina and unobstructed coronary arteries). J Am Coll Cardiol. 2012;59(7):655–62.
Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373–6.
Yasue H, Horio Y, Nakamura N, Fujii H, Imoto N, Sonoda R, et al. Induction of coronary artery spasm by acetylcholine in patients with variant angina: possible role of the parasympathetic nervous system in the pathogenesis of coronary artery spasm. Circulation. 1986;74(5):955–63.
Rosenthal SJ, Ginsburg R, Lamb IH, Baim DS, Schroeder JS. Efficacy of diltiazem for control of symptoms of coronary arterial spasm. Am J Cardiol. 1980 Dec;46(6):1027–32.
Antman E, Muller J, Goldberg S, MacAlpin R, Rubenfire M, Tabatznik B, et al. Nifedipine therapy for coronary-artery spasm. Experience in 127 patients. N Engl J Med. 1980;302(23):1269–73.
Conti CR. Large vessel coronary vasospasm: diagnosis, natural history and treatment. Am J Cardiol. 1985;55(3).
Aizawa T, Ogasawara K, Nakamura F, Hirosaka A, Sakuma T, Nagashima K, et al. Effect of nicorandil on coronary spasm. Am J Cardiol. 1989;63(21).
Marie Robertson R, Wood AJ, Vaughn WK, Robertson D. Exacerbation of vasotonic angina pectoris by propranolol. Circulation. 1982;65:281–5.
Crea F, Lanza GA. Angina pectoris and normal coronary arteries: cardiac syndrome X. Heart. 2004;90(4):457–63.
Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2009;356(8):830–40.
Lundberg JO, Weitzberg E, Gladwin MT. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7(2):156–67.
Kaski JC, Rosano GMC, Collins P, Nihoyannopoulos P, Maseri A, Poole-Wilson PA. Cardiac syndrome X: clinical characteristics and left ventricular function. Long-term follow-up study. J Am Coll Cardiol. 1995;25(4):807–14.
Sütsch G, Oechslin E, Mayer I, Hess OM. Effect of diltiazem on coronary flow reserve in patients with microvascular angina. Int J Cardiol. 1995;52(2):135–43.
Ohba K, Sugiyama S, Sumida H, Nozaki T, Matsubara J, Matsuzawa Y, et al. Microvascular coronary artery spasm presents distinctive clinical features with endothelial dysfunction as nonobstructive coronary artery disease. J Am Heart Assoc. 2012;1(5).
Pauly DF, Johnson BD, Anderson RD, Handberg EM, Smith KM, Cooper-Dehoff RM, et al. In women with symptoms of cardiac ischemia, nonobstructive coronary arteries, and microvascular dysfunction, angiotensin-converting enzyme inhibition is associated with improved microvascular function: a double-blind randomized study from the National Hea. Am Heart J. 2011;162(4):678–84.
Crea F, Pupita G, Galassi AR, El-Tamimi H, Kaski JC, Davies G, et al. Role of adenosine in pathogenesis of anginal pain. Circulation. 1990;81(1):164–72.
Ferrari R, Fox K. Heart rate reduction in coronary artery disease and heart failure. Nat Rev Cardiol. 2016;13(8):493–501.
Guth BD, Heusch G, Seitelberger R, Ross J. Mechanism of beneficial effect of beta-adrenergic blockade on exercise-induced myocardial ischemia in conscious dogs. Circ Res. 1987;60(5):738–46.
Neumann FJ, Sechtem U, Banning AP, Bonaros N, Bueno H, Bugiardini R, et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur Heart J. 2020;41(3):407–77.
Steg PG, De Silva R. Beta-blockers in asymptomatic coronary artery disease no benefit or no evidence? J Am Coll Cardiol. 2014;64(3):253–5.
Borer JS, Le Heuzey JY. Characterization of the heart rate-lowering action of ivabradine, a selective I(f) current inhibitor. Am J Ther. 2008;15(5):461–73.
Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85(3):1093–129.
Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86(5):580–8.
Hasenfuss G, Maier LS. Mechanism of action of the new anti-ischemia drug ranolazine. Clin Res Cardiol. 2008;97(4):222.
Lauder Brunton T. ON THE USE OF NITRITE OF AMYL IN ANGINA PECTORIS. Lancet. 1867;90(2291):97–8.
Srivastava SC, Dewar HA, Newell DJ. Double-blind Trial of Propranolol (Inderal) in Angina of Effort. Br Med J. 1964;2(5411):724 LP–725.
Melville KI, Shister HE, Huq S. Iproveratril: experimental data on coronary dilatation and antiarrhythmic action. Can Med Assoc J. 1964;90(13):761–70.
Mehrotra TN, Bassadone ET. Trimetazidine in the treatment of angina pectoris. Br J Clin Pr. 1967;21(11):553–4.
Sakai K, Shiraki Y, Nabata H. Cardiovascular effects of a new coronary vasodilator N-(2-hydroxyethyl)nicotinamide nitrate (SG-75): comparison with nitroglycerin and diltiazem. J Cardiovasc Pharmacol. 1981;3(1).
Vilaine JP. The discovery of the selective if current inhibitor ivabradine: a new therapeutic approach to ischemic heart disease. Pharmacol Res. 2006;53(5):424–34.
Jain D, Dasgupta P, Hughes LO, Lahiri A, Raftery EB. Ranolazine (RS-43285): a preliminary study of a new anti-anginal agent with selective effect on ischaemic myocardium. Eur J Clin Pharmacol. 1990;38(2):111–4.
Ford TJ, Stanley B, Good R, Rocchiccioli P, McEntegart M, Watkins S, et al. Stratified medical therapy using invasive coronary function testing in angina: the CorMicA trial. J Am Coll Cardiol. 2018;72(23):2841–55.
Klein WW, Jackson G, Tavazzi L. Efficacy of monotherapy compared with combined antianginal drugs in the treatment of chronic stable angina pectoris: a meta-analysis. Coron Artery Dis. 2002;13(8):427–36.
Tardif J-C, Ponikowski P, Kahan T, Investigators AS. Efficacy of the I(f) current inhibitor ivabradine in patients with chronic stable angina receiving beta-blocker therapy: a 4-month, randomized, placebo-controlled trial. Eur Heart J. 2009;30(5):540–8.
Werdan K, Ebelt H, Nuding S, Höpfner F, Hack G, Müller-Werdan U. Ivabradine in combination with beta-blocker improves symptoms and quality of life in patients with stable angina pectoris: results from the ADDITIONS study. Clin Res Cardiol. 2012;101(5):365–73.
Belsey J, Savelieva I, Mugelli A, Camm AJ. Relative efficacy of antianginal drugs used as add-on therapy in patients with stable angina: a systematic review and meta-analysis. Eur J Prev Cardiol. 2015;22(7):837–48.
Torfgård KE, Ahlner J. Mechanisms of action of nitrates. Cardiovasc Drugs Ther. 1994;8(5):701–17.
Parker JD, Parker JO. Nitrate therapy for stable angina pectoris. NEJM. 2009;338(8):520–31.
Andersson KE, Hoglund P. Combination of nitrates with other antianginal drugs. Drugs. 1987;33 Suppl 4(4):43–8.
Harris JR, Hale GM, Dasari TW, Schwier NC. Pharmacotherapy of vasospastic angina. J Cardiovasc Pharmacol Ther. 2016;21(5):439–51.
Lanza GA, Manzoli A, Bia E, Crea F, Maseri A. Acute effects of nitrates on exercise testing in patients with syndrome X. Clinical and pathophysiological implications. Circulation. 1994;90(6):2695–700.
Wei J, Wu T, Yang Q, Chen M, Ni J, Huang D. Nitrates for stable angina: a systematic review and meta-analysis of randomized clinical trials. Int J Cardiol. 2011;146(1):4–12.
Kojima S, Matsui K, Sakamoto T, Ishihara M, Kimura K, Miyazaki S, et al. Long-term nitrate therapy after acute myocardial infarction does not improve or aggravate prognosis. Circ J. 2007;71(3):301–7.
Takahashi J, Nihei T, Takagi Y, et al. Prognostic impact of chronic nitrate therapy in patients with vasospastic angina: multicentre registry study of the Japanese coronary spasm association. Eur Heart J. 2015;36(4):228–37.
Ural D, Kandemir AŞ, Karaüzüm K, Baydemir C, Karaüzüm İY, Bozyel S, et al. Effect of oral nitrates on all-cause mortality and hospitalization in heart failure patients with reduced ejection fraction: a propensity-matched analysis. J Card Fail. 2017;23(4):286–92.
Münzel T, Daiber A, Mülsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97(7):618–28.
Sekiya M, Sato M, Funada J, Ohtani T, Akutsu H, Watanabe K. Effects of the long-term administration of nicorandil on vascular endothelial function and the progression of arteriosclerosis. J Cardiovasc Pharmacol. 2005;46(1):63–7.
Suryapranata H, Serruys PW, De Feyter PJ, Verdouw PD, Hugenholtz PG. Coronary vasodilatory action after a single dose of nicorandil. Am J Cardiol. 1988;61(4):292–7.
Ogino K, Osaki S, Noguchi N, Kitamura H, Omodani H, Kato M, et al. Nicorandil suppressed myocardial purine metabolism during exercise in patients with angina pectoris. Eur J Clin Pharmacol. 1995;48(3–4):189–94.
Cheng K, Alhumood K, El Shaer F, De Silva R. The role of nicorandil in the management of chronic coronary syndromes in the gulf region. Adv Ther. 2021;38(2):925–48.
Kaski JC. Management of vasospastic angina--role of nicorandil. Cardiovasc Drugs Ther. 1995;9(2 Supplement):221–7.
Jia Q, Shi S, Yuan G, Shi J, Shi S, Wei Y, et al. The effect of nicorandil in patients with cardiac syndrome X: a meta-analysis of randomized controlled trials. Medicine (Baltimore). 2020;99(37):e22167.
Hirohata A, Yamamoto K, Hirose E, Kobayashi Y, Takafuji H, Sano F, et al. Nicorandil prevents microvascular dysfunction resulting from PCI in patients with stable angina pectoris: a randomised study. EuroIntervention. 2014;9(9):1050–8.
Dargie HJ. Effect of nicorandil on coronary events in patients with stable angina: the Impact Of Nicorandil in Angina (IONA) randomised trial. Lancet (London, England). 2002;359(9314):1269–75.
Abernethy DR, Schwartz JB. Calcium-antagonist drugs. NEJM. 2008;341(19):1447–57.
Godfraind T. Discovery and development of calcium channel blockers. Front Pharmacol. 2017;8:286.
Ezekowitz MD, Hossack K, Mehta JL, Thadani U, Weidler DJ, Kostuk W, et al. Amlodipine in chronic stable angina: results of a multicenter double-blind crossover trial. Am Heart J. 1995;129(3):527–35.
Chahine RA, Feldman RL, Giles TD, Nicod P, Raizner AE, Weiss RJ, et al. Randomized placebo-controlled trial of amlodipine in vasospastic angina. Amlodipine Study 160 Group. J Am Coll Cardiol. 1993;21(6):1365–70.
Cannon RO, Watson RM, Rosing DR, Epstein SE. Efficacy of calcium channel blocker therapy for angina pectoris resulting from small-vessel coronary artery disease and abnormal vasodilator reserve. Am J Cardiol. 1985;56(4):242–6.
Ong P, Athanasiadis A, Sechtem U. Pharmacotherapy for coronary microvascular dysfunction. Eur Hear J Cardiovasc Pharmacother. 2015;1(1):65–71.
Leon MB, Rosing DR, Bonow RO, Epstein SE. Combination therapy with calcium-channel blockers and beta blockers for chronic stable angina pectoris. Am J Cardiol. 1985;55(3):69B–80B.
Frishman WH, Glasser S, Stone P, Deedwania PC, Johnson M, Fakouhi TD. Comparison of controlled-onset, extended-release verapamil with amlodipine and amlodipine plus atenolol on exercise performance and ambulatory ischemia in patients with chronic stable angina pectoris. Am J Cardiol. 1999;83(4):507–14.
Poole-Wilson PPA, Lubsen PJ, Kirwan BA, Van Dalen FJ, Wagener G, Danchin PN, et al. Effect of long-acting nifedipine on mortality and cardiovascular morbidity in patients with stable angina requiring treatment (ACTION trial): randomised controlled trial. Lancet (London, England). 2004;364(9437):849–57.
Nissen SE, Tuzcu EM, Libby P, Thompson PD, Ghali M, Garza D, et al. Effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure: the CAMELOT study: a randomized controlled trial. JAMA. 2004;292(18):2217–26.
Talajic M, Nattel S. Frequency-dependent effects of calcium antagonists on atrioventricular conduction and refractoriness: demonstration and characterization in anesthetized dogs. Circulation. 1986;74(5):1156–67.
Schroeder JS, Feldman RL, Giles TD, Friedman MJ, DeMaria AN, Kinney EL, et al. Multiclinic controlled trial of diltiazem for Prinzmetal’s angina. Am J Med. 1982;72(2):227–32.
Zhang X, Li Q, Zhao J, Li X, Sun X, Yang H, et al. Effects of combination of statin and calcium channel blocker in patients with cardiac syndrome X. Coron Artery Dis. 2014;25(1):40–4.
The effect of diltiazem on mortality and reinfarction after myocardial infarction. N Engl J Med. 1988;319(7):385–92.
Hansen JF, Mellemgaard K, Pedersen-Bjergaard O, Rasmussen B, Launbjerg J, Fruergaard P, et al. Effect of verapamil on mortality and major events after acute myocardial infarction (the Danish Verapamil Infarction Trial II--DAVIT II). Am J Cardiol. 1990;66(10):779–85.
Weintraub WS, Spertus JA, Kolm P, Maron DJ, Zhang Z, Jurkovitz C, et al. Effect of PCI on quality of life in patients with stable coronary disease. NEJM. 2008;359(7):677–87.
Al-Lamee R, Thompson D, Dehbi H-M, Sen S, Tang K, Davies J, et al. Percutaneous coronary intervention in stable angina (ORBITA): a double-blind, randomised controlled trial. Lancet. 2018;391(10115):31–40.
Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol. 2008;153(8):1589–601.
Buck JD, Hardman HF, Warltier DC, Gross GJ. Changes in ischemic blood flow distribution and dynamic severity of a coronary stenosis induced by beta blockade in the canine heart. Circulation. 1981;64(4):708–15.
Matsuzaki M, Patritti J, Tajimi T, Miller M, Kemper WS, Ross J. Effects of beta-blockade on regional myocardial flow and function during exercise. Am J Physiol Circ Physiol. 1984;247(1):H52–60.
Tham TC, Guy S, McDermott BJ, Shanks RG, Riddell JG. The dose dependency of the alpha- and beta-adrenoceptor antagonist activity of carvedilol in man. Br J Clin Pharmacol. 1995;40(1):19–23.
Seitelberger R, Guth BD, Heusch G, Lee JD, Katayama K, Ross J. Intracoronary alpha 2-adrenergic receptor blockade attenuates ischemia in conscious dogs during exercise. Circ Res. 1988;62(3):436–42.
Bowman AJ, Chen CP, Ford GA. Nitric oxide mediated venodilator effects of nebivolol. Br J Clin Pharmacol. 1994;38(3):199–204.
Safi S, Sethi NJ, Nielsen EE, Feinberg J, Jakobsen JC, Gluud C. Beta-blockers for suspected or diagnosed acute myocardial infarction. Cochrane Database Syst Rev. 2019;12(12):CD012484.
COMMIT (ClOpidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Early intravenous then oral metoprolol in 45852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366(9497):1622–32.
Dondo TB, Hall M, West RM, Jernberg T, Lindahl B, Bueno H, et al. β-blockers and mortality after acute myocardial infarction in patients without heart failure or ventricular dysfunction. J Am Coll Cardiol. 2017;69(22):2710–20.
Thollon C, Cambarrat C, Vian J, Prost JF, Peglion JL, Vilaine JP. Electrophysiological effects of S 16257, a novel sino-atrial node modulator, on rabbit and guinea-pig cardiac preparations: comparison with UL-FS 49. Br J Pharmacol. 1994;112(1):37–42.
DiFrancesco D, Ferroni A, Mazzanti M, Tromba C. Properties of the hyperpolarizing-activated current (if) in cells isolated from the rabbit sino-atrial node. J Physiol. 1986;377:61–88.
Brown H, Difrancesco D. Voltage-clamp investigations of membrane currents underlying pace-maker activity in rabbit sino-atrial node. J Physiol. 1980;308:331–51.
Heusch G, Skyschally A, Gres P, van Caster P, Schilawa D, Schulz R. Improvement of regional myocardial blood flow and function and reduction of infarct size with ivabradine: protection beyond heart rate reduction. Eur Heart J. 2008;29(18):2265–75.
Bucchi A, Baruscotti M, DiFrancesco D. Current-dependent block of rabbit sino-atrial node I(f) channels by ivabradine. J Gen Physiol. 2002;120(1):1–13.
Savelieva I, Camm A. Novel If current inhibitor ivabradine: safety considerations. In: Advances in cardiology. 2006. p. 79–96.
Fox K, Ford I, Steg PG, Tardif J-C, Tendera M, Ferrari R. Ivabradine in stable coronary artery disease without clinical heart failure. N Engl J Med. 2014;371(12):1091–9.
Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R, et al. Relationship between ivabradine treatment and cardiovascular outcomes in patients with stable coronary artery disease and left ventricular systolic dysfunction with limiting angina: a subgroup analysis of the randomized, controlled BEAUTIFUL trial. Eur Heart J. 2009;30(19):2337–45.
Villano A, Di Franco A, Nerla R, Sestito A, Tarzia P, Lamendola P, et al. Effects of ivabradine and ranolazine in patients with microvascular angina pectoris. Am J Cardiol. 2013;112(1):8–13.
Swedberg K, Komajda M, Böhm M, Borer JS, Ford I, Dubost-Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376(9744):875–85.
Tuunanen H, Engblom E, Naum A, Någren K, Scheinin M, Hesse B, et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118(12):1250–8.
Koylan N, Bilge AK, Adalet K, Mercanoglu F, Buyukozturk K. Comparison of the effects of trimetazidine and diltiazem on exercise performance in patients with coronary heart disease. The Turkish trimetazidine study (TTS). Acta Cardiol. 2004;59(6):644–50.
Lopaschuk GD, Barr R, Thomas PD, Dyck JRB. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2003;93(3):e33–7.
Szwed H, Sadowski Z, Elikowski W, Koronkiewicz A, Mamcarz A, Orszulak W, et al. Combination treatment in stable effort angina using trimetazidine and metoprolol. Results of a randomized, double-blind, multicentre study (TRIMPOL II). Eur Heart J. 2001;22(24):2267–74.
Vitale C, Spoletini I, Malorni W, Perrone-Filardi P, Volterrani M, Rosano GMC. Efficacy of trimetazidine on functional capacity in symptomatic patients with stable exertional angina; The VASCO-angina study. Int J Cardiol. 2013;168(2):1078–81.
Manchanda SC, Krishnaswami S. Combination treatment with trimetazidine and diltiazem in stable angina pectoris. Heart. 1997;78(4):353–7.
Ciapponi A, Pizarro R, Harrison J. WITHDRAWN: trimetazidine for stable angina. Cochrane Database Syst Rev. 2017;3(3):CD003614.
Danchin N, Marzilli M, Parkhomenko A, Ribeiro JP. Efficacy comparison of trimetazidine with therapeutic alternatives in stable angina pectoris: a network meta-analysis. Cardiology. 2011;120(2):59–72.
Peng S, Zhao M, Wan J, Fang Q, Fang D, Li K. The efficacy of trimetazidine on stable angina pectoris: a meta-analysis of randomized clinical trials. Int J Cardiol. 2014;177(3):780–5.
Nalbantgil S, Altintiğ A, Yilmaz H, Nalbantgil I, Önder R. The effect of trimetazidine in the treatment of microvascular angina. Int J Angiol. 1999;8(1):40–3.
Ferrari R, Ford I, Fox K, Challeton JP, Correges A, Tendera M, et al. Efficacy and safety of trimetazidine after percutaneous coronary intervention (ATPCI): a randomised, double-blind, placebo-controlled trial. Lancet. 2020;396(10254):830–8.
McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996;93(1):135–42.
Chaitman BR. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation. 2006;113(20):2462–72.
Ohman EM. Chronic stable angina. N Engl J Med. 2016;374:1167–76.
Chaitman BR, Skettino SL, Parker JO, Hanley P, Meluzin J, Kuch J, et al. Anti-ischemic effects and long-term survival during ranolazine monotherapy in patients with chronic severe angina. J Am Coll Cardiol. 2004;43(8):1375–82.
Stone PH, Gratsiansky NA, Blokhin A, Huang I-Z, Meng L. Antianginal efficacy of ranolazine when added to treatment with amlodipine: the ERICA (Efficacy of Ranolazine in Chronic Angina) trial. J Am Coll Cardiol. 2006;48(3):566–75.
Chaitman BR, Pepine CJ, Parker JO, Skopal J, Chumakova G, Kuch J, et al. Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. JAMA. 2004;291(3):309–16.
Salazar CA, Basilio Flores JE, Veramendi Espinoza LE, Mejia Dolores JW, Rey Rodriguez DE, Loza Munárriz C. Ranolazine for stable angina pectoris. Cochrane Database Syst Rev. 2017;2017(2).
Kosiborod M, Arnold SV, Spertus JA, McGuire DK, Li Y, Yue P, et al. Evaluation of ranolazine in patients with type 2 diabetes mellitus and chronic stable angina: results from the terisa randomized clinical trial (Type 2 Diabetes Evaluation of Ranolazine in Subjects With Chronic Stable Angina). J Am Coll Cardiol. 2013;61(20):2038–45.
Weisz G, Généreux P, Iñiguez A, Zurakowski A, Shechter M, Alexander KP, et al. Ranolazine in patients with incomplete revascularisation after percutaneous coronary intervention (RIVER-PCI): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10014):136–45.
Bairey Merz CN, Handberg EM, Shufelt CL, Mehta PK, Minissian MB, Wei J, et al. A randomized, placebo-controlled trial of late Na current inhibition (ranolazine) in coronary microvascular dysfunction (CMD): impact on angina and myocardial perfusion reserve. Eur Heart J. 2015;37(19):1504–13.
Mehta S, Liu PP, Fitzgerald FS, Allidina YK, Douglas Bradley T. Effects of continuous positive airway pressure on cardiac volumes in patients with ischemic and dilated cardiomyopathy. AJRCCM. 2012:13–5.
Morrow DA, Scirica BM, Karwatowska-Prokopczuk E, Murphy SA, Budaj A, Varshavsky S, et al. Effects of ranolazine on recurrent cardiovascular events in patients with non-ST-elevation acute coronary syndromes: the MERLIN-TIMI 36 randomized trial. JAMA. 2007;297(16):1775–83.
Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non-ST-segment–elevation acute coronary syndrome. Circulation. 2007;116(15):1647–52.
Salazar CA, Basilio Flores JE, Veramendi Espinoza LE, Mejia Dolores JW, Rey Rodriguez DE, Loza Munárriz C. Ranolazine for stable angina pectoris. Cochrane Database Syst Rev. 2017;2(2):CD011747.
Rayner-Hartley E, Sedlak T. Ranolazine: a contemporary review. J Am Heart Assoc. 2016;5(3):e003196.
Kumar K, Nearing BD, Bartoli CR, Kwaku KF, Belardinelli L, Verrier RL. Effect of ranolazine on ventricular vulnerability and defibrillation threshold in the intact porcine heart. J Cardiovasc Electrophysiol. 2008;19(10):1073–9.
Chong C-R, Ong GJ, Horowitz JD. Emerging drugs for the treatment of angina pectoris. Expert Opin Emerg Drugs. 2016;21(4):365–76.
Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-kinase in the cardiovascular system. Circ Res. 2016;118(2):352–66.
Liu GJ, Wang ZJ, Wang YF, Xu LL, Wang XL, Liu Y, et al. Systematic assessment and meta-analysis of the efficacy and safety of fasudil in the treatment of cerebral vasospasm in patients with subarachnoid hemorrhage. Eur J Clin Pharmacol. 2012;68(2):131–9.
Mohri M, Shimokawa H, Hirakawa Y, Masumoto A, Takeshita A. Rho-kinase inhibition with intracoronary fasudil prevents myocardial ischemia in patients with coronary microvascular spasm. J Am Coll Cardiol. 2003;41(1):15–9.
Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. Efficacy and safety of fasudil in patients with stable angina: a double-blind, placebo-controlled, phase 2 trial. J Am Coll Cardiol. 2005;46(10):1803–11.
Yoo SY, Song SG, Lee JH, Shin ES, Kim J-S, Park YH, et al. Efficacy of cilostazol on uncontrolled coronary vasospastic angina: a pilot study. Cardiovasc Ther. 2013;31:179–85.
Kim JH, Shin ES, Lee JH, Yoo SY, Park YW, Hong YJ, et al. A randomized multicenter double-blind placebo-controlled trial to evaluate the efficacy and safety of cilostazol in patients with vasospastic angina (STELLA trial). Eur Heart J. 2013;34(suppl_1).
Lopaschuk GD. Metabolic modulators in heart disease: past, present, and future. Can J Cardiol. 2017;33(7):838–49.
Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase and CD36-mediated pathways. J Lipid Res. 2009;50(Suppl):S86–90.
Coort S, Willems J, Coumans W, van der Vusse G, Bonen A, Glatz J, et al. Sulfo-N-succinimidyl esters of long chain fatty acids specifically inhibit fatty acid translocase (FAT/CD36)-mediated cellular fatty acid uptake. In: Glatz JFC, editor. Cellular lipid binding proteins. Developments in molecular and cellular biochemistry. Springer; 2002. p. 213–9.
Makrecka-Kuka M, Liepinsh E, Murray AJ, Lemieux H, Dambrova M, Tepp K, Puurand M, Käämbre T, Han WH, de Goede P, O’Brien KA, Turan B, Tuncay E, Olgar Y, Rolo AP, Palmeira CM, Boardman NT, Wüst RCI, Larsen T. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol (Oxf). 2020;228(3):e13430.
Stanley WC, Morgan EE, Huang H, McElfresh TA, Sterk JP, Okere IC, et al. Malonyl-CoA decarboxylase inhibition suppresses fatty acid oxidation and reduces lactate production during demand-induced ischemia. Am J Physiol Circ Physiol. 2005;289(6):H2304–9.
Ussher JR, Lopaschuk GD. Targeting malonyl CoA inhibition of mitochondrial fatty acid uptake as an approach to treat cardiac ischemia/reperfusion. Basic Res Cardiol. 2009;104(2):203–10.
Dyck JRB, Cheng J-F, Stanley WC, Barr R, Chandler MP, Brown S, et al. Malonyl coenzyme A decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. 2004;94(9):e78–84.
Lopaschuk GD, Karwi QG, Ho KL, Pherwani S, Ketema EB. Ketone metabolism in the failing heart. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(12):158813. https://www.sciencedirect.com/science/article/pii/S1388198120302055.
Kolwicz SC Jr, Airhart S, Tian R. Ketones step to the plate: a game changer for metabolic remodeling in heart failure? Circulation. 2016;133(8):689–91.
Ho KL, Zhang L, Wagg C, Al Batran R, Gopal K, Levasseur J, et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc Res. 2019;115(11):1606–16.
Cowie MR, Fisher M. SGLT2 inhibitors: mechanisms of cardiovascular benefit beyond glycaemic control. Nat Rev Cardiol. 2020;17(12):761–72.
Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care. 2016;39(7):1108 LP–1114.
Azam MA, Chakraborty P, Si D, Du B, Massé S, Lai PFH, et al. Anti-arrhythmic and inotropic effects of empagliflozin following myocardial ischemia. Life Sci. 2021;276:119440.
Davies A, Fox K, Galassi AR, Banai S, Ylä-Herttuala S, Lüscher TF. Management of refractory angina: an update. Eur Heart J. 2021;42(3):269–83.
Takeshita S, Zheng LP, Brogi E, Kearney M, Pu LQ, Bunting S, et al. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J Clin Invest. 1994;93(2):662–70.
Losordo DW, Vale PR, Symes JF, Dunnington CH, Esakof DD, Maysky M, et al. Gene therapy for myocardial angiogenesis. Circulation. 1998;98(25):2800–4.
Ylä-Herttuala S, Bridges C, Katz MG, Korpisalo P. Angiogenic gene therapy in cardiovascular diseases: dream or vision? Eur Heart J. 2017;38(18):1365–71.
Hartikainen J, Hassinen I, Hedman A, Kivelä A, Saraste A, Knuuti J, et al. Adenoviral intramyocardial VEGF-DΔNΔC gene transfer increases myocardial perfusion reserve in refractory angina patients: a phase I/IIa study with 1-year follow-up. Eur Heart J. 2017;38(33):2547–55.
Mathur A, Fernández-Avilés F, Dimmeler S, Hauskeller C, Janssens S, Menasche P, et al. The consensus of the Task Force of the European Society of Cardiology concerning the clinical investigation of the use of autologous adult stem cells for the treatment of acute myocardial infarction and heart failure: update 2016. Eur Heart J. 2017;38(39):2930–5.
Bassetti B, Rurali E, Gambini E, Pompilio G. Son of a lesser god: the case of cell therapy for refractory angina. Front Cardiov Med. 2021;8:818.
Henry TD, Schaer GL, Traverse JH, Povsic TJ, Davidson C, Lee JS, et al. Autologous CD34+ cell therapy for refractory angina: 2-year outcomes from the ACT34-CMI study. Cell Transplant. 2016;25(9):1701–11.
Velagapudi P, Turagam M, Kolte D, Khera S, Hyder O, Gordon P, et al. Intramyocardial autologous CD34+ cell therapy for refractory angina: a meta-analysis of randomized controlled trials. Cardiovasc Revasc Med. 2019;20(3):215–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Giannoni, A., Gentile, F., Borrelli, C. (2023). Pharmacological Treatment of Ischemic Heart Disease. In: Concistrè, G. (eds) Ischemic Heart Disease. Springer, Cham. https://doi.org/10.1007/978-3-031-25879-4_19
Download citation
DOI: https://doi.org/10.1007/978-3-031-25879-4_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-25878-7
Online ISBN: 978-3-031-25879-4
eBook Packages: MedicineMedicine (R0)