
Abstract
Subjects with diabetes mellitus have a high prevalence and rapid progression of coronary artery, peripheral vascular, and cerebral vascular disease secondary in part to (1) increased platelet reactivity; (2) increased thrombotic activity reflecting increased concentrations and activity of coagulation factors and decreased activity of antithrombotic factors; and (3) decreased fibrinolytic system capacity resulting from overexpression of plasminogen activator inhibitor type-1 (PAI-1) by hepatic, arterial, and adipose tissue in response to hyperinsulinemia, hypertriglyceridemia, and hyperglycemia. In addition, macrovascular disease appears to be accelerated by an insulin-dependent imbalance in proteo(fibrino)lytic system activity within walls of arteries predisposing to accumulation of extracellular matrix and paucity of migration of vascular smooth muscle cells during the evolution of atheroma predisposing toward the development of plaques vulnerable to rupture. Therapy designed to reduce insulin resistance decreases concentrations in blood not only of insulin but also of PAI-1. Thus, the treatment of subjects with diabetes, and particularly type 2 diabetes, should focus not only on improved metabolic control but also on reduction of insulin resistance and hyperinsulinemia. Treatment designed to address both the hormonal and metabolic abnormalities of diabetes is likely to reduce hyperactivity of platelets, decrease the intensity of the prothrombotic state, and normalize activity of the fibrinolytic system in blood and in vessel walls thereby reducing the rate of progression of macrovascular disease and its sequelae.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Diabetes
- Prothrombosis
- Platelet reactivity
- Fibrinolysis
- Insulin resistance
- Plasminogen activator inhibitor type-1
- Myocardial infarction
Diabetes and Vascular Disease
Complications of macrovascular disease are responsible for 50% of the deaths in patients with type 2 diabetes mellitus, 27% of the deaths in patients with type 1 diabetes for 35 years or less, and 67% of the deaths in patients with type 1 diabetes for 40 years or more [1, 2]. The rapid progression of macroangiopathy in patients with type 2 diabetes may reflect diverse phenomena; some intrinsic to the vessel wall; angiopathic factors such as elevated homocysteine [3], uncoupled nitric oxide synthase and superoxide generation [4], and hyperlipidemia; deleterious effects of insulin resistance [5] and dysinsulinemia, and excessive or persistent microthrombi secondary to a prothrombotic and antifibrinolytic state with consequent acceleration of vasculopathy secondary to clot-associated mitogens [6, 7]. As a result of these phenomena, cardiovascular mortality is as high as 15% in the 10 years after the diagnosis of diabetes mellitus becomes established [8]. Because more than 90% of patients with diabetes have type 2 diabetes and because macrovascular disease is the cause of death in most patients with type 2 as opposed to type 1 diabetes, type 2 diabetes will be the focus of this chapter. In addition to coronary artery disease, patients with type 2 diabetes have a high prevalence and rapid progression of peripheral arterial disease, cerebral vascular disease, and complications of percutaneous coronary intervention including restenosis [9].
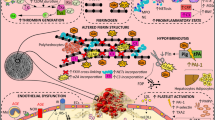
Diabetes mellitus is associated with diverse derangements in platelet function, the coagulation, and the fibrinolytic system, all of which can contribute to prothrombotic state (Tables 5.1 and 5.2). Some are clearly related to metabolic derangements, particularly hyperglycemia. Others appear to be related to insulin resistance and associated hyperinsulinemia. In the material to follow, we will consider mechanisms exacerbating thrombosis as pivotal factors in the progression of atherosclerosis and their therapeutic implications.
Thrombosis and Atherosclerosis
Thrombosis appears to be a major determinant of the progression of atherosclerosis. In early atherosclerosis, microthrombi present on the luminal surface of vessels [10, 11] can potentiate progression of atherosclerosis by exposing the vessel wall to clot-associated mitogens. In later stages of atherosclerosis, mural thrombosis is associated with growth of atherosclerotic plaques and progressive luminal occlusion, exacerbated in diabetes because of impaired compensatory vascular remodeling [12].
The previously conventional view that high-grade occlusive, stenotic coronary lesions represent the final step in a continuum that begins with fatty streaks and culminates in high-grade stenosis has given way to a different paradigm because of evidence that thrombotic occlusion is frequently the result of repetitive rupture of minimally stenotic plaques. Thus, as many as two thirds of lesions responsible for acute coronary syndromes are minimally obstructive (less than 50% stenotic) at a time immediately before plaque rupture [13, 14]. Multiple episodes of disruption of lipid-rich plaques and subsequent thrombosis appear to be responsible for intermittent plaque growth that underlies occlusive coronary syndromes [15, 16]. While plaque erosion has been recognized as an important cause of acute coronary syndromes [17, 18], characterization of plaque morphology in patients with diabetes plus acute coronary syndrome demonstrates a greater incidence of thin-capped atheroma with a large lipid core none to be prone to plaque rupture [18].
The extent of thrombosis in response to plaque rupture depends upon factors potentiating thrombosis (prothrombotic factors), factors limiting thrombosis (antithrombotic factors), and the local capacity of the fibrinolytic system reflecting a balance between activity of plasminogen activators and their primary physiologic inhibitor, and plasminogen activator inhibitor type-1 (PAI-1). Activity of plasminogen activators leads to the generation of plasmin, an active serine proteinase, from plasminogen, an enzymatically inert circulating zymogen present in high concentration (~2 μM) in blood. The activity of plasmin is limited by inhibitors such as α2 macroglobulin.
When only limited thrombosis occurs because of active plasmin-dependent fibrinolysis at the time of rupture of a plaque, plaque growth may be clinically silent. When thrombosis is exuberant because of exaggerated thrombin generation, enhanced platelet activation typical of diabetes [19], and limited fibrinolysis, an occlusive thrombus can give rise to an acute coronary syndrome (acute myocardial infarction, unstable angina, or sudden cardiac death).
The principle components of thrombi are fibrin and platelets. Other plasma proteins and white blood cells are incorporated to a variable extent. The rupture of an atherosclerotic plaque initiates coagulation and adhesion of platelets because of exposure to blood of surfaces denuded of endothelium and to constituents of the vessel wall such as collagen. Coagulation is initiated in the tissue factor pathway, activated by hyperinsulinemia and hyperglycemia [20], by tissue factor, a cell membrane-bound glycoprotein [21,22,23]. Membrane-bound tissue factor binds circulating coagulation factor VII/VIIa to form the coagulation factor “tenase” complex that activates both circulating coagulation factors IX and X expressed on activated macrophages, monocytes, fibroblasts, and endothelium in response to cytokines in the region of the ruptured plaque. Subsequent assembly of the “prothrombinase” complex on platelet and other phospholipid membranes leads to generation of thrombin. Availability of platelet factor Va is a key constituent of the initial prothrombinase complex. Subsequently, thrombin activates coagulation factor V in blood to form Va. Thrombin, in turn, cleaves fibrinogen to form fibrin. The generation of thrombin is sustained and amplified initially by its activation of circulating coagulation factors VIII and V. Thrombin generation is sustained by activation of other components in the intrinsic pathway including factor XI. Platelets are activated by thrombin, and activated platelets markedly amplify generation of thrombin.
A complex feedback system limits generation of thrombin. The tissue factor pathway becomes inhibited by tissue factor pathway inhibitor (TFPI) previously called lipoprotein-associated coagulation inhibitor (LACI). Furthermore, thrombin attenuates coagulation by binding to thrombomodulin on the surface of endothelial cells. The complex activates protein C (to yield protein Ca) that, in combination with protein S, cleaves (inactivates) coagulation factors Va and VIIIa.
Exposure of platelets to the subendothelium after plaque rupture leads to their adherence mediated by exposure to both collagen and multimers within the vessel wall of von Willebrand factor [24, 25]. The exposure of platelets to agonists including collagen, von Willebrand factor, ADP (released by damaged red blood cells and activated platelets), and thrombin leads to further platelet activation. Activation is a complex process that entails shape change (pseudopod extension that increases the surface area of the platelet); activation of the surface glycoprotein IIb/IIIa; release of products from dense granules such as calcium, ADP, and serotonin and from alpha granules such as fibrinogen, factor V, growth factors, and platelet factor 4 that inhibits heparin; and a change in the conformation of the platelet membrane that promotes binding to phospholipids and assembly of coagulation factors.
Activation of surface glycoprotein IIb/IIIa results in a conformational change that exposes a binding site for fibrinogen on the activated conformer [26]. Each molecule of fibrinogen can bind two platelets, thereby leading to aggregation. Surface expression of P-selectin leads to the formation of platelet–leukocyte aggregates and to the activation of monocytes and neutrophils [27].
After activation, the plasma membranes of platelets express negatively charged phospholipids on the outer surface that facilitate the assembly of protein constituents and subsequently activity of the tenase and prothrombinase complexes [23]. Thus, platelets are participated in thrombosis by (1) forming a hemostatic plug (shape change, adherence to the vascular wall and aggregation with other platelets and with leukocytes); (2) supplying coagulation factors and calcium (release of alpha and dense granule contents); (3) providing a surface for the assembly of coagulation factor complexes; and (4) simulating vasoconstriction by releasing thromboxane and other vasoactive substances.
As noted previously, thrombosis-complicating plaque rupture can occlude the lumen entirely or, when limited, contribute in a stepwise fashion over time to progressive stenosis. Mechanisms by which thrombi can contribute to plaque growth include incorporation of an organized thrombus into the vessel wall [28]. Exposure of vessel wall constituents to clot-associated mitogens and cytokines can accelerate neointimalizaiton and migration and proliferation of vascular smooth muscle cells in the media. Fibrin and fibrin degradation products promote the migration of vascular smooth muscle cells and are chemotactic for monocytes [29]. Thrombin itself and growth factors released from platelet alpha granules such as platelet-derived growth factor and transforming growth factor beta activate smooth muscle cells potentiating their migration and proliferation [30,31,32,33]. The powerful role of platelets has been demonstrated by a reduction in the proliferation of smooth muscle cells after mechanical arterial injury in thrombocytopenic rabbits with atherosclerosis [34].
Both local and systemic factors can influence the extent of thrombosis likely to occur in association with plaque rupture. The morphology and biochemical composition of the plaque influence thrombogenic potential. Atheromatous plaques with substantial lipid content are particularly prone to initiate thrombosis in contrast to the antithrombotic characteristics of the luminal surface of the normal vessel wall [35].
Both the severity of vascular injury and the extent of plaque rupture influence the extent to which blood is exposed to subendothelium and consequently to thrombogenicity. The balances between the activity of prothrombotic factors and antithrombotic factors in blood and between thrombogenicity and fibrinolytic system capacity are important determinants of the nature and extent of a thrombotic response to plaque rupture. In subjects with type 2 diabetes, the balances between determinants are shifted toward potentiation and persistence of thrombosis and, hence, toward acceleration of atherosclerosis [36]. The same is true in patients with syndromes of insulin resistance (the metabolic syndrome) [37].
Platelet Function in Subjects with Diabetes Mellitus
The activation of platelets and their participation in a thrombotic response to rupture of an atherosclerotic plaque are critical determinants of the extent of thrombosis, incremental plaque growth, and the development of occlusive thrombi. Increased adherence of platelet to vessel walls manifesting early atherosclerotic changes and the release of growth factors from alpha granules can exacerbate the evolution of atherosclerosis. Evidence of increased activity of platelets in patients with diabetes is reflected by increased concentrations in blood of soluble CD40 (a platelet released mediator of thrombosis and inflammation) and P-selectin (reflecting platelet activation) [36]. Patients with diabetes, particularly those with macrovascular disease, have an increased circulating platelet mass secondary to increased ploidy of megakaryocytes [37]. Activation of platelets is increased with type 2 diabetes, mediated in part by increased VLDL and remnant lipoprotein particles [38, 39]. This is reflected by increased concentrations in urine of a metabolite of thromboxane A2, thromboxane B2, and by the spontaneous aggregation of platelets [40,41,42] in blood. The prevalence of spontaneous aggregation of platelets correlates with the extent of elevation of concentrations of HbA1c [41]. Stringent glycemic control decreases concentrations in urine of thromboxane B2 [40, 42]. In addition, platelets isolated from the blood of subjects with diabetes exhibit impaired vasodilatory capacity [43], apparently mediated by release of a short-acting platelet-derived substance(s) that interferes with the ADP-induced dilatory response seen in normal vessels with intact endothelium [44].
Evidence of Increased Platelet Reactivity
Platelets from subjects with both type 1 and 2 diabetes are hyperreactive [45,46,47,48,49] and in normal subjects subjected to combined hyperglycemia and hyperinsulinemia [50]. Platelet aggregometry performed with platelet-rich plasma and with suspensions of washed platelets in buffers from people with diabetes and control subjects has demonstrated increased aggregation of platelets in response to agonists such as ADP, epinephrine, collagen, arachidonic acid, and thrombin. In addition, spontaneous (in the absence of added agonists) aggregation of platelets from subjects with diabetes is increased compared with aggregation of those from nondiabetic subjects [48], mediated in part by increased expression of platelet FcgammaRIIa receptor [51,52,53,54] and by the collagen receptor, glycoprotein VI [55, 56].
Platelets from subjects with diabetes exhibit increased degranulation in response to diverse stimuli. The capacity to promote growth of smooth muscle cells in vitro is greater as shown by exposure of vascular smooth muscle cells to platelets from subjects with poorly controlled compared with well controlled diabetes [57, 58]. Because alpha granules contain growth factors, the enhanced growth promoting activity of platelets from subjects with poorly controlled diabetes appears likely to be secondary to increased alpha granule degranulation.
The threshold for induction of release of substances residing in dense granules in response to thrombin is lower in platelets from diabetic compared with nondiabetic subjects [59]. In addition, the procoagulant capacity of platelets from subjects with diabetes mellitus is increased [60, 61]. Thus, the generation of coagulation factor Xa and of thrombin is increased by three- to sevenfold in samples of blood containing platelets from diabetic compared with those from nondiabetic subjects [61].
In patients with diabetes, adhesion of platelets is increased because of increased surface expression of glycoprotein Ib-IX [62]. The binding of von Willebrand factor multimers expressed on endothelial cells to glycoprotein Ib-IX mediates adherence and promotes subsequent activation of platelets. Adherence is promoted also by increased concentrations of and activity of von Willebrand factor [62, 63]. Circulating von Willebrand factor stabilizes the coagulant activity of circulating coagulation factor VIIIa [64].
An altered cellular distribution of guanine nucleotide binding proteins (G-proteins) appears to contribute to the increased reactivity of platelets in people with diabetes mellitus [65]. Platelet reactivity would be expected to be increased by the decreased concentrations of inhibitory G-proteins that have been reported [66]. In addition, platelet reactivity would be increased by the greater turnover of phosphoinositide and consequent intraplatelet release of calcium that have been seen [67, 68].
As noted above, activation of platelets leads to the expression of specific conformers of specific glycoproteins. Determination of the percentage of platelets expressing activation-dependent markers with flow cytometry can be used to delineate the extent of platelet activation that has occurred in vivo. Increased surface expression of CD63 (a marker of lysosomal degranulation), thrombospondin (a marker of alpha granule degranulation), and CD62 (also called P-selectin), another marker of alpha granule degranulation, has been observed with platelets isolated from patients with newly diagnosed diabetes and those with advanced diabetes regardless of whether or not overt macrovascular complications were present [69, 70]. The increased plasminogen activator type-1 inhibitor (PAI-1) in plasma (see below) in patients with diabetes is associated with a paradoxically decreased platelet content of PAI-1 [71], consistent with the possibility that release of PAI-1 from the platelets may contribute to the increased PAI-1 in blood.
Platelet survival is reduced in subjects with diabetes. The reduction is most pronounced in those with clinical evidence of vascular disease [72]. Thus, it appears to be more closely correlated with the severity of vascular disease [73] than with the presence of diabetes per se. Accordingly, the decreased survival of platelets may be both a marker of extensive vascular disease and a determinant of its severity.
Adherence of platelets to vessel walls early after injury resulting in de-endothelialization is similar in diabetic and nondiabetic animals [74]. By contrast, increased adherence of platelets to injured arterial segments 7 days after injury occurs in diabetic BB Wistar rats compared with that in control animals. A continued interaction of platelets with the vessel wall after injury is likely to be related to a decreased rate of healing and re-endothelialization in diabetic animals rather than to an increased propensity for adherence per se [75]. Regardless, continued interaction of platelets with the vessel wall and continued exposure of the vessel wall to growth factors released from alpha granules of platelets are likely to accelerate and exacerbate atherosclerosis.
Mechanisms Responsible for Hyperreactivity of Platelets in People with Diabetes
Increased expression of the surface glycoproteins Ib and IIb/IIIa has been observed in platelets from subjects with both type 1 and type 2 diabetes [62]. Glycoprotein Ib/IX binds to von Willebrand factor in the subendothelium and is responsible for adherence of platelets at sites of vascular injury. Interaction between glycoprotein Ib/IX and von Willebrand factor leads to activation of platelets. Activation of glycoprotein IIb/IIIa leads to the binding of fibrinogen and aggregation of platelets. Thus, increased expression of either or both of these two surface glycoproteins is likely to contribute to the increased reactivity that has been observed platelets from people with diabetes. Overexpression of the FcgammaRIIa [51,52,53,54,55,56] and increased GP VI signaling [55, 56] may contribute as well.
Winocour and his colleagues have shown an association between decreased membrane fluidity and hypersensitivity of platelets to thrombin [76]. Reduced membrane fluidity may be a reflection of increased glycation of membrane proteins. A reduction in membrane fluidity occurs following incubation of platelets in media containing concentrations of glucose similar to those seen in blood from subjects with poorly controlled diabetes. Because membrane fluidity is likely to alter membrane receptor accessibility by ligands, reduced membrane fluidity may contribute to hypersensitivity of platelets. Accordingly, improved glycemic control would be expected to decrease glycation of membrane proteins, increase membrane fluidity, and decrease hypersensitivity.
Intracellular mobilization of calcium is critical in several steps involved in the activation of platelets. Platelets from subjects with type 2 diabetes exhibit increased basal concentrations of calcium [77]. Increased phosphoinositide turnover, increased inositide triphosphate production, and increased intracellular mobilization of calcium are evident in response to exposure to thrombin of platelets from subjects with type 2 diabetes [78]. The increased concentrations of several second messengers may contribute to the hypersensitivity seen in platelets from patients with diabetes. In addition, increased production of thromboxane A2 may contribute to the increased platelet reactivity [43, 45].
We have found that the osmotic effect of increased glucose concentrations increases directly platelet reactivity [79]. Exposure of platelets in vitro to increased concentrations of glucose is associated with increased activation of platelets in the absence and presence of added agonist. Exposure of platelets to isotonic concentrations of glucose or mannitol increases platelet reactivity to a similar extent [79]. Thus, the osmotic effect of hyperglycemia on platelet reactivity may contribute to the greater risk of death and re-infarction that has been associated with hyperglycemia in patients with diabetes and myocardial infarction [80,81,82].
Insulin alters reactivity of platelets [83]. Exposure of platelets to insulin decreases platelet aggregation in part by increasing synthesis of nitric oxide that, in turn, increases intraplatelet concentrations of the cyclic nucleotides, cyclic guanosine monophosphate, and cyclic adenosine monophosphate. Both of these cyclic nucleotides are known to inhibit activation of platelets. Thus, it is not surprising that an insulin concentration-dependent increase in nitric oxide production exerts anti-aggregatory effects. Insulin deficiency typical of type 1 diabetes and seen in advanced stages of type 2 diabetes may contribute to increased platelet reactivity by decreasing the tonic inhibition of platelet reactivity otherwise induced by insulin. Furthermore, abnormal insulin signaling may contribute in subjects with type 2 diabetes. Insulin decreases platelet aggregation and adhesion in patients without diabetes, an effect not seen in those with type 2 diabetes [84]. Accordingly, the increased resistance to insulin typical of type 2 diabetes may contribute to increased platelet reactivity by decreasing tonic inhibition of platelets that would have been induced otherwise by the high prevailing concentration of insulin.
Constitutive synthesis of nitric oxide is reduced in platelets from subjects with both type 1 and type 2 diabetes [85]. The oxidative stress associated with diabetes and its uncoupling of nitric oxide synthase from arginine can exacerbate endothelial dysfunction and activation of platelets secondary to generation of superoxide [4, 86]. Thus, tonic inhibition of platelets as well as insulin-dependent suppression of reactivity may be reduced in subjects with diabetes.
Antiplatelet Therapy and Diabetes
While several studies have identified beneficial cardiovascular effects of aspirin for primary prevent in people with diabetes, a meta-analysis that included results from 33,679 demonstrated that the use of aspirin for primary prevention of cardiovascular disease in patients with diabetes mellitus increases the risk of total bleeding without reducing the risk of major adverse cardiovascular outcomes [87]. Considered together, data acquired in vitro and in vivo suggest that platelets from subjects with diabetes are hypersensitive to diverse agonists. Unfortunately, currently available antiplatelet therapy does not restore normal responsiveness to platelets from subjects with diabetes. In animal preparations simulating selected aspects of diabetes, platelets remain hypersensitive to thrombin despite administration of aspirin [88]. Furthermore, responses to aspirin are suboptimal in people with diabetes [89,90,91]. These observations suggest that the hypersensitivity is not a reflection of generation of thromboxane A2, and that the treatment of subjects with diabetes with aspirin (as is being done often inferentially) is unlikely to decrease platelet reactivity to the level typical of that seen with platelets from nondiabetic subjects. Because hyperglycemia per se appears to increase platelet reactivity, improved glycemic control is a critical component of the antithrombotic regimen.
Platelet hyperreactivity has been observed before and during treatment with clopidogrel [48, 92, 93]. This increased platelet reactivity has been associated with a greater risk of subsequent cardiovascular events [92]. Higher doses of clopidogrel suppress platelet reactivity in patients with diabetes to a greater extent, but the effect remains heterogeneous and uniform suppression of platelet hyperreactivity was not seen [94]. Furthermore, withdrawal of cloplidogrel is associated with proinflammatory and prothrombotic effects [95]. These results suggest that patients with diabetes are likely to benefit from more powerful antiplatelet regimens. Ticagrelor, a more powerful P2Y12 antagonist was shown to be more effective than clopidogrel (both agents were used in combination with aspirin) in preventing recurrent heart attack, stroke, and death in patients with acute coronary syndromes, and the beneficial effects of ticagrelor were similar in patients with and without diabetes [96]. Prasugrel plus aspirin was more effective than clopidogrel plus aspirin in patients with acute coronary syndromes, and the beneficial effects were maintained in patients with diabetes [97]. Accordingly, for patients with diabetes and acute coronary syndrome, dual-antiplatelet therapy with aspirin plus either ticagrelor or prasugrel is preferred.
The Coagulation System and Diabetes Mellitus
Activation of the coagulation system leads to the generation of thrombin and thrombin-mediated formation of fibrin from fibrinogen. The generation of thrombin depends on activation of procoagulant factors. It is limited by antithrombotic factors and inhibitors. Fibrinopeptide A (FPA) is released when fibrinogen is cleaved by thrombin. It has a very short half-life in the circulation and is cleared promptly by the kidneys. Elevated concentrations in blood are indicative of thrombin activity in vivo [98]. Subjects with diabetes mellitus (both types 1 and 2) have increased concentrations of FPA in blood and in urine compared with corresponding concentrations in nondiabetic subjects [99,100,101,102]. The highest concentrations are observed in patients with clinically manifest vascular disease [100, 102].
The increased concentrations of FPA seen in association with diabetes reflect an altered balance between prothrombotic and antithrombotic determinants in subjects with diabetes mellitus favoring thrombosis. This interpretation is consistent with other observations suggesting that generation of thrombin is increased with diabetes resulting in increased concentrations in blood of thrombin-antithrombin complexes [103]. The steady-state concentration of thrombin-antithrombin complexes in blood is a reflection of the rate of formation of thrombin being generated over time.
The increased generation of thrombin in people with diabetes is likely to be dependent on increased activity of factor Xa. This has been observed in patients with type 1 diabetes [104]. Factor Xa, a major component of the prothrombinase complex, is formed from components including circulating coagulation factor X assembled on phospholipid membranes in association with the tissue factor VIIa complex. Thrombin is generated by the prothrombinase complex comprising factors Xa, Va, and II assembled on phospholipid membranes. The activity of this complex is reflected by prevailing concentrations in blood of prothrombin fragment 1 + 2, a cleavage product of factor II (prothrombin). Increased concentrations of prothrombin fragment 1 + 2 in blood from patients with type 1 diabetes have been observed, consistent with the presence of a prothrombotic state.
Mechanisms Responsible for a Prothrombotic State Associated with Diabetes
Patients with diabetes mellitus have increased concentrations in blood of the prothrombotic factors fibrinogen, von Willebrand factor, and factor VII coagulant activity [105,106,107]. Among the three coagulation factors, fibrinogen has been most strongly associated with the risk of development of cardiovascular disease [108]. Although the mechanisms responsible for increased concentrations of fibrinogen and von Willebrand factor have not yet been fully elucidated, elevated concentrations in blood of insulin and proinsulin may be determinants in people with type 2 diabetes. This possibility is suggested by the close correlation between concentrations of fibrinogen with those of insulin and proinsulin in healthy subjects [109]. Because prediabetic subjects and people with early stages of diabetes have marked insulin resistance that leads to a compensatory increase in the concentrations in blood of insulin and proinsulin [110,111,112], the hyper(pro)insulinemia of type 2 diabetes is likely to underlie, at least in part, the typically increased concentrations of fibrinogen. Improvement in metabolic control per se (euglycemia and amelioration of hyperlipidemia) has not been associated with normalization of the increased concentrations in blood of fibrinogen, von Willebrand factor, or factor VII coagulant activity [107]. By the same token, the extent of elevation of concentrations in blood of prothrombin fragment 1 + 2 is not closely correlated with the concentration of hemoglobin A1c, a marker of glycation of proteins [113]. First-degree nondiabetic relatives of subjects with type 2 diabetes exhibit increased concentrations of fibrinogen and factor VII coagulant activity in blood compared with values in age-matched controls [114]. Thus, the increases in fibrinogen and factor VII coagulant activity are associated with other, presumably independent features of insulin resistance. Consistent with this observation, controlling hyperglycemia reduces tissue factor expression in patients with type 2 diabetes [115]. Infusion of insulin increased tissue factor procoagulant activity, and the combination of hyperinsulinemia plus hyperglycemia increased tissue factor procoagulant activity to the greatest extent [115]. Accordingly, increased concentrations of prothrombotic factors seen typically in subjects with type 2 diabetes mellitus are likely to reflect the combination of metabolic derangements typical of the diabetic state in combination with insulin resistance and hyperinsulinemia. In fact, hormonal abnormalities, particularly insulin resistance and hyper(pro)insulinemia, appear to underlie the prothrombotic state [109,110,111,112,113,114].
As mentioned in the preceding section on platelet function, procoagulant activity is increased in platelets from subjects with diabetes. Procoagulant activity of monocytes is increased as well [116]. The negatively charged phospholipid surface of platelets and monocytes catalyzes both formation and activity of the tenase and prothrombinase complexes. Thus, increased procoagulant activity of platelets and monocytes can potentiate thrombosis.
Decreased activity of antithrombotic factors in blood can potentiate thrombosis. Of note, concentrations in blood of protein C and activity of antithrombin are decreased in diabetic subjects [117,118,119,120], although not universally [103]. Unlike changes in concentrations of prothrombotic factors, altered concentrations and activity of antithrombotic factors appear to be reflections of the metabolic state typical of diabetes, either type 1 or type 2, especially hyperglycemia. Thus, decreased antithrombotic activity has been associated with nonenzymatic glycation of antithrombin.
To recapitulate, functional activities of the prothrombinase complex and of thrombin itself are increased consistently in blood of people with diabetes. The increased activity is likely to be a reflection of increased procoagulant activity of platelets and monocytes in association with increased concentrations of fibrinogen, von Willebrand factor, and factor VII. Diminished activity in blood of antithrombotic factors secondary to glycation of antithrombin and protein C may contribute to the prothrombotic state. To the extent that glycation of proteins contributes to a prothrombotic state, optimal glycemic control should attenuate it. Accordingly, the most effective mechanism available to attenuate a prothrombotic state is normalization of the hormonal and metabolic abnormalities in patients with diabetes. Results in the DCCT trial are consistent with this interpretation. Despite the fact that the trial focused on microvascular complications of diabetes, known to be influenced by hyperglycemia, a trend toward reduction of macrovascular events was seen with stringent and glycemic control [121]. This trend is consistent with reduction of the intensity of the prothrombotic state and hence attenuation of atherogenesis, determinants of its sequela, or both.
A prespecified analysis of the Cardiovascular Outcomes for People Using Anticoagulation Strategies (COMPASS) compared the effects of rivaroxaban (2.5 mg twice daily) plus aspirin (100 mg daily) versus placebo plus aspirin in patients with diabetes mellitus versus without diabetes mellitus in preventing major vascular events [122]. A consistent and similar relative risk reduction was seen for benefit of rivaroxaban plus aspirin (n = 9152) versus placebo plus aspirin (n = 9126) in patients both with (n = 6922) and without (n = 11,356) diabetes for the primary efficacy end point (hazard ratio, 0.74, p = 0.002; and hazard ratio, 0.77, p = 0.005, respectively, pinteraction = 0.77) and all-cause mortality (hazard ratio, 0.81, p = 0.05; and hazard ratio, 0.84, p = 0.09, respectively; pinteraction = 0.82). Despite the prothrombotic state associated with diabetes, the incidence of bleeding was similar in patients with and without diabetes [122]. In the treatment of atrial fibrillation, results show similar efficacy and safety of direct-acting anticoagulants (DOACs) compared with warfarin in patients with or without diabetes. Treatment with DOACS in patients with diabetes has been associated with a significant relative reduction in vascular death compared to warfarin [123].
Diabetes and Fibrinolysis
Decreased fibrinolytic system capacity is observed consistently in blood from patients with diabetes mellitus, particularly those with type 2 diabetes [124,125,126,127]. It has been known for many years that obesity is associated with impaired fibrinolysis [128]; that elevated blood triglycerides and other hallmarks of hyperinsulinemia are associated with increased activity of PAI-1 [129]; and that elevated PAI-1 is a marker of increased risk of acute myocardial infarction as judged from its presence in survivors compared with age-matched subjects who had not experienced any manifestations of overt coronary artery disease [130]. Recently, the 4G/5G PAI-1 polymorphism has been found to increase the risk of recurrence of myocardial infarction in nonhyperlipidemic subjects [131] and that increased PAI-1 is associated with increasing concentrations of glucose within the normal range in nondiabetic subjects [132]. We have found that impaired fibrinolysis in subjects with type 2 diabetes mellitus, not only under baseline conditions but also in response to physiologic challenge, was attributable to augmented concentrations in blood of circulating PAI-1. Furthermore, obese diabetic subjects exhibited threefold elevations of PAI-1 in blood compared with values in nondiabetic subjects despite tissue-type plasminogen activator (t-PA) values that were virtually the same. The observation of an impairment of fibrinolysis not only under basal conditions but also in response to physiologic stress implicates the pathophysiologic import of the abnormality [125]. Subsequently, we found that precursors of insulin including proinsulin and des[30,31]- and des[63,64]proinsulin-induced time- and concentration-dependent elevation in expression of PAI-1 by human hepatoma cells in culture [133]. In addition, we found that concentrations of PAI-1 can be elevated in blood in normal subjects rendered hyperglycemic, hyperinsulinemic, and hyperlipidemic [134]. Furthermore, women with the polycystic ovarian syndrome, known to be associated with hyperinsulinemia, have increased concentrations of PAI-1 in blood that can be reduced by administration of troglitazone, an insulin sensitizer [135].
Thus, people with type 2 diabetes exhibit a decreased fibrinolytic system capacity secondary to increased PAI-1 in blood. Similar derangements are evident in association with other states of insulin resistance and compensatory hyperinsulinemia in conditions such as obesity [125, 128], hypertension [136], and the polycystic ovarian syndrome [135, 137, 138].
Because the endogenous fibrinolytic system influences the evolution of thrombosis and the rapidity and extent of lysis of thrombi when vascular damage is repaired, overexpression of PAI-1 is likely to exacerbate development and the persistence of thrombi. Results in transgenic mice deficient in PAI-1 compared with wild-type animals are consistent with this hypothesis. Thus, 24 h after arterial injury, persistence of thrombosis and the residual thrombus burden were greater than in wild-type mice that were not deficient in PAI-1 [139]. Thus, increased expression of PAI-1 typical of that seen with the metabolic changes present in type 2 diabetes [140] is likely to be a determinant of increased and persistent thrombosis. Consistent with this observation, higher concentrations of PAI-1 in blood have been independently associated with a greater risk of coronary heart disease [141].
Mechanisms Responsible for the Overexpression of PAI- in Diabetes
Increased expression of PAI-1 in diabetes is undoubtedly multifactorial. A direct effect of insulin on the expression of PAI-1 has been suggested by a positive correlation between the concentration of insulin and PAI-1 in vivo [124, 125, 129, 136,137,138, 142]. Triglycerides and their constituents (fatty acids) appear to contribute to the overexpression of PAI-1 in view of the fact that both insulin and triglycerides independently increase expression of PAI-1 by human hepatoma cells in vitro [140, 143,144,145]. Liver steatosis is another determinant of elevated concentrations of PAI-1, perhaps indicative of the response of both to derangements in the TNF-signaling pathway [146]. Insulin and triglycerides exert a synergistic increase in accumulation of PAI-1 in conditioned media when both are present in pathophysiologic concentrations [140]. Analogous results are obtained with insulin in combination with VLDL-triglyceride, emulsified triglycerides, or albumin-bound-free (nonesterified) fatty acids. Thus, the combination of hyperinsulinemia and hypertriglyceridemia increases expression of PAI-1 consistent with the possibility that the combination is a determinant of the increased PAI-1 in blood in vivo in people with diabetes. Furthermore, because elevated concentrations of glucose increase expression of PAI-1 by endothelial cells and vascular smooth muscle cells in vitro [147, 148], the metabolic state typical of diabetes may elevate concentrations of PAI-1 in blood emanating from release of PAI-1 from vessel wall cells.
A combination of hyperinsulinemia, hypertriglyceridemia, and hyperglycemia increases the concentration of PAI-1 in blood in normal subjects [134]. Although neither the infusion of insulin with euglycemia maintained by euglycemic clamping nor the infusion of triglycerides without induction of hyperinsulinemia in normal subjects increases the concentration of PAI-1 in blood, the induction of hyperglycemia, hypertriglyceridemia, and hyperinsulinemia by infusion of glucose plus emulsified triglycerides plus heparin (to elevate blood free fatty acids) does increase concentrations of PAI-1 in blood. Of note, the infusion of insulin under euglycemic clamp conditions results in a marked decrease in the concentration of blood triglycerides and free fatty acids. Thus, results obtained in the infusion studies demonstrate that the combination of hyperinsulinemia, hyperglycemia, and hypertriglyceridemia is sufficient to increase expression of PAI-1 in healthy subjects. However, results in these studies do not answer the question of whether, as in the case in vitro, insulin increases expression of PAI-1 when concentrations of glucose, triglycerides, and free fatty acids are all maintained within normal ranges. What is clear is that a combination of hormonal (hyperinsulinemia) and metabolic (particularly hypertriglyceridemia) derangements typical of type 2 diabetes mellitus elevate the concentration of PAI-1 in blood. The elevations of PAI-1 may subject people with diabetes to double jeopardy because the ratio of PAI-1 activity to the concentration of PAI-1 protein increases when the latter is high. This appears to reflect a slower rate of loss of PAI-1 activity associated with higher concentrations of PAI-1 protein [149].
Adipose tissue is another potential source of the increased blood PAI-1 in subjects with type 2 diabetes mellitus. Studies performed on genetically obese mice demonstrated that PAI-1 mRNA expression was increased four- to fivefold in mature adipocytes [150]. The injection of insulin into lean mice increased expression of PAI-1 in adipocytes, an effect seen also with 3T3-L1 adipocytes in vitro. We have found that elaboration of PAI-1 from adipocytes is increased by TGF-β, known to be released from activated platelets [151] secondary to increased transcription and furthermore that caloric restriction per se lowers elevated PAI-1 in blood in obese, nondiabetic human subjects [152]. Thus, the elevated concentrations of PAI-1 in blood seen in subjects with type 2 diabetes appear to be secondary to effects of hyperinsulinemia, particularly in combination with hypertriglyceridemia, and to effects of other mediators implicated in the prothrombotic state seen with diabetes on expression of PAI-1 by hepatic, arterial, and adipose tissue.
In addition to elevated PAI-1 in blood, expression of PAI-1 in vessel walls with subsequent elaboration into blood is increased by insulin [153]. Pathophysiologic concentrations of insulin increase the expression of PAI-1 by human arteries in vitro [154], an effect seen in both arterial segments that appear to be grossly normal and those that exhibit atherosclerotic changes [155]. The increased expression of PAI-1 is seen in arterial segments from subjects with or without insulin-resistant states. Augmented expression of PAI-1 is seen in response to insulin with vascular smooth muscle cells in culture [156] and with co-cultured endothelial cells and smooth muscle cells [153]. Insulin increases expression of PAI-1 by vascular tissue in vivo. Local elaboration of PAI-1 follows perfusion with insulin in forearm vascular beds of healthy human subjects [157].
With the use of a co-culture system, one mechanism by which insulin increases arterial wall expression of PAI-1 has been characterized [153]. In vivo, insulin present in the luminal blood is known to be transported from the luminal to the abluminal surface of endothelial cells. In vitro, smooth muscle cells exposed to insulin have been shown to release a soluble factor(s) that increases endothelial cell expression of PAI-1. Thus, it appears likely that insulin in vivo alters expression of PAI-1 in arterial walls through a direct effect on vascular smooth muscle cells that, in turn, increases endothelial cell expression of PAI-1 in a paracrine fashion.
Therapy designed to reduce insulin resistance, the resultant hyperinsulinemia, or both have been shown to reduce PAI-1 in blood as well. Thus, treatment of women with the polycystic ovarian syndrome with metformin or troglitazone decreased concentrations in blood of insulin and of PAI-1 [138]. Changes in the concentrations of PAI-1 in blood correlated significantly with those of insulin [135]. Treatment of patients with type 2 diabetes with an insulin secretagogue, repaglinide, was associated with greater concentrations in blood of PAI-1 compared with treatment with metformin [158]. Similarly, treatment of patients with type 2 diabetes with pioglitazone decreases the concentration of PAI-1 [159]. One mechanism by which thiazolidinediones may decrease expression of PAI-1 is through induction of adiponectin [160]. The effect of rosiglitazone on expression of PAI-1 correlates positively with changes in the concentration of adiponectin and the effect of rosiglitazone of concentrations of PAI-1 in blood is attenuated in mice genetically deficient in adiponectin [160]. The concordance supports the view that insulin contributes to the increased PAI-1 expression seen in vivo. Despite meta analyses that spanned intense controversy [161,162,163], there is no convincing evidence that rosiglitazone increases mortality [164,165,166,167,168], and there is evidence that another thiazolidinedione, pioglitazone, diminishes it [169].
Human subjects who participate in relatively large amounts of leisure time physical activity have low levels of PAI-1 activity in blood [170]. After adjustment for variables indicative of syndromes of insulin resistance such as high body mass index and waist:hip ratio in addition to advanced age and elevated concentrations of triglycerides, the association of PAI-1 activity with physical activity was no longer significant. This observation, particularly in combination with the results seen after therapy with troglitazone and metformin in women with the polycystic ovarian syndrome, demonstrates that interventions designed to attenuate insulin resistance will lower concentrations of PAI-1 in blood and increase fibrinolytic system capacity.
The exposure of human hepatoma cells to gemfibrozil decreases basal and insulin-stimulated secretion of PAI-1 [171]. This inhibitory effect has been observed in vitro but not in vivo [172, 173] despite reductions in vivo in the concentration of triglycerides in blood by 50–60%. No changes in insulin sensitivity or concentrations of insulin in the blood were seen after treatment of patients with gemfibrozil. Thus, unlike therapy with agents that reduce insulin resistance and lower concentrations of insulin, therapy with gemfibrozil that reduces triglycerides without affecting concentrations of insulin does not lower PAI-1 in vivo. These observations support the likelihood that insulin is the critical determinant of altered expression of PAI-1 in subjects with insulin resistance such as those with type 2 diabetes mellitus. As judged from results in studies in which human hepatoma cells were exposed to insulin and triglycerides in vitro, modest elevations in the concentrations of triglycerides and free fatty acids in the setting of hyperinsulinemia may be sufficient to augment expression of PAI-1. Thus, although the concentration of triglycerides in patients treated with gemfibrozil was decreased by 50%, the prevailing concentration of triglycerides may have been sufficient to lead to persistent elevation of PAI-1 in blood in the setting of hyperinsulinemia. Recent results in studies with several statins including atorvastatin fail to show concordant changes in PAI-1 in blood, consistent with this possibility [174].
Fibrinolysis and Arterial Mural Proteolysis
In addition to their role in blood, plasminogen activators and PAI-1 appear to be involved in the evolution of macroangiography in the arterial wall itself [175]. Intramural plasminogen activators and PAI-1 influence proteolytic activity of matrix metalloproteinases (MMPs) that are activated from zymogens by plasmin. Cell surface plasmin-dependent proteolytic activation of MMPs promotes migration of smooth muscle cells and macrophages into the neointima and tunica media. Activation of MMPs appears to be a determinant of plaque rupture in complex atheroma and advanced atherosclerotic lesions, particularly in the vulnerable acellular shoulder regions of plaques [176].
Conversely, overexpression of PAI-1, by inhibiting intramural proteolysis and turnover of matrix, may contribute to accumulation of extracellular matrix particularly in early atheromatous lesions. Overexpression of PAI-1 and the resultant accumulation of extracellular matrix have been implicated as a substrate for activation and migration of smooth muscle cells, chemotaxis of macrophages, and hence, acceleration of early atherosclerosis. Analogously increased expression of PAI-1 has been observed in zones of early vessel wall injury after fatal pulmonary thromboembolism [177].
Taken together, these observations imply that an imbalance between the activity of plasminogen activators and the activity of PAI-1 can contribute to progression of atherosclerosis in diverse directions under diverse conditions. In early lesions, excess activity of PAI-1 may potentiate accumulation of matrix and its consequences. In complex lesions and late atherosclerosis, excess activity of plasminogen activators may exacerbate plaque rupture. Our observations regarding the relative amounts of plasminogen activators and of PAI-1 in association with the severity of atherosclerosis are consistent with both [178]. The tissue content of PAI-1 is increased in early atherosclerotic lesions exemplified by fatty streaks. By contrast, the tissue content of plasminogen activators is increased in more complex lesions at a time when smooth muscle cell proliferation is prominent.
The effects of PAI-1 in vessel wall repair have been clarified in animals genetically modified to be deficient in PAI-1 (PAI-1 knockout mice). Removal of noncellular debris and migration of smooth muscle cells are accelerated after mechanical or electrical injury of arteries in PAI-1-deficient mice [179]. However, clot burden and persistence are increased. Thus, it appears likely that excess of either plasminogen activator or PAI-1 activity in the vessel wall may potentiate atherosclerosis. Excess PAI-1 may potentiate mural thickening secondary to accumulation of extracellular matrix and noncellular debris with diminished migration into the neointima of vascular smooth muscle cells during evolution of plaques destined to be vulnerable to rupture. Excess plasminogen activator activity may potentiate degradation of matrix and plaque rupture [175] in mature, vulnerable plaques. Consistent with this view, we have found increased immunoassayable PAI-1 and decreased urokinase plasminogen activator (u-PA) in atherectomy specimens from occlusive coronary lesions in patients with diabetes with or without restenosis compared with values in corresponding specimens from nondiabetic subjects [154]. Conversely, immunoassayable urokinase in the atheroma was markedly diminished in association with diabetes.
It has been demonstrated that people with type 2 diabetes are remarkably prone not only to primary coronary lesions but also to restenosis after angioplasty [9, 180, 181]. Our observations with extracted atheroma suggest that restenosis, especially that following iatrogenic injury to vessel walls associated with percutaneous coronary intervention, may develop, in part, because of increased expression of PAI-1. Although increased PAI-1 attenuates cell migration, it augments proliferation and inhibits apoptosis [182]. Thus, restenosis may be exacerbated by increased PAI-1 resulting in increased proliferation and decreased apoptosis of smooth muscle cells within the arterial wall [183].
Therapeutic Implications
Consideration of the derangements in platelet function, the coagulation system and the fibrinolytic system, and their contributions to exacerbation of macrovascular disease in type 2 diabetes gives rise to several therapeutic approaches. Because many of the derangements contributing to a prothrombotic state in diabetes are caused by hyperglycemia, rigorous glycemic control is essential. Accordingly, the use of diet, exercise, oral hypoglycemic agents, insulin sensitizers, and if necessary insulin itself are appropriate to lower hemoglobin A1c to <7%. Because other derangements contributing to a prothrombotic state such as attenuation of fibrinolysis appear to be related to insulin resistance and hyper(pro)insulinemia, the use of insulin sensitizers as adjuncts to therapy with insulin or with other oral hypoglycemic agents is likely to be helpful.
Agents that enhance sensitivity to insulin and thereby promote glycemic control but limit hyperinsulinemia merit particular emphasis. Thiazolinediones lower elevated PAI-1 in patients with hyperinsulinemia by attenuating insulin resistance, increasing peripheral glucose disposal, and modifying transcription of genes with protein products that are involved in carbohydrate and lipid metabolism as well as in fibrinolytic system activity. This class of agents exerts favorable effects on intimal medial thickness of carotid arteries in people with type 2 diabetes [184, 185] and appears to reduce the progression of macrovascular disease [169].
Use of metformin may attenuate abnormalities in the fibrinolytic system as well as improving glycemic control, although the primary mechanism of action of the drug differs from that of troglitazone. Metformin and its congeners decrease hepatic glucose output thereby normalizing carbohydrate and lipid metabolism and reducing requirements for insulin and lowering circulating endogenous insulin levels. Side effects are usually minor gastrointestinal disturbances, but lactic acidosis can be encountered particularly in patients with renal dysfunction, congestive heart failure, liver disease, or any condition predisposing to metabolic acidosis including diabetic ketoacidosis or excessive consumption of alcohol. Metformin should be discontinued temporarily when contrast agents are used (e.g., coronary angiography) to avoid lactic acidosis. In contrast to thiazolidinediones, metformin can produce hypoglycemia, particularly when it is used with sulfonylureas.
Sodium-glucose cotransporter 2 (SGLT2) inhibitors favorably affect cardiovascular outcomes. SGLT2 inhibitors function through a novel mechanism of reducing renal tubular glucose reabsorption, producing a reduction in blood glucose without stimulating insulin release. Other benefits may include favorable effects on blood pressure and weight. A meta-analysis [186] that assessed cardiovascular outcomes of all four available SGLT2 inhibitors in patients with type 2 diabetes demonstrated that SGLT2 inhibitors were associated with a reduced risk of major adverse cardiovascular events (hazard ratio 0.90; 95% confidence interval 0.85–0.95) as well as hospitalization for heart failure and cardiovascular death (hazard ratio 0.78; 95% confidence interval 0.73–0.84). Based on this convincing class effect, SGLT2 inhibitors should be considered for all patients with type 2 diabetes, particularly those with cardiovascular disease.
Glucagon-like peptide 1 (GLP-1) receptor agonists stimulate glucose-dependent insulin release from the pancreatic islets and slow gastric emptying, inhibit inappropriate post-meal glucagon release, and reduce food intake. A meta-analysis [187] demonstrated that GLP-1 receptor agonist treatment reduced the incidence of major adverse cardiovascular events by 12% (hazard ratio 0.88, 95% confidence interval 0.82–0.94; p < 0.0001). Consistent reductions were apparent in death from cardiovascular cause (hazard ratio 0.88; 95% confidence interval 0.81–0.96; p = 0.003), fatal or non-fatal stroke (hazard ratio 0.84; confidence interval 0.76–0.93; p < 0.0001), and fatal or non-fatal myocardial infarction (hazard ratio 0.91; confidence interval 0.84–1.00; p = 0.043).
While aspirin is no longer recommended for primary prevention [87], it should be used in patients with established coronary artery disease (secondary prevention). Patients with diabetes and acute coronary syndrome should be treated with dual-antiplatelet therapy that combines aspirin plus either ticagrelor or prasugrel [96, 97]. The combination of aspirin plus low-dose rivaroxaban reduced cardiovascular events in the COMPASS trial and should be considered for long-term treatment [122].
Several complications and concomitants of diabetes can exacerbate a prothrombotic state and accelerate vascular disease. Thus, hypertriglyceridemia, hypertension, and hyperglycemia must be ameliorated. A target-driven, long-term, intensified intervention aimed at multiple risk factors in patients with type 2 diabetes and microalbuminuria reduces the risk of cardiovascular and microvascular events by about 50% [188].
Hypertension should be treated vigorously, generally with ACE inhibitors because of the demonstrated reduction of progression of renal disease accompanying their use. An alternative may be angiotensin receptor-blocking agents. Despite the ominous portent of macrovascular disease in type 2 diabetes, nephropathy continues to be a dominant life-threatening complication with an extraordinarily high incidence. Its occurrence is clearly related to hyperglycemia and may contribute to a prothrombotic state and acceleration of macrovascular disease through diverse mechanisms. Accordingly, rigorous glycemic control is essential.
Life-style modifications including implementation of a regular exercise program, reduction of obesity through dietary measures, and avoidance or cessation of cigarette smoking should be implemented to reduce the intensity of a prothrombotic state and the progression of macrovascular disease.
References
Geiss LS, Herman WH, Smith PJ. Mortality in non-insulin-dependent diabetes. In: Harris MI, Cowie CC, Stern MP, Boyko, Reiber GE, Bennet PH, editors. Diabetes in America. Washington, DC: U.S. Government Printing Office; 1995. p. 233–57, Chap 11, DHHS NIH Publ no. 95-1468.
Portuese E, Orchard T. Mortality in insulin-dependent diabetes. In: Harris MI, Cowie CC, Stern MP, Boyko, Reiber GE, Bennet PH, editors. Diabetes in America. Washington, DC: U.S. Government Printing Office; 1995. p. 221–32, Chap 10, DHHS NIH Publ no. 95-1468.
Alexandru N, Jardin I, Popov D, Simionescu M, Garcia-Estan J, Salido GM, Rosado JA. Effect of homocysteine on calcium mobilisation and platelet function in type 2 diabetes mellitus. J Cell Mol Med. 2007;12:2015.
Sasaki N, Yamashita T, Takaya T, Shinohara M, Shiraki R, Takeda M, Emoto N, Fukatsu A, Hayashi T, Ikemoto K, Nomura T, Yokoyama M, Hirata K-I, Kawashima S. Augmentation of vascular remodeling by uncoupled endothelial nitric oxide synthase in a mouse model of diabetes mellitus. Arterioscler Thromb Vasc Biol. 2008;28:1068–76.
Jeppesen J, Hansen TW, Rasmussen S, Ibsen H, Torp-Pedersen C, Madsbad S. Insulin resistance, the metabolic syndrome, and risk of incident cardiovascular disease. J Am Coll Cardiol. 2007;49:2112–9.
Sobel BE. Coronary artery disease and fibrinolysis: from the blood to the vessel wall. Thromb Haemost. 1999;82:8–13.
Schneider DJ, Sobel BE. Determinants of coronary vascular disease in patients with type II diabetes mellitus and their therapeutic implications. Clin Cardiol. 1997;20:433–40.
Uusitupa MIJ, Niskanen LK, Siitonen O, Voutilainen E, Pyorala K. Ten-year cardiovascular mortality in relation to risk factors and abnormalities in lipoprotein composition in type 2 (non-insulin-dependent) diabetic and non-diabetic subjects. Diabetologia. 1993;36:1175–84.
Kornowski R, Mintz GS, Kent KM, Pichard AD, Satler LF, Bucher TA, Hong MK, Popma JJ, Leon MB. Increased restenosis in diabetes mellitus after coronary interventions is due to exaggerated intimal hyperplasia. Circulation. 1997;95:1366–9.
Velican C, Velican D. The precursors of coronary atherosclerotic plaques in subjects up to 40 years old. Atherosclerosis. 1980;37:33–46.
Spurlock BO, Chandler AB. Adherent platelets and surface microthrombi of the human aorta and left coronary artery: a scanning electron microscopy feasibility study. Scanning Microsc. 1987;1:1359–65.
Nicholls S, Tuzcu E, Kalidindi S, Wolski K, Moon K-W, Sipahi K, Schoenhagen P, Nissen S. Effect of diabetes on progression of coronary atherosclerosis and arterial remodeling. J Am Coll Cardiol. 2008;52:255–62.
Ambrose JA, Tannenbaum AM, Alexpoulos D, Hjemdahl-Monsen CE, Leavy J, Weiss M, Borrico S, Gorling R, Fuster V. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12:56–62.
Little WC, Constantinescu M, Applegate RJ, Kutcher MA, Burrows MT, Kahl FR, Santamore WP. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild-to-moderate coronary artery disease? Circulation. 1988;78:1157–66.
Davies MJ, Richardson PD, Woolf N, Kratz DR, Mann J. Risk of thrombosis in human atherosclerotic plaques role of extracellular lipid, macrophage, and smooth muscle content. Br Heart J. 1993;69:377–81.
Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–71.
Dai J, Xing L, Jia H, Zhu Y, Zhang S, Hu S, Lin L, Ma L, Liu H, Xu M, Ren X, Yu H, Li L, Zou Y, Zhang S, Mintz GS, Hou J, Yu B. In vivo predictors of plaque erosion in patients with ST-segment elevation myocardial infarction: a clinical, angiographical, and intravascular optical coherence tomography study. Eur Heart J. 2018;39:2077–85.
Sugiyama T, Yamamoto E, Bryniarski K, Xing L, Fracassi F, Lee H, Jang IK. Coronary plaque characteristics in patients with diabetes mellitus who presented with acute coronary syndromes. J Am Heart Assoc. 2018;7:e009245.
Fateh-Moghadam S, Li Z, Ersel S, Reuter T, Htun P, Plockinger U, Bocksch W, Dietz R, Gawaz M. Platelet degranulation is associated with progression of intima-media thickness of the common carotid artery in patients with diabetes mellitus type 2. Arterioscler Thromb Vasc Biol. 2005;25:1299–303.
Boden G, Rao AK. Effects of hyperglycemia and hyperinsulinemia on the tissue factor pathway of blood coagulation. Curr Diab Rep. 2007;7:223–7.
Nemerson Y. Tissue factor and hemostasis. Blood. 1988;71:1–8.
Rand MD, Lock JB, Veer CV, Gaffney DP, Mann KG. Blood clotting in minimally altered whole blood. Blood. 1996;88:3432–45.
Monroe DM, Roberts HR, Hoffman M. Platelet procoagulant complex assembly in a tissue factor-initiated system. Br J Haemotol. 1994;88:364–71.
Staatz WD, Rajpara SM, Wayner EA, Carter WG, Santoro SA. The membrane glycoprotein Ia-IIa (VLA-2) complex mediates the Mg+2-dependent adhesion of platelets to collagen. J Cell Biol. 1989;108:1917–21.
Kroll MH, Harris TS, Moake JL, Handin RI, Schafer AI. Von Willebrand Factor binding to platelet GP Ib initiates signals for platelet activation. J Clin Invest. 1991;88:1568–73.
Sims PJ, Ginsberg MH, Plow EF, Shattil SJ. Effect of platelet activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J Biol Chem. 1991;266:7345–52.
Palabrica T, Lobb R, Furie BC, Aronovitz M, Benjamin C, Hsu YM, Sajer SA, Furie B. Leukocyte accumulation promoting fibrin deposition is mediated by P-selectin on adherent platelets. Nature. 1992;359:848–51.
Schwartz CJ, Valente AJ, Kelley JL, Sprague EA, Edwards EH. Thrombosis and the development of atherosclerosis: Roditansky revisited. Semin Thromb Hemost. 1988;14:189–95.
Stirk CM, Kochhar A, Smith EB, Thompson WD. Presence of growth-stimulating fibrin-degradation products containing fragment E in human atherosclerotic plaques. Atherosclerosis. 1993;103:159–69.
Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9.
Scharf RE, Harker LA. Thrombosis and atherosclerosis: regulatory role of interactions among blood components and endothelium. Blut. 1987;55:131–44.
Bar-Shavit R, Hruska KA, Kahn AJ, Wilner GD. Hormone-like activity of human thrombin. Ann N Y Acad Sci. 1986;485:335–48.
Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest. 1992;89:507–11.
Friedman RJ, Stemerman MB, Wenz B, Moore S, Gauldie J, Gent M, Tiell ML, Spaet TH. The effect of thrombocytopenia on experimental arteriosclerotic lesion formation in rabbits. Smooth muscle proliferation and re-endothelialization. J Clin Invest. 1977;60:1191–201.
Fernandez-Ortiz AJ, Badimon JJ, Falk E, Fuster V, Meyer B, Mailhac A, Weng D, Shah PK, Badimon LL. Characterization of relative thrombogenicity of atherosclerotic plaque components: implications for consequences of plaque rupture. J Am Coll Cardiol. 1994;23:1562–9.
Grant PJ. Diabetes mellitus as a prothrombotic condition. J Intern Med. 2007;262:157–72.
Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis and thrombosis. Thromb Haemost. 2008;99:995–1000.
Lim HS, Blann AD, Lip GY. Soluble CD40 ligand, soluble P-selectin, interleukin-6, and tissue factor in diabetes mellitus: relationships to cardiovascular disease and risk factor intervention. Circulation. 2004;109:2524–8.
Brown AS, Hong Y, de Belder A, Beacon H, Beeso J, Sherwood R, Edmonds M, Mrtin JF, Erusalimsky JD. Megakaryoctye ploidy and platelet changes in human diabetes and atherosclerosis. Arterioscler Thromb Vasc Biol. 1997;17:802–7.
Olufadi R, Byrne CD. Effects of VLDL and remnant particles on platelets. Pathophysiol Haemost Thromb. 2006;35:281–91.
Koga H, Sugiyama S, Kugiyama K, Fukushima H, Watanabe K, Sakamoto T, Yoshimura M, Jinnouchi H, Ogawa H. Elevated levels of remnant lipoproteins are associated with plasma platelet microparticles in patients with type-2 diabetes mellitus without obstructive coronary artery disease. Eur Heart J. 2006;27:817–23.
Mayfield RK, Halushka PV, Wohltmann HJ, Lopes-Virella M, Chambers JK, Loadholt CB, Colwell JA. Platelet function during continuous insulin infusion treatment in insulin-dependent diabetic patients. Diabetes. 1985;34:1127–33.
Iwase E, Tawata M, Aida K, Ozaki Y, Kume S, Satoh K, Qi R, Onaya T. A cross-sectional evaluation of spontaneous platelet aggregation in relation to complications in patients with type II diabetes mellitus. Metabolism. 1998;47:699–705.
Davi G, Catalano I, Averna M, Notarbartolo A, Strano A, Ciabattoni G, Patrono C. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N Engl J Med. 1990;322:1769–74.
Winocour PD, Watala C, Kinlough-Rathbone RL. Membrane fluidity is related to the extent of glycation of proteins, but not to alterations in the cholesterol to phospholipid molar ratio in isolated platelet membranes from diabetic and control subjects. Thromb Haemost. 1992;67:567–71.
Ishii H, Umeda F, Nawata H. Platelet function in diabetes mellitus. Diabetes Metab Rev. 1992;8:53–66.
Hendra T, Betteridge DJ. Platelet function, platelet prostanoids and vascular prostacyclin in diabetes. Prostaglandins Leukot Essent Fat Acids. 1989;35:197–212.
Menys VS, Bhatnagar D, Mackness MI, Durrington PN. Spontaneous platelet aggregation in whole blood is increased in non-insulin-dependent diabetes mellitus and in female but not male patients with primary dyslipidemia. Atherosclerosis. 1995;112:115–22.
Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramirez C, Sabate M, Jimenez-Quevedo P, Hernandez R, Moreno R, Escaned J, Alfonso F, Banuelos C, Costa MA, Bass TA, Macaya C. Platelet function profiles in patients with type 2 diabetes and coronary artery disease on combined aspirin and clopidogrel treatment. Diabetes. 2005;54:2430–5.
Vaidyula VR, Boden G, Rao AK. Platelet and monocyte activation by hyperglycemia and hyperinsulinemia in healthy subjects. Platelets. 2006;17:577–85.
Calverley DC, Hacker MR, Loda KA, Brass E, Buchanan TA, Tsao-Wei DD, Groshen S. Increased platelet Fc receptor expression as a potential contributing cause of platelet hypersensitivity to collagen in diabetes mellitus. Br J Haematol. 2003;121:139–42.
Calverley DC, Baldermann LV, Moran K, Chen NN, McFann K. Platelet FcgammaRIIA expression is associated with the alpha2 integrin C807T gene polymorphism in type 2 diabetes. Platelets. 2006;17:78–83.
Schneider DJ, McMahon SR, Chava S, Taatjes-Sommer HS, Meagher S, Ehle GL, Brummel-Ziedins KE. FcγRIIa: a new cardiovascular risk marker. J Am Coll Cardiol. 2018;72:237–8.
Schneider DJ, McMahon SR, Ehle GL, Chava S, Taatjes-Sommer HS, Meagher S. Assessment of cardiovascular risk by the combination of clinical risk scores plus platelet expression of FcγRIIa. Am J Cardiol. 2020;125:670–2.
Cabeza N, Li Z, Schulz C, Kremmer E, Massberg S, Bultmann A, Gawaz M. Surface expression of collagen receptor Fc receptor-gamma/glycoprotein VI is enhanced on platelets in type 2 diabetes and mediates release of CD40 ligand and activation of endothelial cells. Diabetes. 2004;53:2117–21.
Arthur JF, Jandeleit-Dahm K, Andrews RK. Platelet hyperreactivity in diabetes: focus on GPVI signaling-are useful drugs already available? Diabetes. 2017;66:7–13.
Sugimoto H, Franks DJ, Lecavalier L, Chiasson JL, Hamet P. Therapeutic modulation of growth-promoting activity in platelets from diabetics. Diabetes. 1987;36:667–72.
Koschinsky T, Bunting CR, Rutter R, Gries FA. Vascular growth factors and the development of macrovascular disease in diabetes mellitus. Diabetes Metab. 1987;13:318–25.
Winocour PD, Bryszewska M, Watala C, Rand ML, Epand RM, Kinlough-Rathbone RL, Packham MA, Mustard JF. Reduced membrane fluidity in platelets from diabetic patients. Diabetes. 1990;39:241–4.
Rao AK, Goldberg RE, Walsh PN. Platelet coagulation activity in diabetes mellitus. Evidence for relationship between platelet coagulant hyperactivity and platelet volume. J Lab Clin Med. 1984;103:82–92.
Lupu C, Calb M, Ionescu M, Lupu F. Enhanced prothrombin and intrinsic factor X activation on blood platelets from diabetic patients. Thromb Haemost. 1993;70:579–83.
Tschoepe D, Roesen P, Kaufmann L, Schauseil S, Kehrel B, Ostermann H, Gries FA. Evidence for abnormal platelet glycoprotein expression in diabetes mellitus. Eur J Clin Investig. 1990;20:166–70.
Romano M, Pomilio M, Vigneri S, Falco A, Chiesa PL, Chiarelli F, Davi G. Endothelial perturbation in children and adolescents with type 1 diabetes: association with markers of the inflammatory reaction. Diabetes Care. 2001;24:1674–8.
Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;23(1):17. (abstract).
Bastyr EJ III, Lu J, Stowe R, Green A, Vinik AI. Low molecular weight GTP-binding proteins are altered in platelet hyperaggragation in IDDM. Oncogene. 2003;8:515–8.
Livingstone C, McLellan AR, McGregor MA, Wilson A, Connell JM, Small M, Milligan G, Paterson KR, Houslay MD. Altered G-protein expression and adenylate cyclase activity in platelets of non-insulin-dependent diabetic (NIDDM) male subjects. Biochim Biophys Acta. 1991;1096:127–33.
Ishii H, Umeda F, Hashimoto T, Nawata H. Changes in phosphoinositide turnover, Ca2+ mobilization, and protein phosphorylation in platelets from NIDDM patients. Diabetes. 1990;39:1561–8.
Schaeffer G, Wascher TC, Kostner GM, Graier WF. Alterations in platelet Ca2+ signalling in diabetic patients is due to increased formation of superoxide anions and reduced nitric oxide production. Diabetologia. 1999;42:167–76.
Tschoepe D, Roesen P, Esser J, Schwippert B, Nieuwenhuis K, Kehrel B, Gries FA. Large platelets circulate in an activated state in diabetes mellitus. Semin Thromb Hemost. 1991;17:433–8.
Tschoepe D, Driesch E, Schwippert B, Nieuwenhuis K, Gries FA. Exposure of adhesion molecules on activated platelets in patients with newly diagnosed IDDM is not normalized by near-normoglycemia. Diabetes. 1995;44:890–4.
Torr-Brown SR, Sobel BE. Plasminogen activator inhibitor is elevated in plasma and diminished in platelets in patients with diabetes mellitus. Thromb Res. 1994;75:473–7.
Colwell JA. Vascular thrombosis in type II diabetes mellitus. Diabetes. 1993;42:8–11.
Kinlough-Rathbone RL, Packham MA, Mustard JF. Vessel injury, platelet adherence, and platelet survival. Arteriosclerosis. 1983;3:529–46.
Winocour PD, Richardson M, Kinlough-Rathbone RL. Continued platelet interaction with de-endothelialized aortae of spontaneously diabetic BB Wistar rats is associated with slow re-endothelialization and extensive intimal hyperplasia. Int J Exp Pathol. 1993;74:603–13.
Winocour PD, Watala C, Perry DW, Kinlough-Rathbone RL. Reduced fluidity and increased glycation of membrane proteins of platelets from diabetic subjects are not associated with increased platelet adherence to glycated collagen. J Lab Clin Med. 1992;120:921–8.
Oskarsson HJ, Hofmeyer TG. Platelets from patients with diabetes mellitus have impaired ability to mediate vasodilatation. J Am Coll Cardiol. 1996;27:1464–70.
Tschoepe D, Roesen P, Gries FA. Increase in the cytosolic concentration of calcium in platelets of diabetics type II. Thromb Res. 1991;62:421–38.
Ishi H, Umeda F, Hashimoto T, Nawata H. Changes in phosphoinositide turnover, Ca2+ mobilization, and protein phosphorylation in platelets from NIDDM patients. Diabetes. 1990;39:1561–8.
Keating FK, Sobel BE, Schneider DJ. Effects of increased concentrations of glucose on platelet reactivity in healthy subjects and in patients with and without diabetes. Am J Cardiol. 2003;92:1362–5.
Malmberg K, Norhammar A, Wedel H, Ryden L. Glycometabolic state at admission: important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation. 1999;99:2626–32.
Fava S, Aquilina O, Azzopardi J, Agius Muscat H, Fenech FF. The prognostic value of blood glucose in diabetic patients with acute myocardial infarction. Diabet Med. 1996;13:80–3.
Wahab NN, Cowden EA, Pearce NJ, Gardner MJ, Merry H, Cox JL. Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era? J Am Coll Cardiol. 2002;40:1748–54.
Trovati M, Anfossi G, Massucco P, Mattiello L, Costamagna C, Piretto V, Mularoni E, Cavalot F, Bosia A, Ghigo D. Insulin stimulates nitric oxide synthesis in human platelets and, through nitric oxide, increases platelet concentrations of both guanosine-3′,5′-cyclic monophosphate and adenosine-3′,5′-cyclic monophosphate. Diabetes. 1997;46:742–9.
Ferreira IA, Mocking AI, Feijge MA, Gorter G, van Haeften TW, Heemskerk JW, Akkerman JW. Platelet inhibition by insulin is absent in type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2006;26:417–22.
Marina V, Bruno GA, Trucco F, Zumpano E, Tagliabue M, Di Bisceglie C, Pescarmona G. Platelet cNOS activity is reduced in patients with IDDM and NIDDM. Thromb Haemost. 1998;79:520–2.
Anfossi G, Trovati M. Pathophysiology of platelet resistance to anti-aggregating agents in insulin resistance and type 2 diabetes: implications for anti-aggregating therapy. Cardiovasc Hematol Agents Med Chem. 2006;4:111–28.
Khan SU, Ul Abideen Asad Z, Khan MU, Talluri S, Ali F, Shahzeb Khan M, Lone AN, Mookadam F, Krasuski RA, Kaluski E. Aspirin for primary prevention of cardiovascular outcomes in diabetes mellitus: an updated systematic review and meta-analysis. Eur J Prev Cardiol. 2020;27:2034.
Winocour PD, Kinlough-Rathbone RL, Mustard JF. Pathways responsible for platelet hypersensitivity in rats with diabetes. II. Spontaneous diabetes in BB Wistar rats. J Lab Clin Med. 1986;109:154–8.
Yan Y, Phillips DR. Aspirin response and failure in diabetic patients with cardiovascular disease. Curr Opin Pharmacol. 2005;5:190–7.
Takahashi S, Ushida M, Komine R, Shimizu A, Uchida T, Ishihara H, Shibano T, Watanabe G, Ikeda Y, Murata M. Increased basal platelet activity, plasma adiponectin levels, and diabetes mellitus are associated with poor platelet responsiveness to in vitro effect of aspirin. Thromb Res. 2007;119:517–24.
DiChiara J, Bliden KP, Tantry US, Hamed MS, Antonino MJ, Suarez TA, Bailon O, Singla A, Gurbel PA. The effect of aspirin dosing on platelet function in diabetic and nondiabetic patients: an analysis from the aspirin-induced platelet effect (ASPECT) study. Diabetes. 2007;56:3014–9.
Angiolillo DJ, Bernardo E, Sabaté M, Jimenez-Quevedo P, Costa MA, Palazuelos J, Hernández-Antolin R, Moreno R, Escaned J, Alfonso F, Bañuelos C, Guzman LA, Bass TA, Macaya C, Fernandez-Ortiz A. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol. 2007;50:1541–7.
Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost. 2008;100:76–82.
Angiolillo DJ, Shoemaker SB, Desai B, Yuan H, Charlton RK, Bernardo E, Zenni MM, Guzman LA, Bass TA, Costa MA. Randomized comparison of a high clopidogrel maintenance dose in patients with diabetes mellitus and coronary artery disease: results of the Optimizing Antiplatelet Therapy in Diabetes Mellitus (OPTIMUS) study. Circulation. 2007;115:708–16.
Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramirez C, Sabate M, Jimenez-Quevedo P, Hernandez R, Moreno R, Escaned J, Alfonso F, Banuelos C, Costa MA, Bass TA, Macaya C. Clopidogrel withdrawal is associated with proinflammatory and prothrombotic effects in patients with diabetes and coronary artery disease. Diabetes. 2006;55:780–4.
Franchi F, James SK, Ghukasyan Lakic T, Budaj AJ, Cornel JH, Katus HA, Keltai M, Kontny F, Lewis BS, Storey RF, Himmelmann A, Wallentin L, Angiolillo DJ, PLATO Investigators. Impact of diabetes mellitus and chronic kidney disease on cardiovascular outcomes and platelet P2Y12 receptor antagonist effects in patients with acute coronary syndromes: insights from the PLATO trial. J Am Heart Assoc. 2019;8:e011139.
Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, Riesmeyer J, Weerakkody G, Gibson CM, Antman EM, TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15.
Scharfstein JS, Abendschein DR, Eisenberg PR, George D, Cannon CP, Becker RC, Sobel BE, Cupples A, Braunwald D. Loscalzo J for the TIMI-5 Investigators. Usefulness of fibrinogenolytic and procoagulant markers during thrombolytic therapy in predicting clinical outcomes in acute myocardial infarction. Am J Cardiol. 1996;78:503–10.
Jones RL. Fibrinopeptide-A in diabetes mellitus. Relation to levels of blood glucose, fibrinogen disappearance, and hemodynamic changes. Diabetes. 1985;34:836–43.
Librenti MC, D’Angelo A, Micossi P, Garimberti B, Mannucci PM, Pozza G. Beta-thromboglobulin and fibrinopeptide A in diabetes mellitus as markers of vascular damage. Acta Diabetol Lat. 1985;22:39–45.
Marongiu F, Conti M, Mameli G, Sorano GG, Cossu E, Cirillo R, Balestrieri A. Is the imbalance between thrombin and plasmin activity in diabetes related to the behaviour of antiplasmin activity. Thromb Res. 1990;58:91–9.
Pszota HM, Kugler RK, Szigeti G. Fibrinopeptide-A as thrombotic risk marker in diabetic and atherosclerotic coronary vasculopathy. J Med. 1992;23:93–100.
Morishita E, Asakura H, Jokaji H, Saito M, Uotani C, Kumabashiri I, Yamazaki M, Aoshima K, Hashimoto T, Matsuda T. Hypercoagulability and high lipoprotein (a) levels in patients with type II diabetes mellitus. Atherosclerosis. 1996;120:7–14.
Myrup B, Rossing P, Jensen T, Gram J, Kluft C, Jespersen J. Procoagulant activity and intimal dysfunction in IDDM. Diabetologia. 1995;38:73–8.
Kannel WB, D’Agostino RB, Wilson PW, Belanger AJ, Gagnon DR. Diabetes, fibrinogen, and risk of cardiovascular disease: the Framingham experience. Am Heart J. 1990;120:672–6.
Lufkin EG, Fass DN, O’Fallon WM, Bowie EJW. Increased von Willebrand factor in diabetes mellitus. Metabolism. 1979;28:63–6.
Kannel WB, Wolf PA, Wilson PWF, D’Agostino RB. Fibrinogen and risk of cardiovascular disease. JAMA. 1987;258:1183–6.
Knobl P, Schernthaner G, Schnack C, Pietschmann P, Proidl S, Prager R, Vukovich T. Haemostatic abnormalities persist despite glycaemic improvement by insulin therapy in lean type 2 diabetic patients. Thromb Haemost. 1994;71:692–7.
Eliasson M, Roder ME, Dinesen B, Evrin PE, Lindahl B. Proinsulin, intact insulin, and fibrinolytic variables and fibrinogen in healthy subjects. Diabetes Care. 1997;20:1252–5.
Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med. 1990;113:909–15.
Ward WK, LaCava EC, Paquette TL, Beard JC, Wallum BJ, Porte D. Disproportionate elevation of immunoreactive proinsulin in type 2 (non-insulin-dependent) diabetes mellitus and in experimental insulin resistance. Diabetologia. 1987;30:698–702.
Nagi DK, Hendra TJ, Ryle AJ, Cooper TM, Temple RC, Clark PMS, Schneider AE, Hales CN, Yudkin JS. The relationships of concentrations of insulin, intact proinsulin and 32-33 split proinsulin with cardiovascular risk factors in type 2 (non-insulin-dependent) diabetic subjects. Diabetologia. 1990;33:532–7.
Marongiu F, Mascia F, Mameli G, Cirillo R, Balestrieri A. Prothrombin fragment F 1 + 2 levels are high in NIDDM patients independently of the Hb A1 c. Thromb Haemost. 1995;74:805–6.
Mansfield MW, Heywood DM, Grant PJ. Circulating levels of factor VII, fibrinogen, and von Willebrand factor and features of insulin resistance in first-degree relatives of patients with NIDDM. Circulation. 1996;94:2171–6.
Boden G, Vaidyula VR, Homko C, Cheung P, Rao AK. Circulating tissue factor procoagulant activity and thrombin generation in patients with type 2 diabetes: effects of insulin and glucose. J Clin Endocrinol Metab. 2007;92:4352–8.
Jude B, Watel A, Fontaine O, Fontaine P, Cosson A. Distinctive features of procoagulant response of monocytes from diabetic patients. Haemostasis. 1989;19:95–73.
Ceriello A, Russo PD, Zucotti C, Florio A, Nazzaro S, Pietrantuono C, Rosato GB. Decreased antithrombin III activity in diabetes may be due to non-enzymatic glycosylation: a preliminary report. Thromb Haemost. 1983;50:633–4.
Brownlee M, Vlassara H, Cerami A. Inhibition of heparin-catalyzed human antithrombin III activity by nonenzymatic glycosylation. Diabetes. 1984;33:532–5.
Ceriello A, Giugliano D, Quatraro A, Stante A, Consoli G, Dello Russo P, D’Omoferio R. Daily rapid blood glucose variations may condition antithrombin III biologic activity but not its plasma concentration in insulin-dependent diabetes: a possible role for labile on-enzymatic glycation. Diabetes Metab. 1987;13:16–9.
Ceriello A, Quatraro A, Dello Russo P, Marchi E, Barbanti M, Millani MR, Giugliano D. Protein C deficiency in insulin dependent diabetes: a hyperglycemia-related phenomenon. Thromb Haemost. 1990;65:104–7.
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–86.
Bhatt DL, Eikelboom JW, Connolly SJ, Steg PG, Anand SS, Verma S, Branch KRH, Probstfield J, Bosch J, Shestakovska O, Szarek M, Maggioni AP, Widimský P, Avezum A, Diaz R, Lewis BS, Berkowitz SD, Fox KAA, Ryden L, Yusuf S. COMPASS steering committee and investigators. role of combination antiplatelet and anticoagulation therapy in diabetes mellitus and cardiovascular disease: insights from the COMPASS trial. Circulation. 2020;141:1841–54.
Pomero F, Dentali F, Mumoli N, Salomone P, Tangianu F, Desideri G, Mastroiacovo D. The continuous challenge of antithrombotic strategies in diabetes: focus on direct oral anticoagulants. Acta Diabetol. 2019;56:1247–58.
Auwerx J, Bouillon R, Collen D, Geboers J. Tissue-type plasminogen activator antigen and plasminogen activator inhibitor in diabetes mellitus. Arteriosclerosis. 1988;8:68–72.
McGill JB, Schneider DJ, Arfken CL, Lucore CL, Sobel BE. Factors responsible for impaired fibrinolysis in obese subjects and NIDDM patients. Diabetes. 1994;43:104–9.
Soares AL, Sousa MO, Dusse LM, Fernandes AP, Lasmar MC, Novelli BA, Lages Gde F, Carvalho MG. Type 2 diabetes: assessment of endothelial lesion and fibrinolytic system markers. Blood Coagul Fibrinolysis. 2007;18:395–9.
The BARI 2D Investigators. Baseline characteristics of patients with diabetes and coronary artery disease enrolled in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Am Heart J. 2008;156:1–9.
Vague P, Juhan-Vague I, Aillaud MF, Badier C, Viard R, Alessi MC, Collen D. Correlation between blood fibrinolytic activity, plasminogen activator inhibitor level, plasma insulin level and relative body weight in normal and obese subjects. Metabolism. 1986;35:250–3.
Juhan-Vague I, Vague P, Alessi MC, Badier C, Valadier J, Aillaud MF, Atlan C. Relationships between plasma insulin, triglyceride, body mass index, and plasminogen activator inhibitor 1. Diabetes Metab. 1987;13:331.
Keber I, Keber D. Increased plasminogen activator inhibitor activity in survivors of myocardial infarction is associated with metabolic risk factors of atherosclerosis. Haemostasis. 1992;22:187.
Corsetti JP, Ryan D, Moss AJ, Rainwater DL, Zareba W, Sparks CE. Plasminogen activator inhibitor-1 polymorphism (4G/5G) predicts recurrence in nonhyperlipidemic postinfarction patients. Arterioscler Thromb Vasc Biol. 2008;28:548–54.
Heidgaard PE, Sidelmann JJ, Hindsberger C, Olivarius Nde F, Henriksen JE, Gram J. Relationship of glucose concentrations with PAI-1 and t-PA in subjects with normal glucose tolerance. Diabet Med. 2006;23:887–93.
Nordt TK, Schneider DJ, Sobel BE. Augmentation of the synthesis of plasminogen activator inhibitor type-1 by precursors of insulin: a potential risk factor for vascular disease. Circulation. 1994;89:321–30.
Calles-Escandon J, Mirza S, Sobel BE, Schneider DJ. Induction of hyperinsulinemia combined with hyperglycemia and hypertriglyceridemia increases plasminogen activator inhibitor type-1 (PAI-1) in blood in normal human subjects. Diabetes. 1998;47:290–3.
Ehrmann DA, Schneider DJ, Sobel BE, Cavaghan MK, Imperial J, Rosenfeld RL, Polonsky KS. Troglitazone improves defects in insulin action, insulin secretion, ovarian steroidogenesis, and fibrinolysis in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82:2108–16.
Jansson JH, Johansson B, Boman K, Nilsson TK. Hypo-fibrinolysis in patients with hypertension and elevated cholesterol. J Intern Med. 1991;229:309–16.
Sampson M, Kong C, Patel A, Unwin R, Jacobs HS. Ambulatory blood pressure profiles and plasminogen activator inhibitor (PAI-1) activity in lean women with and without the polycytic ovary syndrome. Clin Endocrinol. 1996;45:623–9.
Velazquez EM, Mendoza SG, Wang P, Glueck CJ. Metformin therapy is associated with a decrease in plasma plasminogen activator inhibitor-1, lipoprotein (a), and immunoreactive insulin levels in patients with the polycystic ovary syndrome. Metab Clin Exp. 1997;46:454–7.
Farrehi PM, Ozaki CK, Carmeliet P, Fay WP. Regulation of arterial thrombolysis by plasminogen activator inhibitor-1 in mice. Circulation. 1998;97:1002–8.
Schneider DJ, Sobel BE. Synergistic augmentation of expression of PAI-1 induced by insulin, VLDL, and fatty acids. Coron Artery Dis. 1996;7:813–7.
Brazionis L, Rowley K, Jenkins A, Itsiopoulos C, O’Dea K. Plasminogen activator inhibitor-1 activity in type 2 diabetes: a different relationship with coronary heart disease and diabetic retinopathy. Arterioscler Thromb Vasc Biol. 2008;28:786–91.
Pandolfi A, Giaccari A, Cilli C, Alberta MM, Morviducci L, De Filippis EA, Buongiorno A, Pellegrini G, Capani F, Consoli A. Acute hyperglycemia and acute hyperinsulinemia decrease plasma fibrinolytic activity and increase plasminogen activator inhibitor type 1 in the rat. Acta Diabetol. 2001;38(2):71–6.
Chen Y, Billadello JJ, Schneider DJ. Identification and localization of a fatty acid response region in human plasminogen activator inhibitor-1 gene. Arterioscler Thromb Vasc Biol. 2000;20:2696–701.
Chen Y, Sobel BE, Schneider DJ. Effect of fatty acid chain length and thioesterification on the augmentation of expression of plasminogen activator inhibitor-1. Nutr Metab Cardiovasc Dis. 2002;12:325–30.
Chen Y, Schneider DJ. The independence of signaling pathways mediating increased expression of plasminogen activator inhibitor type 1 in HepG2 cells exposed to free fatty acids or triglycerides. Int J Exp Diab Res. 2002;3:109–19.
Alessi M-C, Bastelica D, Mavri A, Morange P, Berthet B, Grino M, et al. Plasma PAI-1 levels are more strongly related to liver steatosis than to adipose tissue accumulation. Arterioscler Thromb Vasc Biol. 2003;23:1262–8.
Nordt TK, Klassen KJ, Schneider DJ, Sobel BE. Augmentation of synthesis of plasminogen activator inhibitor type-1 in arterial endothelial cells by glucose and its implications for local fibrinolysis. Arterioscler Thromb. 1993;13:1822.
Chen YQ, Su M, Walia RR, Hao Q, Covington JW, Vaughan DE. Sp1 sites mediate activation of the plasminogen activator inhibitor-1 promoter by glucose in vascular smooth muscle cells. J Biol Chem. 1998;273:8225–31.
Sobel BE, Neimane D, Mack WJ, Hodis HN, Buchanan TA. The ratio of plasminogen activator inhibitor type-1 activity to the concentration of plasminogen activator inhibitor type-1 protein in diabetes: adding insult to injury. Coron Artery Dis. 2002;13:275–81.
Samad F, Loskutoff DJ. Tissue distribution and regulation of plasminogen activator inhibitor-1 in obese mice. Mol Med. 1996;2:568–82.
Lundgren CH, Sawa H, Brown SL, Nordt T, Sobel BE, Fujii S. Elaboration of type-1 plasminogen activator inhibitor from adipocytes: a potential pathogenetic link between obesity and cardiovascular disease. Circulation. 1996;93:106–10.
Calles-Escandon J, Ballor D, Harvey-Berino J, Ades P, Tracy R, Sobel BE. Amelioration of the inhibition of fibrinolysis in obese elderly subjects by moderate caloric restriction. Am J Clin Nutr. 1996;64:7–11.
Schneider DJ, Absher PM, Ricci MA. The dependence of augmentation of arterial endothelial cell expression of plasminogen activator inhibitor type 1 by insulin on soluble factors released from vascular smooth muscle cells. Circulation. 1997;96:2868–76.
Sobel BE, Woodcock-Mitchell J, Schneider DJ, Holt RE, Marutsuka K, Gold H. Increased plasminogen activator inhibitor type-1 in coronary artery atherectomy specimens from type 2 diabetic compared with nondiabetic patients: a potential factor predisposing to thrombosis and its persistence. Circulation. 1998;97:2213–21.
Pandolfi A, Iacoviello L, Capani F, Vitalonna E, Donati MB, Consoli A. Glucose and insulin independently reduce the fibrinolytic potential of human vascular smooth muscle cells in culture. Diabetologia. 1996;39:1425–31.
Nordt TK, Peter K, Bode C, Sobel BE. Differential regulation by troglitazone of plasminogen activator inhibitor type 1 in human hepatic and vascular cells. J Clin Endocrinol Metab. 2000;85:1563–8.
Carmassi F, Morale M, Ferrini L, Dell’Omo G, Ferdeghini M, Pedrinelli R, De Negri F. Local insulin infusion stimulates expression of plasminogen activator inhibitor-1 and tissue-type plasminogen activator in normal subjects. Am J Med. 1999;107(4):344–50.
Lund SS, Tarnow L, Stehouwer CD, Schalkwijk CG, Teerlink T, Gram J, Winther K, Frandsen M, Smidt UM, Pedersen O, Parving HH, Vaag AA. Impact of metformin versus repaglinide on non-glycaemic cardiovascular risk markers related to inflammation and endothelial dysfunction in non-obese patients with type 2 diabetes. Eur J Endocrinol. 2008;158:631–41.
Igarashi M, Hirata A, Yamaguchi H, Jimbu Y, Tominaga M. Pioglitazone reduces atherogenic outcomes in type 2 diabetic patients. J Atheroscler Thromb. 2008;15:34–40.
Hoo RL, Chow WS, Yau MH, Xu A, Tso AW, Tse HF, Fong CH, Tam S, Chan L, Lam KS. Adiponectin mediates the suppressive effect of rosiglitazone on plasminogen activator inhibitor-1 production. Arterioscler Thromb Vasc Biol. 2007;27:2777–82.
Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71.
Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–8.
Singh S, Loke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA. 2007;298:1189–95.
American Diabetes Association. Intense blood glucose control yields no significant effect on cardiovascular disease reduction. n.d.. Accessed 12 Jun 2008.
ACCORD Study Group. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59.
Home PD, Pocock SJ, Beck-Nielsen H, Gomis R, Hanefeld M, Jones NP, Komajda M. McMurray JJV for the RECORD Study Group. Rosiglitazone evaluated for cardiovascular outcomes – an interim analysis. N Engl J Med. 2007;357:28–38.
Diamond GA, Bax L, Kaul S. Uncertain effects of rosiglitazone on the risk for myocardial infarction and cardiovascular death. Ann Intern Med. 2007;147:578–81.
Mulrow CD, Cornell JE, Localio AR. Rosiglitazone: a thunderstorm from scarce and fragile data. Ann Intern Med. 2007;147:585–7.
Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L, Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J. PROactive Investigators. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial in macroVascular Events): a randomized controlled trial. Lancet. 2005;366:1279–89.
Eliasson M, Asplund K, Evrin PE. Regular leisure time physical activity predicts high activity of tissue plasminogen activator: the northern Sweden MONICA study. Int J Epidemiol. 1996;25:1182–8.
Nordt TK, Kornas K, Peter K, Fujii S, Sobel BE, Kubler W, Bode C. Attenuation by gemfibrozil of expression of plasminogen activator inhibitor type 1 induced by insulin and its precursors. Circulation. 1997;95:677–83.
Broijersen A, Eriksson M, Wiman B, Angelin B, Hjemdahl P. Gemfibrozil treatment of combined hyperlipoproteinemia. No improvement of fibrinolysis despite marked reduction of plasma triglyceride levels. Arterioscler Thromb Vasc Biol. 1996;16:511–6.
Asplund-Carlson A. Effects of gemfibrozil therapy on glucose tolerance, insulin sensitivity and plasma plasminogen activator inhibitor activity in hypertriglyceridemia. J Cardiovasc Risk. 1996;3:385–90.
Rosenson RS, Tangney CC. Antiatherothrombotic properties of statins. Implications for cardiovascular event reduction. JAMA. 1998;279:1643–50.
Sobel BE, Taatjes DJ, Schneider DJ. Intramural plasminogen activator inhibitor type-1 and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1979–89.
Libby P. Molecular bases of the acute coronary syndromes. Circulation. 1995;91:2844–50.
Lang IM, Moser KM, Schleef RR. Elevated expression of urokinase-like plasminogen activator and plasminogen activator inhibitor type 1 during the vascular remodeling associated with pulmonary thromboembolism. Arterioscler Thromb Vasc Biol. 1998;18:808–15.
Schneider DJ, Ricci MA, Taatjes DJ, Baumann PQ, Reese JC, Leavitt BJ, Absher PM, Sobel BE. Changes in arterial expression of fibrinolytic system proteins in atherogenesis. Arterioscler Thromb Vasc Biol. 1997;17:3294–301.
Carmeliet P, Moons L, Lijnen R, Janssens S, Lupu F, Collen D, Gerard RD. Inhibitory role of plasminogen inhibitor-1 in arterial wound healing and neointimal formation: a gene targeting and gene transfer study in mice. Circulation. 1997;96:3180–91.
Sobel BE. Potentiation of vasculopathy by insulin: implications from an NHLBI Clinical Alert. Circulation. 1996;93:1613–5.
Carrozza JP, Kuntz RE, Fishman RF, Baim DS. Restenosis after arterial injury caused by coronary stenting in patients with diabetes mellitus. Ann Intern Med. 1993;118:344–9.
Chen Y, Kelm RJ Jr, Budd RC, Sobel BE, Schneider DJ. Inhibition of apoptosis and caspace-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1. J Cell Biochem. 2004;92:178–88.
Schneider DJ, Chen Y, Sobel BE. The effect of plasminogen activator inhibitor type 1 on apoptosis. Thromb Haemost. 2008;100:1037.
Minamikawa J, Tanaka S, Yamauchi M, Inoue D, Koshiyama H. Potent inhibitory effect of troglitazone on carotid arterial wall thickness in type 2 diabetes. J Clin Endocrinol Metab. 1998;83:1818–20.
Koshiyama H, Shimono D, Kuwamura N, Minamikawa J, Nakamura Y. Rapid communication: inhibitory effect of pioglitazone on carotid arterial wall thickness in type 2 diabetes. J Clin Endocrinol Metab. 2001;86:3452–6.
McGuire DK, Shih WJ, Cosentino F, Charbonnel B, Cherney DZI, Dagogo-Jack S, Pratley R, Greenberg M, Wang S, Huyck S, Gantz I, Terra SG, Masiukiewicz U, Cannon CP. Association of SGLT2 inhibitors with cardiovascular and kidney outcomes in patients with type 2 diabetes: a meta-analysis. JAMA Cardiol. 2021;6:e204511.
Kristensen SL, Rørth R, Jhund PS, Docherty KF, Sattar N, Preiss D, Køber L, Petrie MC, McMurray JJV. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019;7:776–85.
Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med. 2003;348(5):383–93.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Schneider, D.J. (2023). Diabetes and Thrombosis. In: Johnstone, M., Veves, A. (eds) Diabetes and Cardiovascular Disease. Contemporary Cardiology. Humana, Cham. https://doi.org/10.1007/978-3-031-13177-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-031-13177-6_5
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-031-13176-9
Online ISBN: 978-3-031-13177-6
eBook Packages: MedicineMedicine (R0)