
Abstract
Bronchiectasis is a significant cause of morbidity and mortality. It is the end point of a pathological process. We should be aiming to identify at risk patients before they develop bronchiectasis and treat them aggressively to prevent disease progression. With improved social conditions and health care, infective causes of bronchiectasis have diminished in higher-income countries, and genetic causes are therefore relatively more common. The underlying cause of bronchiectasis should always be sought and readdressed, for example as discoveries of innate immune defects are made. ‘Idiopathic bronchiectasis’ should be a diagnosis of last resort. This chapter reviews potential genetic causes of bronchiectasis and suggests a plan for investigating the underlying aetiology. Management is discussed but it is important to note that suggested treatment strategies are often extrapolated from evidence in bronchiectasis associated with cystic fibrosis; this is likely to be inappropriate in diseases of differing pathophysiology. Rare lung diseases need to be moved out of the ‘orphan’ category by instigating multi-centre, multi-national clinical trials and producing disease-specific evidence-based guidelines.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Bronchiectasis
- Genetic
- Cystic fibrosis
- Primary ciliary dyskinesia
- Primary immunodeficiency
- Diagnosis
- Management
- Pathophysiology
Introduction
Bronchiectasis is a disorder of cough, sputum production and persistent or recurrent bronchial infection associated with damaged and dilated bronchi. Although historically considered irreversible, increased use of high-resolution computed tomography (HRCT) scanning has identified ‘mild bronchiectasis’ which is reversible if treated aggressively at an early stage. Moreover, the radiological disease can be present in some asymptomatic patients, particularly the elderly. European Respiratory Society (ERS) Guidelines for the management of adult bronchiectasis specifically define the disorder as a permanent dilatation of the bronchi with associated chronic symptoms [1]. In recent years, the international prevalence of bronchiectasis has markedly increased in both children and adults [2,3,4].
Reported causes of bronchiectasis vary depending on health care infrastructure. Diffuse bronchiectasis in countries with good diagnostic resources is typically attributed to cystic fibrosis (CF), primary ciliary dyskinesia (PCD), immune deficiency or abnormal bronchial anatomy. Bronchiectasis is more likely to be termed ‘idiopathic’ or post-infectious in settings where exploration of an underlying diagnosis is limited. Post-infectious bronchiectasis is an imprecise diagnosis, and it is usually difficult to confirm causality. However, it is recognised that infections such as pulmonary tuberculosis or severe adenovirus infection predispose individuals to bronchiectasis. In older patients, coexistence of bronchiectasis is commonly reported with COPD or asthma. The relationship of these different airway conditions is poorly understood, and is perhaps partially explained by misdiagnoses due to overlapping symptoms.
Importantly, bronchiectasis is the end-point of a heterogeneous group of diseases and pathological mechanisms [5]. Identifying bronchiectasis is just the starting point to diagnosing the underlying cause. Although some management strategies are shared (e.g. airway clearance therapy), some diseases require specific therapies. The failure of most clinical trials to show positive outcomes possibly reflects the need for future trials targeting specific diseases [6]. Moreover, knowledge of the underlying cause is necessary to provide personalised counselling regarding prognosis, associated morbidities and genetic counselling.
Pathophysiology
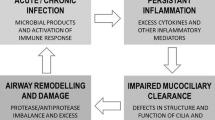
Bronchiectasis is a description of permanent airway dilatation associated with cough, sputum production and bronchial infection; bronchiectasis is usually divided into cystic fibrosis and non-cystic fibrosis related disease. A number of diseases including cystic fibrosis, primary ciliary dyskinesia and immunodeficiency disorders predispose patients to recurrent or chronic infection. Bronchiectasis can also be triggered by tuberculosis or following a severe respiratory infection. These triggers can result in airway plugging, recruitment of inflammatory cells and release of inflammatory mediators and cytokines. In turn, this can lead to the destruction of the elastic and muscular elements of the bronchial walls, causing airway dilatation, and damage to the motile cilia which line the airways [7, 8]. Accumulation of purulent viscous secretions ensues, resulting in the persistence of infection and inflammation. In the 1980s Cole proposed the ‘vicious cycle’ mechanism of ongoing and self-perpetuating damage in bronchiectasis [9]. An environmental insult, often on a background of genetic susceptibility, impairs muco-ciliary clearance resulting in persistence of microbes. The microbial infection causes chronic inflammation resulting in tissue damage and impaired cilia function. This leads to further infection with a cycle of progressive inflammation causing lung damage. Inflammation tends to be neutrophilic, with increased levels of neutrophil proteases such as neutrophil elastase leading to airway damage. Recent data shows this protease release may be related to formation of neutrophil extracellular traps. Eosinophilic inflammation can also be present in up to 30% patients [10, 11]. More recently, it has been appreciated that these interactions are far more complex with each pathophysiological step contributing to all others, “a vortex” rather than a simple circle (Fig. 25.1) [12].

A genetic condition (e.g. PCD or CF) or triggering event (e.g. severe infection) can result in a persistent and progressive cycle or ‘vortex’ of processes that eventually lead to bronchiectasis. Impaired mucociliary clearance and retention of airway secretions render the airways susceptible to recurrent and persistent infection. This process, in turn, incites an inflammatory response causing airways damage, which further impairs the clearance of mucus
Clinical Presentation
Depending on the underlying cause, bronchiectasis can develop at any stage of life. Early investigation of persistent cough is essential if interventions are to be implemented before irreversible bronchiectasis ensues. Persistent wet or productive cough failing to respond to 4 weeks of antibiotic treatment is the key feature that suggests bronchiectasis, or a pre-bronchiectasis syndrome [13]. Most patients with established bronchiectasis expectorate sputum daily, with highly variable volumes. Reduced exercise tolerance and breathlessness may be present, and are generally associated with advanced disease [14]. Haemoptysis is relatively rare in children from affluent settings [15] but is more common in children from low-income settings [16], and in adults. Thoracic pain is relatively common. Individuals with bronchiectasis may wheeze but in contrast to people with asthma this is almost invariably associated with wet cough; asthma can also co-exist with bronchiectasis.
The examination may be normal or may include crackles, wheeze, or chest deformity. Clubbing is reported to occur in some patients with bronchiectasis. Whilst it is an important sign it is not a sensitive one. In general, patients in less affluent settings have more severe diseases, presumably associated with later diagnosis and less aggressive/targeted management [16].
Epidemiology
The introduction of immunisations against pertussis in the 1950s and measles in the 1960s contributed greatly to the decline of post-infective bronchiectasis, as has the decline of pulmonary tuberculosis and the general improvement in social circumstances. However, the genetic and idiopathic disease has proportionally increased in recent times and disease prevalence has risen by more than 40% over the past 15 years. In the UK recent studies suggest a prevalence of 566 per 100,000 in women and 485 per 100,000 in men making bronchiectasis the third most common lung condition behind asthma and COPD [4]. One of the highest prevalence worldwide has recently been reported in the USA with an average of 701 patients per 100,000 people [2].
Bronchiectasis can occur at any age from early childhood, however, the average age in Western cohorts is 60–70 years [14]. It is more prevalent at a younger age in countries where there is a high incidence of pulmonary tuberculosis [17] or in certain indigenous subpopulations, including Pacific Islanders, Indigenous Australians and the Inuit community in North America [18,19,20]. Poor access to antibiotics and immunisations may partially explain these differences, although it is likely that genetic propensity may also play a role [21, 22]. Certainly, children of consanguineous parents are at a disproportionately high risk of genetic causes of bronchiectasis [23]. Moreover, although environmental, immune or anatomical factors may explain the observation that non-CF bronchiectasis is more common in females, this too may have a genetic basis [24]. Differences between nations may partly reflect genetic and environmental discrepancies; however, it is likely that the diagnosis of bronchiectasis in children is often delayed or never considered, making true prevalence difficult to establish.
The most common genetic cause of diffuse bronchiectasis is cystic fibrosis (CF), the incidence of which is estimated to be 1 in 2500 births in white Caucasians. Reports of primary ciliary dyskinesia (PCD) prevalence in European populations have varied greatly from older estimates of 1:40,000 to more recent estimates based on genetic data of 1:7500 [25, 26]. As with most orphan diseases this variation is likely to reflect a lack of awareness of the disease amongst clinicians, absence of a gold standard test, and lack of facilities for investigation, all leading to considerable under-diagnosis. A survey by a European PCD Taskforce suggested that PCD in children is under-diagnosed and diagnosed late, particularly in countries with low health expenditures [27]. The prevalence of other genetic causes of bronchiectasis is low and is considered individually later in this chapter.
Genetic Causes of Bronchiectasis
Bronchiectasis associated with genetic mutations is usually a consequence of recurrent or persistent pulmonary infection caused by disorders of mucociliary clearance or primary immunodeficiency (Table 25.1).
Disorders of Mucociliary Clearance
Ciliated respiratory epithelium lines the airways. The cilia, bathed in periciliary fluid, beat in a coordinated fashion, to propel the overlying mucus along with particles and bacteria to the oropharynx where it can be swallowed or expectorated (Fig. 25.2). Diseases affecting ciliary function, or that change the composition of the periciliary fluid and mucus can impair mucociliary clearance, leading to recurrent infections and inflammation which predispose to bronchiectasis.

In healthy persons, respiratory cilia beat in a coordinated sweeping pattern, which moves mucus and debris, including pathogens towards the oropharynx for swallowing or expectorating. In PCD, immotile or dyskinetic cilia do not beat effectively, and mucus and debris persist in the airways. In CF the inefficient mucociliary clearance (MCC) is due to an abnormal periciliary fluid layer compromising ciliary beating and viscous mucus which is resistant to clearance. (Image provided by Robert Scott)
Cystic Fibrosis
Cystic fibrosis (CF) is an autosomal recessive disorder and is the commonest inherited disease in white populations, with an estimated incidence of 1 per 2500 live births [28]. It is caused by mutations in the cystic fibrosis trans-membrane conductance regulator (CFTR) gene which is located on chromosome 7 and encodes for the CFTR chloride channel which sits in the cell membrane on the apical surface of the cell.
Mutations lead to abnormal ion transport regulation across the cell membrane which, in the lungs, results in abnormal airway fluid. Dehydrated mucus is characteristically highly viscoelastic, and adheres to the cilia and airway cells, causing airway plugging. The reduced-volume periciliary fluid layer does not adequately support and lubricate the cilia, and results in defects of ciliary function (Fig. 25.2). Adherent mucus and impaired ciliary function both contribute to reduced airway clearance, chronic infection and biofilm formation. Eventually, the chronic infection and inflammation lead to bronchiectasis, which can develop very early in life [29]. Bronchiectasis in CF predominantly affects the upper lobes initially, spreading to all lobes over time; the disparity with PCD, which tends to have worse disease in the middle lobe, is difficult to explain [30].
Since the CFTR gene was first sequenced in 1989, [31] our understanding of the underlying pathophysiology of CF has developed rapidly. At the time of writing, over 2000 mutations in CFTR have been described, of which about 350 are thought to be pathogenic (https://www.cftr2.org/welcome). These mutations have been grouped into classes, dependent on their effect on the CFTR protein (Fig. 25.3). For example, class 2 mutations, which include the most common p.Phe508del mutation, lead to a failure of the correct folding of the protein which is then rapidly broken down and hence not expressed on the apical surface of the cell; whereas with class 3 mutations the protein is correctly folded and is present on the apical surface but the channel is blocked closed, termed ‘gating’ mutations. Our understanding of the effects of mutations in CFTR has been fundamental in recent ground-breaking advances in the treatment of CF. It is now possible to correct CFTR dysfunction in patients with specific classes of mutations, and therapies for other mutations are in late-phase trials (see Novel therapies for managing CF).

Mutations causing cystic fibrosis can be grouped into classes, dependent on their effect on the CFTR protein
Although respiratory disease accounts for the majority of morbidity and mortality, [32] CF is a multisystem disorder with manifestations including meconium ileus, pancreatic insufficiency leading to steatorrhea and failure to thrive, liver disease, diabetes, nasal polyposis, sinusitis and infertility in men due to congenital bilateral absence of the vas deferens . Since the widespread use of newborn screening (NBS) for CF, measuring immuno-reactive trypsin (IRT) levels in the blood at about 7 days of life, most cases of CF are diagnosed in infancy. However later presentation, even in adulthood, is not unheard of, particularly where individuals were born prior to initiation of NBS programmes and who carry mutations other than p.Phe508del, hence are more likely to be pancreatic sufficient [33]. The diagnosis is confirmed by assessing the function of the CFTR channel by measuring sweat chloride levels, with a level > 60 mmol/L being diagnostic. Whilst not essential for diagnosis, given the advent of novel therapies based on CFTR mutation class, it is recommended that CF patients go on to be genotyped. Importantly, due to the fact that not all mutations in CFTR are pathogenic, any individual first identified by genotyping (i.e. having two bi-allelic CFTR mutations) should go on to have a confirmatory functional CFTR assessment by sweat test. In difficult diagnostic cases measurements of nasal potential difference can be helpful.
Primary Ciliary Dyskinesia
Primary ciliary dyskinesia (PCD) is a rare, genetically heterogeneous disorder, usually transmitted in an autosomal recessive pattern [34, 35]. Mutations of PCD-causing genes effect the genesis, structure and/or function of motile cilia leading to impaired mucociliary clearance (Fig. 25.2). Cilia dysmotility in the airways classically leads to unexplained neonatal respiratory distress in term infants, daily wet cough from early infancy, bronchiectasis, chronic rhinosinusitis, and conductive hearing impairment [36]. PCD is estimated to affect approximately 1 in 7750 people [26, 37], but many people are undiagnosed or diagnosed late in life, and the true prevalence is unknown [38]. Whilst data from international consortia and large clinics are improving our understanding of disease progression, information concerning morbidity and mortality remain sparse. The International PCD Cohort (iPCD) has reported that lung function impairment during childhood is similar to that found in CF, but by adulthood forced expiratory volume in 1 s (FEV1) is worse in CF [39]. Recent observations from large adult clinics concur that pulmonary disease is heterogeneous in severity [40, 41]. Several studies have reported impaired growth in children with PCD, [42, 43] which may be associated with worse lung function [39, 42, 43].
Neonates typically present with respiratory distress of unknown cause and some have rhinitis [44]. Infants continue to have a persistent wet cough, and usually develop recurrent respiratory tract infections, rhinitis, and serous otitis media associated with conductive hearing difficulty. Respiratory symptoms continue into later childhood and adulthood. Bronchiectasis has been described in pre-school children and is almost universal by early adulthood (Fig. 25.4); in contrast to CF, disease typically affects the middle and lower lobes, with relative sparing of the upper lobes [45,46,47,48]. Bacterial pathogens isolated from the airways are similar to those described in CF, namely Haemophilus influenza, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pneumoniae and Moraxella catarrhalis [49,50,51,52,53].

(a) HRCT of the chest in a 49-years old man with primary ciliary dyskinesia and Kartagener syndrome at the time of evaluation for lung transplantation, demonstrating severe bronchiectasis and consolidation in the anterior lateral segment of the left lower lobe, and (b) bilateral bronchiectasis in the lung bases (associated with centrilobular nodules and tree-in-bud pattern suggestive of bronchiolitis). Note the presence of situs inversus
Motile cilia are important in organs besides the respiratory tract, such as the Eustachian tubes, embryonic node, sperm flagella, the female reproductive tract, and ependyma of the brain and spinal cord. Extra-pulmonary symptoms caused by dysmotile cilia are therefore common, for example, serous otitis media, infertility and rarely hydrocephalus. Embryonic node motile cilia are responsible for left-right asymmetry, and 50% of people with PCD have situs inversus (Kartagener syndrome) or situs ambiguous (Fig. 25.4); associated congenital heart disease is relatively common [34, 54, 55].
Whilst the individual symptoms found in PCD are non-specific, a combination of symptoms indicates a need for prompt referral for PCD diagnostic testing (Table 25.2). Patients are symptomatic from birth or early infancy, yet the mean age of diagnosis is approximately 5 years. Large numbers of patients, particularly adults, have not been investigated and are therefore inappropriately labelled ‘idiopathic bronchiectasis’. The variability in diagnostic rates between countries is considerable probably reflecting clinical knowledge amongst physicians as well as geographical access to diagnostic facilities [27, 58].
Recent European and North American Guidelines concur that diagnosis of PCD requires specialist investigation using a combination of tests [56, 59]. Extremely low levels of nasal nitric oxide support the diagnosis of PCD [60]. However, some patients with PCD have normal nitric oxide levels, and levels can be low in other conditions including CF. Nasal nitric oxide measurement is therefore used as part of the diagnostic algorithm, but can neither confirm nor refute the diagnosis with certainty [25]. High-speed video microscopy can identify ciliary dysfunction (e.g. static, hyperfrequent, or circling cilia). Both nasal nitric oxide and cilia pattern observed by a high-speed video are highly predictive of PCD, and since analyses are available on the day of testing, counselling and treatment can be based on these provisional results [61]. However, confirmation of diagnosis by transmission electron microscopy or genetic testing is required for a definitive diagnosis [56, 59, 62]. Most, patients with PCD have diagnostic abnormalities of ciliary ultrastructure on transmission electron microscopy, and “hallmark abnormalities” confirm the diagnosis [63]. Electron microscopy used to be considered the ‘gold standard’ investigation but it is now recognised that approximately 15–20% of patients with PCD have normal ciliary ultrastructure. Similarly, immunofluorescence labelling to detect and localise intra-ciliary proteins (e.g. DNAH5) has excellent specificity but limited sensitivity to diagnose PCD [64].
Mutations in over 40 genes have been associated with PCD to date (reviewed in [34, 56], and summarised in Fig. 25.5). Bi-allelic pathogenic mutation or hemizygous X-linked mutation in a known PCD gene can confirm a diagnosis, [34, 56, 59] and approximately 70% of PCD cases diagnosed by other methods can be genetically confirmed. The diagnostic sensitivity should continue to improve as new genes are identified. The number and size of the genes results in a large number of variants, many of which are not pathogenic. To avoid false positive diagnoses, it is therefore important to ensure that the reported genotype correlates with phenotypic ciliary ultrastructure and function (Fig. 25.5b) [56]. For example, disease causing genetic variants in DNAH5 are associated with absence of the outer dynein arms and static cilia. Mutations in DNAH11 are associated with normal ciliary ultrastructure by transmission electron microscopy, and the cilia have a hyperfrequent vibratory pattern [34].


(a) Diagram of the transverse section of a respiratory cilium as seen by transmission electron microscopy. Motile cilia have a “9 + 2” arrangement with nine peripheral microtubule doublets surrounding a central pair of single microtubules running the length of the ciliary axoneme. Nexin links and radial spokes maintain the organised structure. Attached to the peripheral microtubules are inner and outer dynein arms. Dynein is a mechanochemical ATPase responsible for generating the force for ciliary beating, hence abnormalities of the dynein arms affect ciliary beating. (b) Examples of transmission electron microscopy from patients with PCD. Their genetic cause predicts the ultrastructural findings
Whatever the genetic defect, people with PCD generally have severely impaired mucociliary clearance leading to the previously described features. However, only some genes are associated with laterality defects or infertility, and some genes are associated with more severe pulmonary disease [55, 65, 66].
There is no cure for PCD and pulmonary management, therefore, aims to optimise health, social and psychological well-being whilst preventing the progression of lung damage. There have been few clinical trials in PCD and management is usually empirically based on evidence from CF. Given the differing underlying pathomechanisms and prognoses, evidence for PCD-specific treatments is urgently needed. Lack of evidence for treatment leads to the disparity of care between countries, and likely detrimental effects on exacerbation frequency, future lung function and health care costs [67]. Recent consensus statements provide guidance for the care of patients with PCD [30, 68]. Since patients with PCD also have extra-pulmonary disease, multidisciplinary care is required by a team including pulmonologists, physiotherapists, audiologists, ENT, cardiologists and fertility experts [68, 69]. General care for bronchiectasis is considered later in this chapter.
Other Ciliopathies
Non-motile or ‘primary’ cilia are found on the surface of many cells in the body. An increasing number of diseases are attributed to abnormal motile or primary ciliary function, collectively known as ciliopathies (http://www.ciliopathyalliance.org). For example, dysfunction of primary cilia in the eye can cause retinitis pigmentosa, and in the kidney can cause autosomal dominant polycystic kidney disease (ADPKD) or nephronophthisis.
OFD1 is an X-linked gene associated with several overlapping ciliopathies including oral-facial-digit syndrome and Joubert syndrome. Respiratory defects appear to be highly variable in males carrying OFD1 mutations. Where there is a motile cilia defect the condition is termed Simpson–Golabi–Behmel syndrome. These individuals suffer from bronchiectasis, PCD symptoms, overgrowth and can also have abnormally large kidneys, liver and spleen [70]. There are also several case reports of patients with PCD-like disease in association with retinitis pigmentosa caused by X-linked mutations in retinitis pigmentosa GTPase regulator (RPGR).
Autosomal dominant polycystic kidney disease (ADPKD) is an example of renal ciliopathy associated with bronchiectasis. It affects between 1 in 400 and 1 in 1000 people [71]. The disease most commonly manifests in adulthood and is caused by defective ciliary function in renal epithelial cells. Two genes, PKD1 and PKD2 coding for proteins known as polycystins have been implicated in the pathogenesis of ADPKD. In ADPKD, impaired primary cilial sensing results in abnormal intracellular signalling, cell hyperproliferation, and cyst formation [72]. A predisposition to bronchiectasis is recognised, although the mechanism for this is not fully understood [73].
Dysfunction of motile cilia and bronchiectasis are also described in some but not all cases of Bardet Biedl syndrome (BBS). A mutagenic syndrome characterised by retinal degeneration, obesity, polydactyly, cognitive impairment, kidney anomalies and hypogonadism [74, 75]. Bronchiectasis in BBS has been associated with the gene NPHP10.
Whereas one non-motile, primary cilium is found per cell there are multiple motile cilia in the respiratory tract. Defects in genes coding for proteins in the pathway responsible for the generation of multiple motile cilia can lead to a condition termed ‘reduced generation of multiple motile cilia’ (RGMMC). Assessment of airway cells reveals only a few motile cilia per cell, resulting in significant impairment of the mucociliary escalator. Individuals present with PCD like symptoms of rhinitis, neonatal respiratory distress, otitis media, and recurrent chest infections. In CCNO and MCIDAS bronchiectasis usually occurs in childhood and can be severe. Due to the multiple motile cilia of the brain being affected, hydrocephalus is also common [76,77,78]. In contrast to PCD, RGMMC is not associated with situs inversus since the cilia in the embryonic node are not affected. Recently defects in one of the master regulators of ciliogenesis FOXJ1 have also been described to cause bronchiectasis in an autosomal dominant inheritance pattern.
Primary Immunodeficiency Disorders
Primary immunodeficiency disorders (PID) account for a significant proportion of cases of bronchiectasis in developed countries. Series suggest as many as 7% of adults [79] and 20% of paediatric cases in higher-income countries may be attributable to PID [80]. PID includes a heterogeneous group of disorders of immune development or function affecting innate or adaptive immunity. Common variable immunodeficiency (CVID), X-linked agammaglobulinemia (XLA) and chronic granulomatous disease (CGD) are the most common immunodeficiencies found in association with bronchiectasis [81].
Common Variable Immunodeficiency
Common variable immunodeficiency (CVID) has an estimated prevalence of 1 per 25–50,000 population and is the commonest PID. Presentation is usually in young adulthood but diagnosis may be delayed [82]. Patients affected by CVID display a defective antibody response to protein and polysaccharide antigens and low levels of immunoglobulin (Ig) G, IgA and/or IgM. Affected individuals are at risk of recurrent bacterial infection, autoimmune disease and malignancy [83]. Clinical features vary, perhaps reflecting heterogeneity of underlying molecular defects or disease-modifying factors. To date, a monogenic cause has been identified in only 2–10% of cases [84]. Many disease genes have been identified in CVID, including those encoding receptors, ligands and intracellular signalling molecules.
Possibly as a consequence of delayed diagnosis, the risk of bronchiectasis is greater in patients with CVID than in those with X-linked agammaglobulinemia (XLA) and may approach 70% [85]. The additional immune dysregulation associated with CVID might also contribute to this high rate of bronchiectasis. One prospective study of CVID and XLA patients on immunoglobulin replacement therapy found older age and lower IgA levels to be risk factors for bronchiectasis in CVID, in contrast to XLA where the incidence of pneumonia was the major risk factor [86].
X-Linked Agammaglobulinemia
X-linked agammaglobulinemia (XLA) accounts for 85% of congenital agammaglobulinemia and is estimated to affect 1–2 per million people in the UK [87, 88]. It is characterised by an almost complete absence of circulating B lymphocytes and of all immunoglobulin [89]. Mutations of the Bruton tyrosine kinase (BTK) gene cause an incomplete block of B cell development at the pre-B cell stage [87]. Affected individuals are susceptible to infection by encapsulated bacteria and Mycoplasma. Autosomal recessive forms of agammaglobulinemia have been identified that are caused by mutations in genes that encode for other components of the pre-B cell receptor and its signalling pathway [90]. Pulmonary infections can occur shortly after birth but generally become noticeable beyond 6 months of age following the disappearance of maternal IgG. The most common age for diagnosis is under a year but presentations as late as 5 years have been known [91, 92]. Whilst the risk of developing significant lung disease increases over time, there is some evidence that severity can be reduced by early detection and treatment [93].
Chronic Granulomatous Disease and Other Disorders of Neutrophil Function
Chronic granulomatous disease (CGD) results from impaired function of NADPH oxidase. This enzyme is required for the effective functioning of the phagocytic respiratory burst and for superoxide production. Impaired NADPH oxidase is generally transmitted by X-linked inheritance but autosomal recessive variants are also recognised [94]. Mean age at presentation in autosomal recessive disease is 10 years, slightly later than X-linked disease where the mean is 5 years, suggestive of a more severe phenotype [95]. Patients are vulnerable to recurrent and severe bacterial and fungal infections, including Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia and Aspergillus spp.
A similar pattern of recurrent pneumonia and lung aspergillosis may also be observed in patients with severe congenital neutropenia [96]. Most commonly this disorder occurs as a consequence of mutations in the gene encoding for neutrophil elastase [97] but it can also be attributable to mutations affecting a mitochondrial protein thought to be involved in protecting myeloid cells from apoptosis [98] or an endosomal protein involved in intracellular signalling [99]. The characteristic feature of this disease is low levels of circulating neutrophils and hence vulnerability to bacterial and fungal pathogens.
Other Immunodeficiency Diseases Associated with Bronchiectasis
It is currently difficult to identify many causes of innate immunodeficiency, and it is likely that as new defects are discovered, many individuals currently described as suffering from ‘idiopathic bronchiectasis’ will be found to have an underlying disorder, some involving defects of innate immunity. Rare instances of bronchiectasis in association with deficiency of C2, mannose-binding lectin or l-ficolin have been reported [100]. Deficiencies affecting the complement cascade are also known to affect the severity of bronchiectatic disease in CVID [101] and in CF [102].
Ataxia telangiectasia is an autosomal recessive multisystem disorder resulting from mutation of the Ataxia telangiectasia mutated (ATM) gene, characterised by the development of telangiectasia and cerebellar ataxia. It affects 1 in 150,000 people in Europe [103]. It is the most common of the DNA repair disorders and is associated with chromosomal instability and cellular radiosensitivity, rendering sufferers susceptible to cancer and to infection. The ATM gene is involved in antibody class switch recombination and defects in this process may underlie the increased susceptibility of ataxia telangiectasia patients to bacterial infections [104]. The most common humoral immunological defects are diminished or absent serum IgA and IgG2, and impaired antibody responses to vaccines [105]. Protection against immune deficiency can be conferred by small amounts of preserved ATM function; mutations resulting in complete loss of ATM kinase activity have been shown to be more likely to be associated with recurrent respiratory infections than those associated with residual kinase activity [106]. Ataxia telangiectasia leads to thymic hypoplasia and variable T cell deficiency. It is likely that recurrent aspiration due to swallowing impairment also contributes to respiratory disease [107]. Bronchiectasis is reported in 10–50% of patients, becoming established by late childhood. The overwhelming bronchopulmonary disease can be fatal and the median survival is 19 years [108].
Bronchiectasis, alongside severe types of pneumonia, empyemas and pneumatoceles, is common in children with hyper-IgE or Job syndrome, which is associated with a wide variety of lymphocyte and humoral function defects as well as very high levels of serum IgE [109]. Job syndrome is inherited in an autosomal dominant pattern; most often due to a loss of function mutation of the signal transducer and activator of transcription 3 or STAT3 gene, [110] although in some cases the responsible mutation is unknown. STAT3 is a transcription factor that influences the expression of a variety of genes and plays a key role in many cellular processes such as growth and apoptosis. An autosomal recessive form of hyper IgE syndrome is recognised, this is less common than the autosomal dominant form and less likely to have respiratory complications. Bronchiectasis is also seen in association with Shwachman-Bodian-Diamond syndrome, caused by deficiency in DOCK8 which is involved in actin polymerisation and cytoskeletal rearrangement. This rare autosomal recessive disorder is characterised by exocrine pancreatic insufficiency, bone marrow dysfunction, leukemia predisposition, and skeletal abnormalities. In most cases, bone marrow dysfunction results in neutropenia in the majority of patients and may be accompanied by defects in neutrophil mobility, migration, and chemotaxis. Where neutropenia is less prominent a clinical phenotype similar to that of CVID can arise [111].
Other Genetic Disorders Predisposing to Bronchiectasis
Although the majority of genetic causes of bronchiectasis are related to defects of mucociliary clearance or PID, congenital abnormalities affecting the structure of the bronchial wall can predispose to bronchiectasis. For example, Marfan syndrome is a rare hereditary disorder characterised by skeletal, cardiovascular and ocular abnormalities. It follows an autosomal dominant inheritance with a variable expression which results in a defect in Type 1 collagen. Pulmonary abnormalities occur in approximately 10% of patients, the commonest being spontaneous pneumothorax and emphysema. Rarely cases of bronchiectasis have been described in adults and children with Marfan syndrome.
Epithelial sodium channels (ENaC) are important in the airways for regulating the osmolarity of periciliary fluid, and maintaining the periciliary fluid volume necessary for mucociliary clearance. The role of mutations encoding ENaC proteins (SCNN1A, SCNN1B, SCNN1G) in bronchiectasis is currently unclear. It is proposed that polymorphisms in ENaC genes, as well as ENaC mutations in trans with CFTR mutations might be risk factors for bronchiectasis, but it is not invariably associated with disease [112, 113].
Alpha-1 antitrypsin (AAT) deficiency is an autosomal co-dominant disorder that is caused by SERPINA1 mutations and leads to emphysema. Bronchiectasis has been reported, most frequently, in association with severe PiZZ genotypes, this is largely seen in adulthood. A study of 74 patients with severe AAT deficiency found high-resolution CT scan evidence of bronchiectasis in 70 subjects, this was judged clinically significant in 20 [114]. AAT deficiency alleles are over-represented in patients with bronchiectasis and asthma combined, suggesting that bronchiectasis may occur as a consequence of airway obstruction, in turn reducing airway clearance [115]. In individuals with humoral immunodeficiency lower AAT levels have been found in those with bronchiectasis, suggesting that where infection causes persistently elevated neutrophil elastase activity higher levels of AAT might be necessary to protect the lower airways [116].
Several autoimmune diseases with predominant symptoms outside the airways are associated with bronchiectasis, most notably rheumatoid arthritis and inflammatory bowel disease (IBD). Whilst causative genes have not been identified for these conditions, associated genetic risk loci have been reported in IBD and variations in human leukocyte antigen (HLA) genes, especially the HLA-DRB1 gene, are a risk factor for rheumatoid arthritis.
Young syndrome, is a poorly defined clinical diagnosis characterised by bronchiectasis, chronic sinusitis and impaired fertility. The cause of Young syndrome is not known, but it has been suggested to have an unresolved genetic cause, whilst others speculate that it could be due to mercury exposure. Young’s syndrome has overlapping symptomatology, with PCD and CF, and since there is little to support the existence of Young syndrome as an entity in its own right, perhaps most patients in fact have one of these conditions.
The cause of bronchiectasis in some syndromes is likely to be multifactorial. For example, Down syndrome may be associated with immunodeficiency, uncoordinated swallowing, gastro-oesophageal reflux and tracheomalacia; all of these may contribute to a predisposition to bronchiectasis.
Idiopathic Bronchiectasis
ERS guidelines for the management of bronchiectasis suggest a minimal set of aetiological investigations upon diagnosis [1]. These include differential blood count, serum immunoglobulins (total IgG, IgA, IgM) and testing for allergic bronchopulmonary aspergillosis (ABPA). Additional tests are recommended where specific clinical features are present or in patients with severe or rapidly progressive disease. Despite thorough investigation, an underlying cause cannot be found in at least 30% of patients with bronchiectasis; these cases are referred to as idiopathic [18, 79,80,81, 117]. A typical patient with the idiopathic disease is a non-smoking, post-menopausal female, although any age and both genders can be affected. Increasingly in the USA individuals with the idiopathic disease have chronic infection with non-tuberculous mycobacteria (NTM).
Some cases of idiopathic bronchiectasis are familial and it is likely that a number of patients have unrecognised impairment of the innate immune system, or have one of the increasingly recognised CFTR mutations associated with milder CF phenotypes associated with isolated lung disease. CFTR mutations have been found to be overrepresented in individuals identified as suffering from idiopathic bronchiectasis who do not have a full CF phenotype. The 5 T CFTR mutation in particular has been found at high frequency in this patient group. Recent data suggest that the 5 T polythymidine tract sequence of intron 8 on specific haplotype backgrounds (TG12 and M470V) may underlie low levels of full-length functional CFTR protein and cause CF-like lung disease [118]. Furthermore a significantly higher number of patients with pulmonary NTM have been found to have low-frequency, protein-affecting variants in immune, CFTR, cilia, and connective tissue genes [119].
The idiopathic disease is often not as severe as CF or PCD. A comparison of data in the European bronchiectasis registry (EMBARC) between individuals with the idiopathic disease compared to PCD and immune deficiency showed that individuals with bronchiectasis due to PCD had increased disease severity, measured by a bronchiectasis severity index 7.5 (±4.9), when compared to age- and gender-matched cohorts with idiopathic disease 5.7 (±5.2) or immune deficiency 5.9 (±4.7). Analysis of components contributing to the bronchiectasis severity index score revealed an average 10% reduction in FEV1% in PCD and an increased incidence of Pseudomonas isolation from sputum in PCD patients (46%) [120].
Diagnosis of Bronchiectasis
Whilst recent evidence-based guidelines recommend approaches to identify and investigate adults for bronchiectasis, paediatric practice is based on expert opinion and personal practice [16, 121, 122]. The criteria for diagnosing bronchiectasis, based on the guidelines and opinion documents are summarised in Box 25.1. Ideally, patients at risk of bronchiectasis should be identified before irreversible damage develops. For example, it is widely accepted that newborn screening for CF reduces long-term pulmonary morbidity because of the early introduction of airway clearance physiotherapy, treatment of infections and pancreatic enzyme therapy. Similarly, in PCD, observational data suggest that lung function decline is stabilised and can even be reversed following diagnosis and instigation of appropriate pulmonary management. We expect that onset of bronchiectasis might also be delayed. Moreover, patients and their families believe that an early diagnosis is beneficial [58].
Guidelines suggest that bronchiectasis should be considered in patients with persistent production of mucopurulent sputum, particularly if they have risk factors [1, 121]. The investigation should be considered in patients with rheumatoid arthritis or inflammatory bowel disease if they have a chronic productive cough; and in patients with COPD who are having frequent exacerbations and a previous sputum culture for Pseudomonas aeruginosa [121]. It should be considered in otherwise healthy adults with a cough that persists for more than 8 weeks, particularly if the cough is productive of sputum [121]. Children with a chronic wet cough failing to respond to 4 weeks of oral antibiotics, or recurrent episodes of protracted bacterial bronchitis are at high risk for bronchiectasis [13, 16]. The diagnosis should also be considered in patients with positive sputum culture for organisms such as Pseudomonas aeruginosa, people with persistent chest signs, haemoptysis or finger clubbing [16, 121].
A baseline chest x-ray should be performed, and an HRCT should then be used to confirm the diagnosis [121]. Dilated airways with thickened walls are sometimes visible on chest X-ray as parallel ‘tram tracks’ or ‘ring shadows’, and fluid-filled bronchi may be visible as ‘gloved-finger’ shadows. Situs inversus might direct investigations for PCD. However, a chest X-ray is not sensitive to detect the structural changes associated with bronchiectasis. HRCT has very good sensitivity and specificity to detect bronchiectasis not identified by chest X-ray (Fig. 25.4). Characteristically bronchiectasis is defined by bronchial dilation seen on HRCT as one or more of the following: broncho-arterial ratio >1, lack of tapering, airway visibility within 1 cm of the costal pleural surface or touching mediastinal pleura [121]. The following are also associated with bronchiectasis: bronchial wall thickening, mucus impaction and air trapping on an expiratory scan [123]. HRCT should not be performed during acute respiratory exacerbations as bronchial dilation is difficult to assess in the presence of consolidated lung, whilst pulmonary collapse can cause misleading ‘traction bronchiectasis’ by pulling on neighbouring bronchi [124]. In conditions without biological markers to identify which patients will go on to develop significant lung disease repeat CT assessments might be required. Caution is advised particularly in patients with PID who have genetic defects affecting DNA recombination and DNA repair. Lung MRI may become a possible alternative to CT scanning [125].
In addition to imaging, further tests are important to confirm the disease severity, and to identify co-morbidities or underlying aetiology. Lower airway samples should be sent from all patients with bronchiectasis for routine and mycobacterial culture [16, 121]. Measures of lung function are non-specific and non-sensitive in bronchiectasis, but contribute to the assessment of disease severity. FEV1 is often normal in early disease although a reduced FEV1 in the presence of normal functional vital capacity is common. The lung clearance index is a measure that has been suggested to be a good monitor of disease in CF [126] and is being evaluated as an early marker of lung disease in other bronchiectatic diseases such as PCD [127,128,129,130].
Fibreoptic bronchoscopy can be considered to assess airway structure and calibre and exclude pathology such as severe tracheomalacia, bronchomalacia or tracheal bronchi which may contribute to bronchiectatic change. The bronchoscopic examination can also provide lavage fluid for evidence of chronic aspiration measured by pepsin, amylase or fat-laden macrophages, and for culture and microscopy.
Once a diagnosis of bronchiectasis has been made, an investigation of the underlying cause should be sought. The clinical history should direct investigations which will usually include investigation for CF, PCD and PID [1, 121].
Management of Patients with Bronchiectasis
Several guidelines have recently been published for the management of bronchiectasis. For CF, guidelines include evidence-based documents from NICE [131], the CF Trust and CF Foundation. There is the paucity of evidence for treating other causes of bronchiectasis. At best, recommendations for non-CF-bronchiectasis and PCD are conditional and based on the low or very low quality of evidence, but are mostly based on the consensus of experts [1, 6, 30, 68, 69, 121]. No treatments have been licenced worldwide for the treatment of non-CF bronchiectasis. The underlying aetiology may impact the treatment plan; in children, it is estimated that identification of a specific cause of bronchiectasis prompts a management change in over 50% of cases [132].
The overall aim is to delay progression of bronchiectasis, and maximise lung function, exercise tolerance, quality of life and nutrition. A multifaceted approach is needed to treat infections and inflammation, whilst promoting mucus clearance (Fig. 25.1).
Pulmonary exacerbations are a cause of significant morbidity and need to be prevented, recognised and treated in an attempt to prevent further lung damage caused by the infection and inflammation (Fig. 25.1) [123, 133]. Epidemiological, clinical and laboratory evidence suggest that bacterial and viral infections are major causes of pulmonary exacerbations; environmental pollution might also contribute. Some patients do not recover the accompanying reduction in lung function despite aggressive treatment of the episode with antibiotics and physiotherapy, and in CF patients with higher exacerbation rates have an increased rate of decline in lung function [134,135,136]. Pulmonary exacerbations are related to neutrophilic inflammation, and neutrophil serine proteases, including neutrophil elastase, are increased in the sputum of patients at baseline and increase further during exacerbations. Brensocatib, an inhibitor of neutrophil serine proteases, reduced exacerbations in a 24 week phase 2 trial [137].
Management includes promoting clearance of secretions and the use of antibiotic therapies both to prevent and to treat the recurrent infection. In addition to routine vaccination schedules, patients should receive pneumococcal and influenza vaccinations and avoid exposure to tobacco smoke. Some CVID patients have been shown to raise antibodies in response to the influenza antigens but XLA patients do not. Family members of people with PID should also be vaccinated [138].
As with many Orphan Diseases, evidence for efficacy of treatments (e.g. PCD) is often extrapolated from studies of more common disorders such as CF, and may not be appropriate for other forms of bronchiectasis [30, 67]. Indeed, a number of trials have confirmed that efficacious drugs for CF do not necessarily benefit those with non-CF bronchiectasis [6, 139,140,141]. Randomised controlled trials (RCTs) are urgently needed for individual diseases, which by the necessity for rare diseases will be multicentre and likely require international collaboration. The results of the only multinational RCT for PCD recently confirmed that azithromycin prophylaxis is efficacious and safe to reduce pulmonary exacerbations [142, 143]. An important step in designing the RCTs includes careful consideration of disease-appropriate endpoints which might also differ for bronchiectasis of different causes. Exacerbation frequency or time to exacerbation is a clinically important endpoint, and consensus definitions are now in place for CF, PCD and non-CF bronchiectasis [123, 133, 144]. Quality of life instruments are also in place, but responsiveness to treatment still needs to be established for QOL-B and QOL-PCD [145,146,147,148]. There is an urgent need for disease-specific endpoints for trials in bronchiectasis.
Airway Clearance Therapy (ACT)
A variety of physiotherapy techniques are available to assist airway clearance, including chest percussion, postural drainage, breathing exercises and mechanical interventions such as cough-assist and positive expiratory pressure (PEP) adjuncts and oscillatory-PEP [149, 150]. The aims of ACT include mobilising and aiding clearance of secretions to optimise sputum expectoration, relieve symptoms, and improve well-being. Although of proven benefit in CF, there are few studies demonstrating the efficacy of physiotherapy in non-CF bronchiectasis that can guide frequency or choice of therapy. Generally, it is agreed that all patients should be taught and encouraged to conduct regular ACT [1, 30, 68, 69, 121, 149,150,151].
The choice of technique will depend in part on the age of the patient, their clinical state, and patient acceptability. Often a combination of techniques is employed. A commonly used ACT, particularly in adults, is the active cycle of breathing technique; this is based upon deep breaths followed by ‘huffs’ and ‘coughs’ to aid sputum clearance interspersed with periods of relaxed controlled breathing. The active cycle of breathing can be combined with postural drainage and manual techniques. Positive end expiratory pressure (PEP) techniques using oscillating PEP devices, such as a Flutter valve or Acapella, can be combined with postural drainage or forced expiration techniques. A further option is the technique of autogenic drainage in which a sequence of controlled breaths at low then progressively higher lung volumes is used to collect and expectorate sputum.
Nebulised treatments have been shown to assist mucus clearance in patients with CF, but efficacy differs in patients with bronchiectasis or other causes. For example, recombinant human DNase (rhDNase) lyses neutrophil DNA which originates mainly from decaying neutrophils at sites of airway inflammation. There is good evidence for its use in CF [152]. However, a large study of patients with non-CF bronchiectasis showed a faster decline in FEV1 and more frequent exacerbations in patients who received rhDNase in comparison to those treated with placebo [153]. rhDNase is therefore not generally recommended outside CF [1, 30, 121].
Agents such as hypertonic saline and mannitol are often used to assist with mucus clearance as an adjunct to airway clearance therapy. Nebulised hypertonic saline and mannitol often benefit patients with CF, particularly older children and adults when used as regular therapy, or during exacerbations [154, 155]. However, the benefits for bronchiectasis of other causes are less clear, perhaps due to the heterogeneity of the studies and populations [1, 139, 156,157,158,159].
Associations between physical exercise and lung function in CF are inconsistent. However, the benefits of exercise are multifactorial and exercise is generally considered an important component of management with the aim to improve exercise tolerance and quality of life. Exercise programmes have been beneficial in CF inpatient, outpatient and community settings. Moreover, exercise programmes impact exercise capacity in adults with non-CF bronchiectasis, with benefits achieved in 6–8 weeks [1]. Exercise is therefore advocated for all patients with bronchiectasis [1, 30, 68, 69].
Management of Infections
Antibiotic therapy can be prescribed either as long-term prophylaxis or in response to infections.
Guidelines recommend offering long-term antibiotics to adults with bronchiectasis who have three or more exacerbations per year [1, 121]. Long-term macrolide treatment, for example with azithromycin, is beneficial to reduce the number of pulmonary exacerbations [143, 160,161,162]. Macrolides have anti-inflammatory effects beyond their anti-bacterial actions which may be useful in the context of bronchiectasis [163]. In patients with chronic P. aeruginosa infection, long-term inhaled antibiotics e.g. nebulised colistin, is recommended [1, 121]. There is a concern that long-term use of antibiotics may accelerate the development of antibiotic resistance; regular surveillance of sputum by culture and sensitivity is essential.
Pulmonary exacerbations require prompt treatment with antibiotics. Antibiotic choice should reflect results of sputum culture and sensitivities when available, and empirical antibiotics can be started whilst awaiting microbiology results [1, 68, 69, 121]. Sputum culture is possible only in adults and older children who are able to expectorate. In younger children, cough swabs or nasopharyngeal aspirates can be taken for bacterial culture, although upper airway specimens are inferior and broncho-alveolar lavage specimens may be required. In general, antibiotic courses for 14 days are prescribed, but the duration should be individualised depending on the patient’s condition. Intravenous antibiotics are sometimes required when patients are unwell or do not respond to oral therapy, particularly following a second course..
Based on the poor clinical outcomes of patients with P. aeruginosa infection, eradication therapy is recommended for patients with CF and new growth of the pathogen [164]. Eradication is similarly recommended for new isolates in non-CF cases, although the evidence base is low [1, 69, 123].
Anti-Inflammatory Management
As already discussed, the anti-inflammatory effects of macrolide therapy may be beneficial in patients with bronchiectasis, but the risks of antibiotic resistance and other side-effects need consideration. There is no evidence for the use of inhaled corticosteroids in patients with bronchiectasis, except where patients have co-existing asthma or COPD [1, 69, 121]. Similarly, there is insufficient evidence to recommend statins as an anti-inflammatory agent for bronchiectasis [1, 121]. A number of approaches to target neutrophil-driven inflammation are under investigation (e.g. neutrophil elastase or cathepsin C inhibition).
Immune Therapy
Efforts to reconstitute the immune system can be effective in PID. Recombinant IFNγ therapy, for example, is effective in reducing the number of severe infections in CGD [165] and recombinant granulocyte stimulating factor can be used in severe congenital neutropenia [96]. Systemically administered AAT augmentation therapy can be used to treat the imbalance between elastases and inhibitors within the lung and, in the future, inhaled AAT might be possible. This would reduce costs since less protein would be necessary to target the lungs directly [166].
Patients with CVID benefit from immunoglobulin substitution therapy [167]. Subcutaneous infusions are better tolerated by patients than intravenous infusions and may achieve more stable trough levels with a lower risk of adverse reactions [168]. Doses may need to be increased in the presence of active lung disease as this increases immunoglobulin turnover [169]. Subcutaneous immunoglobulin infusion has been demonstrated to reduce the frequency of exacerbations and to slow bronchiectasis progression [170]. However, lung involvement can progress despite immunoglobulin replacement. Risks of viral contamination are low, although a finite risk exists, particularly of prion-related disease. Previous guidelines recommended a trough IgG level of at least 5 g/L, however in more recent studies higher and individualised levels have been recommended.
Newborn screening for primary immune deficiencies might be possible to allow treatment before bronchiectasis develops. One approach would be based on the detection of kappa-deleting recombination excision circles (KRECs). KRECs are produced during immunoglobulin gene rearrangement throughout B-lymphocyte maturation, patients with B-lymphocyte defects will have low levels of or absent KRECs regardless of the exact aetiology of the defect [171].
The only curative treatment at present for many immune deficiencies is matched stem cell transplantation; patients must receive antimicrobial cover, particularly for the organisms they are known to be colonised with for the duration of immunosuppression related to this procedure [172, 173]. Since the majority of affected individuals will not have a healthy HLA-matched sibling donor, techniques including T-cell depletion were developed to facilitate donations from unrelated donors or HLA-mismatched parental donations. Low-toxicity pre-conditioning regimens using targeted busulfan doses or treosulfan in combination with fludarabine have demonstrated excellent engraftment and survival [174].
Gene therapy provides an alternative strategy with which to cure or alleviate select inherited diseases. A corrected copy of a gene is transferred to the somatic cells of affected individuals. Correction of genetic defects is limited to either terminally differentiated, long-lived post-mitotic cells or easily accessible stem cells. To treat haematopoietic system stem cells, implicated in primary immunodeficiency, a viral vector capable of integrating within the host genome is necessary for gene delivery. Barriers to the success of this treatment strategy are: (1) low protein expression due to poor gene transfer [175], (2) risk of insertional mutagenesis, [176] and (3) immunogenicity of the vector or transgene product [177]. Nevertheless gene-modified autologous bone marrow transplantation represents a promising treatment, free from the immunological complications associated with transplantation from an HLA-mismatched donor.
In order to replenish the peripheral pool of immune cells with cells containing the transduced gene, the transduced cells must have a selective advantage. For example in adenosine deaminase (ADA) deficiency (a severe combined immunodeficiency associated with pneumocystis and other bacterial, viral and fungal respiratory infection although not commonly bronchiectasis) genetically modified T lymphocyte precursors are able to metabolise toxic purine products and have a selective advantage over unmodified lymphocytes [178]. Similarly, rare spontaneous partial phenotypic correction of severe T cell immunodeficiencies has been observed in which clonal expansion of one or several T cell precursors carrying a wild-type sequence of the disease-causing gene can differentiate into mature, functional T cells capable of supporting normal immunity [179]. For haematopoietic disorders in which wild-type gene expression is essential to the function of terminally differentiated cells, chances of success can be improved by mild myelosuppressive treatment prior to gene-therapy which leads to a higher proportion of transduced progenitor cells due to better engraftment.
Following the success of gene therapy treatments for severe combined immune deficiency these techniques have been applied to a number of monogenic immune deficiencies, including X-linked CGD [180]. Initially trials using gamma-retroviruses were complicated by clonal expansion of myeloid cells, thought to be due to insertions near cellular proto-oncogenes. More recently gene delivery vectors have been developed, derived from the lentivirus class of retroviruses. These can be produced with enhancer elements that “self-inactivate” reducing the chances of turning on cellular genes near their integration sites. Conditions such as CVID which are caused by one of several genes, present a challenge for gene therapy. Correcting each gene defect will require a separate development process. Moreover, gene therapy will need to be very effective and safe before it is considered preferable to current best medical therapy.
For genes involved in processes such as cell activation and intracellular signalling it is important to preserve normal expression patterns in addition to restoring function. An alternative solution for genes such as BTK, involved in XLA, is to repair the disease-causing gene in its native chromosomal site. Gene editing relies on systems such as homing endonucleases, zinc finger nucleases, Transcription activator-like effector nucleases (TALENs), Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR associated protein 9 (CRISPR/Cas9). A repair in the target DNA is then directed by providing single or double-stranded DNA copies of the target sequence. Recent pre-clinical work using CRISPR/Cas9 has demonstrated successful repair of defects in stem cells from patients with CGD [181]. New conditioning regimens using monoclonal antibodies to haemopoietic stem cells proteins might increase safety and permit use of gene editing in milder immune deficiencies such as XLA.
Surgery
Surgical intervention is generally not recommended, although it can be considered in patients with localised non-functioning lung segments, where it is anticipated that lobectomy may prevent infection of adjacent healthy areas [1]. Close collaboration between the surgeon and pulmonologist is essential throughout the work-up and peri-operative period, with particular attention to pre-operative nutrition and pulmonary rehabilitation.
Transplantation should be considered in patients with untreatable end-stage respiratory failure. Particular planning is needed for PCD patients with situs anomalies.
Novel Therapies for Managing Cystic Fibrosis
Over recent years ground-breaking small molecule therapies that improve and can normalise CFTR function have been developed and come into widespread clinical use in CF patients. The first of these, a ‘potentiator’ called ivacaftor causes the CFTR channel to be ‘wedged open’ and was demonstrated to cause significant improvements in FEV1 (10.6%) and body mass index, alongside normalising of sweat chloride levels in patients over 12 years with p.Gly551Asp mutations (a class 3 ‘gating’ mutation; Fig. 25.3) [182]. Further studies demonstrated similar improvements in younger children, and it is licensed in Europe for people with susceptible mutations from 6 months of age [183,184,185]. However ivacaftor had mixed results with class 4 mutations, ion transport defects (Fig. 25.3), with improvements in FEV1 seen in adults but not in children and did not work in class 2 mutations, protein folding defects that include far the most common mutation p.Phe508del [186].
Therefore, in order to impact on CFTR function in the majority of CF patients with class 2 mutations, the same manufacturer developed ‘corrector’ molecules that act to correct the abnormal folding of the CFTR protein, hence allowing it to be transported successfully to the apical surface of the cell. The first ‘corrector’ developed was lumacaftor, which was combined with ivacaftor a marketed drug. However, whilst a large RCT showed reasonable improvements in exacerbation rates and severity, the improvement of FEV1 was small, (2.6–4.0%) in homozygous p.Phe508del patients over 12 years of age [187]. Subsequent trials in younger patients have demonstrated similar modest benefits [188]. The manufacturer has since released a further combination therapy, swapping lumacaftor for an alternative ‘corrector’ tezacaftor. Whilst this drug seems to have a similar effect on lung function to lumacaftor/ivacaftor it has an advantageous side effect profile [189, 190].
However, most recently, data on a triple combination therapy has been published. This combines the two medicines tezacaftor/ivacaftor) with a second ‘corrector’, elexacaftor. This triple therapy led to good improvements in FEV1 in p.Phe508del. homozygotes and heterozygotes (10–11% and 13.8% respectively, over and above the beneficial effect tezacaftor/ivacaftor), [191, 192] and may be a game-changer for the management of the majority of patients with CF.
However, whilst these medicines have a dramatic effect on CFTR function they come at a very high cost. All the medicines described here have been marketed at well over £100,000 per patient per year, with the list price for triple therapy at over $300,000, making them unaffordable to many who would benefit most from them. The price tag for the new triple therapy is, as yet, unknown.
It is also important to mention the efforts made to pursue an alternative treatment strategy to correct CFTR dysfunction. This was led by the CF Gene Therapy Consortium in the UK, which developed a nebulised gene therapy. This had the potential advantage of correcting all classes of CF mutations and was assessed in a phase 2b RCT. However, whilst establishing the proof of principle of this complex science, the actual clinical improvements seen in CF patients were small, with only a 3.7% difference in FEV1 between the treatment and placebo arms [193].
Vignette
A patient was referred for PCD diagnostic testing at 4 years of age (see PCD details above). She had been born at term and was noted to have nasal congestion and tachypnoea from shortly after birth, but did not require medical intervention. Throughout infancy, she had recurrent chest infections and a daily wet productive cough. She also had glue ear treated with grommets which resulted in otorrhea and no improvement in hearing. She had normal cardiac situs and her parents are white Caucasian and non-consanguineous. Her sister has a glue ear but there is otherwise no family history of note.
Using high-speed video analysis it was impossible to obtain an accurate beat frequency on two separate occasions. The cilia demonstrated stiff vibrating movements rather than the usual coordinated sweeping motion. Electron microscopy demonstrated normal ultrastructure (Fig. 25.5) with the normal arrangement of microtubules radial spokes and outer dynein arms. In view of the normal transmission electron microscopy, genetic analysis was undertaken, confirming mutations in DNAH11 gene; this gene had previously been reported as a cause of PCD with normal ciliary ultrastructure [194].
Since diagnosis, she has commenced twice daily airways clearance (physiotherapy) and is aware of the need for prompt treatment of any intercurrent infection. In addition to the usual childhood vaccinations, she has influenza cover annually. She is reviewed by a multidisciplinary team that includes a respiratory pediatrician, ENT consultant, physiotherapist and respiratory nurse 4 monthly. She also has audiology reviews annually to monitor the need for hearing aids.
Learning Points from the Case
-
PCD often presents in the neonatal period but diagnosis is often delayed until later childhood [27].
-
Diagnostic evaluation is often complicated and requires specialist expertise [56, 195].
-
Although 50% of patients have situs inversus, the diagnosis should be suspected in patients with situs solitus if other symptoms are present.
-
Management of non-pulmonary disease necessitates the involvement of a multidisciplinary team, which may include ENT, audiology, cardiology, and fertility specialists.
-
Consensus statements are available to provide guidelines for management of PCD [30, 68, 69]. However evidence is extrapolated from more prevalent diseases, mostly CF; this is almost certainly inappropriate.
Summary
It is likely that the true incidence of bronchiectasis with a genetic basis is underestimated. As our understanding of innate immunity, cilia biology and ion-transport disorders are better characterised, the aetiology of many cases currently labeled as ‘idiopathic’ will become better understood. As with other Orphan Diseases, the diagnosis and management of patients with bronchiectasis are largely determined by local interests and provision, and many patients find it difficult to access appropriate care. Most doctors have little experience with rarer causes of bronchiectasis and will base management on evidence from CF. Indeed, the evidence base for managing non-CF bronchiectasis is poor. The CF community has made substantial advances in recent decades, resulting in improved morbidity and mortality. Whilst these advances have been beneficial to the care of patients with non-CF bronchiectasis, disease-specific treatments are urgently needed.
Box 25.1
Diagnostic criteria for bronchiectasis in adults and children (adapted from British Thoracic Society guidelines for adults 2019 [121] and an expert statement for children [16]) |
Bronchiectasis is defined as thin-section CT scan showing one or more of the following: • Broncho-arterial ratio > 1 • Lack of bronchial tapering • Airways visible within the lung periphery Other CT features commonly associated with bronchiectasis include: • Bronchial wall thickening • Mucus impaction • Mosaic perfusion/air trapping on expiratory CT Following a diagnosis of bronchiectasis, investigate the underlying cause. Consider: • Cystic fibrosis • Primary ciliary dyskinesia • Immune deficiency • Rheumatoid arthritis • Chronic obstructive pulmonary disease (COPD) • Inflammatory bowel disease |
References
Polverino E, Goeminne PC, McDonnell MJ, Aliberti S, Marshall SE, Loebinger MR, et al. European Respiratory Society guidelines for the management of adult bronchiectasis. Eur Respir J. 2017;50(3):1700629.
Henkle E, Chan B, Curtis JR, Aksamit TR, Daley CL, Winthrop KL. Characteristics and health-care utilization history of patients with bronchiectasis in US Medicare enrollees with prescription drug plans, 2006 to 2014. Chest. 2018;154(6):1311–20.
McCallum GB, Binks MJ. The epidemiology of chronic suppurative lung disease and bronchiectasis in children and adolescents. Front Pediatr. 2017;5:27.
Quint JK, Millett ER, Joshi M, Navaratnam V, Thomas SL, Hurst JR, et al. Changes in the incidence, prevalence and mortality of bronchiectasis in the UK from 2004 to 2013: a population-based cohort study. Eur Respir J. 2016;47(1):186–93.
Traversi L, Miravitlles M, Martinez-Garcia MA, Shteinberg M, Bossios A, Dimakou K, et al. ROSE: radiology, obstruction, symptoms and exposure—a Delphi consensus definition of the association of COPD and bronchiectasis by the EMBARC Airways Working Group. ERJ Open Res. 2021;7(4):00399-2021. www.ncbi.nlm.nih.gov/pmc/articles/PMC8607072/.
Chalmers JD, Chotirmall SH. Bronchiectasis: new therapies and new perspectives. Lancet Respir Med. 2018;6(9):715–26.
Shum DK, Chan SC, Ip MS. Neutrophil-mediated degradation of lung proteoglycans: stimulation by tumor necrosis factor-alpha in sputum of patients with bronchiectasis. Am J Respir Crit Care Med. 2000;162(5):1925–31.
Gaga M, Bentley AM, Humbert M, Barkans J, O’Brien F, Wathen CG, et al. Increases in CD4+ T lymphocytes, macrophages, neutrophils and interleukin 8 positive cells in the airways of patients with bronchiectasis. Thorax. 1998;53(8):685–91.
Cole P. The damaging role of bacteria in chronic lung infection. J Antimicrob Chemother. 1997;40(Suppl A):5–10.
Keir HR, Shoemark A, Dicker AJ, Perea L, Pollock J, Giam YH, et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: an international, observational, multicohort study. Lancet Respir Med. 2021;9(8):873–84. https://pubmed.ncbi.nlm.nih.gov/33609487/.
Shoemark A, Shteinberg M, De Soyza A, Haworth CS, Richardson H, Gao Y, et al. Characterization of eosinophilic bronchiectasis: a European multicohort study. Am J Respir Crit Care Med. 2022;205(8):894–902. https://pubmed.ncbi.nlm.nih.gov/35050830/.
Flume PA, Chalmers JD, Olivier KN. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet. 2018;392(10150):880–90.
Goyal V, Grimwood K, Marchant J, Masters IB, Chang AB. Does failed chronic wet cough response to antibiotics predict bronchiectasis? Arch Dis Child. 2014;99(6):522–5.
Chalmers JD, Goeminne P, Aliberti S, McDonnell MJ, Lonni S, Davidson J, et al. The bronchiectasis severity index. An international derivation and validation study. Am J Respir Crit Care Med. 2014;189(5):576–85.
Tsao PC, Lin CY. Clinical spectrum of bronchiectasis in children. Acta Paediatr Taiwan. 2002;43(5):271–5.
Chang AB, Bush A, Grimwood K. Bronchiectasis in children: diagnosis and treatment. Lancet. 2018;392(10150):866–79.
Dhar R, Singh S, Talwar D, Mohan M, Tripathi SK, Swarnakar R, et al. Bronchiectasis in India: results from the European multicentre bronchiectasis audit and research collaboration (EMBARC) and respiratory research network of India registry. Lancet Glob Health. 2019;7(9):e1269–e79.
Twiss J, Metcalfe R, Edwards E, Byrnes C. New Zealand national incidence of bronchiectasis “too high” for a developed country. Arch Dis Child. 2005;90(7):737–40.
Chang AB, Grimwood K, Mulholland EK, Torzillo PJ. Bronchiectasis in indigenous children in remote Australian communities. Med J Aust. 2002;177(4):200–4.
Singleton R, Morris A, Redding G, Poll J, Holck P, Martinez P, et al. Bronchiectasis in Alaska native children: causes and clinical courses. Pediatr Pulmonol. 2000;29(3):182–7.
Waite DA, Wakefield SJ, Moriarty KM, Lewis ME, Cuttance PC, Scott AG. Polynesian bronchiectasis. Eur J Respir Dis Suppl. 1983;127:31–6.
Griese EU, Ilett KF, Kitteringham NR, Eichelbaum M, Powell H, Spargo RM, et al. Allele and genotype frequencies of polymorphic cytochromes P4502D6, 2C19 and 2E1 in aborigines from western Australia. Pharmacogenetics. 2001;11(1):69–76.
O’Callaghan C, Chetcuti P, Moya E. High prevalence of primary ciliary dyskinesia in a British Asian population. Arch Dis Child. 2010;95(1):51–2.
Morrissey BM, Harper RW. Bronchiectasis: sex and gender considerations. Clin Chest Med. 2004;25(2):361–72.
Lucas JSA, Walker WT, Kuehni CE, Lazor R. Primary ciliary dyskinesia. Eur Respir Monogr. 2011;54:201–17.
Hannah WB, Seifert BA, Truty R, Zariwala MA, Ameel K, Zhao Y, et al. The global prevalence and ethnic heterogeneity of primary ciliary dyskinesia gene variants: a genetic database analysis. Lancet Respir Med. 2022;10(5):459–68. https://pubmed.ncbi.nlm.nih.gov/35051411/.
Kuehni CE, Frischer T, Strippoli MP, Maurer E, Bush A, Nielsen KG, et al. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur Respir J. 2010;36(6):1248–58.
Davies JC, Alton EW, Bush A. Cystic fibrosis. BMJ. 2007;335(7632):1255–9.
Stick SM, Brennan S, Murray C, Douglas T, von Ungern-Sternberg BS, Garratt LW, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009;155(5):623–8 e1.
Lucas JS, Alanin MC, Collins S, Harris A, Johansen HK, Nielsen KG, et al. Clinical care of children with primary ciliary dyskinesia. Expert Rev Respir Med. 2017;11(10):779–90.
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–73.
Wood RE, Boat TF, Doershuk CF. Cystic fibrosis. Am Rev Respir Dis. 1976;113(6):833–78.
Rodman DM, Polis JM, Heltshe SL, Sontag MK, Chacon C, Rodman RV, et al. Late diagnosis defines a unique population of long-term survivors of cystic fibrosis. Am J Respir Crit Care Med. 2005;171(6):621–6.
Lucas JS, Davis SD, Omran H, Shoemark A. Primary ciliary dyskinesia in the genomics age. Lancet Respir Med. 2019;8(2):202–16. https://doi.org/10.1016/S2213-2600(19)30374-1.
Wallmeier J, Nielsen KG, Kuehni CE, Lucas JS, Leigh MW, Zariwala MA, et al. Motile ciliopathies. Nat Rev Dis Primers. 2020;6(1):77.
Goutaki M, Meier AB, Halbeisen FS, Lucas JS, Dell SD, Maurer E, et al. Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J. 2016;48(4):1081–95.
Lucas JS, Walker WT, Kuehni CE, Lazor R. Primary ciliary dyskinesia. In: Courdier J-F, editor. Orphan lung diseases (ERS monograph). Sheffield: European Respiratory Society; 2011. p. 201–17.
Shoemark A, Griffin H, Wheway G, Hogg C, Lucas JS, Genomics England Research Consortium, et al. Genome sequencing reveals underdiagnosis of primary ciliary dyskinesia in bronchiectasis. Eur Respir J. 2022:2200176. https://pubmed.ncbi.nlm.nih.gov/35728977/.
Halbeisen F, Goutaki M, Maurer E, Casaulta C, Crowley S, Haarman E, et al. Lung growth in children and young adults with primary ciliary dyskinesia (PCD): an iPCD cohort study. Eur Respir J. 2016;48(suppl 60).
Shah A, Shoemark A, MacNeill SJ, Bhaludin B, Rogers A, Bilton D, et al. A longitudinal study characterising a large adult primary ciliary dyskinesia population. Eur Respir J. 2016;48(2):441–50.
Frija-Masson J, Bassinet L, Honore I, Dufeu N, Housset B, Coste A, et al. Clinical characteristics, functional respiratory decline and follow-up in adult patients with primary ciliary dyskinesia. Thorax. 2017;72(2):154–60.
Goutaki M, Halbeisen FS, Spycher BD, Maurer E, Belle F, Amirav I, et al. Growth and nutritional status, and their association with lung function: a study from the International primary ciliary dyskinesia cohort. Eur Respir J. 2017;50(6):1701659.
Marino LV, Harris A, Johnstone C, Friend A, Newell C, Miles EA, et al. Characterising the nutritional status of children with primary ciliary dyskinesia. Clin Nutr. 2019;38(5):2127–35.
Mullowney T, Manson D, Kim R, Stephens D, Shah V, Dell S. Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics. 2014;134(6):1160–6.
Brown DE, Pittman JE, Leigh MW, Fordham L, Davis SD. Early lung disease in young children with primary ciliary dyskinesia. Pediatr Pulmonol. 2008;43(5):514–6.
Magnin ML, Cros P, Beydon N, Mahloul M, Tamalet A, Escudier E, et al. Longitudinal lung function and structural changes in children with primary ciliary dyskinesia. Pediatr Pulmonol. 2012;47(8):816–25.
Jain K, Padley SP, Goldstraw EJ, Kidd SJ, Hogg C, Biggart E, et al. Primary ciliary dyskinesia in the paediatric population: range and severity of radiological findings in a cohort of patients receiving tertiary care. Clin Radiol. 2007;62(10):986–93.
Tadd K, Morgan L, Rosenow T, Schultz A, Susanto C, Murray C, et al. CF derived scoring systems do not fully describe the range of structural changes seen on CT scans in PCD. Pediatr Pulmonol. 2019;54(4):471–7.
Davis SD, Ferkol TW, Rosenfeld M, Lee HS, Dell SD, Sagel SD, et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med. 2015;191(3):316–24.
Alanin MC, Nielsen KG, von Buchwald C, Skov M, Aanaes K, Hoiby N, et al. A longitudinal study of lung bacterial pathogens in patients with primary ciliary dyskinesia. Clin Microbiol Infect. 2015;21(12):1093.e1–7.
Rogers GB, Carroll MP, Zain NM, Bruce KD, Lock K, Walker W, et al. Complexity, temporal stability, and clinical correlates of airway bacterial community composition in primary ciliary dyskinesia. J Clin Microbiol. 2013;51(12):4029–35.
Maglione M, Montella S, Mollica C, Carnovale V, Iacotucci P, De Gregorio F, et al. Lung structure and function similarities between primary ciliary dyskinesia and mild cystic fibrosis: a pilot study. Ital J Pediatr. 2017;43(1):34.
Wijers CD, Chmiel JF, Gaston BM. Bacterial infections in patients with primary ciliary dyskinesia: comparison with cystic fibrosis. Chron Respir Dis. 2017;14(4):392–406.
Shapiro AJ, Davis SD, Ferkol T, Dell SD, Rosenfeld M, Olivier KN, et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: insights into situs ambiguus and heterotaxy. Chest. 2014;146(5):1176–86.
Best S, Shoemark A, Rubbo B, Patel MP, Fassad MR, Dixon M, et al. Risk factors for situs defects and congenital heart disease in primary ciliary dyskinesia. Thorax. 2019;74(2):203–5.
Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, et al. European respiratory society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. 2017;49(1):1601090.
Behan L, Dimitrov BD, Kuehni CE, Hogg C, Carroll M, Evans HJ, et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J. 2016;47(4):1103–12.
Behan L, Dunn Galvin A, Rubbo B, Masefield S, Copeland F, Manion M, et al. Diagnosing primary ciliary dyskinesia: an International patient perspective. Eur Respir J. 2016;48(4):1096–107.
Shapiro AJ, Davis SD, Polineni D, Manion M, Rosenfeld M, Dell SD, et al. Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2018;197(12):e24–39.
Collins SA, Gove K, Walker W, Lucas JS. Nasal nitric oxide screening for primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J. 2014;44(6):1589–99.
Rubbo B, Shoemark A, Jackson CL, Hirst RA, Thompson J, Hayes J, et al. Accuracy of high-speed video analysis to diagnose primary ciliary dyskinesia. Chest. 2019;155(5):1008–17.
Shoemark A, Boon M, Brochhausen C, Bukowy-Bieryllo Z, De Santi MM, Goggin P, et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary ciliary dyskinesia (BEAT PCD TEM criteria). Eur Respir J. 2020;55(4):1900725.
Shoemark A, Boon M, Brochhausen C, Bukowy-Bieryllo Z, De Santi MM, Goggin P, et al. International consensus guideline for reporting transmission electron microscopy results in the diagnosis of primary ciliary dyskinesia (BEAT PCD TEM Criteria). Eur Respir J. 2020;55(4):1900725. https://pubmed.ncbi.nlm.nih.gov/32060067/.
Shoemark A, Frost E, Dixon M, Ollosson S, Kilpin K, Patel M, et al. Accuracy of immunofluorescence in the diagnosis of primary ciliary dyskinesia. Am J Respir Crit Care Med. 2017;196(1):94–101.
Vanaken GJ, Bassinet L, Boon M, Mani R, Honore I, Papon JF, et al. Infertility in an adult cohort with primary ciliary dyskinesia: phenotype-gene association. Eur Respir J. 2017;50(5):1700314.
Davis SD, Rosenfeld M, Lee HS, Ferkol TW, Sagel SD, Dell SD, et al. Primary ciliary dyskinesia: longitudinal study of lung disease by ultrastructure defect and genotype. Am J Respir Crit Care Med. 2019;199(2):190–8.
Crowley S, Azevedo I, Boon M, Bush A, Eber E, Haarman E, et al. Access to medicines for rare diseases: beating the drum for primary ciliary dyskinesia. ERJ Open Res. 2020;6(3):00377–2020.
Shapiro AJ, Zariwala MA, Ferkol T, Davis SD, Sagel SD, Dell SD, et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr Pulmonol. 2016;51(2):115–32.
Barbato A, Frischer T, Kuehni CE, Snijders D, Azevedo I, Baktai G, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J. 2009;34(6):1264–76.
Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120(2):171–8.
Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287–301.
Grantham JJ. Lillian Jean Kaplan International prize for advancement in the understanding of polycystic kidney disease. Understanding polycystic kidney disease: a systems biology approach. Kidney Int. 2003;64(4):1157–62.
Driscoll JA, Bhalla S, Liapis H, Ibricevic A, Brody SL. Autosomal dominant polycystic kidney disease is associated with an increased prevalence of radiographic bronchiectasis. Chest. 2008;133(5):1181–8.
Shoemark A, Dixon M, Beales PL, Hogg CL. Bardet biedl syndrome: motile ciliary phenotype. Chest. 2015;147(3):764–70.
Schaefer E, Zaloszyc A, Lauer J, Durand M, Stutzmann F, Perdomo-Trujillo Y, et al. Mutations in SDCCAG8/NPHP10 cause Bardet-Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol Syndromol. 2011;1(6):273–81.
Boon M, Wallmeier J, Ma L, Loges NT, Jaspers M, Olbrich H, et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun. 2014;5:4418.
Amirav I, Wallmeier J, Loges NT, Menchen T, Pennekamp P, Mussaffi H, et al. Systematic analysis of CCNO variants in a defined population: implications for clinical phenotype and differential diagnosis. Hum Mutat. 2016;37(4):396–405.
Wallmeier J, Al-Mutairi DA, Chen CT, Loges NT, Pennekamp P, Menchen T, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet. 2014;46(6):646–51.
Shoemark A, Ozerovitch L, Wilson R. Aetiology in adult patients with bronchiectasis. Respir Med. 2007;101(6):1163–70.
Eastham KM, Fall AJ, Mitchell L, Spencer DA. The need to redefine non-cystic fibrosis bronchiectasis in childhood. Thorax. 2004;59(4):324–7.
Nikolaizik WH, Warner JO. Aetiology of chronic suppurative lung disease. Arch Dis Child. 1994;70(2):141–2.
Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–86.
Gathmann B, Mahlaoui N, Gerard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(1):116–26.
Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59.
Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. QJM. 2002;95(10):655–62.
Quinti I, Soresina A, Guerra A, Rondelli R, Spadaro G, Agostini C, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol. 2011;31(3):315–22.
Edgar JD, Buckland M, Guzman D, Conlon NP, Knerr V, Bangs C, et al. The United Kingdom primary immune deficiency (UKPID) registry: report of the first 4 years’ activity 2008-2012. Clin Exp Immunol. 2014;175(1):68–78.
Gathmann B, Grimbacher B, Beaute J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006-2008. Clin Exp Immunol. 2009;157(Suppl 1):3–11.
Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–8.
Conley M. Autosomal recessive agammaglobulinemia. In: Ochs HS, Edvard Smith CI, Puck JM, editors. Primary immunodeficiency diseases: a molecular and genetic approach. 2nd ed. New York: Oxford University Press; 2007. p. 304–12.
Howard V, Greene JM, Pahwa S, Winkelstein JA, Boyle JM, Kocak M, et al. The health status and quality of life of adults with X-linked agammaglobulinemia. Clin Immunol. 2006;118(2-3):201–8.
Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85(4):193–202.
Plebani A, Soresina A, Rondelli R, Amato GM, Azzari C, Cardinale F, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol. 2002;104(3):221–30.
Roos DK, Curnutte JT. Chronic granulomatous disease. In: Ochs HDS, Edvard Smith CI, Puck JM, editors. Primary immunodeficiency disease: a molecular and genetic approach. 2nd ed. New York: Oxford University Press; 2007. p. 525–49.
van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4(4):e5234.
Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, et al. Severe chronic neutropenia: treatment and follow-up of patients in the severe chronic neutropenia International registry. Am J Hematol. 2003;72(2):82–93.
Horwitz MS, Duan Z, Korkmaz B, Lee HH, Mealiffe ME, Salipante SJ. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007;109(5):1817–24.
Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007;39(1):86–92.
Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, Schaffer AA, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007;13(1):38–45.
Kilpatrick DC, Chalmers JD, MacDonald SL, Murray M, Mohammed A, Hart SP, et al. Stable bronchiectasis is associated with low serum L-ficolin concentrations. Clin Respir J. 2009;3(1):29–33.
Fevang B, Mollnes TE, Holm AM, Ueland T, Heggelund L, Damas JK, et al. Common variable immunodeficiency and the complement system; low mannose-binding lectin levels are associated with bronchiectasis. Clin Exp Immunol. 2005;142(3):576–84.
Garred P, Pressler T, Madsen HO, Frederiksen B, Svejgaard A, Hoiby N, et al. Association of mannose-binding lectin gene heterogeneity with severity of lung disease and survival in cystic fibrosis. J Clin Invest. 1999;104(4):431–7.
Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97(11):813–22.
Pan-Hammarstrom Q, Lahdesmaki A, Zhao Y, Du L, Zhao Z, Wen S, et al. Disparate roles of ATR and ATM in immunoglobulin class switch recombination and somatic hypermutation. J Exp Med. 2006;203(1):99–110.
Kraus M, Lev A, Simon AJ, Levran I, Nissenkorn A, Levi YB, et al. Disturbed B and T cell homeostasis and neogenesis in patients with ataxia telangiectasia. J Clin Immunol. 2014;34(5):561–72.
Driessen GJ, Ijspeert H, Weemaes CM, Haraldsson A, Trip M, Warris A, et al. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. J Allergy Clin Immunol. 2013;131(5):1367–75.e9.
Lefton-Greif MA, Crawford TO, Winkelstein JA, Loughlin GM, Koerner CB, Zahurak M, et al. Oropharyngeal dysphagia and aspiration in patients with ataxia-telangiectasia. J Pediatr. 2000;136(2):225–31.
Crawford TO, Skolasky RL, Fernandez R, Rosquist KJ, Lederman HM. Survival probability in ataxia telangiectasia. Arch Dis Child. 2006;91(7):610–1.
Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections—an autosomal dominant multisystem disorder. N Engl J Med. 1999;340(9):692–702.
Levy DE, Loomis CA. STAT3 signaling and the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1655–8.
Khan S, Hinks J, Shorto J, Schwarz MJ, Sewell WA. Some cases of common variable immunodeficiency may be due to a mutation in the SBDS gene of Shwachman-diamond syndrome. Clin Exp Immunol. 2008;151(3):448–54.
Rademacher J, Schulz A, Hedtfeld S, Stanke F, Ringshausen F, Welte T, et al. Nasal potential difference of carriers of the W493R ENaC variant with non-cystic fibrosis bronchiectasis. Eur Respir J. 2016;47(1):322–4.
Guan WJ, Li JC, Liu F, Zhou J, Liu YP, Ling C, et al. Next-generation sequencing for identifying genetic mutations in adults with bronchiectasis. J Thorac Dis. 2018;10(5):2618–30.
Parr DG, Guest PG, Reynolds JH, Dowson LJ, Stockley RA. Prevalence and impact of bronchiectasis in alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2007;176(12):1215–21.
Cuvelier A, Muir JF, Hellot MF, Benhamou D, Martin JP, Benichou J, et al. Distribution of alpha(1)-antitrypsin alleles in patients with bronchiectasis. Chest. 2000;117(2):415–9.
Peppers BP, Zacharias J, Michaud CR, Frith JA, Varma P, Henning M, et al. Association between alpha1-antitrypsin and bronchiectasis in patients with humoral immunodeficiency receiving gammaglobulin infusions. Ann Allergy Asthma Immunol. 2018;120(2):200–6.
Lonni S, Chalmers JD, Goeminne PC, McDonnell MJ, Dimakou K, De Soyza A, et al. Etiology of non-cystic fibrosis bronchiectasis in adults and its correlation to disease severity. Ann Am Thorac Soc. 2015;12(12):1764–70.
Noone PG, Pue CA, Zhou Z, Friedman KJ, Wakeling EL, Ganeshananthan M, et al. Lung disease associated with the IVS8 5T allele of the CFTR gene. Am J Respir Crit Care Med. 2000;162(5):1919–24.
Szymanski EP, Leung JM, Fowler CJ, Haney C, Hsu AP, Chen F, et al. Pulmonary nontuberculous mycobacterial infection. A multisystem, multigenic disease. Am J Respir Crit Care Med. 2015;192(5):618–28.
Shoemark A. PCD in adults with bronchiectasis: data from the EMBARC registry. Eur Respir J. 2018;52.
Hill AT, Sullivan AL, Chalmers JD, De Soyza A, Elborn SJ, Floto AR, et al. British thoracic society guideline for bronchiectasis in adults. Thorax. 2019;74(Suppl 1):1–69.
Al-Jahdali H, Alshimemeri A, Mobeireek A, Albanna AS, Al Shirawi NN, Wali S, et al. The Saudi thoracic society guidelines for diagnosis and management of noncystic fibrosis bronchiectasis. Ann Thorac Med. 2017;12(3):135–61.
Hill AT, Haworth CS, Aliberti S, Barker A, Blasi F, Boersma W, et al. Pulmonary exacerbation in adults with bronchiectasis: a consensus definition for clinical research. Eur Respir J. 2017;49(6):1700051.
Fall A, Spencer D. Paediatric bronchiectasis in Europe: what now and where next? Paediatr Respir Rev. 2006;7(4):268–74.
Milito C, Pulvirenti F, Serra G, Valente M, Pesce AM, Granata G, et al. Lung magnetic resonance imaging with diffusion weighted imaging provides regional structural as well as functional information without radiation exposure in primary antibody deficiencies. J Clin Immunol. 2015;35(5):491–500.
Aurora P, Stanojevic S, Wade A, Oliver C, Kozlowska W, Lum S, et al. Lung clearance index at 4 years predicts subsequent lung function in children with cystic fibrosis. Am J Respir Crit Care Med. 2011;183(6):752–8.
Irving SJ, Ives A, Davies G, Donovan J, Edey AJ, Gill SS, et al. Lung clearance index and high-resolution computed tomography scores in primary ciliary dyskinesia. Am J Respir Crit Care Med. 2013;188(5):545–9.
Green K, Buchvald FF, Marthin JK, Hanel B, Gustafsson PM, Nielsen KG. Ventilation inhomogeneity in children with primary ciliary dyskinesia. Thorax. 2012;67(1):49–53.
Nyilas S, Schlegtendal A, Yammine S, Casaulta C, Latzin P, Koerner-Rettberg C. Further evidence for an association between LCI and FEV1 in patients with PCD. Thorax. 2015;70(9):896.
Boon M, Vermeulen FL, Gysemans W, Proesmans M, Jorissen M, De Boeck K. Lung structure-function correlation in patients with primary ciliary dyskinesia. Thorax. 2015;70(4):339–45.
Walshaw MJ. Cystic fibrosis: diagnosis and management—NICE guideline 78. Paediatr Respir Rev. 2019;31:12–4.
Li AM, Sonnappa S, Lex C, Wong E, Zacharasiewicz A, Bush A, et al. Non-CF bronchiectasis: does knowing the aetiology lead to changes in management? Eur Respir J. 2005;26(1):8–14.
Lucas JS, Gahleitner F, Amorim A, Boon M, Brown P, Constant C, et al. Pulmonary exacerbations in patients with primary ciliary dyskinesia: an expert consensus definition for use in clinical trials. ERJ Open Res. 2019;5(1):00147–2018.
Sunther M, Bush A, Hogg C, McCann L, Carr SB. Recovery of baseline lung function after pulmonary exacerbation in children with primary ciliary dyskinesia. Pediatr Pulmonol. 2016;51(12):1362–6.
Sanders DB, Bittner RC, Rosenfeld M, Hoffman LR, Redding GJ, Goss CH. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am J Respir Crit Care Med. 2010;182(5):627–32.
Sanders DB, Hoffman LR, Emerson J, Gibson RL, Rosenfeld M, Redding GJ, et al. Return of FEV1 after pulmonary exacerbation in children with cystic fibrosis. Pediatr Pulmonol. 2010;45(2):127–34.
Chalmers JD, Haworth CS, Metersky ML, Loebinger MR, Blasi F, Sibila O, et al. Phase 2 trial of the DPP-1 inhibitor Brensocatib in bronchiectasis. N Engl J Med. 2020;383(22):2127–37.
Bijl M, Kallenberg CG, van Assen S. Vaccination of the immune-compromised patients with focus on patients with autoimmune-inflammatory diseases. Neth J Med. 2011;69(1):5–13.
Bilton D, Tino G, Barker AF, Chambers DC, De Soyza A, Dupont LJ, et al. Inhaled mannitol for non-cystic fibrosis bronchiectasis: a randomised, controlled trial. Thorax. 2014;69(12):1073–9.
Haworth CS, Foweraker JE, Wilkinson P, Kenyon RF, Bilton D. Inhaled colistin in patients with bronchiectasis and chronic Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2014;189(8):975–82.
Barker AF, O’Donnell AE, Flume P, Thompson PJ, Ruzi JD, de Gracia J, et al. Aztreonam for inhalation solution in patients with non-cystic fibrosis bronchiectasis (AIR-BX1 and AIR-BX2): two randomised double-blind, placebo-controlled phase 3 trials. Lancet Respir Med. 2014;2(9):738–49.
Kobbernagel HE, Buchvald FF, Haarman EG, Casaulta C, Collins SA, Hogg C, et al. Study protocol, rationale and recruitment in a European multi-centre randomized controlled trial to determine the efficacy and safety of azithromycin maintenance therapy for 6 months in primary ciliary dyskinesia. BMC Pulm Med. 2016;16(1):104.
Kobbernagel HE, Buchvald FF, Haarman EG, Casaulta C, Collins SA, Hogg C, et al. Efficacy and safety of azithromycin maintenance therapy in primary ciliary dyskinesia (BESTCILIA): a multicentre, double-blind, randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2020;8(5):493–505.
Bilton D, Canny G, Conway S, Dumcius S, Hjelte L, Proesmans M, et al. Pulmonary exacerbation: towards a definition for use in clinical trials. Report from the EuroCareCF working group on outcome parameters in clinical trials. J Cyst Fibros. 2011;10(Suppl 2):S79–81.
Lucas JS, Behan L, Dunn Galvin A, Alpern A, Morris AM, Carroll MP, et al. A quality-of-life measure for adults with primary ciliary dyskinesia: QOL-PCD. Eur Respir J. 2015;46(2):375–83.
Dell SD, Leigh MW, Lucas JS, Ferkol TW, Knowles MR, Alpern A, et al. Primary ciliary dyskinesia: first health-related quality-of-life measures for pediatric patients. Ann Am Thorac Soc. 2016;13(10):1726–35.
Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the cystic fibrosis questionnaire-revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest. 2009;135(6):1610–8.
Paredes Aller S, Quittner AL, Salathe MA, Schmid A. Assessing effects of inhaled antibiotics in adults with non-cystic fibrosis bronchiectasis—experiences from recent clinical trials. Expert Rev Respir Med. 2018;12(9):769–82.
Schofield LM, Duff A, Brennan C. Airway clearance techniques for primary ciliary dyskinesia; is the cystic fibrosis literature portable? Paediatr Respir Rev. 2018;25:73–7.
Lee AL, Button BM, Tannenbaum EL. Airway-clearance techniques in children and adolescents with chronic suppurative lung disease and bronchiectasis. Front Pediatr. 2017;5:2.
Schofield LM, Horobin HE. Growing up with primary ciliary dyskinesia in Bradford, UK: exploring patients experiences as a physiotherapist. Physiother Theory Pract. 2014;30(3):157–64.
Jones AP, Wallis C. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. 2010;3:CD001127.
O’Donnell AE, Barker AF, Ilowite JS, Fick RB. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase study group. Chest. 1998;113(5):1329–34.
Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev. 2018;9:Cd001506.
Nevitt SJ, Thornton J, Murray CS, Dwyer T. Inhaled mannitol for cystic fibrosis. Cochrane Database Syst Rev. 2018;2:Cd008649.
Paff T, Daniels JM, Weersink EJ, Lutter R, Vonk Noordegraaf A, Haarman EG. A randomised controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia. Eur Respir J. 2017;49(2):1601770.
Nicolson CH, Stirling RG, Borg BM, Button BM, Wilson JW, Holland AE. The long term effect of inhaled hypertonic saline 6% in non-cystic fibrosis bronchiectasis. Respir Med. 2012;106(5):661–7.
Kellett F, Robert NM. Nebulised 7% hypertonic saline improves lung function and quality of life in bronchiectasis. Respir Med. 2011;105(12):1831–5.
Bilton D, Daviskas E, Anderson SD, Kolbe J, King G, Stirling RG, et al. Phase 3 randomized study of the efficacy and safety of inhaled dry powder mannitol for the symptomatic treatment of non-cystic fibrosis bronchiectasis. Chest. 2013;144(1):215–25.
Chalmers JD, Boersma W, Lonergan M, Jayaram L, Crichton ML, Karalus N, et al. Long-term macrolide antibiotics for the treatment of bronchiectasis in adults: an individual participant data meta-analysis. Lancet Respir Med. 2019;7(10):845–54.
Kelly C, Chalmers JD, Crossingham I, Relph N, Felix LM, Evans DJ, et al. Macrolide antibiotics for bronchiectasis. Cochrane Database Syst Rev. 2018;3:Cd012406.
Smith D, Du Rand IA, Addy C, Collyns T, Hart S, Mitchelmore P, et al. British thoracic society guideline for the use of long-term macrolides in adults with respiratory disease. BMJ Open Respir Res. 2020;7(1):e000489.
Crosbie PA, Woodhead MA. Long-term macrolide therapy in chronic inflammatory airway diseases. Eur Respir J. 2009;33(1):171–81.
Mogayzel PJ Jr, Naureckas ET, Robinson KA, Brady C, Guill M, Lahiri T, et al. Cystic fibrosis foundation pulmonary guideline. Pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann Am Thorac Soc. 2014;11(10):1640–50.
Gallin JI, Alling DW, Malech HL, Wesley R, Koziol D, Marciano B, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med. 2003;348(24):2416–22.
Brand P, Schulte M, Wencker M, Herpich CH, Klein G, Hanna K, et al. Lung deposition of inhaled alpha1-proteinase inhibitor in cystic fibrosis and alpha1-antitrypsin deficiency. Eur Respir J. 2009;34(2):354–60.
Eijkhout HW, van Der Meer JW, Kallenberg CG, Weening RS, van Dissel JT, Sanders LA, et al. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial. Ann Intern Med. 2001;135(3):165–74.
Nicolay U, Kiessling P, Berger M, Gupta S, Yel L, Roifman CM, et al. Health-related quality of life and treatment satisfaction in North American patients with primary immune deficiency diseases receiving subcutaneous IgG self-infusions at home. J Clin Immunol. 2006;26(1):65–72.
Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenous immunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet. 1987;1(8541):1075–7.
Gregersen S, Aalokken TM, Mynarek G, Fevang B, Holm AM, Ueland T, et al. Development of pulmonary abnormalities in patients with common variable immunodeficiency: associations with clinical and immunologic factors. Ann Allergy Asthma Immunol. 2010;104(6):503–10.
Nakagawa N, Imai K, Kanegane H, Sato H, Yamada M, Kondoh K, et al. Quantification of kappa-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J Allergy Clin Immunol. 2011;128(1):223–5.e2.
Pai SY, DeMartiis D, Forino C, Cavagnini S, Lanfranchi A, Giliani S, et al. Stem cell transplantation for the Wiskott-Aldrich syndrome: a single-center experience confirms efficacy of matched unrelated donor transplantation. Bone Marrow Transplant. 2006;38(10):671–9.
Seger RA, Gungor T, Belohradsky BH, Blanche S, Bordigoni P, Di Bartolomeo P, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985-2000. Blood. 2002;100(13):4344–50.
Rousso SZ, Shamriz O, Zilkha A, Braun J, Averbuch D, Or R, et al. Hematopoietic stem cell transplantations for primary immune deficiencies: 3 decades of experience from a tertiary medical center. J Pediatr Hematol Oncol. 2015;37(5):e295–300.
Jiang H, Pierce GF, Ozelo MC, de Paula EV, Vargas JA, Smith P, et al. Evidence of multiyear factor IX expression by AAV-mediated gene transfer to skeletal muscle in an individual with severe hemophilia B. Mol Ther. 2006;14(3):452–5.
Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–9.
Aubert D, Menoret S, Chiari E, Pichard V, Durand S, Tesson L, et al. Cytotoxic immune response blunts long-term transgene expression after efficient retroviral-mediated hepatic gene transfer in rat. Mol Ther. 2002;5(4):388–96.
Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360(5):447–58.
Wada T, Schurman SH, Otsu M, Garabedian EK, Ochs HD, Nelson DL, et al. Somatic mosaicism in Wiskott—Aldrich syndrome suggests in vivo reversion by a DNA slippage mechanism. Proc Natl Acad Sci U S A. 2001;98(15):8697–702.
Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12(4):401–9.
De Ravin SS, Reik A, Liu PQ, Li L, Wu X, Su L, et al. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat Biotechnol. 2016;34(4):424–9.
Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72.
De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–80.
Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219–25.
Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4(2):107–15.
Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind, randomised controlled trial. Lancet Respir Med. 2015;3(7):524–33.
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–31.
Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5(7):557–67.
Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor-Ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377(21):2024–35.
Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, et al. Tezacaftor-Ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–23.
Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1612–20.
Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019;394(10212):1940–8.
Alton E, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3(9):684–91.
Schwabe GC, Hoffmann K, Loges NT, Birker D, Rossier C, de Santi MM, et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat. 2008;29(2):289–98.
Kuehni CE, Lucas JS. Diagnosis of primary ciliary dyskinesia: summary of the ERS task force report. Breathe (Sheff). 2017;13(3):166–78.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lucas, J.S., Pike, K.C., Walker, W.T., Shoemark, A. (2023). Diffuse Bronchiectasis of Genetic or Idiopathic Origin. In: Cottin, V., Richeldi, L., Brown, K., McCormack, F.X. (eds) Orphan Lung Diseases. Springer, Cham. https://doi.org/10.1007/978-3-031-12950-6_25
Download citation
DOI: https://doi.org/10.1007/978-3-031-12950-6_25
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-12949-0
Online ISBN: 978-3-031-12950-6
eBook Packages: MedicineMedicine (R0)