
Abstract
Objective
Ataxia telangiectasia (AT) is a rare genetic, multi-system disorder characterized by neurodegeneration, chromosome instability, B and T cell immunodeficiency and a predisposition to cancer. We examined immunologic parameters reflecting cell development and proliferation and their relevancy to the clinical phenotype in affected individuals.
Patients and Methods
AT patients from the AT National Clinic in Israel underwent immunological investigation. Their T and B cell workup included lymphocyte subset counts, immunoglobulin levels, responses to mitogenic stimulations, TCR-Vβ families and BCR immunoglobulin heavy chain spectratyping, TCR rearrangement excision circles (TRECs) and Kappa-deleting recombination excision circles (KRECs).
Results
Thirty-seven AT patients (median age 12.7 years, range 4.2–25.1) were evaluated. CD20 B and CD3 T lymphocytes were decreased in 67 % and 64 % of the patients, respectively, while only 33 % of the patients had reduced lymphoproliferative responses. Almost all AT patients displayed extremely low TRECs and KRECs levels, irrespective of their age. Those levels were correlated to one another and to the amounts of CD3+ and CD20+ cells, respectively. Abnormal TCR-Vβ repertoires were found with different degrees of clonality or reduced expression in these AT patients. There was no clear clustering of expansions to specific TCR-Vβ genes. PCR spectratyping analysis of the FR2 IgH BCR gene rearrangements in peripheral blood was abnormal in 50 % of the patients.
Conclusion
The immunodeficiency associated with AT is combined, remains low over time and not progressive. It is characterized by low TREC and KREC copies suggestive of abnormal T and B cell neogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ataxia telangiectasia (AT) is a rare, multi-systemic, autosomal recessive disorder characterized by neurodegeneration, chromosome instability, radiosensitivity, immunodeficiency and a predisposition to cancer. The defective gene in AT, AT mutated (ATM), is a Ser/Thr key protein kinase that is involved in the DNA damage response (1). A variation of both B and T cell immunodeficiencies has long been recognized in AT patients. B cell immunodeficiency is characterized by decreased or absent serum IgA, IgE, and IgG2 and impaired antibody responses. T cell immunodeficiency includes lymphopenia with diminished numbers of CD4 T lymphocytes, impaired lymphoproliferative responses to mitogens and antigens, and anergy (2). Absence or a rudimentary appearance of a thymus gland, the main organ for T cell development, is also a well-described immune abnormality associated with AT (3). It is widely accepted that ATM deficiency results in abnormal surface T cell receptor (TCR) expression in CD4 + CD8+ cells and inefficient thymocyte selection (4). Functional T and B cell receptors (TCRs and BCRs, respectively) are created through the ligation of coding joint (cj) recombination sites. During these processes, the byproducts, TCR rearrangement excision circles (TRECs) and Kappa-deleting recombination excision circles (KRECs), are formed through signal joint (sj) recombination sites (5). TRECs and KRECs can be used as DNA markers of thymic and bone marrow outputs, respectively, since they are present exclusively in naïve T and B cells that have recently emigrated from these organs (6). Consequently, the cj and sj ratio of these recombination sites reflects T and B cell homeostasis. Both sj-TRECs and sj-KRECs levels are extremely low in the peripheral blood of AT patients (7). In addition, quantitation of TRECs and KRECs by a single Guthrie card punch was recently used in original samples obtained from AT patients: it revealed marked reductions of both markers and reflected reduced development of T and B cell immunity (8). Indeed, T cell lymphopenia in newborns was recently found to be a feature of AT, as revealed by TREC screening followed by exome sequencing (9). Measurements of TRECs and KRECs, together with assessment of TCR and BCR repertoires reflect T and B cell homeostasis (10). These markers are affected by thymic and bone marrow cell production, respectively, as well as by their peripheral reduction due to cell expansions (5). A systematic evaluation of the levels of these markers in the peripheral blood of AT patients, specifically with regard to their clinical disease and associated malignant and infectious complications, was conducted in our study, in order to understand the immunologic mechanisms involved in AT.
Methods
Patients
Thirty-seven patients followed at the Ataxia Telangiectasia National Clinic at the Edmond and Lily Safra Children Hospital, Sheba Medical Center in Israel were enrolled in this study which was approved by the Institutional Review Board. The diagnosis of AT was made in according to typical clinical findings plus one of the following: (1) a proven mutation in the ATM gene, (2) deficient ATM protein proven by Western blotting, and (3) elevated α-fetoprotein, cerebellar atrophy on magnetic resonance imaging (MRI), and immune deficiency/chromosomal breakage/T-cell lymphoreticular malignancy. Parents provided signed informed consent to obtain blood from their children. The medical records of the patients were retrospectively evaluated. Recurrent infections in patients were defined as two or more sinopulmonary infections per year.
Immune function
Cell surface markers of peripheral blood mononuclear cells (PBMCs) were determined by immunofluorescent staining and flow cytometry (Epics V; Coulter Electronics, Hialeah, FL) with antibodies purchased from Coulter Diagnostics. Lymphocyte proliferation in response to phytohemagglutinin and anti-CD3 was determined by tritiated thymidine incorporation as previously described (11). Age-adjusted normal values for the cell surface markers were obtained from Shearer et al. (12). For lymphocyte proliferation studies, the cells were harvested 3 days after collection, and samples were counted in a liquid scintillation counter. All assays were performed in triplicates, and counts per minutes (cpm) were obtained. Serum concentration of immunoglobulins was measured by nephelometry.
Quantification of TRECs and KRECs
The amount of TRECs was determined by real-time quantitative (RQ)-PCR. Reactions were performed by using 0.5 μg genomic DNA extracted from the patient’s PBMCs. PCRs contained TaqMan universal PCR master mix (Applied Biosystems), specific primers (900 nM), and FAM-TAMRA probes (250 nM) as previously described (13). RQ-PCR was carried out in an ABI PRISM 7900 Sequence Detector System (Applied Biosystems). The standard curve was constructed by using serial dilutions of a known TREC plasmid (generously provided by Dr. Daniel Douek, Vaccine Research Center, National Institute of Allergy and Infectious Diseases, Bethesda, MD). The dilutions contained 103 to 106 copies of the sj-TREC plasmid. The number of TRECs in a given sample was automatically calculated by comparing the obtained Ct value of a patient’s sample to the standard curve using an absolute quantification algorithm. The amount of signal and coding joint of KRECs was determined by RQ-PCR as described above for TRECs. Amplification of RNAseP (TaqMan assay, Applied Biosystems) served as a quality control to verify similar amounts of genomic DNA that were used in the assays for both the TREC and KREC analyses. The specific primers and probe combinations for KREC detection were as previously described (10). Thirty-six age-matched normal individuals were used as controls. Each experiment was performed in triplicate, and the threshold for Ct determination was positioned at the same level each time.
BCR and TCR gene rearrangement analyses
Representatives of specific TCR-Vβ families were detected and quantified using flow cytometry (Becton-Dickinson, Calibur) according to the manufacturer’s instructions (Beckman Coulter) and as previously described (10). Normal control values were obtained from the IOTest Beta Mark-Quick Reference Card (Beckman Coulter, Fullerton, CA). The BCR gene rearrangement analyses were performed on genomic DNA obtained from the patient’s PBMCs without specific cell enrichment, as previously described (6). Briefly, the immunoglobulin heavy chain (IGH) genes were amplified by PCR using fluorescence-labeled Vh primers according to the standardized Biomed 2 protocol (14). Fluorescence-labeled PCR products (1 μl of each) were added to a mixture of 8.5 ml deionized formamide and 0.5 μl GeneScan 500TM Rox internal lane standard (PE Applied Biosystem, Germany), and separated using the 3 500 Genetic Analyzer (Applied Biosystem). Results were analyzed using the GeneMapper software (Applied Biosystem). BCR repertoire profiles were characterized as “oligoclonal” if they contained 4 or fewer peaks, as “polyclonal skewed” if they had more than 4 peaks but were not Gaussian-like, and as “polyclonal” if they had more than 4 peaks in a Gaussian-like distribution.
Statistics
Data were analyzed by the SPSS (version 21) program. A p value <0.05 was considered significant. Categorical variables were compared using the Chi-square test, and continuous variables by the t-test (presented as means with standard deviations [SD]) or by the Mann Whitney test (medians with interquartile range [IQR]). Correlations between continuous variables were evaluated using the Spearman's rho test.
Results
Clinical findings
The study cohort consisted of 37 AT patients (median age 12.7 years, range 4.2–25.1 years; 22 males). The diagnosis of AT was confirmed by mutation analysis in 29 patients, molecular tests (e.g., abnormal ATM protein expression or abnormal chromosomal breakage) in 4 patients, or based solely upon clinical presentation in 4 patients. There was a family history of AT in 24 patients and parental consanguinity in 28. The main presenting symptom, which became evident at a median age of 3.3 years (range 1.4–17 years), was ataxia in 30 patients. Twenty-seven patients had recurrent infections consisting mainly of pneumonia (20 patients). There were 21 patients who were on intravenous immunoglobulin (IVIG) replacement therapy and 10 were on prophylactic antibiotics. The median age at IVIG initiation was 7 years (range 1.8–14). Twenty-one patients required the use of a wheelchair. Ten patients developed malignancies that included T-cell acute lymphoblastic leukemia, germinoma, renal cell carcinoma and teratoma. The clinical and genetic findings of the reported patients are summarized in Table 1.
Lymphocyte number and in vitro T cell function
Lymphocyte subsets and immunoglobulin levels were determined in 34 of the patients (Table 2). IgG levels were decreased in 6 of them (6/34, 18 %), and 20 patients with normal IgG levels were on IVIG replacement treatment during the period of data collection. IgA levels were decreased or absent in 21 of these patients (21/34, 62 %), and 8 of them (8/32, 25 %) had high IgM levels. Twenty-one of the AT patients were lymphopenic (21/34, 62 %). CD20+ B lymphocytes were decreased in 23 patients (23/34, 67 %: Fig. 1a), and CD3+ T lymphocytes were decreased in 22 patients (22/34, 64 %: Fig. 1b). There was a significant correlation between the CD20+ B and CD3+ T lymphocytes (Rs = 0.39, p = 0.02). Functional evaluations of T lymphocytes were performed in 32 patients. Reduced lymphoproliferative responses were found in 12 patients (12/32, 37 %: Fig. 2a) following PHA mitogenic stimulation, and in 10 patients (10/32, 31 %: Fig. 2b) following CD3 mitogenic stimulation.

Lymphocyte subset analysis. The cell surface markers CD20+ a and CD3+ b of patient’s cells were detected by flow cytometry using immunofluorescent staining. The results are presented in correlation with patient’s age. The dashed line represents the age-dependent normal values of CD20 a and CD3 b according to Shearer et al. (12)

In vitro T cell function. Lymphocyte proliferation was detected in response to PHA6 a and anti-CD3 b mitogenic stimulation using tritiated thymidine incorporation. The results are presented in correlation with patient’s age. The dashed line represents the age-dependent normal values of mitogenic response to PHA6 a and anti-CD3 b in 40 healthy controls
TREC and KREC levels
Thymic and bone marrow outputs were quantified by measuring sj-TREC and sj-KREC levels (Table 2). Almost all AT patients displayed extremely low levels of TRECs and KRECs in comparison with age-matched healthy individuals (Fig. 3a). These findings were age-independent (Fig. 3b and c). The TREC and KREC levels correlated significantly (Rs = 0.49, p = 0.003), indicating concomitant combined T and B cell immunodeficiency. The median TREC levels were very low in the AT patients (23.2 copies/0.5 μg DNA, range 0–338.7 copies/0.5 μg DNA) compared to the healthy controls (1065.7 copies/0.5 μg DNA, range 298.2-4676.3 copies/0.5 μg DNA). TREC levels did not correlate with the lymphocyte numbers (Rs = 0.26, p = 0.13), nor with the response to the PHA (Rs = 0.14, p = 0.43) or anti-CD3 (Rs = 0.2, p = 0.28) mitogenic stimulations. They did, however, correlate with CD3+ cell numbers (R = 0.33, p = 0.05). Patients with lymphopenia (defined as a lymphocyte count <1 500/mm3) had lower TREC levels compared to patients with a lymphocyte count >1 500/mm3 (mean=30.5 and 90.16 copies/0.5 μg DNA, respectively, p = 0.038). TREC levels were significantly associated with the use of a monthly IVIG replacement therapy regimen (p = 0.002) and prophylactic antibiotics (p = 0.04), but not with recurrent infections (p = 0.104) (Fig. 4).

sj-TREC and sj-KREC copies in AT patients. a sj-TREC and sj-KREC copies were simultaneously detected on genomic DNA isolated from PBMCs obtained from 26 AT patients (closed circles) and 36 age-matched healthy controls (open circles). Quantifications were performed by RQ-PCR in TaqMan assays. Results are shown in log scale. The dashed line represents the cutoff values for normal TRECs and KRECs. b, c sj-TREC b and sj-KREC c copies were detected by RQ-PCR in TaqMan assays on genomic DNA isolated from PBMCs obtained from 32 AT patients. Results are presented in correlation with patient’s age

Correlation of sj-TREC copies with the presence of recurrent infection and treatment. AT patients were grouped according to the presence or absence of recurrent infection and according to their relevant treatments (prophylactic antibiotics or IVIG replacement therapy), and copies of genomic sj-TRECs were detected. SPSS statistical analysis was performed. The graph indicates the interquartile range (IQR) of the measurements. The horizontal lines represent the median values. Including recurrent infections: median=21.6 (IQR = 8.9-31.7), n = 26; excluding recurrent infections: median = 40.8 (IQR = 13.3-75.9), n = 8, p = 0.11; including prophylactic antibiotics: median=10.3 (IQR = 2–29.2), n = 9; excluding prophylactic antibiotics: median = 24 (IQR = 16.7-47.7), n = 25, p = 0.04; including IVIG treatment: median = 16.9 (IQR = 6.4-23.2), n = 19; excluding IVIG treatment: median = 38 (IQR = 23.3-84), n = 15, p = 0.001
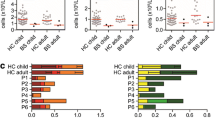
The median sj-KREC levels for the AT patients was 34.5 copies/0.5 μg DNA (range 0–1018.2 copies/0. 5 μg DNA) compared to 447.8 copies/0.5 μg DNA for the healthy controls (range 86.2-2471.9 copies/0. 5 μg DNA). The median cj-KREC levels for the AT patients was 1061.1 copies/0.5 μg DNA (range 132–9998.9 copies/0.5 μg DNA), compared to 2083.1 copies/0.5 μg DNA for the healthy controls (range 590.7-5281.7 copies/0. 5 μg DNA). The sj-KREC levels correlated significantly with the CD20+ cell numbers (Rs = 0.46, p = 0.007), but not with the total lymphocyte numbers or with the use of IVIG (p = 0.12), prophylactic antibiotics (p = 0.73) or recurrent infections (p = 0.11). The ratio between genomic sj-KRECs and cj-KRECs reflects both B cell neogenesis and peripheral replication of B lymphocytes. This ratio in our cohort study was significantly lower in the AT patients (median=2.8 %, IQR = 0.67-8.1 %) compared to the healthy controls (median = 21.3 %, IQR = 14.6-28.8, p ≥0.0001, Fig. 5). This finding is suggestive of either reduced B cell neogenesis or increased peripheral proliferation of B lymphocytes.

The ratio between sj-KRECs and cj-KRECs. Copies of genomic sj-KRECs and cj-KRECs were detected in PBMCs of AT patients and age-matched healthy controls. Their ratios were calculated and SPSS statistical analysis was performed. The graph indicates the interquartile range (IQR) of the measurements. The horizontal lines represent the median values. Controls: median = 21.3 (IQR = 14.6-28.8), n = 31, p < 0.0001; Patients: median = 2.8 (IQR = 0.6-8.1), n = 34, p < 0.0001
TCR and BCR repertoires
Flow cytometry analysis of the TCR-Vβ repertoire on total lymphocytes obtained from 19 patients (data not shown) revealed abnormal TCR-Vβ repertoires with different degrees of clonality or reduced expression in all of those patients. Significant abnormalities (i.e., above or below the highest and lowest normal values) were observed in 41 % of the 24 TCR-Vβ genes investigated in each patient. There was no clear clustering of expansions to certain TCR-Vβ genes, however, the TCRs expression tended to have the lowest values with increased age. Thus, clonality seemed to decrease with age, while reduced expression of different TCRs seemed to increase with age (Fig. 6).

TCR-Vβ repertoire: age-dependent clonality and expression analyses. Flow cytometer analysis was performed on the TCR-Vβ repertoire on total lymphocytes obtained from 19 AT patients. The number of normal (upper panel), high (middle panel) or low (lower panel) expression of the various TCR-Vβ clones were counted and presented as being age-dependent (grey bars) in each patient sample. The linear regression in each panel is depicted as a dashed line
PCR spectra-typing analysis of the FR2 IgH BCR gene rearrangements in peripheral blood was performed on 30 patients, and abnormal results (oligoclonal or polyclonal skewed) were detected in 15 of them (Fig. 7). An abnormal IgH BCR repertoire was not associated with sj-KREC levels, presence of malignancies or patient’s age.

BCR rearrangement analysis. FR2 IgH BCR gene rearrangement was detected in 30 AT patient’s PBMCs by PCR using fluorescence-labeled Vh primers followed by Gene Scan analysis. One of the three possible patterns (polyclonal, polyclonal skewed, oligoclonal) of the IgH BCR repertoire was obtained for each patient. P polyclonal; S polyclonal skewed; O oligoclonal
Discussion
AT is a well-defined combined type of immunodeficiency involving both T and B cell immunity. Here, we show low TREC and KREC levels in AT patients, irrespective of their age, reflecting impaired T and B cell neogenesis. Significant clonal abnormalities of both TCR and BCR were observed in most of the patients. Thus, our findings suggest that a developmental defect is the central mechanism underlying the immunodeficiency of AT.
Increased susceptibility to infections could be expected to be a dominant characteristic of AT as a result of a variety of cellular and humoral immunodeficiencies. The immune system of AT patients is considered as being abnormal from birth, however, and they do not have increased frequency or greater severity of immune abnormalities with age (2). Recurrent sinopulmonary infections may occur during the first 2 years of life (15). Indeed, 73 % of our young patients displayed recurrent infections, similar to the frequency reported by others (16). Importantly, this finding could not be attributed to a progression of their immunodeficiency insofar as it remained stable. Recurrent pulmonary infections and progression in lung disease, therefore, could be secondary to ineffective ability to cough, abnormal airway secretion clearance, oropharyngeal dysphagia and recurrent aspiration, and even to ATM abnormal function in pulmonary epithelial cells (17). In addition, some patients are wheelchair-bound at an early age and their neurological disease may contribute to further respiratory deterioration. Indeed, interstitial lung disease is found in a significant number of AT patients even in the absence of overt immune deficiency (18). Moreover, mycobacteria and opportunistic pathogens (such as fungi or pneumocystis) that characterize severe immunodeficiency rarely cause pulmonary infection in AT (2). In our study, not finding any correlation between both TREC and KREC levels and the occurrence of recurrent infections reinforces the above notion that mechanisms other than low TREC and KREC levels are responsible for the increased numbers of recurrent infections in AT patients. Interestingly, while KREC levels were not associated with the different therapeutic interventions (IVIG replacement therapy or prophylactic antibiotics) in AT patients, we found that lower TREC levels were significantly associated with these therapeutic interventions. This suggests that lower TREC levels are not necessarily an indicator for these therapeutic interventions but may rather only reflect the low CD3+ cells which are apparently not the cause of recurrent infections in these patients.
We observed steadily low TREC and KREC levels irrespective of the patient’s age. These markers were highly correlated, thus suggestive of combined immunodeficiency. They were also in correlation with CD3+ and CD20+ cell counts, respectively, reflecting mainly reduced production of T and B cells. Since these may be early markers of the disease (8), AT patients might be diagnosed through neonatal screening programs using TREC and KREC measurements. In fact, recent results of exome sequencing and analysis of 2 newborns who screened positively for severe combined immunodeficiency led to the diagnosis of AT (9). Moreover, 7 samples of 13 older AT patients for whom newborn dried blood spots had been stored tested positive for severe immunodeficiency under the criteria of that newborn screening program (9).
Two possible mechanisms can explain the immunological defects in AT patients, abnormal T and B cell development or impaired peripheral activation of T and B cells. In T cells, thymic hypoplasia is characteristic of both human patients and ATM −/− mutant mice (3, 4), pointing to a primary developmental defect. Indeed, there is delayed TCR alpha coding joint formation that results in a reduction of alpha/betaTCR-expressing immature cells and leads to inefficient thymocyte selection in ATM deficiency (4). Moreover, this deficiency disrupts TCR alpha locus integrity and the maturation of CD4 + CD8+ thymocytes (19), and ATM-deficient mouse thymocytes are altered when passing through the β- or γδ-selection checkpoint, suggesting early thymic developmental failure of T cells (20). It has recently been shown that ATM plays an important role in the development of T cells, including the TCR beta rearrangements, generation of the TCR beta CDR3 repertoires and efficient TCR beta-dependent T cell development (21). This is reinforced by the presence of terminally differentiated effector memory cells and normal numbers of circulating memory and effector CD4+ and CD8+ T cells, indicating prior activation as well as dramatic reduction of thymus output content (22, 23). On the other hand, it was found that abnormal intracellular signal transduction leading to reduced responsiveness to antigenic stimulation accounts for the reduced T cell activation in AT patients (23). Our current findings support a T cell developmental defect rather than an activation defect since there were both low TREC levels and reduced immune repertoire diversities in the patients of our cohort. Furthermore, most of our patients displayed normal responses to mitogenic stimulations. Similarly, a normal T cell response to PHA was observed in the 2 young patients who were diagnosed through newborn screening (9). The presence of reduced TRECs and skewed but not clonal diversity of the alpha/beta T cell repertoire, even in babies with AT, further supports that thymic activity is affected in AT patients (7). Thus, our findings of low TREC levels, relatively normal responses to mitogenic stimulations and a skewed TCR repertoire point to a predominant developmental abnormality rather than an abnormal peripheral cell activation and clonal expansion as being responsible for the T cell immunodeficiency in AT patients.
The research on the development of B cells in AT patients has been less extensive despite the known B cell immunodeficiencies. The application of a murine model for investigating B cell function in human ATM deficiency is less valuable given that ATM−/−mice display preserved B cell functions and AT patients do not (24). On the other hand, most AT patients have a restricted IgH repertoire (23) and a marked reduction of sj-KRECs in dried blood spots obtained from AT samples on Guthrie cards (8). AT patients were recently shown to display disturbed naive B-cell homeostasis, as evidenced by low cell numbers, increased proliferation, a large proportion of CD21, low CD38, low anergic B cells, and decreased antigen receptor repertoire diversity (22). A reduction of transitional and naive mature B cell counts is the hallmark of the abnormal naive B cell homeostasis detected in AT patients. These latter studies suggest that a developmental defect plays a critical role in the immunodeficiency of B cells in AT patients, and the results of our current study support this hypothesis. While the detection of unchanging low numbers of B cells and sj-KREC copies, regardless of the patient’s age, together with the abnormal repertoire of the BCRs clearly point to a developmental defect, the low numbers of sj-KRECs could rather be the consequence of peripheral B cell activation and secondary dilution or low bone marrow output. Indeed, the low ratio between sj-KREC and cj-KREC displayed by our patients could reflect both abnormal B cell neogenesis and a high replication history of the B lymphocyte subsets, all of which suggest that B cell activation also plays an important role in the B cell immunodeficiency in AT.
Taken together, our findings revealed disturbed B cell and T cell homeostasis in AT patients. The predominant reduction of T and B cell neogenesis, as reflected by reduced TREC and KREC levels, respectively, was unrelated to the patient’s age. As such, it follows that these abnormalities can be detected by newborn screening assays for T and B cell immunodeficiency. We therefore suggest that a developmental defect rather than activation and proliferation defects in T cells predominate in the immunodeficiency in AT. We also propose that both of those mechanisms in B cells underlie the immunodeficiency in AT.
Abbreviations
- AT:
-
Ataxia telangiectasia
- ATM:
-
AT mutated
- BCR:
-
B-cell receptor
- CJ:
-
Coding joints
- CPM:
-
Counts per minutes
- IGH:
-
Immunoglobulin heavy chain
- IVIG:
-
Intravenous immunoglobulin
- KREC:
-
Kappa-deleting recombination excision circles
- ND:
-
Not done
- PMBC:
-
Peripheral blood mononuclear cell
- RQ-PCR:
-
Real-time quantitative polymerase chain reaction
- SJ:
-
Signal joints
- TCR:
-
T-cell receptor
- TREC:
-
T-cell receptor excision circle
References
Mavrou A, Tsangaris GT, Roma E, Kolialexi A. The ATM gene and ataxia telangiectasia. Anticancer Res. 2008;28(1B):401–5. Epub 2008/04/04.
Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. 2004;144(4):505–11. Epub 2004/04/08.
Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9(10):759–69. Epub 2008/09/25.
Vacchio MS, Olaru A, Livak F, Hodes RJ. ATM deficiency impairs thymocyte maturation because of defective resolution of T cell receptor alpha locus coding end breaks. Proc Natl Acad Sci U S A. 2007;104(15):6323–8. Epub 2007/04/05.
Somech R. T-cell receptor excision circles in primary immunodeficiencies and other T-cell immune disorders. Curr Opin Allergy Clin Immunol. 2011;11(6):517–24. Epub 2011/10/06.
Somech R, Lev A, Simon AJ, Hanna S, Etzioni A. T- and B-cell defects in a novel purine nucleoside phosphorylase mutation. The Journal of allergy and clinical immunology. 2012;130(2):539–42. Epub 2012/05/15.
Micheli R, Pirovano S, Calandra G, Valotti M, Plebani A, Albertini A, et al. Low thymic output and reduced heterogeneity of alpha/beta, but not gamma/delta, T lymphocytes in infants with ataxia-telangiectasia. Neuropediatrics. 2003;34(3):165–7. Epub 2003/08/12.
Borte S, von Dobeln U, Fasth A, Wang N, Janzi M, Winiarski J, et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood. 2012;119(11):2552–5. Epub 2011/12/02.
Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, Tang LF, et al. Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol. 2013;33(3):540–9. Epub 2012/12/25.
Lev A, Simon AJ, Bareket M, Bielorai B, Hutt D, Amariglio N, et al. The kinetics of early T and B cell immune recovery after bone marrow transplantation in RAG-2-deficient SCID patients. PLoS One. 2012;7(1):e30494. Epub 2012/02/02.
Amariglio N, Lev A, Simon A, Rosenthal E, Spirer Z, Efrati O, et al. Molecular assessment of thymus capabilities in the evaluation of T-cell immunodeficiency. Pediatr Res. 2010;67(2):211–6. Epub 2009/10/28.
Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. The Journal of allergy and clinical immunology. 2003;112(5):973–80. Epub 2003/11/12.
Lev A, Simon AJ, Broides A, Levi J, Garty BZ, Rosenthal E, et al. Thymic function in MHC class II-deficient patients. The Journal of allergy and clinical immunology. 2013;131(3):831–9. Epub 2012/12/12.
van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–317. Epub 2003/12/13.
Bott L, Lebreton J, Thumerelle C, Cuvellier J, Deschildre A, Sardet A. Lung disease in ataxia-telangiectasia. Acta Paediatr. 2007;96(7):1021–4. Epub 2007/05/26.
Demuth I, Dutrannoy V, Marques Jr W, Neitzel H, Schindler D, Dimova PS, et al. New mutations in the ATM gene and clinical data of 25 AT patients. Neurogenetics. 2011;12(4):273–82. Epub 2011/10/04.
Berkun Y, Vilozni D, Levi Y, Borik S, Waldman D, Somech R, et al. Reversible airway obstruction in children with ataxia telangiectasia. Pediatr Pulmonol. 2010;45(3):230–5. Epub 2010/02/11.
Schroeder SA, Swift M, Sandoval C, Langston C. Interstitial lung disease in patients with ataxia-telangiectasia. Pediatr Pulmonol. 2005;39(6):537–43. Epub 2005/03/25.
Matei IR, Gladdy RA, Nutter LM, Canty A, Guidos CJ, Danska JS. ATM deficiency disrupts Tcra locus integrity and the maturation of CD4 + CD8+ thymocytes. Blood. 2007;109(5):1887–96. Epub 2006/11/02.
Isoda T, Takagi M, Piao J, Nakagama S, Sato M, Masuda K, et al. Process for immune defect and chromosomal translocation during early thymocyte development lacking ATM. Blood. 2012;120(4):789–99. Epub 2012/06/20.
Hathcock KS, Bowen S, Livak F, Hodes RJ. ATM influences the efficiency of TCRbeta rearrangement, subsequent TCRbeta-dependent T cell development, and generation of the pre-selection TCRbeta CDR3 repertoire. PLoS One. 2013;8(4):e62188. Epub 2013/04/30.
Driessen GJ, Ijspeert H, Weemaes CM, Haraldsson A, Trip M, Warris A, et al. Antibody deficiency in patients with ataxia telangiectasia is caused by disturbed B- and T-cell homeostasis and reduced immune repertoire diversity. The Journal of allergy and clinical immunology. 2013;131 (5):1367–75 e9. Epub 2013/04/10.
Giovannetti A, Mazzetta F, Caprini E, Aiuti A, Marziali M, Pierdominici M, et al. Skewed T-cell receptor repertoire, decreased thymic output, and predominance of terminally differentiated T cells in ataxia telangiectasia. Blood. 2002;100(12):4082–9. Epub 2002/10/24.
Xu Y. ATM in lymphoid development and tumorigenesis. Adv Immunol. 1999;72:179–89. Epub 1999/06/11.
Acknowledgments
The Jeffrey Modell Foundation (JMF), the Legacy Heritage Biomedical Science Partnership Program of the Israel Science Foundation and the Chief Scientist Office of the Ministry of Health for their support of Dr. Somech. Esther Eshkol is thanked for editorial assistance. This work was performed in partial fulfillment of the M.D. thesis requirements of the Sackler Faculty of Medicine, Tel Aviv University (M.K.).
The authors have no financial relationships relevant to this article to disclose.
The authors have no conflicts of interest relevant to this article to disclose.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Matan Kraus and Atar Lev contributed equally
Rights and permissions
About this article

Cite this article
Kraus, M., Lev, A., Simon, A.J. et al. Disturbed B and T cell homeostasis and neogenesis in patients with ataxia telangiectasia. J Clin Immunol 34, 561–572 (2014). https://doi.org/10.1007/s10875-014-0044-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-014-0044-1