
Abstract
We report on a large family in which a novel X-linked recessive mental retardation (XLMR) syndrome comprising macrocephaly and ciliary dysfunction co-segregates with a frameshift mutation in the OFD1 gene. Mutations of OFD1 have been associated with oral–facial–digital type 1 syndrome (OFD1S) that is characterized by X-chromosomal dominant inheritance and lethality in males. In contrast, the carrier females of our family were clinically inconspicuous, and the affected males suffered from severe mental retardation, recurrent respiratory tract infections and macrocephaly. All but one of the affected males died from respiratory problems in infancy; and impaired ciliary motility was confirmed in the index patient by high-speed video microscopy examination of nasal epithelium. This family broadens the phenotypic spectrum of OFD1 mutations in an unexpected way and sheds light on the complexity of the underlying disease mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
X-linked mental retardation (XLMR) is a clinically and genetically heterogeneous condition that affects approximately 1.8 in 1,000 males (Herbst and Miller 1980). To date, more than 140 different syndromic forms of XLMR have been described. The underlying genetic defect has been identified in approximately half of these cases, and 20 different genes have been implicated in non-syndromic XLMR (Ropers and Hamel 2005).
Oral–facial–digital type 1 syndrome (OFD1S, OMIM 311200) is an X-linked dominant, male-lethal disorder characterized by craniofacial malformations, digital abnormalities, mental retardation and polycystic kidney disease. OFD1S is caused by mutations of the OFD1 gene (formerly known as CXORF5) (Ferrante et al. 2001), the gene product of which is required for the formation of primary cilia (Ferrante et al. 2005).
Ciliary dysfunction has been associated, among many other disorders, with mental retardation (Gerdes and Katsanis 2005). Motile cilia, which are characterized by a 9 + 2 microtubule component, are found in respiratory epithelial cells, in oviducts, in testicular ducts and sperm cells, in the ependymal lining of brain ventricles, and in the embryonic node. Dysfunction of these cilia, as in primary ciliary dyskinesia, leads to respiratory problems, infertility, hydrocephalus and randomization of left/right body asymmetry (Eley et al. 2005; Van’s Gravesande and Omran 2005). Primary (non-motile) cilia, on the other hand, have a 9 + 0 microtubule composition. They are found, with few exceptions such as lymphocytes, on nearly all cell types of the body. Elucidation of their functions has attracted increased attention since causative links were established to cystic kidney disease (Hildebrandt and Otto 2005), retinitis pigmentosa (Khanna et al. 2005), sensorineural deafness (Whitlon 2004), anosmia (Kulaga et al. 2004) and mental retardation (Gerdes and Katsanis 2005).
Here, we report on a family with a novel syndrome comprising X-linked recessive mental retardation, ciliary dyskinesia and macrocephaly, and on the detection of an OFD1 frameshift mutation in the affected males and obligate carrier females of this family.
Subjects and methods
Patients
Nine males in a large Polish family with apparent X-linked recessive inheritance suffered from developmental delay, respiratory problems and macrocephaly (Fig. 1).

Pedigree of the family and haplotype data from the region Xp22.32-Xp21.3. Affected males are denoted by black squares, carrier females by dots within empty circles. Arrows indicate recombination events encompassing the linkage interval between flanking markers DXS1060 and DXS1214
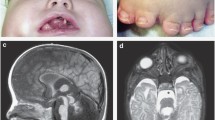
The index patient (IV: 1) was an 11-year-old son of healthy, non-consanguineous parents. He was born at term (40th week) after an uncomplicated pregnancy with a birth weight of 3,850 g (∼50th percentile), body length of 51 cm (∼50th percentile), head circumference of 36 cm (> 90th percentile), and chest circumference of 35 cm (50th percentile). APGAR score was 3–6. The boy suffered from recurrent upper and lower airway infections, which necessitated frequent hospital treatments during the first 5 years. At the age of eleven, macrocephaly was present [head circumference 62 cm (≫ 97th percentile)], but no hydrocephalus or other brain malformation was detected by CT and MRI brain scans. He was severely mentally retarded (IQ 20). Dysmorphic features included high-arched palate, low-set ears, broad thumbs, short fingers and obesity (Fig. 2). Penis and testicles were of normal size. Ultrasound examination of the kidneys and routine metabolic tests showed no abnormalities.

Photographs of the index patient (IV: 1) at the age of 11 years. Note large head, obesity and short fingers and toes
The younger brother (IV: 3) of the index patient was born in the 38th week of pregnancy by Caesarean section with a birth weight of 4,120 g (> 97th percentile), body length of 54 cm (> 97th percentile), head circumference of 39 cm (≫ 97th percentile), and chest circumference of 34 cm (50th percentile). APGAR score was 5–7–9–9. He had unilateral post-axial hexadactyly on the right hand. Postpartum he suffered from respiratory distress syndrome and recurrent respiratory tract infections. Ultrasound examination of the head and abdomen showed no abnormalities. His development was very poor and he died at the age of 18 months.
Another male in this family (IV: 11) was born in the 35th week of pregnancy by Caesarean section with a birth weight of 1,915 g (∼10th percentile), head circumference of 36 cm (> 90th percentile), and body length of 46 cm (3rd percentile). APGAR score was 4–7. Neurological examination revealed psychomotor retardation and decreased muscle tone at that time. Ultrasound examination of the head showed ventricular dilatation, but this did not necessitate surgical intervention. He had bilateral inguinal hernias. Apart from short fingers, no other dysmorphic signs were reported. He died at the age of three years as a result of pulmonary insufficiency.
The other affected males (II: 3, III: 9, III: 10, III: 11, III: 12) also suffered from chronic respiratory tract infections according to the reports of the family, and at least some of them were said to have had large head circumferences. The carrier females II: 2, II: 5, II: 7, III: 2, III: 4, and III: 8 were examined clinically, by abdominal ultrasound and urine and blood tests. No cognitive, oral, facial, digital or renal abnormalities were detected.
Routine chromosome analyses in two affected males (IV: 1 and IV: 11, Fig. 1) revealed normal male karyotypes; and trinucleotide repeat expansion in the FMR1 gene and CFTR gene mutations were excluded in IV: 1.
Genotyping and linkage analysis
Blood samples were collected from 17 family members after informed consent. Genomic DNA was extracted from venous blood according to standard procedures (Sambrook et al. 1989). For linkage mapping, 68 X-chromosomal single nucleotide polymorphism (SNP) markers were employed (SNP Assays on Demand, Applera, Norwalk, USA.). SNP typing was performed according to the manufacturer’s protocol.
Two- and multi-point linkage analyses were performed using the MLINK and LINKMAP programs, respectively, both from the LINKAGE package, version 5.2 (Lathrop and Lalouel 1984). Allele frequencies of individual SNPs in the Caucasian population were taken from the Celera database.
Comparative genomics analysis
The ciliary phenotype in this family prompted us to search for cilia-specific genes in the linkage interval by a comparative genomics approach similar to a method described previously (Avidor-Reiss et al. 2004; Li et al. 2004). We looked for orthologues of these genes in the genomes of Chlamydomonas reinhardtii, a ciliated organism, and Arabidopsis thaliana, a non-ciliated organism, by BLASTP alignment (E ≤ 10−10).
DHPLC screening and sequencing
Mutation screening of functional and positional candidate genes was performed by denaturing high performance liquid chromatography (DHPLC with the WAVE® system, Transgenomic, Omaha, USA) and subsequent sequencing in the male index patient, in obligate heterozygotes, and in a panel of 167 normal male controls. For all selected genes, coding sequences, together with approximately 50 nucleotides from neighboring intronic regions, were amplified from genomic DNA in independent PCR reactions (conditions available on request). Primers were designed using the Primer3 program (http://www.genome.wi.mit.edu/genome_software/other/primer3.html/). Sequencing reactions were performed in both directions using the BigDye Terminator kit (PE Biosystems, Rockville, USA) and subsequently visualized using a 3730xl DNA Analyzer (Applied Biosystems, Rockville, USA).
An X chromosome inactivation analysis was performed at the androgen receptor (AR) locus as described previously (Allen et al. 1992).
RNA isolation and northern blot hybridization
RNA was isolated from lymphoblastoid cell lines using TRIzol (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s protocol. For each sample, poly-A+ RNA was obtained from 100 μg of total RNA using Dynabeads (Invitrogen, Karlsruhe, Germany). Samples were separated in a formaldehyde-containing gel in 1 × MOPS buffer [3-(N-morpholinol) propanesulfonic acid, EDTA, pH 7.0], transferred to a Hybond N+ membrane, and cross-linked by UV. cDNA probes for hybridization were generated by reverse transcription of 1 μg of total RNA, using Superscript III reverse transcriptase (Life Technologies BRL, Rockville, USA) in the presence of 20 units of RNAse inhibitor (Promega, Mannheim, Germany) and 25 nM dNTPs. The reaction was performed at 50°C for one hour and was terminated by a 15 min incubation at 70°C. The cDNA was used as a template for synthesis of the probe as follows: 2 μl of cDNA was amplified using Long Template Enzyme (Roche, Mannheim, Germany) and Premix J (Epicentre, Madison, USA) under conditions for long PCR products. To amplify OFD1 transcripts, the following primers were used: 5′-GAC AGA GGC AGG GTT CTG AG-3′ and 5′-TTA GAA GCT GGG CAG CAA AC-3′. Fragments of 3.2 kb and 3.8 kb, which represent the two isoforms of the OFD1 gene, were generated. Products were separated in a 1% agarose gel, purified with the QIAquick gel extraction kit (Qiagen, Hilden, Germany) and verified by sequence analysis. Subsequently, the 3.2 kb fragment was labeled with 32[P]dCTP and hybridized to the northern blot in ULTRAhyb buffer (Ambion, Austin, USA). The blot was washed in accordance with the manufacturer’s protocol and was exposed to Fuji medical X-ray films at −80°C for 24 h using an intensifying screen. Autoradiograms were scanned using an Epson Expression 1680 Pro scanner.
Evaluation of ciliary motility
Since the respiratory tract infections of the affected males were suggestive of primary ciliary dyskinesia, we studied the cilia function in the index patient (IV: 1, Fig. 1). Respiratory epithelial cells were obtained by trans-nasal brushings (Cytobrush Plus, Medscand, Malmö, Sweden). Cells were immediately suspended in cell culture medium (Gibco, Karlsruhe, Germany) and spread onto glass slides. The biopsy material was then viewed with a Zeiss inverted phase contrast microscope equipped with a Redlake ES-310 Turbo monochrome high-speed video camera (Redlake, San Diego, USA) set at 125 frames per second. Ciliary beat frequency was assessed with the SAVA system (Sisson-Ammons Video Analysis) (Sisson et al. 2003). Ciliary movements were evaluated on slow motion playbacks as described previously (Fliegauf et al. 2005).
Results
The genetic defect maps to Xp22.32-Xp21.2
The highest LOD score in two-point analysis was obtained for DXS8019 (Z max = 2.57 at θ = 0.00) at Xp22.13. Ten adjacent markers also showed positive LOD scores. The linkage interval spanned 26 cM (25 Mb) and was flanked by markers DXS1214 and DXS1060. Upon multi-point analysis, significant evidence for linkage was found across the entire Xp22.32-Xp21.2 region, with a maximum LOD score of 2.99 at DXS8019. Haplotype analysis confirmed these findings (Fig. 1).
A frameshift mutation in the OFD1 gene co-segregates with the disorder
In-silico analysis of the 25 Mb linkage interval in Xp22 revealed 164 known protein-coding genes. By comparative genome analysis we identified four genes (NLGN4X, HCCS, MOSPD2, OFD1) with orthologues in Chlamydomonas but not in Arabidopsis, and we performed mutation analysis in these genes.
We detected a duplication of four nucleotides (2122-2125dupAAGA) in exon 16 of the OFD1 gene (GenBank accession number NM_003611) in DNA from the index patient (IV: 1), his affected cousin (IV: 11) and all obligate carrier females (II: 2, II: 5, II: 7, III: 2, III: 4, III: 8) (Fig. 3). This change was not found in 168 healthy male controls.

Sequence chromatogram showing the mutation (2,122–2,125dupAAGA) in the index patient (a), and the wild-type sequence in a normal male control (b). c Predicted amino acid sequence of the patient shows a premature stop codon (TGA, asterisk)
The OFD1 gene comprises 34,590 bp and has two main splice variants: OFD1a (Cxorf5-1, GenBank Y16355, 3,615 bp), and OFD1b (Cxorf5-2, GenBank Y15164, 4,278 bp) (de Conciliis et al. 1998). The 2,122–2,125dupAAGA mutation is expected to introduce a premature stop codon in the open reading frame of the OFD1a transcript (Fig. 4).

a Exon–intron structure of the two main splice variants of OFD1: OFD1a (3,615 bp) and OFD1b (4,278 bp) (not to scale). Untranslated regions are indicated by diagonal stripes. b Structure of the corresponding OFD1 isoforms. The position of the 2,122-2,125dupAAGA mutation is indicated by arrows
X chromosome inactivation analysis revealed absence of skewing in the informative carrier females III: 2, III: 4 and III: 8 (data not shown).
Expression analysis of OFD1 in the index patient
Northern blot analysis using a 3,185 bp cDNA fragment complementary to the OFD1a transcript as a probe revealed two bands representing the two OFD1 transcripts in controls. A decrease in expression of OFD1a (which harbors a premature termination codon in its open reading frame) in comparison to OFD1b was detected in the patient (Fig. 5).

Results of northern blot analysis. In the index patient (P), the relative amount of OFD1a transcript (3.6 kb) is reduced compared to the OFD1b transcript (4.3 kb). C1–C4 = RNA from four male controls
Respiratory cilia of the index patient exhibit a dyskinetic beating pattern
Functional evaluation of respiratory cilia using high-speed video analysis revealed a severely disorganized ciliary beating pattern with lack of coordination (see supplementary videos online), while the ciliary beat frequency was not reduced. The observation of dyskinetic cilia by direct light microscopy confirmed the diagnosis of primary ciliary dyskinesia in this family.
Discussion
In the course of a long-term project to elucidate gene defects in families with X-linked mental retardation, we mapped a family with a novel XLMR syndrome comprising ciliary dyskinesia and macrocephaly to Xp22.13, and identified a novel mutation in OFD1.
To our knowledge, only one family with a similar X-linked phenotype has been described before. Brzustowicz et al. (1999) reported a family in which the affected males suffered from macrocephaly, neurological impairment, and gastrointestinal and genitourinary malfunction; and all died from pneumonia and sepsis within the first 8 weeks of life (Brzustowicz et al. 1999). Linkage analysis in this family revealed a locus in Xp22 that includes the OFD1 gene. Considering the phenotypic similarity to our family (macrocephaly and respiratory problems), we speculate that an OFD1 mutation might be the underlying defect also in this family.
The presence of an OFD1 mutation in the family we report here is remarkable because alterations of this gene have so far been associated exclusively with the X-linked dominant oral–facial–digital type 1 syndrome (Ferrante et al. 2001). Affected individuals in this family bear no phenotypic resemblance to those with OFD1S, and, also importantly, the disease exhibits an X-linked recessive mode of inheritance in this family.
Explanations for the strikingly different phenotypic consequences between this mutation and previously reported OFD1 mutations remain speculative at present. The mutation in our family exclusively affects the open reading frame of OFD1a, but several other mutations have been reported that likewise spare the open reading frame of OFD1b in female patients with a classical OFD1S phenotype (Ferrante et al. 2001; Rakkolainen et al. 2002; Thauvin-Robinet et al. 2006). In the index patient of our family, we detected a reduction in the expression of OFD1a compared to that of OFD1b, which is probably due to nonsense-mediated decay of the OFD1a transcript. It is, however, difficult to assess the relevance of this finding as northern blot investigations were not reported in studies on other OFD1 mutations. Furthermore, we cannot rule out the existence of additional transcripts in other tissues, which might be affected differently by specific mutations.
The human OFD1 protein(s) and their mouse orthologue Ofd1 are localized at the centrosome and basal body of primary cilia (Ferrante et al. 2005; Romio et al. 2004). Absence of cilia at the embryonic node was observed in male Ofd1 knock-out mice; also, epithelial cells of kidney cysts in heterozygous female Ofd1 knock-outs lacked cilia (Ferrante et al. 2005). These findings indicate that Ofd1 is required for cilia assembly, probably by participation in intraflagellar transport (Ferrante et al. 2005). However, there were no reports of abnormalities in the motile cilia of respiratory epithelial cells in OFD1 syndrome patients or in Ofd1 knock-out mice. The finding of a severely disorganized beating pattern of respiratory cilia in our index patient supports the role of OFD1 in ciliary function. Bearing in mind that OFD1 escapes X-inactivation in humans, the absence of respiratory problems in OFD1S females might be explicable by the presence of one functional copy of OFD1 (de Conciliis et al. 1998).
Severe mental retardation is the most prominent feature in the affected males of our family. We speculate that this might be caused by impaired function of primary cilia, which are found also on neurons (Fuchs and Schwark 2004). The function of these neuronal cilia is still largely elusive, but they almost certainly act as sensory organs of the cells, and they might play a role in cell proliferation and differentiation (Fuchs and Schwark 2004; Hildebrandt and Otto 2005; Hirokawa and Takemura 2004). Mental retardation is also a prominent feature in other ciliary disorders, e.g. Bardet–Biedl syndrome (OMIM 209901, 606151, 600151, 600374, 603650, 604896, 607590, 608132), Joubert syndrome (OMIM 608629, 608894), Alstrom syndrome (OMIM 203800), and in approximately 40% of females with OFD type 1 syndrome (Odent et al. 1998).
In summary, the family reported here presents with a novel XLMR syndrome that is allelic to OFD type 1 syndrome. This observation broadens the spectrum of OFD1-related disorders considerably, and, as a practical consequence, OFD1 mutation analysis should no longer be restricted to female patients with classical OFD1S phenotypes. Further investigations are needed to elucidate the mechanisms leading to such diverse phenotypic consequences of OFD1 mutations.
References
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW (1992) Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 51: 1229–1239
Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS (2004) Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117: 527–539
Brzustowicz LM, Farrell S, Khan MB, Weksberg R (1999) Mapping of a new SGBS locus to chromosome Xp22 in a family with a severe form of Simpson-Golabi–Behmel syndrome. Am J Hum Genet 65: 779–783
de Conciliis L, Marchitiello A, Wapenaar MC, Borsani G, Giglio S, Mariani M, Consalez GG, Zuffardi O, Franco B, Ballabio A, Banfi S (1998) Characterization of Cxorf5 (71–7A), a novel human cDNA mapping to Xp22 and encoding a protein containing coiled-coil alpha-helical domains. Genomics 51: 243–250
Eley L, Yates LM, Goodship JA (2005) Cilia and disease. Curr Opin Genet Dev 15: 308–314
Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, Sylvie O, Bernard L, Malcolm S, Winter R, Ballabio A, Franco B (2001) Identification of the gene for oral–facial–digital type I syndrome. Am J Hum Genet 68: 569–576
Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dolle P, Franco B (2005) Oral–facial–digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet 38(1):112–117
Fliegauf M, Olbrich H, Horvath J, Wildhaber JH, Zariwala MA, Kennedy M, Knowles MR, Omran H (2005) Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am J Respir Crit Care Med 171: 1343–1349
Fuchs JL, Schwark HD (2004) Neuronal primary cilia: a review. Cell Biol Int 28: 111–118
Gerdes JM, Katsanis N (2005) Microtubule transport defects in neurological and ciliary disease. Cell Mol Life Sci 62: 1556–1570
Herbst DS, Miller JR (1980) Nonspecific X-linked mental retardation II: the frequency in British Columbia. Am J Med Genet 7: 461–469
Hildebrandt F, Otto E (2005) Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet 6: 928–940
Hirokawa N, Takemura R (2004) Molecular motors in neuronal development, intracellular transport and diseases. Curr Opin Neurobiol 14: 564–573
Khanna H, Hurd TW, Lillo C, Shu X, Parapuram SK, He S, Akimoto M, Wright AF, Margolis B, Williams DS, Swaroop A (2005) RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J Biol Chem 280: 33580–33587
Kulaga HM, Leitch CC, Eichers ER, Badano JL, Lesemann A, Hoskins BE, Lupski JR, Beales PL, Reed RR, Katsanis N (2004) Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet 36: 994–998
Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36: 460–465
Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, Li H, Blacque OE, Li L, Leitch CC, Lewis RA, Green JS, Parfrey PS, Leroux MR, Davidson WS, Beales PL, Guay-Woodford LM, Yoder BK, Stormo GD, Katsanis N, Dutcher SK (2004) Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 117: 541–552
Odent S, Le Marec B, Toutain A, David A, Vigneron J, Treguier C, Jouan H, Milon J, Fryns JP, Verloes A (1998) Central nervous system malformations and early end-stage renal disease in oro-facio-digital syndrome type I: a review. Am J Med Genet 75: 389–394
Rakkolainen A, Ala-Mello S, Kristo P, Orpana A, Jarvela I (2002) Four novel mutations in the OFD1 (Cxorf5) gene in Finnish patients with oral–facial–digital syndrome 1. J Med Genet 39: 292–296
Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA (2004) OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol 15: 2556–2568
Ropers HH, Hamel BC (2005) X-linked mental retardation. Nat Rev Genet 6: 46–57
Sambrook J MT, Fritsch EF (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Sisson JH, Stoner JA, Ammons BA, Wyatt TA (2003) All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc 211: 103–111
Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, Goldenberg A, Mortier G, Heron D, Sagot P, Bouvier AM, Huet F, Cusin V, Donzel A, Devys D, Teyssier JR, Faivre L (2006) Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral–facial–digital syndrome type 1: a French and Belgian collaborative study. J Med Genet 43: 54–61
Van’s Gravesande KS, Omran H (2005) Primary ciliary dyskinesia: clinical presentation, diagnosis and genetics. Ann Med 37: 439–449
Whitlon DS (2004) Cochlear development: hair cells don their wigs and get wired. Curr Opin Otolaryngol Head Neck Surg 12: 449–454
Acknowledgments
We would like to thank the patients and their families for their participation in the studies. This work was supported by the EU (grant no. QLRT-2001-01810) and by a ‘Partner Group’ grant from the Max Planck Society (to H.H.R and A.L.-B.). H.O. and M.F. were supported by grants from the Deutsche Forschungsgemeinschaft (DFG Om 6/2; SFB592).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Budny, B., Chen, W., Omran, H. et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral–facial–digital type I syndrome . Hum Genet 120, 171–178 (2006). https://doi.org/10.1007/s00439-006-0210-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-006-0210-5