
Abstracts
Growth failure is a hallmark in children suffering from chronic kidney disease (CKD). It is most pronounced in children with congenital CKD requiring dialysis treatment. Optimal conservative management including adequate nutrition and growth hormone treatment, as well as early (preemptive) transplantation, is mandatory in order to reduce height deficit before kidney transplantation. After transplantation the use of efficacious immunosuppressive strategies for an optimized graft function and early withdrawal, or even complete avoidance, of steroids are required to improve final height.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Growth failure
- Chronic kidney disease
- Nutrition
- Puberty
- Growth hormone
- Transplantation
- Dialysis
- Immunosuppressive treatment
Introduction
Maintaining optimum growth is one of the most challenging problems in the management of children with chronic kidney disease (CKD): reports published in 2013 showed that approximately 50% of children requiring kidney replacement therapy (KRT) before their 13th birthday had a final height below the normal range [1,2,3,4]. Short stature is a marker for increased mortality and hampers the psychosocial integration of pediatric CKD patients [5,6,7]. There is, however, evidence that, alongside advancements in the medical and technical management of CKD and kidney replacement therapy (KRT), height prognosis has substantially improved over the past decades [1, 2, 4, 8,9,10,11]. In 799 pre-dialysis children with mostly mild to moderate CKD (median age 11 years, estimated glomerular filtration rate (eGFR) 50 mL/min/1.73 m2) followed in the Chronic Kidney Disease in Children (CKiD) Study, the median height standard deviation score (SDS) was −0.55. Among 594 patients from 12 European countries with CKD stage 3–5 (median eGFR 30 mL/min/1.73 m2) who were followed prospectively in the 4C Study, mean height SDS was −1.57, with 36% of patients having a height below the third percentile. In 1001 children in the International Pediatric Peritoneal Dialysis Network (IPPN), the mean height SDS at initiation of dialysis was −1.97 [10]. Hence, these data from contemporaneous large cohort studies demonstrate that growth failure progressively occurs with decreasing eGFR. In the 2020 USRDS report the percentage of incident end-stage kidney disease (ESKD) patients with short stature was lowest in the oldest patients age groups, being 20.3% at 18–19.9 years, 25.8% at 14–17, 20.1% at 10–13, 38.8% at 5–9 and 51.9% at 0–4 years of age [11, 12].
There appears to be a positive trend over time: e.g. in Germany, the mean standardized height in children on KRT has increased over the past 20 years from −3.0 SDS to −1.8 SDS [1]. Yet this is not the case in all parts of the world, particularly in those with inadequate local resources, where height prognosis remains dismally low [2, 13]. Even across Europe, the 4C Study observed growth failure to vary widely between countries, from 7% to 44% [9].
There is no single cause of failing growth in children with CKD (Fig. 56.1). The two most important influences are the severity of CKD and young age at its onset, so that children with CKD due to congenital kidney abnormalities manifesting in infancy are usually more severely affected than those with acquired conditions developing in later childhood. This review summarizes the current knowledge of the phenotype, pathophysiology and therapeutic options for children with failing growth resulting from CKD.

The etiology of growth failure in CKD is multifactorial and includes intrauterine growth restriction (IUGR), genetic factors such as parental height and primary renal disease, prematurity, and malnutrition which especially limits growth in children with congenital CKD. Mineral and bone disorder (CKD-MBD), metabolic acidosis, anemia, loss of electrolytes and water, and disturbances of the somatotropic and gonadotropic hormone axes are additional factors. CKD is a state of growth hormone (GH) insensitivity, characterized by deficiency of functional insulin-like growth factor I (IGF-I), to reduced GH receptor expression in target organs like the liver, disturbed GH receptor signaling via the Janus kinase - signal transducers, activators of transcription (JAK2-STAT5) due to inflammation induced SOCS (suppressor of cytokine signalling), and increased IGF-binding capacity due to excess of IGFBPs. Finally, reduced release of hypothalamic gonadotropin-releasing hormone (GnRH), due to uremia-related inhibitory factors such as angiotensin II (AngII), and steroid treatment may result in decreased circulating levels of bioactive luteinizing hormone (LH), hypogonadism and reduced pubertal growth spurt. PTH, parathyroid hormone; FSH, follicle stimulating hormone; IGFBP, insulin-like growth factor binding proteins. [Reproduced with permission of [30])
Normal Growth
The physiological growth pattern can be divided into fetal, infantile, mid-childhood and pubertal phases (Fig. 56.2) [14]. During fetal life, nearly 30% of final height has already been achieved, so low birth weight and prematurity can substantially influence subsequent growth and final height attainment: although many otherwise normal infants born prematurely grow normally, and those who are small for gestational age (SGA) catch up in the first 6 months of life, around 10%, particularly if SGA, remain below the normal range for height into adulthood. A further one third to one half of total postnatal growth occurs during the first 2 years of life, and 20% during the pubertal phase. Throughout each postnatal phase the predominant influences on growth are different. Whereas in infancy, growth mainly depends on nutritional intake, growth in mid-childhood is driven by the somatotropic hormone axis. During puberty, the gonadotropic hormone axis stimulates growth via increased proliferation of growth plate chondrocytes and modulation of growth hormone (GH) secretion from the pituitary gland, resulting in the pubertal growth spurt (Fig. 56.2) [14].

Typical growth pattern in congenital chronic kidney disease. Relative loss in the nutrient-dependent infantile and gonadal hormone-dependent puberty phases, and percentile-parallel growth in the mainly GH-dependent growth period in mid-childhood are shown. The shaded area represents the normal range, third to 97th centiles. (Reproduced with permission of [14])
Effect of CKD on the Phases of Growth
The classical growth pattern for a child born with CKD was described in 1974 [15]. Length at birth is usually already below the mean (Fig. 56.2) [16]. As with any chronic disease, height velocity is most affected during periods of rapid growth. Marked growth retardation occurs during the first 2 years of life, followed by percentile parallel growth in mid-childhood, but catch-up growth is unusual. In the pre-pubertal years, the appearance of secondary sexual characteristics is delayed and the growth rate again decreases disproportionately [17, 18]. The pubertal growth spurt is later than normal and its magnitude impaired, resulting in further loss of growth potential and reduced final height (Fig. 56.2) [14]. Over the last 20 years, although these basic principles remain, this classic description has been reassessed as there have been new concepts and treatments for most patients in all postnatal phases of growth.
The Fetal Phase
Reduced fetal growth was described as a feature of the classic growth pattern of the child with CKD in 1994, and has been demonstrated in several studies since [16, 19,20,21,22]. Both prematurity and low birth weights are common. The incidence is particularly high in infants on dialysis but perhaps more surprisingly is high in children with less severe CKD as well [23]. Registry data do not always distinguish between infants who do or do not have co morbidities (such infants often have below normal mean birth weight and length) but it has been shown that of just over 400 children with a mean eGFR of around 40 mL/min/1.73 m2 in the US CKD registry, low birth weight (LBW, <2500 g) occurred in 17%, prematurity (gestational age < 36 weeks) in 12% and small for gestational age (SGA, birth weight < tenth percentile for gestational age) in 14%. It has been hypothesized that poor intrauterine growth conditions, e.g. maternal malnutrition and smoking, could cause both intrauterine growth retardation and kidney dysplasia [24, 25]. Likewise genetic abnormalities, e.g. dysregulation of Wilms’ tumor suppressor gene, could cause both short stature and renal hypodysplasia [26]. Interestingly, in the US CKD registry 40% of patients had needed intensive care (ICU) at birth. The comparable overall incidence of abnormal birth history in the US population is 7–8%. Low birth weight, prematurity, SGA and requirement for ICU were all risk factors for poor growth outcomes, independent of renal function [21]. Likewise, intrauterine growth retardation, neonatal distress and parental height were shown to be important independent predictors of poor growth outcome in a cohort of 509 German children with CKD stage 3–5 [27].
The Infantile Phase
It is not surprising that adverse effects on growth are most intense during the infantile phase, and in particular the first 6 months of life, as the rate of growth is as high as 25 cm per year at birth, 18 cm per year at 6 months of age, and still 12 cm per year at 12 months of age. These figures are higher than at any other time during childhood and adolescence. Such growth challenges require nutrient intakes that are relatively higher than at any other age. As the infantile phase is predominantly dependent on nutrition, inadequate intake at this time can have a dramatic influence on growth. Indeed, any circumstances leading to decreased growth rates in this phase result in severe growth retardation and a potentially irreversible loss of growth potential [28, 29]. The decrease in mean standardized height can amount to 0.6 SD per month in infants with CKD stage 5. In recent years the increasing acceptance of KRT for all infants, including those with associated comorbidities, has increased the challenge to achieve normal growth in this age group [20]. Malnutrition in young children with CKD is due to inadequate nutritional intake and frequently recurrent vomiting as well. In addition, catabolic episodes due to infections, loss of water and electrolytes, and CKD-mineral and bone disorder (CKD-MBD) are major contributing factors to growth impairment in this period. If these disturbances are adequately controlled, severe stunting can be avoided in the majority of patients without untreatable comorbidities [19, 20, 30]. However, most infants suffering from severe CKD need supplementary feeding in order to provide adequate nutrient, water and electrolyte intake [31].
The Childhood Phase
During this phase, the somatotropic hormone axis becomes the most important influence on growth. Growth is closely correlated with the degree of kidney dysfunction in this period. Although there is no critical threshold, growth patterns are typically stable if the GFR remains above 25 mL/min/1.73 m2 and tend to diverge from the percentiles below this level [23]. Sequels of CKD such as anemia, metabolic acidosis and malnutrition seem to be less important in mid-childhood. However, even a growth rate that parallels the centiles may not be ‘normal’, as children with good renal function and steroid-free immunosuppression following transplantation exhibit significant catch-up, compatible with the concept of continued suppression of an intrinsic catch-up growth potential in the uremic state [32, 33].
The Pubertal Phase
Pubertal Development
Delayed onset and progression of pubertal development was a common feature when KRT programs for children started [17]. Studies of the timing of pubertal onset have been hampered by the fact that bone age is only a crude marker for assessment in CKD. Indeed, the distribution of bone age at pubertal onset varies at least as much as the distribution of chronological age in these patients. However, data from the late eighties demonstrated a delay of pubertal onset by 2–2.5 years in children with ESKD [17]. Menarche occurred after the upper limit of the normal age range (i.e. 15 years) in almost half of the girls treated by dialysis or transplantation [34]. Moreover, despite the achievement of pubertal stage IV or V, a substantial proportion of dialysis patients presented with permanently impaired reproductive function [35]. Fortunately, in the last twenty years most children requiring KRT before pubertal age present with normal or only slightly delayed pubertal onset. In two recent studies, mean age at pubertal onset as well as age at menarche did not differ between children on KRT and healthy children; and the serum levels of pubertal reproductive hormones were normal in the great majority of patients [1, 36]. Bone maturation in patients on KRT continues to be delayed by approximately 1.4 years compared to healthy children, although this does not negatively impact on pubertal development [1]. The age at onset of puberty has been positively correlated with the age at transplantation. Thus, early renal transplantation is a prerequisite for prevention of pubertal delay in children with stage 5 CKD [18, 36]. A recent analysis of the CKiD cohort including children with CKD stage 3–5 revealed delayed menarche in 10% of adolescent patients which was associated with African American race, lower eGFR, ever corticosteroid use, and longer duration of CKD [18, 37]. Delayed menarche was strongly associated with reduced height (<−2.0 SDS). Thus, delayed puberty is an important contributor to short stature in female patients even prior to dialysis. Patients who show delayed puberty—defined in boys by a testicular volume < 4 mL at the age of 14 years and in girls by a breast stage <B2 at age 13.5 years—should undergo work up by a pediatric endocrinologist including potential induction of puberty [30].
Pubertal Growth
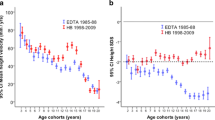
During the last two decades in parallel with the improvement in sexual maturation has been an improvement in pubertal height gain [1, 2, 17, 18]. Longitudinal growth in 384 German children on KRT who were followed between 1998 and 2009 was compared with 732 children enrolled in the European Dialysis and Transplant Association (EDTA) Registry between 1985 and 1988 (Fig. 56.3) [1]. In line with previous studies, the pubertal growth spurt in the EDTA patients was delayed by approximately 2.5 years. In many patients no clear pubertal growth spurt was present, and consequently standardized height decreased during pubertal age. In contrast, a clear pubertal growth spurt was present and the onset of the pubertal growth spurt was within the normal range in the majority of the patients followed-up more recently. Consequently, standardized height even improved during puberty and until adult height. A strong negative correlation between total pubertal height gain and age at transplantation was reported in two studies a [18, 36]. Thus, whereas 20 years ago a loss of about 1.0 SD was expected during puberty, nowadays normal or only slightly reduced pubertal growth spurt can be expected if long-term dialysis is avoided.

(a) Mean height velocity of European children with renal replacement therapy in the EDTA study 1985–1988 (blue lines) versus the Hannover/Berlin (H/B) pediatric population cohort 1998–2009 (red lines) in different age cohorts. (b) Age-dependent height standard deviation score of European children on kidney replacement therapy 1985–1988 (EDTA study, n = 732, blue error bars) and in the HB group (n = 384, red error bars). EDTA European Dialysis and Transplant Association, CI Confidence interval. Reproduced with permission of [1]
Segmental Growth
It has been postulated that during malnutrition there is preferential preservation of growth of vital organs at the expense of less vital tissues such as the limbs, so that malnutrition during childhood results in disproportionate stunting with impairment of leg growth, and preserved trunk and head growth [38]. Consequently, relative leg length is increasingly used as a biomarker of childhood nutrition in epidemiological studies [39, 40]. Information pertaining to segmental growth has been collected in the CKD Growth and Development Study, in which more than 800 pediatric CKD patients before and after transplantation have been enrolled since 1998 [41,42,43]. Patients with a long-term history of CKD and KRT showed an age related disproportionate growth pattern [41,42,43]. Growth impairment and disproportionality was most obvious in early childhood. Sitting height was mostly preserved, whereas growth of the legs and arms was most severely affected. This resulted in a markedly elevated sitting height index (ratio of sitting height to total body height). Leg length was more affected in prepubertal patients. Consequently, body disproportion was less pronounced in pubertal patients. In addition to transplant function and steroid exposure, congenital CKD, smallness for gestational age, young age, and use of recombinant human growth hormone (rhGH) in the pre-transplant period were significantly associated with growth outcome (stature and degree of body disproportion. Catch-up growth after kidney transplantation is mainly related to improved trunk growth in children aged less than 4 years and stimulated leg growth in older children resulting in complete normalization of body proportions until attainment of adult height in the vast majority of patients [42, 44].
Adult Height and Height Prediction
When interpreting the adult heights of patients treated for CKD in childhood, it has to be remembered that the data obtained at any time will reflect treatment practices spanning the previous two decades. Furthermore, most reports of adult heights do not or incompletely discriminate according to patient characteristics (e.g. diagnoses, ages of onset of CKD, types and durations of KRT), and in particular registries do not separate out children with co morbidities that affect growth in their own right. With that in mind, reduced adult heights have been reported in up to 50% of CKD patients, although there has been a trend for improvement over the past decade [1,2,3,4, 17, 18, 45]. Mean adult heights vary from 148–158 cm for females and 162–168 cm for males (second centiles 151 and 163 cm respectively).
The ESPN/EDTA registry has shown that, after adjustment for age and period of start of KRT, final height increased significantly from −1.93 SDS in children who started KRT before 1990, to −1.78 in children in 1990–1999, and to −1.61 in those starting KRT after 1999 (p < 0.001). While 55% of patients attained an adult height within the normal range between 1990 and 19,995, this figure had risen to 62% between 2006–2011 [2]. The improvement in adult height over time was independent of age at the start of KRT (Fig. 56.4). Poorest growth outcomes were associated with earlier start and longer duration of dialysis or a diagnosis of a metabolic disorder such as cystinosis and hyperoxaluria; whereas those with a longer time spent with a renal transplant and those treated with rhGH did the best [2,3,4, 46,47,48,49].

Changes in final height SDS over time according to age and period of start of KRT (n = 981). The horizontal line in the middle of the box represents the median, the bottom and top of the box represent the lower and upper quartiles, respectively, and the ends of the whiskers represent the tenth and the 90th percentiles. (Reproduced with permission of [2])
There is evidence that over the years, final height post transplantation is improving [3]. This is likely to be due to a combination of factors such as better growth attained pre transplant, e.g. by adequate nutrition and rhGH therapy, pre-emptive transplantation thus avoiding dialysis, and to the development of protocols that minimize the use of corticosteroids. European data show an improvement in final height from −2.06 SDS in children who reached adulthood in 1990–1995 to −1.33 SDS in 2006–2011[2]. In the 2014 NAPRTCS report, the mean height SDS of those >19 years of age was −1.37. Twenty-five percent of these patients had a height SDS of −2.2 or worse, and 10% were more than 3.2 SD below the mean. This has improved considerably as adult height SDS was −1.93 with the 1987–1991 cohort, −1.51 for 1992–1996 cohort; −1.06 for the 1997–2001 cohort, −0.98 for the 2002–2006 cohort and −0.89 for the most recent cohort (https://naprtcs.org/system/files/2014_Annual_Transplant_Report.pdf).
Older age at start of KRT, starting KRT more recently, cumulative time with a transplant, and greater height SDS at initiation of KRT were independently associated with a higher adult height SDS. Most impressively, recent results of the avoidance of steroids post transplant altogether are excellent, with mean final heights of 177 cm and 175 cm in males transplanted prepubertally and postpubertally respectively, with similar figures of 165 cm and 162 cm for females [4].
The application of adult height prediction methods in children suffering from CKD is not recommended. In several validation studies final height was overpredicted by 3–10 cm [17, 18, 46]. Most likely this reflects the complexity and thus unpredictability of growth and development under the conditions of chronic uremia, with highly variable and dynamic impacts of disease progression, medications, bone disease, KRT modalities, skeletal maturation, and pubertal timing [18, 46].
Causes of Growth Failure in CKD
Growth failure in CKD is due to a complex interplay of many different factors, with varying effects at different ages and stages of CKD (Fig. 56.1). While some factors, such as nutritional and hormonal abnormalities, and hematological and metabolic derangements such as acidosis, electrolyte imbalance, and CKD-MBD are potentially correctable, the effects of others, such as birth parameters, associated syndromes, race and parental heights, are not [27]. There is a substantial global variation of the degree of growth failure in children on KRT which is at least partly explained by differences in economics. In a recent survey of the IPPN network gross national income was a strong independent predictor of standardized height, adding to the impact of other well-known factors, e.g. congenital CKD, anuria, and dialysis vintage as outlined below [50]. Likewise the country of residence was an independent predictor of growth outcome in a large European cohort of children with CKD stage 3–5, which may be related to differences in the timing of diagnosis and/or referral to a center specialized for children with CKD—which in turn may have economic causes [9].
Cause of Renal Disease
Congenital Abnormalities of the Kidneys and Urinary Tract (CAKUT)
Renal tubular sodium and bicarbonate losses are common in children with CAKUT, and can cause salt depletion and acidosis both of which can contribute to growth failure [51]. Supplementation with salt and bicarbonate may be necessary, along with free access to water.
Glomerulopathies
Growth may be affected in children with glomerulopathies, even in early CKD [52]. The nephrotic state per se and glucocorticoid treatment are known risk factors. Prolonged high corticosteroid doses lead to severe growth failure. Although partial catch-up growth can be seen after cessation of glucocorticoid treatment, this is usually restricted to young (prepubertal) patients [53]. Congenital nephrotic syndrome is often associated with severe growth retardation during the first months of life, even in patients with preserved kidney function, and seems to be secondary to persistent edema, recurrent infections, losses of peptide and protein-bound hormones in the urine, and/or protein-calorie malnutrition [54, 55]. In the Finnish-type nephrotic syndrome adequate nutritional support is vital and bilateral nephrectomies and initiation of dialysis may be necessary to stabilize growth. In less severe types of congenital nephrotic syndrome, unilateral nephrectomy and/or treatment with prostaglandin synthesis inhibitors and RAS antagonists can reduce proteinuria and thereby stabilize growth and the overall clinical condition [54,55,56].
Tubular and Interstitial Nephropathies
Tubular dysfunction characterized by losses of electrolytes, bicarbonate, and water can lead to severe growth failure even in the presence of normal glomerular function. The growth suppressive effects of isolated tubular defects are illustrated by the severe growth failure typically seen in patients suffering from renal tubular acidosis, Bartter syndrome, and nephrogenic diabetes insipidus [57]. Supplementation with electrolytes, water and bicarbonate may be able to prevent growth failure or even induce catch-up growth [58].
The most severe growth failure, which may be very difficult to treat, occurs in patients suffering from complex tubular disorders such as Fanconi syndrome [58,59,60,61]. In these patients, only partial catch-up growth can usually be achieved even with vigorous water and electrolyte supplementation. Systemic metabolic disorders (such as cystinosis, primary hyperoxaluria and mitochondrial cytopathies) resulting in complex tubular dysfunction, progressive loss of kidney function, and involvement of other vital organs (e.g. liver, bone, and brain) also lead to severe growth failure [61,62,63]. In children with nephropathic cystinosis, growth failure occurs already in infancy when glomerular function is typically not yet compromised. Progressive growth failure is further sustained by generalized deposition of cystine crystals altering the function of the growth plate, bone marrow, hypothalamus, and pituitary and thyroid glands. Early initiation of treatment with cystine depleting agents (cysteamine) results in an improvement of growth rates and substantial delay in the development of ESKD [61, 64]. Nevertheless, in a recent European study mean standardized height in children suffering from nephropathic cystinosis with CKD 2–5 was 1.0 SDS lower compared to that in children with other causes of CKD at comparable age and degree of CKD, compatible with an additional impact of an underlying osteoblast defect in this disease [65, 66]. In patients with primary hyperoxaluria, supplementary treatment with citrate and pyridoxine can delay the progression of CKD in some patients, and possibly improve longitudinal growth [62]. However real catch-up growth after combined liver-kidney transplantation is rarely observed even in prepubertal oxalosis patients [48]. New ribonucleic acid interference (RNAi) therapies are expected to become the standard of treatment in these children, so that end-stage kidney disease, KRT and consecutive growth failure may be avoidable in the vast majority of cases in the future [67].
Stage of CKD and Dialysis
Even moderate reduction of GFR has been reported to result in impaired growth. The principal registry providing data on the epidemiology of growth in conservatively managed CKD is the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS). The 2006 report covers the 10 years between 1994–2004 and includes a very large cohort of over 5000 children with GFRs of up to 75 mL/min/1.73 m2. As expected, the most growth retarded were the youngest children, with a mean standardized height for under 2 years of age of −2.3 SD, but mean height SDS was reduced at all ages (−1.7, −1.4, −1.0 at 2–6, 6–12 and > 12 years respectively), with over one third overall being below the third centile for height. Standardized height worsened with progression of CKD, so that there was a strong correlation between creatinine clearance and height SDS (−3.2, −1.9, −1.5, and −0.9 for GFR <10, 10–25, 25–50 and > 50 mL/min/1.73 m2 respectively) [68]. This means that many children, and particularly the very young, are already short at the time of entry to KRT programmes [69]. This has been confirmed more recently in a cohort of 42 children followed during the first 6 years of life, when the mean height SDS was normal at CKD stage 1–2, approximately −0.5 at CKD stage 3–5 and much less, at −1.5 SDS, for those on dialysis [70].
That short stature becomes more common in children on dialysis has been confirmed by the United States Renal Data System (USRDS), a registry collecting data on patients on KRT programs in the US. The 2007 report shows that the height and weight of approximately half of children on dialysis were below the 20th centile for the normal population [71]. Comparison of 2007 and 2016 data demonstrates that the prevalence of short stature in the incident pediatric ESKD population has not improved over the past 10 years [12]. The British Association for Paediatric Nephrology (BAPN) report for height SDS of prevalent patients on dialysis at the end of 2017 was −2 with interquartile range of −1 to −3, having been −1.3 (−0.3 to −2.4) SDS at the start of KRT (UK Renal Registry (2021) UK Renal Registry 23rd Annual Report – data to 31/12/2019, Bristol, UK. Available from renal.org/audit-research/annual-report). Children on dialysis were shorter than their transplanted peers whose mean Height SDS was −1.0 (−0.2 to −2), although both groups are below the heights of the normal age matched population. The International Pediatric Dialysis Registry collects data from more than 3000 children on peritoneal dialysis (PD) from around the world, and is, therefore, able to provide comparisons of all aspects of PD according to region in the largest cohort of children on PD to date. Currently, the mean standardized height on commencing PD is −2.35 SDS, and is below normal worldwide, but there is a large variation, ranging in 21 countries from −1.3 in the UK, to −3.5 in Brazil. Regional variations in resources are likely to contribute to these differences [31]. Given our knowledge that in the majority of reports standardized height declines with increasing time on dialysis, the obvious key to prevention of growth deterioration is pre-emptive transplantation [72].
Protein-Calorie Malnutrition
Anorexia is a common symptom of CKD, due to a combination of altered taste sensation, decreased clearance of cytokines that affect appetite and satiety, obligatory losses of salt and water leading to a preference for salty foods and large volumes of water, and the need for multiple medications. Vomiting is also common, particularly in infants, whose diet is liquid and therefore high volume, and because gastro-esophageal reflux is frequent and elevated polypeptide hormones result in abnormal gastrointestinal motility. PD results in raised intra-abdominal pressure, which may affect both appetite and cause vomiting and constipation, and dietary and fluid restrictions may result in an inadequate diet. Peritoneal dialysate losses of protein and sodium may be high. Finally, co-morbidities may cause poor feeding in their own right [31, 73]. Nutritional deficiency is, therefore, one of the most frequent and important factors contributing to growth failure. The Pediatric Renal Nutrition Taskforce has produced guidelines for the nutritional management of children with CKD stages 2–5 and on dialysis (vide infra) [74, 75].
Protein-Energy Wasting
Protein-energy wasting (PEW) is characterized by maladaptive responses including anorexia, elevated basic metabolic rate, wasting of lean body tissue, and under-utilization of fat tissue for energy [76]. It differs from malnutrition in which appetite is maintained, and weight loss is associated with protective metabolic responses such as a decreased basic metabolic rate and preservation of lean body mass at the expense of fat mass. Malnutrition can usually be overcome by nutritional supplementation or changes in the composition of the diet, whereas PEW can only be partially reversed by increased nutrition. Why some children develop PEW is unknown, but inflammation is likely to play a role [77]. However, growth failure is one of the main manifestations of PEW in children with CKD. New understandings of the pathophysiology of PEW in CKD have the potential for novel therapeutic strategies such as ghrelin agonists and melanocortin antagonists [31].
Obesity
At this stage it is important to mention obesity, which is emerging as a growing problem for children with CKD. In the ESPN/ERA-EDTA registry including 25 countries, of 5199 patients below the age of 18 years the prevalence of underweight was 4.3%, while 19.6% and 11.2% were overweight or obese respectively [78]. Receiving steroid therapy and living with a kidney transplant were independent risk factors for overweight. The incidence of obesity parallels that around the world in the normal population. The IPPN database demonstrates this regional variation, with BMI-SDS ranging from a mean of 0.8 in the US to −1.4 in India in children of all ages [31]. Twenty six percent of infants were obese in the US and 50% malnourished in Turkey [31, 58]. In North America, the frequency of obesity is increasing in the CKD population both before as well as at CKD stage 5 [79]. Obesity is a particularly a problem after kidney transplantation. This has been studied in the NAPRTCS database in a retrospective cohort study of 4326 children transplanted between 1995 and 2006, and followed up to 2007. Median BMI increased by 11% at 6 months but with no substantial changes thereafter [80]. In Europe, children with the lowest BMI and those over 5 years of age at transplant showed the greatest increases in BMI post-transplant [78]. UK 2017 data shows that the mean BMI of transplanted children was 1 SDS (0–1.8), whereas it was 0 (−0.8–1.1) for children on dialysis (UK Renal Registry (2021) UK Renal Registry 23rd Annual Report—data to 31/12/2019, Bristol, UK. Available from renal.org/audit-research/annual-report). The use of steroid sparing regimens may mitigate post transplant obesity [4]. Important to note, among transplanted recipients, a very short stature (OR: 1.64, 95% CI: 1.40–1.92) and glucocorticoid treatment (OR: 1.23, 95% CI: 1.03–1.47) were associated with a higher risk of being overweight/obese. Hence, at least in post-transplant patients, obesity is also a risk factor for poor growth.
Metabolic Acidosis
Metabolic acidosis (serum bicarbonate <22 mEq/L) usually begins when the GFR falls below 50% of normal, and is associated with decreased longitudinal growth and increased protein breakdown [81, 82]. Metabolic acidosis is also associated with endocrine consequences: in experimental uremia there is increased glucocorticoid production, increased protein degradation, and profound effects on the somatotropic hormone axis. The latter is characterized by down regulation of spontaneous GH secretion by the pituitary gland, decreased expression of the GH-receptor and insulin like growth factor I (IGF-I) receptor in target organs, and decreased IGF I serum concentrations [83]. Hence, metabolic acidosis induces a state of GH insensitivity, which is likely to contribute to impaired longitudinal growth in CKD patients.
Disturbances of Water and Electrolyte Balance
Although the relationship between salt loss and growth failure has not been formally proven in CKD, children with isolated tubular disorders resulting in urinary salt and water losses show severe growth retardation which can be at least partly resolved by adequate salt and water supplementation. The same applies to patients with a reduced chloride diet or with familial chloride diarrhea [84]. Growth impairment in diabetes insipidus supports the concept that polyuria may also contribute to growth failure in CKD patients [85].
CKD-Mineral and Bone Disorder (CKD-MBD)
It is widely accepted that skeletal deformities due to CKD-MBD contribute to uremic growth failure [86, 87]. Pronounced secondary hyperparathyroidism can interfere with longitudinal growth by destruction of the growth plate architecture, epiphyseal displacement and metaphyseal fractures. Severe destruction of the metaphyseal bone architecture may result in complete growth arrest. Treatment with 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) improves growth in uremic rats, but this not been demonstrated in children with CKD [88, 89].
PTH levels primarily reflect osteoblast activity. Therefore, it has been speculated that low PTH levels may be associated with poor bone and statural growth, and conversely high PTH levels might be expected in well growing children; and similarly, that poor growth might be expected with adynamic bone disease and better growth in its absence. Information on this issue is conflicting. Diminished growth rates have been shown in four dialyzed patients who had adynamic bone disease on bone biopsy, and PTH levels were positively correlated with the annual change in standardized height [90]. However, the proportion of patients showing adynamic bone disease in this population, where all subjects received high-dose intermittent calcitriol treatment, was rather high (25%). Therefore, this does not represent patients treated nowadays [91]. Indeed, low bone turnover was noted in only 4% of pediatric patients on dialysis in a recent study. In addition, there was no relationship between PTH levels and growth rates in 35 prepubertal children on dialysis for more than one year. Moreover, stable growth was seen with PTH levels only slightly above the normal range, and even catch-up growth occurred in children younger than 2 years [92]. In addition, one well-designed direct histomorphometric assessment in children on dialysis revealed no association between low bone turnover and body growth [93]. The IPPN offers the most up to date information from the largest cohort of PD patients: the annual prospective change in standardized height tended to correlate inversely with time-integrated mean PTH levels: patients with mean PTH levels >500 pg/mL (i.e. > 9 times upper limit of normal (ULN)) showed a significant loss in height SDS as compared to children with lower PTH levels (−0.28 versus −0.05 SDS per year; p < 0.05) [94]. Thus, dialyzed children with normal or only slightly elevated PTH levels have the potential for normal growth, whereas patients with high PTH levels (>500 pg/mL) are at increased risk of growth failure.
Anemia
Longstanding anemia in CKD patients has profound systemic consequences including anorexia and catabolism due to altered energy turnover, and multiple dysfunctions of organ systems. Retardation of growth and development is a hallmark of untreated chronic anemias of non-renal origin, such as thalassemia major. Theoretically, anemia may interfere with growth via mechanisms such as poor appetite, undercurrent infections, cardiac complications and reduced oxygen supply to cartilage. The advent of recombinant human erythropoietin in the late 1980s permitted study of the effects of anemia correction on longitudinal growth in CKD. Although short-term stimulatory effects of erythropoietin on longitudinal growth have been reported anecdotally, no persistent catch-up growth could be demonstrated in several multicenter clinical trials [95, 96] Partial correction of anemia has, though, improved exercise capacity and decreased heart rate and resting oxygen consumption [95, 97].
Endocrine Changes
Uremia interferes with the metabolism and regulation of various peptide hormones. This leads to inappropriate concentrations of circulating hormones and/or altered hormone action on target tissues (Fig. 56.1). Distinct alterations of the somatotropic and gonadotropic hormone axes have been identified, which are believed to play an important role in uremic growth failure [98].
Gonadotropic Hormone Axis
Gonadal Hormones
Low or low normal total and free testosterone (T) as well as dihydrotestosterone (DHT) plasma concentrations due to decreased synthesis and/or increased metabolic clearance have been reported in adolescents and adults with long-standing uremia [99]. The reduced conversion of T to DHT secondary to diminished 5α-reductase activity might contribute to the delayed pubertal development seen in some boys on dialysis [100]. Likewise, plasma estradiol levels in women tend to decrease in parallel with GFR reduction, and some adolescent girls show low-normal or decreased estradiol levels in relation to pubertal age [100, 101]. However, these observations were all made more than 20 years ago. At least in transplanted children this issue seems to be resolved nowadays. In a recent study, the majority of transplanted children without prior long-term dialysis had normal estradiol and testosterone levels [36]. This may at least partly explain the improvement of pubertal development in patients on KRT during the last decades [1].
Gonadotropins
Increased plasma concentrations of LH and FSH in combination with decreased or low-normal gonadal hormones suggest a state of compensated hypergonadotropic hypogonadism in patients with CKD stage 5 [102]. However, in patients on dialysis, gonadotrophin secretion may be inadequate relative to the degree of hypogonadism. This is compatible with an additional defect of pituitary gonadotropin release and the analysis of spontaneous pulsatile LH secretion has provided insights into the underlying pathophysiology [103, 104]. In dialyzed patients mean LH plasma levels are elevated despite significantly reduced pituitary LH secretion, due to the markedly impaired kidney metabolic clearance of LH. When kidney function is restored by kidney transplantation, pulsatile LH secretion normalizes and hypergonadotropic FSH and/or LH levels are only rarely observed [36, 103]. Animal studies suggest that a primary hypothalamic defect may contribute to the delayed onset of puberty in patients with uremia. The observed reduced release of hypothalamic gonadotropin-releasing hormone (GnRH) is due to uremia-related inhibitory factors and/or to an increased tone of the inhibitory neurotransmitter gamma-aminobutyric acid [105, 106].
Beyond the quantitative alterations of gonadotropin release, uremia also affects the biological quality of circulating gonadotropins. In pubertal and adult dialysis patients the proportion of bioactive LH in relation to the total immunochemically measurable amount of LH is reduced. This might be due to altered glycosylation and/or accumulation of less active isoforms [105,106,107].
In summary, insufficient activation of the hypothalamic GnRH pulse generator, likely mediated via circulating inhibitors, appears to be the key abnormality underlying delayed puberty and altered sexual functions in patients with CKD stage 5. However, kidney transplantation is able to completely normalize all these alterations in the majority of patients if long periods on dialysis treatment are avoided.
Somatotropic Hormone Axis
Growth Hormone Secretion and Metabolism
In both pediatric and adult CKD patients fasting GH concentrations are normal or even increased, depending on the stage of CKD. GH, a 22-kilodalton protein, is almost freely filtered by the glomerulus (sieving coefficient ~0.82) and thereby ultimately cleared from the circulation [108]. Indeed, a linear relationship between GFR and the metabolic clearance rate of GH has been shown; GH clearance is reduced by approximately 50% in patients with CKD stage 5 [108, 109]. The prolonged plasma half-life of GH, rather than increased endogenous secretion, explains the increased circulating GH concentrations in uremia. Pituitary GH secretion is unaltered in prepubertal patients but decreased in adolescents with CKD, suggesting insufficient stimulation by gonadal steroids during puberty [110, 111]. In addition, malnutrition and metabolic acidosis negatively impact GH secretion rates in children with CKD [112].
Growth Hormone Receptor and GH Signaling
Experimental studies have advanced our understanding of uremic GH resistance. Both, the GH-induced hepatic as well as the growth plate cartilage IGF-I synthesis is diminished, due to either decreased expression of the GH receptor (GH-R) and/or a postreceptor signaling defect [113, 114]. Whereas reduced expression of the GH-R encoding mRNA in liver and growth plate chondrocytes was seen, hepatic and growth plate cartilage GH-R protein levels were comparable in uremic and non-uremic animals when corrected for uremia-associated anorexia by pair feeding [113,114,115]. Thus, whereas a decreased GH-R abundance in the liver and the growth plate cartilage is questionable, a postreceptor GH signaling defect was identified as cause of the diminished hepatic IGF-I secretion upon GH stimulation. In fact, aberrant GH-dependent JAK-STAT signaling has been noted in experimental animals [116]. Binding of GH to its receptor leads to activation of the JAK-STAT cascade by tyrosine phosphorylation, and transcriptional activation of IGF-I synthesis and proteins of the suppressor of cytokine signaling (SOCS) family. The latter are responsible for dephosphorylation of the GH-activated cascade and as such provide a GH-regulated negative feedback loop. However, under the conditions of chronic uremia the equilibrium between GH-induced transcriptional activation of IGF-I and SOCS is shifted towards SOCS overstimulation. Recent studies suggest that the chronic inflammatory state associated with CKD contributes to GH resistance, as SOCS are also induced by inflammatory cytokines [117, 118].
In humans, GH binding protein (GHBP), which enters the circulation by proteolytic cleavage of the extracellular receptor domain, is taken as a measure of GH receptor expression. In line with animal experiments, GHBP plasma levels in CKD patients are decreased and are related to residual kidney function [119].
Insulin-Like Growth Factor Plasma Binding and Tissue Action
Apart from GH resistance, insensitivity to IGF-I is also found in patients with advanced CKD [120,121,122]. While serum concentrations of IGF-I and IGF-II are usually within the normal range in children with CKD stage 1–4, IGF-I levels are slightly reduced and those of IGF-II mildly increased in dialyzed patients [123]. In contrast to the unchanged total amount of circulating immunoreactive IGF, somatomedin bioavailability is reduced in advanced CKD pointing to the existence of circulating inhibitors [124]. A low-molecular weight somatomedin inhibitor (~1 kDa) was reported to circulate in uremic serum in an early study, but this has not been characterized further. Later studies focused on the accumulation of the specific high-affinity IGF-binding proteins (IGFBP1–6), which are normally cleared by the kidneys and are considered the main cause of diminished somatomedin bioactivity in uremia. In particular, the concentrations of IGFBP-1, −2, −3, −4 and − 6 increase as kidney function declines and IGFBP-1, -2 and -6 have been shown to inhibit IGF I bioactivity in-vitro [125, 126]. In contrast, the serum concentration of IGFBP-5 is normal and IGFBP-5 proteins undergo intense proteolytic cleavage in chronic uremia [126]. Likewise, the elevated level of IGFBP-3 is mostly due to the accumulation of proteolytic fragments whereas intact IGFBP-3 is markedly diminished [127]. The molar excess of IGFBPs over IGFs is approximately 150% in children with CKD and 200% in children on dialysis as compared to 25% in children without CKD. An inverse correlation between growth retardation and IGFBP-1, -2, and -4 serum concentrations has been described [128]. Reduced IGF bioactivity can be normalized by removing unsaturated IGFBP [124]. These data are in favor of the concept that serum IGFBPs increase with progression of CKD, and that the greater excess of IGFBPs by CKD stage 5 contributes to the more severe growth failure and reduced response to rhGH therapy in these children. In addition, cellular IGF signaling is impaired in the uremic state; it remains to be elucidated whether a postreceptor mechanism similar to the one observed for GH signaling is responsible for this phenomenon [118, 122, 124].
In summary, the marked deficiency of IGF-I synthesis, the modest elevation of GH levels due to decreased metabolic clearance, and increased IGF plasma binding capacity, strongly support the concept of a multilevel homeostatic failure of the GH-IGF-I system.
Corticosteroid Treatment
Long-term glucocorticoid treatment in patients after transplantation leads to diminished longitudinal growth by impairment of the somatotropic hormone axis. High-dose glucocorticoid treatment suppresses pulsatile GH release from the pituitary gland mainly by reduction of pulse amplitude [129]. The physiologic increase in GH secretion during puberty is reduced in allograft recipients receiving glucocorticoid treatment (≥ 4 mg/m2 per day) and the association between sex steroid plasma concentrations and GH release observed in healthy adolescents is blunted [129]. These changes are mainly due to increased hypothalamic somatostatin release.
In addition to reduced GH release, corticosteroids suppress GH-R mRNA and protein in animals and most likely also in humans [130]. Consequently, hepatic IGF-I mRNA levels are reduced in animals receiving glucocorticoids. However, plasma concentrations of IGF-I in patients treated by glucocorticoids are normal or only slightly reduced. In individual children on corticosteroid treatment impaired longitudinal growth occurs despite normal GH secretion and plasma IGF-I levels, suggesting insensitivity to GH and IGF-I at the level of the growth plate. Indeed, a direct growth inhibiting effect of dexamethasone on the growth plate was shown by local injection in rabbits [131]. In cultured growth plate chondrocytes glucocorticoids decreased DNA synthesis and cell proliferation in a dose-dependent fashion, associated with reduced expression of the GH receptor and diminished paracrine IGF-I synthesis [132]. In addition, pharmacological doses of glucocorticoids also impaired the proliferative response to the calciotropic hormones calcitriol and PTH [132]. This is at least partly related to a diminished release of paracrine IGF-I secretion by these hormones. IGF-I modulates its own activity in cultured rat growth plate chondrocytes by the synthesis of both inhibitory (IGFBP-3) and stimulatory (IGFBP-5) binding proteins. This is modified by glucocorticoid treatment. Therefore, glucocorticoid treatment not only interferes with the somatotropic hormone axis with respect to GH secretion and GH / IGF-I receptor signaling, but also by modulation of paracrine IGF-I synthesis and binding by IGF binding proteins. Even more important, based on in vivo studies in rabbits catch-up growth after glucocorticoid exposure may remain incomplete in general [131]. Consequently, all efforts must be undertaken to reduce steroid exposure in children with CKD before and after kidney transplantation [133].
Treatment of Growth Failure in Chronic Kidney Disease
General Measures
The main measures for prevention and treatment of growth failure in children with CKD are summarized in Table 56.1. Close growth monitoring with intervals depending on previous growth, age and stage of CKD is essential. Early referral to a pediatric nephrology center followed by careful nutritional and metabolic management is vital in the prevention of growth retardation [134]. Growth retardation is clearly correlated to the degree of CKD. Therefore, adequate measures should be undertaken to preserve GFR, and to provide adequate dialysis in those children who require maintenance dialysis [72]. Preservation of kidney function requires treatment of elevated blood pressure, aiming for blood pressure values below the 50th and 75th percentile in proteinuric and non-proteinuric children, respectively [135]. Renin-angiotensin aldosterone system inhibitors, preferentially angiotensin-converting enzyme inhibitors or angiotensin receptor inhibitors, should be used to treat high blood pressure and ameliorate proteinuria in children with CKD [136]. Nephrotoxic medication should be avoided, and urinary tract infections in children with congenital abnormalities of the kidneys and urinary tract (CAKUT) should be treated. Finally, disease specific treatment, e.g. treatment with cysteamine in patients with nephropathic cystinosuis, may be required.
Adequate nutritional management is crucial in infants and young children, who frequently need supplementary feeding via nasogastric tube or, preferably, gastrostomy [75, 137]. In a retrospective analysis of growth in 101 infants and young children with severe CKD it could be demonstrated that persistent catch-up growth can be achieved in the majority of patients when there is good metabolic control and enteral feeding is commenced at the first sign of growth delay (Fig. 56.5) [20]. However, spontaneous growth as well as catch-up growth after initiation of enteral feeding is limited in patients with comorbidities [20, 138]. The use of enteral feeding varies around the world, as has been clearly demonstrated by the IPPN report of 153 infants on PD: gastrostomies are most commonly used in the US, where 80% of infants on PD are gastrostomy fed; 20% have gastrostomies in Europe, but there are very small numbers or none in the rest of the world [13]. Nasogastric feeding is commonest in Europe and Latin America. Gastrostomy feeding, rather than demand or nasogastric tube feeding, is associated with better preservation of linear growth in the infants in the IPPN database (Fig. 56.6)) [13]. This may be related to decreased vomiting with gastrostomies as compared to nasogastric tubes. Catch-up growth in children started on enteral feeding after the age of 2 years may be substantial but is markedly reduced in those on dialysis and strongly negatively correlated with age [139]. The assurance of adequate caloric and energy intake requires the patient and families to be advised by a kidney dietician, especially when supplementary feeding via nasogastric or gastrostomy tube is required [74, 137] In general, the initial prescription for energy intake in children with CKD should approximate that of healthy children of the same age (suggested dietary intake, SDI) [74]. To optimize growth in children with suboptimal weight gain and linear growth, energy intake should be adjusted towards the higher end of the SDI [74]. Caloric intake should account for growth failure and be related to “height age” rather than to chronological age if the child’s height is below the normal range. Calorie intake in excess of 100% of SDI may not improve catch-up growth but rather results in obesity and may thereby negatively contribute to long-term cardiovascular morbidity in CKD patients [140, 141]. If there is excessive weight gain dietary energy intake should be reduced for children on PD to compensate for the energy derived from dialysate glucose, estimated at 8–12 kcal/kg per day. In addition; to promote optimal growth, target protein intake in children with CKD should be at the upper end of the SDI [74]. In patients on PD, a slightly higher intake (+0.2 g/kg/day) is recommended to compensate for dialytic protein losses. The aim is to maintain a normal serum albumin and a urea below 20 mmol/L as far as possible. High protein intakes should be avoided since, despite many attempts, anabolizing or growth promoting effects of high-protein diets have neither been demonstrated in animal models nor in children with CKD. On the contrary, high protein diets may be detrimental by aggravating metabolic acidosis and augmenting the dietary phosphorus load.

Course of mean standardized height and body mass index (BMI) of children presenting within the first 6 months of life with a glomerular filtration rate less than 20 mL/min/1.73 m2 receiving tube feeding in order to provide at least 100% of the recommended daily allowance (RDA) of healthy children. (a) Height SDS and BMI values for all patients. (b) Height SDS and BMI values for patients without comorbidites. (c) Height SDS and BMI values for patients with comorbidities. (Reproduced with permission of [20])

Whereas both nasogastric tube (NGT) and gastrostomy (GS) feeding improve nutritional status, only GS feeding associates with stabilized linear growth in young infants undergoing CPD. The data points represent mean estimates at key time points of postnatal development, (i.e., birth, commencement of CPD, initiation and discontinuation of nasogastric tube or gastrostomy feeding, enrollment to IPPN [study entry], and last available observation). Two-dimensional error bars denote the 95% confidence intervals to mean age and SDS at the respective time point. [Reproduced with permission of [13]]
Metabolic acidosis should be vigorously treated by alkali supplementation aiming for serum bicarbonate levels equal or above 22 mEq/L. This can be assured by treatment with sodium bicarbonate and/or the use of HCO3-based or lactate based dialysis solutions in patients on dialysis [11]. In addition, supplementation of water and electrolytes is essential in patients presenting with polyuria and/or salt losing nephropathies. Supplementation of sodium chloride is also important in young children on PD, since significant amounts of sodium chloride (i.e. 2–5 mmol/kg body weight) may be eliminated via ultrafiltration.
Dialysis and Intensified Dialysis
Although dialysis attenuates the uremic state, longitudinal growth is not usually improved and long-term PD or HD are associated with a gradual loss of standardized height in children and adolescents, and can be as high as 1 SD per year in infants [9, 142,143,144]. Children on dialysis who maintain some residual kidney function have the best growth; indeed residual kidney function may be a better predictor of longitudinal growth than dialytic clearance [145, 146]. However, a recent Italian study in infants on chronic PD reported catch-up growth in 50% of patients [147]. High peritoneal transporter status, a condition associated with increased morbidity and mortality in adults, is associated with poor longitudinal growth in children on chronic PD [148]. This might be due to the putative association of high peritoneal transport with inflammation, which can suppress statural growth by interference with GH signaling (vide supra), or excessive losses of proteins and amino acids important to growth.
It has been suggested that intensified dialysis, achieved by either extended thrice-weekly sessions, daily nocturnal or short daily sessions, might be able to induce catch-up growth [148,149,150]. According to a French study, catch-up growth can be maximized when intensified hemodiafiltration (3 h, 6 times a week) and rhGH therapy are combined [148, 149, 151,152,153,154]. Using this approach in 15 mainly prepubertal children for an average observation time of 21 months, there was an average increase in growth velocity from 3.8 cm/year at baseline to 8.9 cm/year (Fig. 56.7). This resulted in a mean of 1.7 SDS gain of standardized height, representing complete catch-up growth according to the attainment of the target height SDS [153]. A recent non-randomized study demonstrated superior growth in children treated with hemodiafiltration compared to those with conventional hemodialysis (HD) (Fig. 56.8) [155]. From a pathophysiological point of view, intensified hemodiafiltration (HDF) is a better substitute for physiological kidney function and may allow substantially better clearance of uremic toxins, e.g. middle-molecular weight compounds [155]. As a result, microinflammation and metabolic acidosis may be abolished, leading to improved appetite and tissue anabolism. The improved removal of inflammatory cytokines might reverse GH resistance enabling the full therapeutic potential of rhGH [152]. The positive effects of intensified dialysis, particularly home HD, usually outweigh the potential impact on psychosocial integration and treatment costs. Prospective randomized trials appear required to provide definite proof to this promising concept. In particular, the relative contribution of concomitant rhGH therapy to the growth improvement observed with intensified dialysis remains to be defined.

Examples of growth charts (height and weight chart; growth velocity chart in centimeters per year; body mass under chart) of two patients on daily online hemodiafiltration (start indicated by bars) in addition to rhGH treatment . TC on height chart is the familial target height in (centimeters). [Reproduced with permission of [153]]

Improved height SDS in children on hemodiafiltration (HDF) compared to hemodialysis (HD). The figure shows change in height SDS in the HD and HDF arms at baseline and 1-year follow-up. Data are shown as median and interquartile range. Within-group analyses performed by Wilcoxon matched-pairs signed-rank test and HD versus HDF cohorts compared by Mann–Whitney U test. At 12 months the height SDS in the HDF group was higher than in the HD group (P = 0.04). (Reproduced with permission of [155])
Transplantation
Although many of the metabolic and endocrine disorders contributing to uremic growth failure are resolved by kidney transplantation, post-transplant catch-up growth is usually restricted to young children and occurs far from regularly [3, 156]. As well as transplant function, age, severity of stunting at time of transplantation and glucocorticoid dosage are inversely associated with longitudinal growth. One retrospective study found that while standardized height was comparable at time of transplantation, it was significantly higher among living-related donor (LRD) than deceased donor transplant (DDT) recipients 5 years later [157]. This benefit of LRD grafts was independent of GFR arguing for preferential LRD in children with respect to post-transplant growth.
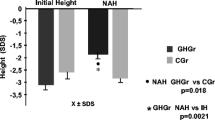
It must be stressed that even low-dose glucocorticoid treatment (<4 mg/m2/day) results in growth suppression in children after transplantation. While complete steroid withdrawal has been associated with unacceptably high rejection rates in children with azathioprine and/ or cyclosporine A medication, withdrawal or even complete steroid avoidance appears much safer with the currently preferred immunosuppressants [158]. In a randomized trial of late steroid withdrawal in patients on treatment with cyclosporine A, mycophenolate mofetil steroid-free patients showed improved growth compared to controls (i.e. change in height SDS; 0.6 ± 0.1 versus −0.2 ± 0.1) within 27 months [159]. However, catch-up growth in pubertal patients was rather limited compared to that in prepubertal patients. It seems logical that if steroids are withdrawn at an early stage, or even completely avoided, a better growth outcome will be observed. Indeed, a retrospective analysis of longitudinal growth in a cohort of 74 children who had been weaned off steroids within 6 months of transplantation showed remarkable results [4, 159]. Mean adult height was −0.5 ± 1.1 SDS and −1.0 ± 1.3 SDS in prepubertal and pubertal patients and was within the normal range (>−2 SD) in 94% and 80% of them respectively (Fig. 56.9). Likewise, early steroid-withdrawal (< 6 weeks) and complete steroid avoidance improved standardized height by approximately 1.0 SDS within 3–5 years post-transplantation [32, 33, 160, 161]. Although experimental data indicate that mTOR inhibitors may interfere with chondrocyte proliferation and/or gonadal hormone synthesis, recent case control studies in transplanted children revealed similar growth rates in patients with and without mTOR inhibitor treatment [162].

Mean standardized height at time of renal transplantation compared to adult height and comparison of adult height with genetic target height in patients with steroid-withdrawal during month 4–6 after transplantation
Mean standardized height at the time of transplantation and final height in prepubertal (n = 36) and pubertal (n = 24) patients. (d) Mean adult height (open bars) and genetic target height (hatched bars) in boys (n = 25) and girls (n = 17). Data in (c) and (d) are given as mean ± SEM. (Reproduced with permission of [4])
In summary, efforts to avoid a height deficit before transplantation, early (preemptive) kidney transplantation, and immunosuppressive strategies characterized by the early withdrawal or even complete avoidance of steroids can improve adult height and normalize body proportions in children after successful transplantation.
Endocrine Therapies
Vitamin D
Calcitriol deficiency is a major cause of secondary hyperparathyroidism and CKD-MBD. Although calcitriol supplementation reverses the biochemical, radiographic, and histological signs of high turnover bone disease, neither experimental nor clinical studies demonstrate consistent improvement in longitudinal growth [89, 162,163,164]. These conflicting results might be due to differences in the mode of administration and to the pleiotropic calcitriol-specific effects on growth plate chondrocytes. In addition, only a week association between parathyroid hormone (PTH) levels and linear growth was reported in children with advanced CKD [94]. In general, minimal PTH suppressive active vitamin D analogues dosages should be used in order to keep PTH levels in the desired target range [165]. Finally, supplementation with cholecalciferol or ergocalciferol should be initiated in children with serum 25-hydroxyvitamin D concentrations below 75 nmol/L (<30 ng/mL) [166].
Calcimimetics
Uncontrolled and controlled studies provide evidence that calcimimetics are an effective therapy for secondary hyperparathyroidism in pediatric dialysis patients [167]. Calcimimetics suppress PTH secretion by activating the calcium-sensing receptor (CaR). The CaR is expressed by epiphyseal chondrocytes; its stimulation stimulates chondrocytic proliferation and differentiation. Thus, calcimimetics may affect longitudinal growth in uremia as well. In fact, the calcimimetic cinacalcet improved food efficiency and body weight gain in uremic rats, but no effects on growth plate morphology and/or longitudinal growth were seen [168]. Likewise, no beneficial or adverse effect on longitudinal growth was noted during calcimimetic treatment periods of up-to three years in children on dialysis [167]. A comprehensive European guideline on the use of cinacalcet in children on dialysis was published recently [169].
Growth Hormone
Pharmacological treatment of children with CKD and growth delay with rhGH actually predated the elucidation of the pathomechanisms that underlie chronic uremic alterations of the GH-IGF-1 axis [114, 169,170,171]. Administration of rhGH markedly stimulates IGF-I synthesis with only a modest effect on IGFBPs, thereby normalizing somatomedin bioactivity and promoting longitudinal growth [172]. The efficacy and safety of long-term treatment with rhGH in children with CKD before and after kidney transplantation is well established and clinical practice recommendations on this topic were recently published in 2019 [30].
Efficacy of rhGH in Prepubertal Children
In prepubertal children with pre-dialysis CKD, rhGH therapy typically doubles height velocity during the first treatment year [173]. Catch-up growth continues asymptotically during extended treatment [174,175,176]. In dialyzed children, the treatment response is significantly attenuated compared to children with pre-dialysis CKD (0.8 SD vs. 1.3 SD) [177]. RhGH responsiveness is similarly poor in children on peritoneal dialysis and standard hemodialysis, but as noted previously, can be markedly improved when dialytic clearance is augmented by daily HDF [153].
Based on the current experience with rhGH in pediatric CKD patients, a model to predict growth response was developed [178]. The prediction model was developed using a cohort of 208 prepubertal children with CKD stage 3-5D followed in a pharmaco-epidemiological survey (KIGS), and validated in an independent group of 67 CKD patients registered at the Dutch Growth Research Foundation. The height velocity during the first rhGH treatment year (PHV) was predicted by the following equation: PHV (centimeters per year) = 13.3 – [age (years) × 0.38 + (weight SDS × 0.39)] – [hereditary renal disorder (0 when absent or 1 when present) × 1.16] + [Ln rhGH dose (milligrams per kilogram per week) × 1.04] + [GFR (milliliters per minute × 1.73 m2) × 0.023]. This equation explains 37% of the overall variability of the growth response. The SE of the estimate or error SD of the prediction model was 1.6 cm and non-responders in the validation group were correctly identified. This model may help in predicting non-responders and in tailoring treatment strategies for growth retarded children with CKD.
Several RCTs have shown the benefit of rhGH therapy in short prepubertal renal transplant recipients. A meta-analysis of five prospective RCTs involving a total of 401 patients showed that patients receiving rhGH therapy had a significantly higher growth velocity 1 year after the initiation of therapy than the control group, with a mean height SDS difference of 0.68 (95% CI 0.25–1.11) [173].
Effects of rhGH on Pubertal Growth and Adult Height
In a study following patients with CKD and ESKD from late prepubertal age to final height, the average height increment in rhGH treated patients was twice that seen in a matched control group [46]. The main benefit for total growth and final height was achieved before the onset of the pubertal growth spurt, whereas no overall effect on pubertal height gain was observed (Fig. 56.10).

(a) Synchronized mean height velocity curves of 32 boys (left panel) and 6 girls (right panel) with CKD during rhGH Treatment (closed circles), as compared with 50 children with CKD not treated with rhGH (open circles) and 232 normal children (thin lines). The dots indicate the time of the first observation, which corresponds to the start of rhGH treatment in the growth hormone-treated children, minimal prespurt height velocity, and the time of the end of the pubertal growth spurt. (Reproduced with permission of [46]). (b) Synchronized mean height curves of 32 boys (left panel) and 6 girls (right panel) with CKD during rhGH treatment (closed circles), as compared with 50 children with CKD not treated with rhGH (open circles). Normal values are indicated by the third, 50th, and 97th percentile. The dots indicate the time of the first observation, which corresponds to the start of rhGH treatment in the growth hormone-treated children, and the time of the end of the pubertal growth spurt. (Reproduced with permission of [46])
Data on adult height are available from 11 non-randomized trials in which rhGH was administered for at least 2 years, comprising a total of 836 patients on various modes of KRT [30]. In five studies a matched historical control group was included. The median change in standardized height until attainment of adult height amounted to 1.1 SDS (range 0.2–1.6 SDS) in rhGH treated patients (p < 0.05 for each final height measurement versus initial height measurement). This change corresponded to a median absolute increase in rhGH treated patients by 7.4 cm (range 1.4–10.8 cm) in boys and 7.0 cm (range 1.3–10.1 cm) in girls, based on European reference values. However, this calculation may underestimate the rhGH effect since in the non-rhGH treated controls adult height was significantly below the initial standardized height indices in all except one study. Height attained at the start of rhGH and throughout the duration of rhGH treatment were positively associated with final height, whereas time spent on dialysis, age at puberty onset, and age of start of rhGH were negatively associated with final height [46, 47]. Taken together, the available studies suggest that rhGH improves adult height in short prepubertal and pubertal CKD patients prior to and after kidney transplantation.
Efficacy of rhGH in Infants
According to standard concepts of the pathophysiology of uremic growth failure, malnutrition and fluid and electrolyte imbalances have a much greater impact on infant growth than alterations of somatotropic hormones. Consequently, correction of the nutritional status has been considered the primary measure to restore normal growth in growth retarded infants, postponing the option of endocrine therapeutic intervention to beyond the second year of life. This concept has been challenged by several reports of initiating rhGH in growth retarded infants with CKD [179,180,181]. A randomized controlled study involving 30 growth retarded infants (mean age: 16 months) with moderate CKD (mean eGFR: 25 mL/min/1.73 m2) revealed excellent catch-up growth from from −3.0 to −1.1 SDS within 24 months of treatment, in contrast to no significant change in controls [179]. Another study reported an increase in height SDS from −3.3 to −2.2 within 12 months in 8 infants with a mean age of 22 months and CKD stage 3–4 [180]. In a randomized study of 16 infants with stage 3–4 CKD who were receiving at least 80% of the recommended daily allowence for calories and of whom seven were enterally fed, those randomized to rhGH showed significantly higher length gains (14.5 versus 9.5 cm/year; delta height SDS +1.43 versus −0.11) [181]. Hence, the results of these studies lend further support to the previous observation that the relative efficacy and cost efficiency of rhGH is actually best when initiated at young age, i.e. during infancy and early childhood [30]. While the provision of adequate nutrition is certainly vital to growth and development of infants with CKD, some children show growth failure despite adequate nutrition. In these patients, any further increases of energy intake typically lead to fat deposition, but not catch-up growth. Early rhGH therapy appears to be an attractive option to accelerate length and weight gain in such infants. The fact that the enhanced growth also helps the infant achieve the body size required for kidney transplantation more expeditiously is another substantial benefit [30].
General rhGH Treatment Strategies
Children from 6 months of age with stage 3–5 CKD or on dialysis should be candidates for rhGH therapy if they have persistent growth failure, defined as a height below the third percentile for age and sex and a height velocity below the 25th percentile, once other potentially treatable risk factors for growth failure have been adequately addressed, and provided the child has growth potential (open epiphysis on X-ray of the wrist) [30]. RhGH therapy should also be considered for children older than 6 months with stage 3–5 CKD or on dialysis who present with a height between the third and tenth percentile but persistent low height velocity (<25th percentile), once other potentially treatable risk factors for growth failure have been adequately addressed. Such early, preventive therapy is probably more cost-effective than starting at a more advanced age when growth retardation has become more evident and higher absolute rhGH doses are required.
Children suffering from nephropathic cystinosis often show severe growth retardation despite somewhat mild reductions in GFR, which is thought to be related to the deleterious effects of Fanconi syndrome, resulting in hypophosphatemic rickets and malnutrition and/or an underlying obsteoblast/osteoclast defect [61]. Therefore, rhGH treatment is recommended in short children with nephropathic cystinosis at all stages of CKD [30].
The growth response to rhGH treatment is positively associated with residual kidney function, target height, initial target height deficit and duration of rhGH treatment, and inversely correlated with the age at start of treatment [46, 47, 174, 177, 178]. Daily dosing is more effective than three doses per week and the optimal dose is 0.045–0.05 mg/kg body weight per day by subcutaneous injections in the evening [30]. Parents and physicians should encourage children from about 8 to 10 years of age to do the rhGH injections on their own if adequate training and adherence can be assured, because this may ultimately improve patient adherence and self-esteem [30]. Whereas discontinuation of rhGH results in catch-down growth in approximately 75% of CKD patients this phenomenon is rarely observed when rhGH treatment is discontinued after kidney transplantation, highlighting the close relationship between kidney function and growth [182]. Furthermore, although the absolute height gain achieved by rhGH is independent of age, the reference range increases with age. Thus, rhGH treatment should be started as early as growth retardation becomes evident (i.e. height below third percentile) [30]. If height velocity in the first year of rhGH treatment is less than 2 cm per year over baseline, it is recommended to assess patient adherence to rhGH therapy through the measurement of serum IGF-I levels and/or the use of an injection pen with downloadable memory function, ensuring administration of the correct weight-adjusted GH dose, and addressing any, possibly subclinical, nutritional and metabolic issues [30].
The primary treatment target should be to return the child’s height into her/his individual genetic percentile channel. Treatment may be suspended once this target is reached, but growth should be monitored closely as outlined above. In patients receiving rhGH while on conservative treatment rhGH should be continued after the initiation of dialysis, but stopped at the time of kidney transplantation. RhGH therapy should, however, subsequently also be considered for pediatric kidney transplant recipients for whom expected catch-up growth cannot be achieved by steroid minimization or for patients in whom steroid withdrawal is not feasible due to high immunological risk, particularly in children with suboptimal graft function (eGFR <50 mL/min/1.73 m2). Growth should be monitored for at least 1 year post-transplantation before rhGH therapy is considered, in order to allow for spontaneous catch-up growth without need for rhGH therapy [30].
Potential Adverse Events Associated with rhGH Therapy
The safety of long-term rhGH treatment in CKD has been evaluated in several clinical studies and registries [30]. RhGH therapy in short children with CKD stage 3–5D and after kidney transplantation was not associated with an increased incidence of malignancy, slipped capital femoral epiphysis, avascular necrosis, glucose intolerance, pancreatitis, CKD progression, acute allograft rejection or fluid retention. Intracranial hypertension (ICH), manifesting in 3 out of 1376 CKD patients, was the only adverse event significantly associated with rhGH therapy [183]. However, in all three instances ICH occurred after discontinuation of rhGH. Due to the potentially increased risk of ICH in CKD, baseline fundoscopy is recommended prior to therapy initiation [30]. Furthermore, hydration should be carefully monitored in CKD patients receiving rhGH since overhydration may be a predisposing factor for ICH. In the presence of symptoms like headache or vomiting, an immediate workup for ICH including fundoscopy should be performed.
Although insulin secretion increases during the first year of rhGH treatment and hyperinsulinemia persists during long-term therapy, normal glucose tolerance is preserved during up to 5 years of rhGH administration in CKD patients on conservative treatment, dialysis, and after kidney tansplantation. Hyperinsulinemia is most pronounced in transplanted patients on concomitant glucocorticoid therapy. Hyperinsulinemia may, at least in theory, contribute to the development of atherosclerosis or induce diabetes mellitus by exhaustion of ß-cells. However, up to now this has not been observed in CKD patients receiving rhGH [183].
Aggravation of secondary hyperparathyroidism has rarely been reported in CKD patients on rhGH treatment, and the underlying pathomechanisms remain to be elucidated [184]. RhGH might have a direct stimulatory effect on the parathyroid gland and/or might have subtle effects on calcium homeostasis which in turn stimulate PTH secretion. Finally, increased longitudinal bone growth by rhGH treatment may unmask preexisting renal osteodystrophy. Therefore, bone metabolism should be evaluated carefully in candidates for rhGH therapy and severe hyperparathyroidism and renal osteodystrophy should be adequately treated before initiation of such therapy in CKD patients. Likewise, rhGH therapy should be stopped in patients with persistent severe secondary hyperparathyroidism (PTH > 500 pg/mL). RhGH may be reinstituted when PTH levels return to the desired PTH target range [30].
Future Perspectives
Despite attention to nutrition and the availability of rhGH therapy, the problem of CKD associated growth failure has not been resolved in the majority of dialysis patients. If early kidney transplantation is not possible, the concept of intensified hemodiafiltration combined with rhGH may be a promising option for patients suffering from growth retardation and GH insensitivity on conventional dialysis therapy. Independent of concomitant rhGH therapy, hemodiafiltration appears to allow improved growth rates than conventional hemodialysis and, if available, should be the preferred extracorporeal dialysis modality in all growing children.
Self-reported nonadherence to rhGH is associated with poorer growth velocity in children with CKD. Therefore, monitoring and optimizing adherence to rhGH therapy is key to satisfactory growth outcomes [185]. Another promising avenue of clinical research may be the provision of recombinant IGF-I administered as monotherapy or in combination with rhGH, as well as targeting of the SOCS2 signaling pathway [186].
A particular challenge is the management of severely diminished pubertal height gain seen in some adolescents with CKD. In such adolescents, pharmacological inhibition of epiphyseal closure may allow an extended duration of the remaining growth period. Since the closure of the epiphyseal growth plate is induced by local estrogen action, inhibition of estrogen synthesis is a principal therapeutic option. Whereas gonadotropin-releasing hormone analogues arrest pubertal progress, the potential growth benefit would come at the psychological disadvantage of delayed sexual maturation. In boys, aromatase inhibitors, which suppress local conversion of testosterone to estradiol, might extend the growth phase without affecting pubertal development and thereby increase the time window for the use of rhGH therapy. Indeed, the combined use of armomatase inhibitors and rhGH was associated with modest increases in adult height in boys with idiopathic short stature.
[187, 188]. However, some important questions about their long-term safety exist which at the moment prevent this from being generally recommended [189]. It would be fascinating to study its efficacy in adolescents on long-term dialysis. Nevertheless, successful early (preemptive) kidney transplantation with minimal steroid exposure is ultimately the best current measure to improve growth and final height in children with CKD stage 5.
References
Franke D, Winkel S, Gellermann J, Querfeld U, Pape L, Ehrich JH, Haffner D, Pavicic L, Zivicnjak M. Growth and maturation improvement in children on renal replacement therapy over the past 20 years. Pediatr Nephrol. 2013;28:2043–51.
Harambat J, Bonthuis M, van Stralen KJ, Ariceta G, Battelino N, Bjerre A, Jahnukainen T, Leroy V, Reusz G, Sandes AR, Sinha MD, Groothoff JW, Combe C, Jager KJ, Verrina E, Schaefer F, for the ESPN/ERA-EDTA Registry. Adult height in patients with advanced CKD requiring renal replacement therapy during childhood. Clin J Am Soc Nephrol. 2013;
Fine RN, Martz K, Stablein D. What have 20 years of data from the north american pediatric renal transplant cooperative study taught us about growth following renal transplantation in infants, children, and adolescents with end-stage renal disease? Pediatr Nephrol. 2010;25:739–46.
Klare B, Montoya CR, Fischer DC, Stangl MJ, Haffner D. Normal adult height after steroid-withdrawal within 6 months of pediatric kidney transplantation: a 20 years single center experience. Transpl Int. 2012;25:276–82.
Wong CS, Gipson DS, Gillen DL, Emerson S, Koepsell T, Sherrard DJ, Watkins SL, Stehman-Breen C. Anthropometric measures and risk of death in children with end-stage renal disease. Am J Kidney Dis. 2000;36:811–9.
Furth SL, Stablein D, Fine RN, Powe NR, Fivush BA. Adverse clinical outcomes associated with short stature at dialysis initiation: a report of the north american pediatric renal transplant cooperative study. Pediatrics. 2002;109:909–13.
Ku E, Fine RN, Hsu CY, McCulloch C, Glidden DV, Grimes B, Johansen KL. Height at first RRT and mortality in children. Clin J Am Soc Nephrol. 2016;11:832–9.
Hartung EA, Furth SL. Growth in children on renal replacement therapy: a shrinking problem? Pediatr Nephrol. 2013;28:1905–8.
Behnisch R, Kirchner M, Anarat A, Bacchetta J, Shroff R, Bilginer Y, Mir S, Caliskan S, Paripovic D, Harambat J, Mencarelli F, Büscher R, Arbeiter K, Soylemezoglu O, Zaloszyc A, Zurowska A, Melk A, Querfeld U, Schaefer F, and the 4C Study Consortium. Determinants of statural growth in european children with chronic kidney disease: findings from the cardiovascular comorbidity in children with chronic kidney disease (4C) study. Front Pediatr. 2019;7:278.
Schaefer F, Benner L, Borzych-Dużałka D, Zaritsky J, Xu H, Rees L, Antonio ZL, Serdaroglu E, Hooman N, Patel H, Sever L, Vondrak K, Flynn J, Rébori A, Wong W, Hölttä T, Yildirim ZY, Ranchin B, Grenda R, Testa S, Drożdz D, Szabo AJ, Eid L, Basu B, Vitkevic R, Wong C, Pottoore SJ, Müller D, Dusunsel R, Celedon CG, Fila M, Sartz L, Sander A, Warady BA, International Pediatric Peritoneal Dialysis Network (IPPN) Registry. Global variation of nutritional status in children undergoing chronic peritoneal dialysis: a longitudinal study of the international pediatric peritoneal dialysis network. Sci Rep. 2019;9:4886-z.
Rodig NM, McDermott KC, Schneider MF, Hotchkiss HM, Yadin O, Seikaly MG, Furth SL, Warady BA. Growth in children with chronic kidney disease: a report from the chronic kidney disease in children study. Pediatr Nephrol. 2014;29:1987–95.
United States Renal Data System. USRDS annual data report: Epidemiology of kidney disease in the United States, vol. 2020. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2020. p. 2021.
Rees L, Azocar M, Borzych D, Watson AR, Buscher A, Edefonti A, Bilge I, Askenazi D, Leozappa G, Gonzales C, van Hoeck K, Secker D, Zurowska A, Ronnholm K, Bouts AH, Stewart H, Ariceta G, Ranchin B, Warady BA, Schaefer F, International Pediatric Peritoneal Dialysis Network (IPPN) Registry. Growth in very young children undergoing chronic peritoneal dialysis. J Am Soc Nephrol. 2011;22:2303–12.
Schaefer F, Mehls O. Endocrine, metabolic and growth disorders. In: Holliday MA, Barrat TM, Avner ED, editors. Pediatric nephrology. 3rd ed. Baltimore: Williams & Wilkins; 1994. p. 1241–86.
Downie AB, Mulligan J, Stratford RJ, Betts PR, Voss LD. Are short normal children at a disadvantage? the wessex growth study. BMJ. 1997;314:97–100.
Karlberg J, Schaefer F, Hennicke M, Wingen AM, Rigden S, Mehls O. Early age-dependent growth impairment in chronic renal failure. european study group for nutritional treatment of chronic renal failure in childhood. Pediatr Nephrol. 1996;10:283–7.
Schaefer F, Seidel C, Binding A, Gasser T, Largo RH, Prader A, Scharer K. Pubertal growth in chronic renal failure. Pediatr Res. 1990;28:5–10.
Nissel R, Brazda I, Feneberg R, Wigger M, Greiner C, Querfeld U, Haffner D. Effect of renal transplantation in childhood on longitudinal growth and adult height. Kidney Int. 2004;66:792–800.
Kari JA, Gonzalez C, Ledermann SE, Shaw V, Rees L. Outcome and growth of infants with severe chronic renal failure. Kidney Int. 2000;57:1681–7.
Mekahli D, Shaw V, Ledermann SE, Rees L. Long-term outcome of infants with severe chronic kidney disease. Clin J Am Soc Nephrol. 2010;5:10–7.
Greenbaum LA, Munoz A, Schneider MF, Kaskel FJ, Askenazi DJ, Jenkins R, Hotchkiss H, Moxey-Mims M, Furth SL, Warady BA. The association between abnormal birth history and growth in children with CKD. Clin J Am Soc Nephrol. 2011;6:14–21.
Franke D, Volker S, Haase S, Pavicic L, Querfeld U, Ehrich JH, Zivicnjak M. Prematurity, small for gestational age and perinatal parameters in children with congenital, hereditary and acquired chronic kidney disease. Nephrol Dial Transplant. 2010;25:3918–24.
Schaefer F, Wingen AM, Hennicke M, Rigden S, Mehls O. Growth charts for prepubertal children with chronic renal failure due to congenital renal disorders. european study group for nutritional treatment of chronic renal failure in childhood. Pediatr Nephrol. 1996;10:288–93.
Cetin I, Mando C, Calabrese S. Maternal predictors of intrauterine growth restriction. Curr Opin Clin Nutr Metab Care. 2013;16:310–9.
Jagadapillai R, Chen J, Canales L, Birtles T, Pisano MM, Neal RE. Developmental cigarette smoke exposure: kidney proteome profile alterations in low birth weight pups. Toxicology. 2012;299:80–9.
Menendez-Castro C, Hilgers KF, Amann K, Daniel C, Cordasic N, Wachtveitl R, Fahlbusch F, Plank C, Dotsch J, Rascher W, Hartner A. Intrauterine growth restriction leads to a dysregulation of wilms' tumour supressor gene 1 (WT1) and to early podocyte alterations. Nephrol Dial Transplant. 2013;28:1407–17.
Franke D, Alakan H, Pavicic L, Gellermann J, Muller D, Querfeld U, Haffner D, Zivicnjak M. Birth parameters and parental height predict growth outcome in children with chronic kidney disease. Pediatr Nephrol. 2013;28:2335–41.
Jones RW, Rigden SP, Barratt TM, Chantler C. The effects of chronic renal failure in infancy on growth, nutritional status and body composition. Pediatr Res. 1982;16:784–91.
Kleinknecht C, Broyer M, Huot D, Marti-Henneberg C, Dartois AM. Growth and development of nondialyzed children with chronic renal failure. Kidney Int Suppl. 1983;15:40.
Drube J, Wan M, Bonthuis M, Wühl E, Bacchetta J, Santos F, Grenda R, Edefonti A, Harambat J, Shroff R, Tönshoff B, Haffner D, European Society for Paediatric Nephrology Chronic Kidney Disease Mineral and Bone Disorders, Dialysis, and Transplantation Working Groups. Clinical practice recommendations for growth hormone treatment in children with chronic kidney disease. Nat Rev Nephrol. 2019;15:577–89.
Rees L, Mak RH. Nutrition and growth in children with chronic kidney disease. Nat Rev Nephrol. 2011;7:615–23.
Sarwal MM, Ettenger RB, Dharnidharka V, Benfield M, Mathias R, Portale A, McDonald R, Harmon W, Kershaw D, Vehaskari VM, Kamil E, Baluarte HJ, Warady B, Tang L, Liu J, Li L, Naesens M, Sigdel T, Waskerwitz J, Salvatierra O. Complete steroid avoidance is effective and safe in children with renal transplants: a multicenter randomized trial with three-year follow-up. Am J Transplant. 2012;12:2719–29.
Delucchi A, Valenzuela M, Lillo AM, Guerrero JL, Cano F, Azocar M, Zambrano P, Salas P, Pinto V, Ferrario M, Rodriguez J, Cavada G. Early steroid withdrawal in pediatric renal transplant: five years of follow-up. Pediatr Nephrol. 2011;26:2235–44.
Rizzoni G, Broyer M, Brunner FP, Brynger H, Challah S, Kramer P, Oules R, Selwood NH, Wing AJ, Balas EA. Combined report on regular dialysis and transplantation of children in europe, XIII, 1983. Proc Eur Dial Transplant Assoc Eur Ren Assoc. 1985;21:66–95.
Burke BA, Lindgren B, Wick M, Holley K, Manivel C. Testicular germ cell loss in children with renal failure. Pediatr Pathol. 1989;9:433–44.
Tainio J, Qvist E, Vehmas R, Jahnukainen K, Holtta T, Valta H, Jahnukainen T, Jalanko H. Pubertal development is normal in adolescents after renal transplantation in childhood. Transplantation. 2011;92:404–9.
Kim HS, Ng DK, Matheson MB, Atkinson MA, Warady BA, Furth SL, Ruebner RL. Delayed menarche in girls with chronic kidney disease and the association with short stature. Pediatr Nephrol. 2020;35:1471–5.
Zivicnjak M, Franke D, Ehrich JH, Filler G. Does growth hormone therapy harmonize distorted morphology and body composition in chronic renal failure? Pediatr Nephrol. 2000;15:229–35.
Gunnell D, Whitley E, Upton MN, McConnachie A, Smith GD, Watt GC. Associations of height, leg length, and lung function with cardiovascular risk factors in the midspan family study. J Epidemiol Community Health. 2003;57:141–6.
Kinra S, Sarma KV, Hards M, Smith GD, Ben-Shlomo Y. Is relative leg length a biomarker of childhood nutrition? long-term follow-up of the hyderabad nutrition trial. Int J Epidemiol. 2011;40:1022–9.
Zivicnjak M, Franke D, Filler G, Haffner D, Froede K, Nissel R, Haase S, Offner G, Ehrich JH, Querfeld U. Growth impairment shows an age-dependent pattern in boys with chronic kidney disease. Pediatr Nephrol. 2007;22:420–9.
Franke D, Thomas L, Steffens R, Pavicic L, Gellermann J, Froede K, Querfeld U, Haffner D, Zivicnjak M. Patterns of growth after kidney transplantation among children with ESRD. Clin J Am Soc Nephrol. 2015;10:127–34.
Franke D, Steffens R, Thomas L, Pavicic L, Ahlenstiel T, Pape L, Gellermann J, Muller D, Querfeld U, Haffner D, Zivicnjak M. Kidney transplantation fails to provide adequate growth in children with chronic kidney disease born small for gestational age. Pediatr Nephrol. 2017;32:511–9.
Grohs J, Rebling RM, Froede K, Hmeidi K, Pavičić L, Gellermann J, Müller D, Querfeld U, Haffner D, Živičnjak M. Determinants of growth after kidney transplantation in prepubertal children. Pediatr Nephrol. 2021;36:1871–80.
Offner G, Latta K, Hoyer PF, Baum HJ, Ehrich JH, Pichlmayr R, Brodehl J. Kidney transplanted children come of age. Kidney Int. 1999;55:1509–17.
Haffner D, Schaefer F, Nissel R, Wuhl E, Tonshoff B, Mehls O. Effect of growth hormone treatment on the adult height of children with chronic renal failure. german study group for growth hormone treatment in chronic renal failure. N Engl J Med. 2000;343:923–30.
Nissel R, Lindberg A, Mehls O, Haffner D, Pfizer International Growth Database (KIGS) International Board. Factors predicting the near-final height in growth hormone-treated children and adolescents with chronic kidney disease. J Clin Endocrinol Metab. 2008;93:1359–65.
Nissel R, Latta K, Gagnadoux MF, Kelly D, Hulton S, Kemper MJ, Ruder H, Soderdahl G, Otte JB, Cochat P, Roquet O, Jamieson NV, Haffner D. Body growth after combined liver-kidney transplantation in children with primary hyperoxaluria type 1. Transplantation. 2006;82:48–54.
Brinkert F, Lehnhardt A, Montoya C, Helmke K, Schaefer H, Fischer L, Nashan B, Bergmann C, Ganschow R, Kemper MJ. Combined liver-kidney transplantation for children with autosomal recessive polycystic kidney disease (ARPKD): Indication and outcome. Transpl Int. 2013;26:640–50.
Schaefer F, Borzych-Duzalka D, Azocar M, Munarriz RL, Sever L, Aksu N, Barbosa LS, Galan YS, Xu H, Coccia PA, Szabo A, Wong W, Salim R, Vidal E, Pottoore S, Warady BA, & IPPN investigators (2012) Impact of global economic disparities on practices and outcomes of chronic peritoneal dialysis in children: Insights from the international pediatric peritoneal dialysis network registry. Perit Dial Int 32: 399–409.
Hiraoka M. Medical management of congenital anomalies of the kidney and urinary tract. Pediatr Int. 2003;45:624–33.
Tullus K, Webb H, Bagga A. Management of steroid-resistant nephrotic syndrome in children and adolescents. Lancet Child Adolesc Health. 2018;2:880–90.
Rees L, Greene SA, Adlard P, Jones J, Haycock GB, Rigden SP, Preece M, Chantler C. Growth and endocrine function in steroid sensitive nephrotic syndrome. Arch Dis Child. 1988;63:484–90.
Dufek S, Holtta T, Trautmann A, Ylinen E, Alpay H, Ariceta G, Aufricht C, Bacchetta J, Bakkaloglu SA, Bayazit A, Cicek RY, Dursun I, Duzova A, Ekim M, Iancu D, Jankauskiene A, Klaus G, Paglialonga F, Pasini A, Printza N, Said Conti V, do Sameiro Faria M, Schmitt CP, Stefanidis CJ, Verrina E, Vidal E, Vondrak K, Webb H, Zampetoglou A, Bockenhauer D, Edefonti A, Shroff R. Management of children with congenital nephrotic syndrome: Challenging treatment paradigms. Nephrol Dial Transplant. 2018;
Dufek S, Ylinen E, Trautmann A, Alpay H, Ariceta G, Aufricht C, Bacchetta J, Bakkaloglu S, Bayazit A, Caliskan S, do Sameiro Faria M, Dursun I, Ekim M, Jankauskiene A, Klaus G, Paglialonga F, Pasini A, Printza N, Conti VS, Schmitt CP, Stefanidis C, Verrina E, Vidal E, Webb H, Zampetoglou A, Edefonti A, Holtta T, Shroff R, ESPN Dialysis Working Group. Infants with congenital nephrotic syndrome have comparable outcomes to infants with other renal diseases. Pediatr Nephrol. 2019;34:649–55.
Boyer O, Schaefer F, Haffner D, Bockenhauer D, Hölttä T, Bérody S, Webb H, Heselden M, Lipska-Zie Tkiewicz BS, Ozaltin F, Levtchenko E, Vivarelli M. Management of congenital nephrotic syndrome: consensus recommendations of the ERKNet-ESPN working group. Nat Rev Nephrol. 2021;17:277–89.
Haffner D, Weinfurth A, Manz F, Schmidt H, Bremer HJ, Mehls O, Scharer K. Long-term outcome of paediatric patients with hereditary tubular disorders. Nephron. 1999;83:250–60.
Lopez-Garcia SC, Emma F, Walsh SB, Fila M, Hooman N, Zaniew M, Bertholet-Thomas A, Colussi G, Burgmaier K, Levtchenko E, Sharma J, Singhal J, Soliman NA, Ariceta G, Basu B, Murer L, Tasic V, Tsygin A, Decramer S, Gil-Pena H, Koster-Kamphuis L, La Scola C, Gellermann J, Konrad M, Lilien M, Francisco T, Tramma D, Trnka P, Yuksel S, Caruso MR, Chromek M, Ekinci Z, Gambaro G, Kari JA, Konig J, Taroni F, Thumfart J, Trepiccione F, Winding L, Wuhl E, Agbas A, Belkevich A, Vargas-Poussou R, Blanchard A, Conti G, Boyer O, Dursun I, Pinarbasi AS, Melek E, Miglinas M, Novo R, Mallett A, Milosevic D, Szczepanska M, Wente S, Cheong HI, Sinha R, Gucev Z, Dufek S, Iancu D, Kleta R, Schaefer F, Bockenhauer D, European dRTA Consortium. Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant. 2019;
Greco M, Brugnara M, Zaffanello M, Taranta A, Pastore A, Emma F. Long-term outcome of nephropathic cystinosis: a 20-year single-center experience. Pediatr Nephrol. 2010;25:2459–67.
Haffner D, Weinfurth A, Seidel C, Manz F, Schmidt H, Waldherr R, Bremer HJ, Mehls O, Scharer K. Body growth in primary de toni-debre-fanconi syndrome. Pediatr Nephrol. 1997;11:40–5.
Hohenfellner K, Rauch F, Ariceta G, Awan A, Bacchetta J, Bergmann C, Bechtold S, Cassidy N, Deschenes G, Elenberg E, Gahl WA, Greil O, Harms E, Herzig N, Hoppe B, Koeppl C, Lewis MA, Levtchenko E, Nesterova G, Santos F, Schlingmann KP, Servais A, Soliman NA, Steidle G, Sweeney C, Treikauskas U, Topaloglu R, Tsygin A, Veys K, Vigier VR, Zustin J, Haffner D. Management of bone disease in cystinosis: Statement from an international conference. J Inherit Metab Dis. 2019;42:1019–29.
Rumsby G, Cochat P. Primary hyperoxaluria. N Engl J Med. 2013;369:2163.
Emma F, Salviati L. Mitochondrial cytopathies and the kidney. Nephrol Ther. 2017;13(Suppl. 1):S23–8.
Kimonis VE, Troendle J, Rose SR, Yang ML, Markello TC, Gahl WA. Effects of early cysteamine therapy on thyroid function and growth in nephropathic cystinosis. J Clin Endocrinol Metab. 1995;80:3257–61.
Ewert A, Leifheit-Nestler M, Hohenfellner K, Büscher A, Kemper MJ, Oh J, Billing H, Thumfart J, Stangl G, Baur AC, Föller M, Feger M, Weber LT, Acham-Roschitz B, Arbeiter K, Tönshoff B, Zivicnjak M, Haffner D. Bone and mineral metabolism in children with nephropathic cystinosis compared with other CKD entities. J Clin Endocrinol Metab. 2020;105:dgaa267. https://doi.org/10.1210/clinem/dgaa267.
Battafarano G, Rossi M, Rega LR, Di Giovamberardino G, Pastore A, D'Agostini M, Porzio O, Nevo N, Emma F, Taranta A, Del Fattore A. Intrinsic bone defects in cystinotic mice. Am J Pathol. 2019;189:1053–64.
Weigert A, Martin-Higueras C, Hoppe B. Novel therapeutic approaches in primary hyperoxaluria. Expert Opin Emerg Drugs. 2018;23:349–57.
Seikaly MG, Salhab N, Gipson D, Yiu V, Stablein D. Stature in children with chronic kidney disease: analysis of NAPRTCS database. Pediatr Nephrol. 2006;21:793–9.
North american pediatric renal trials and collaborative studies. (2005). NAPRTCS annual report 2005. available at: https://Web.emmes.com/study/ped/annlrept/annlrept2005.pdf.
Katsoufis CP, DeFreitas MJ, Infante JC, Castellan M, Cano T, Safina Vaccaro D, Seeherunvong W, Chandar JJ, Abitbol CL. Risk assessment of severe congenital anomalies of the kidney and urinary tract (CAKUT): A birth cohort. Front Pediatr. 2019;7:182.
United states renal data system (USRDS). 2008 report. USRDS coordinating center, minneapolis, pp. 296–297.
Haffner D. Strategies for optimizing growth in children with chronic kidney disease. Front Pediatr. 2020;8:399.
Rees L, Brandt ML. Tube feeding in children with chronic kidney disease: technical and practical issues. Pediatr Nephrol. 2010;25:699–704.
Shaw V, Polderman N, Renken-Terhaerdt J, Paglialonga F, Oosterveld M, Tuokkola J, Anderson C, Desloovere A, Greenbaum L, Haffner D, Nelms C, Qizalbash L, Vande Walle J, Warady B, Shroff R, Rees L. Energy and protein requirements for children with CKD stages 2–5 and on dialysis-clinical practice recommendations from the pediatric renal nutrition taskforce. Pediatr Nephrol. 2020;35:519–31.
Rees L, Shaw V, Qizalbash L, Anderson C, Desloovere A, Greenbaum L, Haffner D, Nelms C, Oosterveld M, Paglialonga F, Polderman N, Renken-Terhaerdt J, Tuokkola J, Warady B, Walle JV, Shroff R, Pediatric Renal Nutrition Taskforce. Delivery of a nutritional prescription by enteral tube feeding in children with chronic kidney disease stages 2–5 and on dialysis-clinical practice recommendations from the pediatric renal nutrition taskforce. Pediatr Nephrol. 2021;36:187–204.
Mak RH, Cheung WW, Zhan JY, Shen Q, Foster BJ. Cachexia and protein-energy wasting in children with chronic kidney disease. Pediatr Nephrol. 2012;27:173–81.
Rees L. Protein energy wasting; what is it and what can we do to prevent it? Pediatr Nephrol. 2019;
Bonthuis M, van Stralen KJ, Verrina E, Groothoff JW, Alonso Melgar A, Edefonti A, Fischbach M, Mendes P, Molchanova EA, Paripovic D, Peco-Antic A, Printza N, Rees L, Rubik J, Stefanidis CJ, Sinha MD, Zagozdzon I, Jager KJ, Schaefer F. Underweight, overweight and obesity in paediatric dialysis and renal transplant patients. Nephrol Dial Transplant. 2013;28(Suppl. 4):iv195–204.
Rees L, Jones H. Nutritional management and growth in children with chronic kidney disease. Pediatr Nephrol. 2013;28:527–36.
Foster BJ, Martz K, Gowrishankar M, Stablein D, Al-Uzri A. Weight and height changes and factors associated with greater weight and height gains after pediatric renal transplantation: a NAPRTCS study. Transplantation. 2010;89:1103–12.
Bailey JL, Wang X, England BK, Price SR, Ding X, Mitch WE. The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J Clin Invest. 1996;97:1447–53.
Boirie Y, Broyer M, Gagnadoux MF, Niaudet P, Bresson JL. Alterations of protein metabolism by metabolic acidosis in children with chronic renal failure. Kidney Int. 2000;58:236–41.
Brungger M, Hulter HN, Krapf R. Effect of chronic metabolic acidosis on the growth hormone/IGF-1 endocrine axis: new cause of growth hormone insensitivity in humans. Kidney Int. 1997;51:216–21.
Grossman H, Duggan E, McCamman S, Welchert E, Hellerstein S. The dietary chloride deficiency syndrome. Pediatrics. 1980;66:366–74.
Sharma S, Ashton E, Iancu D, Arthus MF, Hayes W, Van't Hoff W, Kleta R, Bichet DG, Bockenhauer D. Long-term outcome in inherited nephrogenic diabetes insipidus. Clin Kidney J. 2018;12:180–7.
Schmitt CP, Mehls O. Mineral and bone disorders in children with chronic kidney disease. Nat Rev Nephrol. 2011;7:624–34.
Haffner D, Fischer DC. Bone cell biology and pediatric renal osteodystrophy. Minerva Pediatr. 2010;62:273–84.
Mehls O, Ritz E, Gilli G, Wangdak T, Krempien B. Effect of vitamin D on growth in experimental uremia. Am J Clin Nutr. 1978;31:1927–31.
Mehls O, Knoller N, Oh J, Wesch H, Wunsche B, Schmitt CP. Daily but not pulse calcitriol therapy improves growth in experimental uremia. Pediatr Nephrol. 2000;14:658–63.
Kuizon BD, Goodman WG, Juppner H, Boechat I, Nelson P, Gales B, Salusky IB. Diminished linear growth during intermittent calcitriol therapy in children undergoing CCPD. Kidney Int. 1998;53:205–11.
Bakkaloglu SA, Wesseling-Perry K, Pereira RC, Gales B, Wang HJ, Elashoff RM, Salusky IB. Value of the new bone classification system in pediatric renal osteodystrophy. Clin J Am Soc Nephrol. 2010;5:1860–6.
Cansick J, Waller S, Ridout D, Rees L. Growth and PTH in prepubertal children on long-term dialysis. Pediatr Nephrol. 2007;22:1349–54.
Waller S, Shroff R, Freemont AJ, Rees L. Bone histomorphometry in children prior to commencing renal replacement therapy. Pediatr Nephrol. 2008;23:1523–9.
Borzych D, Rees L, Ha IS, Chua A, Valles PG, Lipka M, Zambrano P, Ahlenstiel T, Bakkaloglu SA, Spizzirri AP, Lopez L, Ozaltin F, Printza N, Hari P, Klaus G, Bak M, Vogel A, Ariceta G, Yap HK, Warady BA, Schaefer F, International Pediatric PD Network (IPPN). The bone and mineral disorder of children undergoing chronic peritoneal dialysis. Kidney Int. 2010;78:1295–304.
Morris KP, Sharp J, Watson S, Coulthard MG. Non-cardiac benefits of human recombinant erythropoietin in end stage renal failure and anaemia. Arch Dis Child. 1993;69:580–6.
Jabs K. The effects of recombinant human erythropoietin on growth and nutritional status. Pediatr Nephrol. 1996;10:324–7.
Martin GR, Ongkingo JR, Turner ME, Skurow ES, Ruley EJ. Recombinant erythropoietin (epogen) improves cardiac exercise performance in children with end-stage renal disease. Pediatr Nephrol. 1993;7:276–80.
Haffner D, Zivicnjak M. Pubertal development in children with chronic kidney disease. Pediatr Nephrol. 2017;32:949–64.
Belgorosky A, Ferraris JR, Ramirez JA, Jasper H, Rivarola MA. Serum sex hormone-binding globulin and serum nonsex hormone-binding globulin-bound testosterone fractions in prepubertal boys with chronic renal failure. J Clin Endocrinol Metab. 1991;73:107–10.
Van Kammen E, Thijssen JH, Schwarz F. Sex hormones in male patients with chronic renal failure. I. the production of testosterone and of androstenedione. Clin Endocrinol. 1978;8:7–14.
Ferraris JR, Domene HM, Escobar ME, Caletti MG, Ramirez JA, Rivarola MA. Hormonal profile in pubertal females with chronic renal failure: before and under haemodialysis and after renal transplantation. Acta Endocrinol. 1987;115:289–96.
Schaefer F, Seidel C, Mitchell R, Scharer K, Robertson WR. Pulsatile immunoreactive and bioactive luteinizing hormone secretion in adolescents with chronic renal failure. the cooperative study group on pubertal development in chronic renal failure (CSPCRF). Pediatr Nephrol. 1991;5:566–71.
Schaefer F, Veldhuis JD, Robertson WR, Dunger D, Scharer K. Immunoreactive and bioactive luteinizing hormone in pubertal patients with chronic renal failure. cooperative study group on pubertal development in chronic renal failure. Kidney Int. 1994;45:1465–76.
Schaefer F, Daschner M, Veldhuis JD, Oh J, Qadri F, Scharer K. In vivo alterations in the gonadotropin-releasing hormone pulse generator and the secretion and clearance of luteinizing hormone in the uremic castrate rat. Neuroendocrinology. 1994;59:285–96.
Daschner M, Philippin B, Nguyen T, Wiesner RJ, Walz C, Oh J, Sandow J, Mehls O, Schaefer F. Circulating inhibitor of gonadotropin releasing hormone secretion by hypothalamic neurons in uremia. Kidney Int. 2002;62:1582–90.
Schaefer F, Vogel M, Kerkhoff G, Woitzik J, Daschner M, Mehls O. Experimental uremia affects hypothalamic amino acid neurotransmitter milieu. J Am Soc Nephrol. 2001;12:1218–27.
Mitchell R, Bauerfeld C, Schaefer F, Scharer K, Robertson WR. Less acidic forms of luteinizing hormone are associated with lower testosterone secretion in men on haemodialysis treatment. Clin Endocrinol. 1994;41:65–73.
Haffner D, Schaefer F, Girard J, Ritz E, Mehls O. Metabolic clearance of recombinant human growth hormone in health and chronic renal failure. J Clin Invest. 1994;93:1163–71.
Schaefer F, Baumann G, Haffner D, Faunt LM, Johnson ML, Mercado M, Ritz E, Mehls O, Veldhuis JD. Multifactorial control of the elimination kinetics of unbound (free) growth hormone (GH) in the human: regulation by age, adiposity, renal function, and steady state concentrations of GH in plasma. J Clin Endocrinol Metab. 1996;81:22–31.
Tonshoff B, Veldhuis JD, Heinrich U, Mehls O. Deconvolution analysis of spontaneous nocturnal growth hormone secretion in prepubertal children with preterminal chronic renal failure and with end-stage renal disease. Pediatr Res. 1995;37:86–93.
Schaefer F, Veldhuis JD, Stanhope R, Jones J, Scharer K. Alterations in growth hormone secretion and clearance in peripubertal boys with chronic renal failure and after renal transplantation. cooperative study group of pubertal development in chronic renal failure. J Clin Endocrinol Metab. 1994;78:1298–306.
Challa A, Chan W, Krieg RJ Jr, Thabet MA, Liu F, Hintz RL, Chan JC. Effect of metabolic acidosis on the expression of insulin-like growth factor and growth hormone receptor. Kidney Int. 1993;44:1224–7.
Troib A, Landau D, Kachko L, Rabkin R, Segev Y. Epiphyseal growth plate growth hormone receptor signaling is decreased in chronic kidney disease-related growth retardation. Kidney Int. 2013;84:940–9.
Schaefer F, Chen Y, Tsao T, Nouri P, Rabkin R. Impaired JAK-STAT signal transduction contributes to growth hormone resistance in chronic uremia. J Clin Invest. 2001;108:467–75.
Edmondson SR, Baker NL, Oh J, Kovacs G, Werther GA, Mehls O. Growth hormone receptor abundance in tibial growth plates of uremic rats: GH/IGF-I treatment. Kidney Int. 2000;58:62–70.
Rabkin R, Sun DF, Chen Y, Tan J, Schaefer F. Growth hormone resistance in uremia, a role for impaired JAK/STAT signaling. Pediatr Nephrol. 2005;20:313–8.
Wiezel D, Assadi MH, Landau D, Troib A, Kachko L, Rabkin R, Segev Y. Impaired renal growth hormone JAK/STAT5 signaling in chronic kidney disease. Nephrol Dial Transplant. 2014;29:791–9.
Tsao T, Fervenza F, Friedlaender M, Chen Y, Rabkin R. Effect of prolonged uremia on insulin-like growth factor-I receptor autophosphorylation and tyrosine kinase activity in kidney and muscle. Exp Nephrol. 2002;10:285–92.
Tonshoff B, Cronin MJ, Reichert M, Haffner D, Wingen AM, Blum WF, Mehls O. Reduced concentration of serum growth hormone (GH)-binding protein in children with chronic renal failure: Correlation with GH insensitivity. the european study group for nutritional treatment of chronic renal failure in childhood. the german study group for growth hormone treatment in chronic renal failure. J Clin Endocrinol Metab. 1997;82:1007–13.
Fouque D. Insulin-like growth factor 1 resistance in chronic renal failure. Miner Electrolyte Metab. 1996;22:133–7.
Powell DR, Liu F, Baker BK, Hintz RL, Lee PD, Durham SK, Brewer ED, Frane JW, Watkins SL, Hogg RJ. Modulation of growth factors by growth hormone in children with chronic renal failure. the southwest pediatric nephrology study group. Kidney Int. 1997;51:1970–9.
Ding H, Gao XL, Hirschberg R, Vadgama JV, Kopple JD. Impaired actions of insulin-like growth factor 1 on protein synthesis and degradation in skeletal muscle of rats with chronic renal failure. evidence for a postreceptor defect. J Clin Invest. 1996;97:1064–75.
Tonshoff B, Blum WF, Wingen AM, Mehls O. Serum insulin-like growth factors (IGFs) and IGF binding proteins 1, 2, and 3 in children with chronic renal failure: Relationship to height and glomerular filtration rate. the european study group for nutritional treatment of chronic renal failure in childhood. J Clin Endocrinol Metab. 1995;80:2684–91.
Blum WF, Ranke MB, Kietzmann K, Tonshoff B, Mehls O. Growth hormone resistance and inhibition of somatomedin activity by excess of insulin-like growth factor binding protein in uraemia. Pediatr Nephrol. 1991;5:539–44.
Powell DR, Liu F, Baker BK, Hinzt RL, Kale A, Suwanichkul A, Durham SK. Effect of chronic renal failure and growth hormone therapy on the insulin-like growth factors and their binding proteins. Pediatr Nephrol. 2000;14:579–83.
Ulinski T, Mohan S, Kiepe D, Blum WF, Wingen AM, Mehls O, Tonshoff B. Serum insulin-like growth factor binding protein (IGFBP)-4 and IGFBP-5 in children with chronic renal failure: Relationship to growth and glomerular filtration rate. the european study group for nutritional treatment of chronic renal failure in childhood. german study group for growth hormone treatment in chronic renal failure. Pediatr Nephrol. 2000;14:589–97.
Lee DY, Park SK, Yorgin PD, Cohen P, Oh Y, Rosenfeld RG. Alteration in insulin-like growth factor-binding proteins (IGFBPs) and IGFBP-3 protease activity in serum and urine from acute and chronic renal failure. J Clin Endocrinol Metab. 1994;79:1376–82.
Tonshoff B, Kiepe D, Ciarmatori S. Growth hormone/insulin-like growth factor system in children with chronic renal failure. Pediatr Nephrol. 2005;20:279–89.
Schaefer F, Hamill G, Stanhope R, Preece MA, Scharer K. Pulsatile growth hormone secretion in peripubertal patients with chronic renal failure. cooperative study group on pubertal development in chronic renal failure. J Pediatr. 1991;119:568–77.
Olney RC. Mechanisms of impaired growth: effect of steroids on bone and cartilage. Horm Res. 2009;72(Suppl. 1):30–5.
Baron J, Klein KO, Colli MJ, Yanovski JA, Novosad JA, Bacher JD, Cutler GB Jr. Catch-up growth after glucocorticoid excess: a mechanism intrinsic to the growth plate. Endocrinology. 1994;135:1367–71.
Klaus G, Jux C, Fernandez P, Rodriguez J, Himmele R, Mehls O. Suppression of growth plate chondrocyte proliferation by corticosteroids. Pediatr Nephrol. 2000;14:612–5.
Haffner D, Leifheit-Nestler M. CKD-MBD post kidney transplantation. Pediatr Nephrol. 2021;36:41–50.
Bertram JF, Goldstein SL, Pape L, Schaefer F, Shroff RC, Warady BA. Kidney disease in children: latest advances and remaining challenges. Nat Rev Nephrol. 2016;12:182–91.
Lurbe E, Agabiti-Rosei E, Cruickshank JK, Dominiczak A, Erdine S, Hirth A, Invitti C, Litwin M, Mancia G, Pall D, Rascher W, Redon J, Schaefer F, Seeman T, Sinha M, Stabouli S, Webb NJ, Wuhl E, Zanchetti A. 2016 European society of hypertension guidelines for the management of high blood pressure in children and adolescents. J Hypertens. 2016;34:1887–920.
van den Belt SM, HJL H, Gracchi V, de Zeeuw D, Wuhl E, Schaefer F, ESCAPE Trial Group. Early proteinuria lowering by angiotensin-converting enzyme inhibition predicts renal survival in children with CKD. J Am Soc Nephrol. 2018;29:2225–33.
Rees L, Shaw V, Qizalbash L, Anderson C, Desloovere A, Greenbaum L, Haffner D, Nelms C, Oosterveld M, Paglialonga F, Polderman N, Renken-Terhaerdt J, Tuokkola J, Warady B, Walle JV, Shroff R, Pediatric Renal Nutrition Taskforce. Delivery of a nutritional prescription by enteral tube feeding in children with chronic kidney disease stages 2–5 and on dialysis-clinical practice recommendations from the pediatric renal nutrition taskforce. Pediatr Nephrol. 2020;
Behnisch R, Kirchner M, Anarat A, Bacchetta J, Shroff R, Bilginer Y, Mir S, Caliskan S, Paripovic D, Harambat J, Mencarelli F, Buscher R, Arbeiter K, Soylemezoglu O, Zaloszyc A, Zurowska A, Melk A, Querfeld U, Schaefer F, and the 4C Study Consortium. Determinants of statural growth in european children with chronic kidney disease: findings from the cardiovascular comorbidity in children with chronic kidney disease (4C) study. Front Pediatr. 2019;7:278.
Marlais M, Stojanovic J, Jones H, Cleghorn S, Rees L. Catch-up growth in children with chronic kidney disease started on enteral feeding after 2 years of age. Pediatr Nephrol. 2020;35:113–8.
Coleman JE, Watson AR. Gastrostomy buttons for nutritional support in children with cystinosis. Pediatr Nephrol. 2000;14:833–6.
Ledermann SE, Shaw V, Trompeter RS. Long-term enteral nutrition in infants and young children with chronic renal failure. Pediatr Nephrol. 1999;13:870–5.
Neu AM, Bedinger M, Fivush BA, Warady BA, Watkins SL, Friedman AL, Brem AS, Goldstein SL, Frankenfield DL. Growth in adolescent hemodialysis patients: data from the centers for medicare & medicaid services ESRD clinical performance measures project. Pediatr Nephrol. 2005;20:1156–60.
Quinlan C, Bates M, Sheils A, Dolan N, Riordan M, Awan A. Chronic hemodialysis in children weighing less than 10 kg. Pediatr Nephrol. 2013;28:803–9.
Rees L, Schaefer F, Schmitt CP, Shroff R, Warady BA. Chronic dialysis in children and adolescents: challenges and outcomes. Lancet Child Adolesc Health. 2017;1:68–77.
Shroff R, Wright E, Ledermann S, Hutchinson C, Rees L. Chronic hemodialysis in infants and children under 2 years of age. Pediatr Nephrol. 2003;18:378–83.
Chadha V, Blowey DL, Warady BA. Is growth a valid outcome measure of dialysis clearance in children undergoing peritoneal dialysis? Perit Dial Int. 2001;21(Suppl. 3):179.
Vidal E, Edefonti A, Murer L, Gianoglio B, Maringhini S, Pecoraro C, Sorino P, Leozappa G, Lavoratti G, Ratsch IM, Chimenz R, Verrina E, Italian Registry of Paediatric Chronic Dialysis. Peritoneal dialysis in infants: the experience of the italian registry of paediatric chronic dialysis. Nephrol Dial Transplant. 2012;27:388–95.
Schaefer F, Klaus G, Mehls O. Peritoneal transport properties and dialysis dose affect growth and nutritional status in children on chronic peritoneal dialysis. mid-european pediatric peritoneal dialysis study group. J Am Soc Nephrol. 1999;10:1786–92.
Tom A, McCauley L, Bell L, Rodd C, Espinosa P, Yu G, Yu J, Girardin C, Sharma A. Growth during maintenance hemodialysis: impact of enhanced nutrition and clearance. J Pediatr. 1999;134:464–71.
Katz A, Bock GH, Mauer M. Improved growth velocity with intensive dialysis. consequence or coincidence? Pediatr Nephrol. 2000;14:710–2.
Fischbach M, Zaloszyc A, Laetitia H, Menouer S, Terzic J. Why does three times per week hemodialysis provide inadequate dialysis for children? Hemodial Int. 2014;18(Suppl 1):39.
Fischbach M, Fothergill H, Seuge L, Zaloszyc A. Dialysis strategies to improve growth in children with chronic kidney disease. J Ren Nutr. 2011;21:43–6.
Fischbach M, Terzic J, Menouer S, Dheu C, Seuge L, Zalosczic A. Daily on line haemodiafiltration promotes catch-up growth in children on chronic dialysis. Nephrol Dial Transplant. 2010;25:867–73.
Fischbach M, Fothergill H, Zaloszyc A, Menouer S, Terzic J. Intensified daily dialysis: the best chronic dialysis option for children? Semin Dial. 2011;24:640–4.
Shroff R, Smith C, Ranchin B, Bayazit AK, Stefanidis CJ, Askiti V, Azukaitis K, Canpolat N, Agbas A, Aitkenhead H, Anarat A, Aoun B, Aofolaju D, Bakkaloglu SA, Bhowruth D, Borzych-Duzalka D, Bulut IK, Buscher R, Deanfield J, Dempster C, Duzova A, Habbig S, Hayes W, Hegde S, Krid S, Licht C, Litwin M, Mayes M, Mir S, Nemec R, Obrycki L, Paglialonga F, Picca S, Samaille C, Shenoy M, Sinha MD, Spasojevic B, Stronach L, Vidal E, Vondrak K, Yilmaz A, Zaloszyc A, Fischbach M, Schmitt CP, Schaefer F. Effects of hemodiafiltration versus conventional hemodialysis in children with ESKD: The HDF, heart and height study. J Am Soc Nephrol. 2019;30:678–91.
Laster ML, Fine RN. Growth following solid organ transplantation in childhood. Pediatr Transplant. 2014;18:134–41.
Pape L, Ehrich JH, Zivicnjak M, Offner G. Growth in children after kidney transplantation with living related donor graft or cadaveric graft. Lancet. 2005;366:151–3.
Grenda R. Steroid withdrawal in renal transplantation. Pediatr Nephrol. 2013;28:2107–12.
Hocker B, Weber LT, Feneberg R, Drube J, John U, Fehrenbach H, Pohl M, Zimmering M, Frund S, Klaus G, Wuhl E, Tonshoff B. Improved growth and cardiovascular risk after late steroid withdrawal: 2-year results of a prospective, randomised trial in paediatric renal transplantation. Nephrol Dial Transplant. 2010;25:617–24.
Webb NJ, Douglas SE, Rajai A, Roberts SA, Grenda R, Marks SD, Watson AR, Fitzpatrick M, Vondrak K, Maxwell H, Jaray J, Van Damme-Lombaerts R, Milford DV, Godefroid N, Cochat P, Ognjanovic M, Murer L, McCulloch M, Tonshoff B. Corticosteroid-free kidney transplantation improves growth: 2-year follow-up of the TWIST randomized controlled trial. Transplantation. 2015;99:1178–85.
Grenda R, Watson A, Trompeter R, Tonshoff B, Jaray J, Fitzpatrick M, Murer L, Vondrak K, Maxwell H, van Damme-Lombaerts R, Loirat C, Mor E, Cochat P, Milford DV, Brown M, Webb NJ. A randomized trial to assess the impact of early steroid withdrawal on growth in pediatric renal transplantation: the TWIST study. Am J Transplant. 2010;10:828–36.
Billing H, Burmeister G, Plotnicki L, Ahlenstiel T, Fichtner A, Sander A, Hocker B, Tonshoff B, Pape L. Longitudinal growth on an everolimus- versus an MMF-based steroid-free immunosuppressive regimen in paediatric renal transplant recipients. Transpl Int. 2013;26:903–9.
Schmitt CP, Ardissino G, Testa S, Claris-Appiani A, Mehls O. Growth in children with chronic renal failure on intermittent versus daily calcitriol. Pediatr Nephrol. 2003;18:440–4.
Sanchez CP, He YZ. Bone growth during daily or intermittent calcitriol treatment during renal failure with advanced secondary hyperparathyroidism. Kidney Int. 2007;72:582–91.
Shroff R, Wan M, Nagler EV, Bakkaloglu S, Cozzolino M, Bacchetta J, Edefonti A, Stefanidis CJ, Vande Walle J, Ariceta G, Klaus G, Haffner D, Schmitt CP, European Society for Paediatric Nephrology Chronic Kidney Disease Mineral and Bone Disorders and Dialysis Working Groups. Clinical practice recommendations for treatment with active vitamin D analogues in children with chronic kidney disease stages 2–5 and on dialysis. Nephrol Dial Transplant. 2017;32:1114–27.
Shroff R, Wan M, Nagler EV, Bakkaloglu S, Fischer DC, Bishop N, Cozzolino M, Bacchetta J, Edefonti A, Stefanidis CJ, Vande Walle J, Haffner D, Klaus G, Schmitt CP, European Society for Paediatric Nephrology Chronic Kidney Disease Mineral and Bone Disorders and Dialysis Working Groups. Clinical practice recommendations for native vitamin D therapy in children with chronic kidney disease stages 2–5 and on dialysis. Nephrol Dial Transplant. 2017;32:1098–113.
Warady BA, Ng E, Bloss L, Mo M, Schaefer F, Bacchetta J. Cinacalcet studies in pediatric subjects with secondary hyperparathyroidism receiving dialysis. Pediatr Nephrol. 2020;35:1679–97.
Nakagawa K, Perez EC, Oh J, Santos F, Geldyyev A, Gross ML, Schaefer F, Schmitt CP. Cinacalcet does not affect longitudinal growth but increases body weight gain in experimental uraemia. Nephrol Dial Transplant. 2008;23:2761–7.
Bacchetta J, Schmitt CP, Ariceta G, Bakkaloglu SA, Groothoff J, Wan M, Vervloet M, Shroff R, Haffner D, European Society for Paediatric Nephrology and the Chronic Kidney Disease-Mineral and Bone Disorders and Dialysis Working Group of the, ERA-EDTA. Cinacalcet use in paediatric dialysis: a position statement from the european society for paediatric nephrology and the chronic kidney disease-mineral and bone disorders working group of the ERA-EDTA. Nephrol Dial Transplant. 2020;35:47–64.
Mehls O, Ritz E, Hunziker EB, Eggli P, Heinrich U, Zapf J. Improvement of growth and food utilization by human recombinant growth hormone in uremia. Kidney Int. 1988;33:45–52.
Kovacs G, Fine RN, Worgall S, Schaefer F, Hunziker EB, Skottner-Lindun A, Mehls O. Growth hormone prevents steroid-induced growth depression in health and uremia. Kidney Int. 1991;40:1032–40.
Powell DR, Durham SK, Liu F, Baker BK, Lee PD, Watkins SL, Campbell PG, Brewer ED, Hintz RL, Hogg RJ. The insulin-like growth factor axis and growth in children with chronic renal failure: a report of the southwest pediatric nephrology study group. J Clin Endocrinol Metab. 1998;83:1654–61.
Hodson EM, Willis NS, Craig JC. Growth hormone for children with chronic kidney disease. Cochrane Database Syst Rev. 2012;(2):CD003264.
Haffner D, Wuhl E, Schaefer F, Nissel R, Tonshoff B, Mehls O. Factors predictive of the short- and long-term efficacy of growth hormone treatment in prepubertal children with chronic renal failure. the german study group for growth hormone treatment in chronic renal failure. J Am Soc Nephrol. 1998;9:1899–907.
Fine RN, Kohaut E, Brown D, Kuntze J, Attie KM. Long-term treatment of growth retarded children with chronic renal insufficiency, with recombinant human growth hormone. Kidney Int. 1996;49:781–5.
Hokken-Koelega A, Mulder P, De Jong R, Lilien M, Donckerwolcke R, Groothof J. Long-term effects of growth hormone treatment on growth and puberty in patients with chronic renal insufficiency. Pediatr Nephrol. 2000;14:701–6.
Wuhl E, Haffner D, Nissel R, Schaefer F, Mehls O. Short dialyzed children respond less to growth hormone than patients prior to dialysis. german study group for growth hormone treatment in chronic renal failure. Pediatr Nephrol. 1996;10:294–8.
Mehls O, Lindberg A, Nissel R, Haffner D, Hokken-Koelega A, Ranke MB. Predicting the response to growth hormone treatment in short children with chronic kidney disease. J Clin Endocrinol Metab. 2010;95:686–92.
Fine RN, Attie KM, Kuntze J, Brown DF, Kohaut EC. Recombinant human growth hormone in infants and young children with chronic renal insufficiency. genentech collaborative study group. Pediatr Nephrol. 1995;9:451–7.
Maxwell H, Rees L. Recombinant human growth hormone treatment in infants with chronic renal failure. Arch Dis Child. 1996;74:40–3.
Santos F, Moreno ML, Neto A, Ariceta G, Vara J, Alonso A, Bueno A, Afonso AC, Correia AJ, Muley R, Barrios V, Gomez C, Argente J. Improvement in growth after 1 year of growth hormone therapy in well-nourished infants with growth retardation secondary to chronic renal failure: Results of a multicenter, controlled, randomized, open clinical trial. Clin J Am Soc Nephrol. 2010;5:1190–7.
Fine RN, Brown DF, Kuntze J, Wooster P, & Kohau7t EE (1996) Growth after discontinuation of recombinant human growth hormone therapy in children with chronic renal insufficiency. the genentech cooperative study group. J Pediatr 129: 883–891.
Fine RN, Ho M, Tejani A, Blethen S. Adverse events with rhGH treatment of patients with chronic renal insufficiency and end-stage renal disease. J Pediatr. 2003;142:539–45.
Picca S, Cappa M, Martinez C, Moges SI, Osborn J, Perfumo F, Ardissino G, Bonaudo R, Montini G, Rizzoni G. Parathyroid hormone levels in pubertal uremic adolescents treated with growth hormone. Pediatr Nephrol. 2004;19:71–6.
Akchurin OM, Schneider MF, Mulqueen L, Brooks ER, Langman CB, Greenbaum LA, Furth SL, Moxey-Mims M, Warady BA, Kaskel FJ, Skversky AL. Medication adherence and growth in children with CKD. Clin J Am Soc Nephrol. 2014;9:1519–25.
Kovacs GT, Oh J, Kovacs J, Tonshoff B, Hunziker EB, Zapf J, Mehls O. Growth promoting effects of growth hormone and IGF-I are additive in experimental uremia. Kidney Int. 1996;49:1413–21.
Mauras N, Gonzalez de Pijem L, Hsiang HY, Desrosiers P, Rapaport R, Schwartz ID, Klein KO, Singh RJ, Miyamoto A, Bishop K. Anastrozole increases predicted adult height of short adolescent males treated with growth hormone: a randomized, placebo-controlled, multicenter trial for one to three years. J Clin Endocrinol Metab. 2008;93:823–31.
Miller BS, Ross J, Ostrow V. Height outcomes in children with growth hormone deficiency and idiopathic short stature treated concomitantly with growth hormone and aromatase inhibitor therapy: data from the ANSWER program. Int J Pediatr Endocrinol. 2020, 2020:19-z. Epub 2020 Oct 6
Saroufim R, Eugster EA. Non-GH agents and novel therapeutics in the management of short stature. Indian J Pediatr. 2021; https://doi.org/10.1007/s12098-021-03824-3. Online ahead of print
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Haffner, D., Rees, L. (2023). Growth and Puberty in Chronic Kidney Disease. In: Schaefer, F., Greenbaum, L.A. (eds) Pediatric Kidney Disease. Springer, Cham. https://doi.org/10.1007/978-3-031-11665-0_56
Download citation
DOI: https://doi.org/10.1007/978-3-031-11665-0_56
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-11664-3
Online ISBN: 978-3-031-11665-0
eBook Packages: MedicineMedicine (R0)