
Abstract
DNA-binding proteins (DBPs) have an important role in various regulatory tasks. In recent years, with developing of deep learning, many fields like natural language processing, computer vision and so on have achieve great success. Some great model, for example DeepBind, brought deep learning to motif discovery and also achieve great success in predicting DNA-transcription factor binding, aka motif discovery. But these methods required integrating multiple features with raw DNA sequences such as secondary structure and their performances could be further improved. In this paper, we propose an efficient and simple neural network-based architecture, DBPCNN, integrating conservation scores and epigenomic data to raw DNA sequences for predicting in-vitro DNA protein binding sequence. We show that conservation scores and epigenomic data for raw DNA sequences can significantly improve the overall performance of the proposed model. Moreover, the automatic extraction of the DBA-binding proteins can enhance our understanding of the binding specificities of DBPs. We verify the effectiveness of our model on 20 motif datasets from in-vitro protein binding microarray data. More specifically, the average area under the receiver operator curve (AUC) was improved by 0.58% for conservation scores, 1.29% for MeDIP-seq, 1.20% for histone modifications respectively, and 2.19% for conservation scores, MeDIP-seq and histone modifications together. And the mean average precision (AP) was increased by 0.62% for conservation scores, 1.46% for MeDIP-seq, 1.27% for histone modifications respectively, and 2.29% for conservation scores, MeDIP-seq and histone modifications together.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Convolution neural network
- Deep learning
- Conversation scores
- Epigenomic data
- One-hot representation
- Motif discovery
- DNA-binding protein
- Transcription factor
1 Introduction
DNA-binding proteins (DBPs), or transcription factors [1], play an important role in cell biological processes including transcription, translation, repair, and replication machinery [2,3,4]. In addition, it has also been reported that some genomic variants in TFBSs are related to serious diseases [5]. Therefore, discovering transcription factor binding site (TFBS), a subsequence of DNA where the binding between the DBPs and its DNA subsequence targets take place, is crucial for further understanding of the transcriptional regulation mechanism in gene expression. A better understanding of protein-DNA binding preferences helps to annotate and study the function of cis-regulatory elements, and identifying in-vitro protein-DNA binding sites is the first step in understanding protein-DNA binding preferences [6].
With the development of high-throughput sequencing technologies, especially protein binding microarrays (PBMs [7]), it provides a large amount of in-vitro binding data to help us study in-vitro protein-DNA binding preferences. The elements in PBMs represent a probability distribution over DNA alphabet {A, C, G, and T} for each position in motif sequence. There are many detection technologies to study protein-DNA binding preferences from raw DNA sequences based on PBMs [8]. However, these methods assume that the nucleotides in the binding site are independently contributed to the calculation of the binding preference and have nothing to do with the nucleotides in other positions. Dependencies between nucleotides can be explicitly encoded by kmers [9, 10], and the result shows that using kmers as encoding rule is better than PBMs. But these methods have some weak points, like having difficulty in handling large-scale data, poor generalization performance and so on. With the rapid development of deep learning in recent years, new computational methods such as convolutional neural networks (CNNs) and recurrent neural networks (RNNs) have shown their superior ability in predicting protein-DNA binding sites [11,12,13,14,15,16,17,18,19,20]. Also, there are some research works by data processing [21,22,23,24,25]. DeepBind is the earliest attempts to apply deep learning to the motif discovery task and has proved to be an effective model. But them only use raw DNA sequences as input data, Various studies showed that transcription factor binding sites are conserved among species [26,27,28,29,30]. Conservation scores [31] and epigenomic data [32] could be a nice data supplement to raw DNA sequences. In other words, integrating conservation scores and epigenomic data to raw DNA sequences can help us study in-vitro protein-DNA binding preferences.
In this paper, we first focus on in-depth exploitation of deep convolution neural network with application on in-vitro motif discovery task in Sect. 2. We call our model DBPCNN, which uses CNNs extract features from input data, i.e. raw DNA sequences, conservation scores and epigenomic data, and then train model to predict DNA-protein binding sites. Then we will show some experiment results in Sect. 3 and discuss the promotion of conservation scores and epigenomic data. At last, we have a concise summary and future outlook for further research.
2 Materials and Methods
In this section, we first introduce the relevant in-vitro DNA protein binding dataset, evolutionary information, epigenomic data and its data preprocessing procedure. Second, architecture of our deep convolution network namely eDeepCNN is presented in detail. Third, we give a briefing of evaluation metric and training hyper-parameters in our experiment.
2.1 Dataset and Preprocessing
2.1.1 DNA Sequence
We downloaded 20 universal protein binding microarrays (uPBMs) datasets from the DREAM5 project [20], which comes from a variety of protein families. Each TF dataset, consisting of ~40,000 unaligned 35-mer probe sequences, comprises a complete set of PBM probe intensities from two distinct microarray designs named HK and ME. These datasets have been normalized according to the total signal intensity.
2.1.2 Evolutionary Information and Epigenomic Data
The evolutionary information was obtained from (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/phyloP100way/) where we used the conservation scores of multiple alignments of 99 vertebrate genomes to the human genome.These scores were obtained from the PHAST package (http://compgen.bscb.cornell.edu/phast/). The values of these scores were scaled to 0–1. In this paper, we use two kinds of data, i.e. MeDIP-seq and histone modifications. The information was obtained from ENCODE Epigenetics database (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/wgEncodeRegMarkH3k27ac/).
2.1.3 Data Preprocessing
To accurately evaluate the performance of our proposed method, five-fold cross-validation strategy was adopted in this paper. Five-fold cross-validation strategy repeated five times in total. Within each time, TF dataset was randomly divided into 5 folds of roughly equal size, and four of them were used as the training data while the rest was used as the test data. During training, we randomly sampled 1/8 of the training set as the validation set.
Each input RNA sequence S = (s1, s2,…, sn) was one-hot encoded. Thus, A, C, G, T, and N were encoded as (1000), (0100), (0010), (0001), and (0000) respectively. The length of the input sequence is n = 101nt. In addition to one-hot encoding, we added conservation (evolutionary) information (Convs), MeDIP-seq (MDS) and histone modifications (HMS) of each nucleotide of the input sequence. Thus, each input sequence S with n nucleotides is encoded as n × 7 such as four channels for one-hot encoding and the other three channels for conservation scores, MeDIP-seq and histone modifications respectively.
2.2 Network Architecture
DeepBind [20] introduced a single layer convolution neural network followed by a max global pooling layer to extract sequence features in motif discovery, which was proved to be a great success.
The length of the transcription factor binding sites in eukaryotes ranges from 5nt to 30nt as reported by Stewart et al. [33]. Therefore, the input length of the proposed models is set to 101nt. Each sequence is centered on the transcription factor binding site and the additional nucleotides were used for providing contextual information.

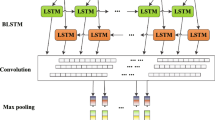
An overview of the DBPCNN model. A raw DNA sequence is first encoded into a one-hot matrix and MDS, HMS, Convs. The first convolutional layer computes a score for all potential local motif. The second convolution layer discovers the interactions between the learned motifs of the first convolution layer. The learned features from the convolution layers go through fully connected layers with a softmax layer at the output for prediction.
Therefore, we proposed a deeper neural network model composed of two convolution layers accompanied by dropout and local pooling strategies, namely DBPCNN. The first convolutional layer computes a score for all potential local motif, which is the same as DeepBind. And we design the second convolutional layers, in the hope that it can capture the interaction pattern in neighboring sequence. The second convolution layer takes the motif score sequence computed by the first convolution layer as input and recognizes the distribution pattern of the motif score sequence, which, in other words, takes the interaction of the local motifs into consideration. Combining multiply convolution layer improves the receptive field of DBPCNN model and allows an overall pattern recognition of the candidate sequence. Each convolution layer is followed by a local max pooling layer and a dropout layer. It should be noticed that dropout strategy plays an important role in our model, in the light of the overfitting risk accompanied by the expanding parameter size and model complexity. A global max pooling layer is used to capture the global context information of DNA sequences and feeds it into a two layer fully connected neural network to obtain final prediction.
The convolution layer is a one-dimensional convolution expressed in Eq. (1). I is the input, o and k are the indices of the output position and the kernels, respectively, and Wk is the weight matrix of S × N shape with S filters and N input channels.
The fully connected layer is expressed in Eq. (2).
The dropout layer is added to switch off certain neurons at training time in order to reduce overfitting. Adding dropout after fully connected layer results in Eq. (3) where mi is sampled form Bernoulli distribution.
The rectified linear unit activation function was used in this design and it is given in Eq. (4). ReLU function introduces non-linear features to DBPCNN model.
The final layer is the softmax layer that normalizes its input vector z into a probability distribution having M probabilities proportional to the exponential of the input numbers, expressed by Eq. (5).
Figure 1 plots a graphical illustration of DBPCNN and the detailed parameter settings including convolution kernel size and number of filters in each layer are listed in Table 1. Input data is (B, 101, 7). It should be mentioned that part of our hyper-parameter settings inherent from classic deep learning methods in motif discovery like DeepBind, which have proved to be optimal choices, while some other parts were chosen from hyper-parameter grid search in training procedure.
2.3 Evaluation Metric
We select positive and negative samples with the ratio 1:1. Our DBPCNN model uses AUC (Area under the Curve of ROC) as metric evaluation. In the binary classification problem, it is generally said that the category which is predicted positive is positive, while the category which is predicted negative is negative. If the prediction is correct, the result is true, and if the prediction is wrong, the result is false (True). For a two-category prediction problem, combining the above four cases, you can get the confusion matrix shown in Table 2. We can draw ROC curve according to confusion matrix.
2.4 Experiment Setting
The learnable parameters (e.g. weights and bias) in neural network were initialized by Glorot uniform initializer [34], and optimized by Adam [35] algorithm with a mini-batch-size of 100. We implemented grid search strategy over some sensitive hyper-parameters, i.e. dropout ratio, L2 weight decay, and momentum in SGD optimizer. An early stopping strategy was also adopted to fight against overfitting problem in our model. Detailed hyper-parameter setting is listed in Table 3.
3 Results and Analysis
3.1 Results Display
In order to verify the effectiveness of conservation scores (Convs), MeDIP-seq (MDS) and histone modifications (HMS), we conduct series of experiments. We use different data as model input, i.e. raw DNA sequences, raw DNA sequences + Convs, raw DNA sequences + MDS, raw DNA sequences + HMS, raw DNA sequences + Convs + MDS + HMS respectively. The result of comparison is illustrated in Fig. 2, and Fig. 3.

A scatter plot comparing the achieved AUC (left) and AP (right) of the proposed model DBPCNN using raw DNA sequences only and by integrating Convs, MDS, HMS respectively to raw DNA sequences.

The comparison of the performance of DBPCNN with different input data in term of average AUC and average AP.
3.2 Effect of Conservation Scores (Convs), MeDIP-seq (MDS), Histone Modifications (HMS)
In order to study the importance of adding evolutionary information, we trained the DBPCNN model using raw DNA sequences only. For a fair comparison, we have searched the best hyper-parameters again in the case of using raw DNA sequences only using similar grid search parameters as shown in Table 3. The average AUC of using raw DNA sequence only was 88.00% while it was 88.58% integrating the conservation scores, 89.29% integrating the MeDIP-seq, and 89.20% integrating the histone modifications to raw DNA sequences respectively. On the other hand, the mean AP of using raw DNA sequences only was 88.45% while it was 89.07% integrating the conservation scores to raw DNA sequences, 89.91% integrating the MeDIP-seq, and 89.72% integrating the histone modifications to raw DNA sequences respectively. Thus, adding conservation scores to the raw DNA sequences improved the performance by 0.58% and 0.62% in terms of AUC and AP respectively, MeDIP-seq by 1.29% and 1.46%, and histone modifications by 1.20% and 1.27%. The Figs. 3 show that AUC and AP scores of all 20 in-vitro uPBM datasets experiments were improved by integrating the conservation scores with raw DNA sequences. Then we conduct experiments integrating the conservation scores, MeDIP-seq, and histone modifications to raw DNA sequences, and the average AUC was 90.19% comparing 88.00% and the average AP was 90.74% comparing 88.45%. There was 2.19% increase to average AUC and 2.29% to average AP.
4 Conclusion and Future Work
Motif discovery is an important process for a better studying of different biological tasks. In this paper, we propose a simple and efficient deep convolution neural network model, namely DBPCNN for predicting in-vitro DNA-protein binding site, integrating the conservation scores, MeDIP-seq, and histone modifications with raw DNA sequences. Integrating three data to DNA sequences respectively can achieve the average AUC and AP, and while including the conservation scores, MeDIP-seq, and histone modifications together to raw DNA sequences, we can get better result comparing only any data.
Although we get outstanding result by integrating the conservation scores, MeDIP-seq, and histone modifications to raw DNA sequences to predict in-vitro DNA-protein binding site, there are many evidences show that shape in local DNA sequence plays an important role in DNA-protein binding process [36,37,38]. And different encoding rules also can influence results [39,40,41,42,43]. As we know, encoding input to embedding vector is a commonly used data-preprocessing way, which can convert sparse vector to dense vector to reduce dimension. Therefore, incorporating the DNA shape information into deep convolution neural network and using embedding method as data-preprocessing way would be a promising method to improve DNA binding site prediction, which would be our future work direction.
References
Lambert, S.A., et al.: The human transcription factors. Cell 172, 650–665 (2018)
Vaquerizas, J.M., Kummerfeld, S.K., Teichmann, S.A., Luscombe, N.M.: A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 10, 252 (2009)
Stormo, G.D.J.B.: DNA binding sites: representation and discovery. Bioinformatics 16, 16–23 (2000)
Lee, T.I., Young, R.A.: Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013)
Zhu, L., Zhang, H.-B., Huang, D.-S.: Direct AUC optimization of regulatory motifs. Bioinformatics 33, i243–i251 (2017)
Tianyin, Z., Ning, et al.: Quantitative modeling of transcription factor binding specificities using DNA shape. Proc. Natl. Acad. Sci. 112–115 (2015)
Berger, M.F., Philippakis, A.A., Qureshi, A.M., et al.: Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24(11), 1429–1435 (2006)
Stormo, G.D., Zhao, Y.: Determining the specificity of protein-DNA interactions. NAT Rev. Genet. 11(11), 751–760 (2010)
Gordân, R., et al.: Genomic regions flanking e-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 3, 1093–1104 (2013)
Fletezbrant, C., Lee, D., Mccallion, A.S., Beer, M.: kmer-SVM: a web server for identifying predictive regulatory sequence features in genomic data sets. Nucleic Acids Res. 41, 544–556 (2013)
Shen, Z., Bao, W., Huang, D.: Recurrent neural network for predicting transcription factor binding sites. Sci. Rep. 8, 15270 (2018)
Zhang, Q., Zhu, L., Bao, W., Huang, D.S.: Weakly-supervised convolutional neural network architecture for predicting protein-DNA binding. IEEE/ACM Trans. Comput. Biol. Bioinform. 17(2), 679–689 (2020)
Zhang, Q., Zhu, L., Huang, D.S.: High-order convolutional neural network architecture for predicting DNA-protein binding sites. IEEE/ACM Trans. Comput. Biol. Bioinform. 16(4), 1184–1192 (2019)
Zhang, Q., Shen, Z., Huang, D.-S.: Modeling in-vivo protein-DNA binding by combining multiple-instance learning with a hybrid deep neural network. Sci. Rep. 9, 8484 (2019)
Xu, W., Zhu, L., Huang, D.S.: DCDE: an efficient deep convolutional divergence encoding method for human promoter recognition. IEEE Trans. NanoBioscience 18(2), 136–145 (2019)
Zhang, H., Zhu, L., Huang, D.S.: DiscMLA: an efficient discriminative motif learning algorithm over high-throughput datasets. IEEE/ACM Trans. Comput. Biol. Bioinform. 15(6), 1810–1820 (2018)
Zhang, H., Zhu, L., Huang, D.S.: WSMD: weakly-supervised motif discovery in transcription factor ChIP-seq data. Sci. Rep. 7 (2017). https://doi.org/10.1038/s41598-017-03554-7
Yu, W., Yuan, C.-A., Qin, X., Huang, Z.-K., Shang, L.: Hierarchical attention network for predicting DNA-protein binding sites. In: Huang, D.-S., Jo, K.-H., Huang, Z.-K. (eds.) ICIC 2019. LNCS, vol. 11644, pp. 366–373. Springer, Cham (2019). https://doi.org/10.1007/978-3-030-26969-2_35
Weirauch, M.T., et al.: Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 31, 126–134 (2013)
Alipanahi, B., Delong, A., Weirauch, M.T., Frey, B.J.: Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015)
Zhu, L., Bao, W.Z., Huang, D.S.: Learning TF binding motifs by optimizing fisher exact test score. IEEE/ACM Trans. Comput. Biol. Bioinform. (2017)
Zhu, L., Zhang, H.-B., Huang, D.S.: LMMO: a large margin approach for optimizing regulatory motifs. IEEE/ACM Trans. Comput. Biol. Bioinform. 15(3), 913–925 (2018)
Zhu, L., Zhang, H.-B., Huang, D.-S.: Direct AUC optimization of regulatory motifs. Bioinformatics 33(14), i243–i251 (2017). https://doi.org/10.1093/bioinformatics/btx255
Zhu, L., Guo, W., Deng, S.-P., Huang, D.S.: ChIP-PIT: Enhancing the analysis of ChIP-Seq data using convex-relaxed pair-wise interaction tensor decomposition. IEEE/ACM Trans. Comput. Biol. Bioinform. 13(1), 55–63 (2016)
Guo, W.L., Huang, D.S.: An efficient method to transcription factor binding sites imputation via simultaneous completion of multiple matrices with positional consistency. Mol. Biosyst. 13, 1827–1837 (2017)
Boffelli, D., et al.: Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299(5611), 1391–1394 (2003)
Bpffelli, D., Nobrega, M.A., Rubin, E.M.: Comparative genomics at the vertebrate extremes. Nat. Rev. Genet. 5(6), 456–465 (2004)
McGuire, A.M., Hughes, J.D., Church, G.M.: Conservation of dna regulatory motifs and discovery of new motifs in microbial genomes. Genome Res. 10(6), 744–757 (2000)
Li, H., Rhodius, V., Gross, C., Siggia, E.D.: Identification of the binding sites of regulatory proteins in bacterial genomes. Proc. Natl. Acad. Sci. 99(18), 11772–11777 (2002)
Woolfe, A., et al.: Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3(1), e7 (2004)
Tayara, H., Chong, K.: Improved predicting of the sequence specificities of RNA binding proteins by deep learning. IEEE/ACM Trans. Comput. Biol. Bioinform. (2020)
Jing, F., Zhang, S.-W., Cao, Z., Zhang, S.: Combining sequence and epigenomic data to predict transcription factor binding sites using deep learning. In: Zhang, F., Cai, Z., Skums, P., Zhang, S. (eds.) ISBRA 2018. LNCS, vol. 10847, pp. 241–252. Springer, Cham (2018). https://doi.org/10.1007/978-3-319-94968-0_23
Stewart, A.J., Hannenhalli, S., Plotkin, J.B.: Why transcription factor binding sites are ten nucleotides long. Genetics 192(3), 973–985 (2012)
Glorot, X., Bengio, Y.: Understanding the difficulty of training deep feedforward neural networks. In: International Conference on Artificial Intelligence and Statistics, pp. 249–256 (2010)
Zeiler, M.D.: ADADELTA: an adaptive learning rate method. arXiv abs/1212.5701 (2012)
Rohs, R., West, S.M., Sosinsky, A., Liu, P., Mann, R.S., Honig, B.: The role of DNA shape in protein–DNA recognition. Nature 461, 1248–1253 (2009)
Zhou, T., et al.: Quantitative modeling of transcription factor binding specificities using DNA shape. Proc. Natl. Acad. Sci. U.S.A. 112, 4654–4659 (2015)
Zhang, Q., Shen, Z., Huang, D.: Predicting in-vitro transcription factor binding sites using DNA sequence + shape. IEEE/ACM Trans. Comput. Biol. Bioinform. 1 (2019)
Tsatsaronis, G., Panagiotopoulou, V.: A generalized vector space model for text retrieval based on semantic relatedness. In: Conference of the European Chapter of the Association for Computational Linguistics, pp. 70–78 (2009)
Wang, J., Huang, P., Zhao, H., Zhang, Z., Zhao, B., Lee, D.L.: Billion-scale commodity embedding for E-commerce recommendation in Alibaba. In: Knowledge Discovery and Data Mining, pp. 839–848 (2018)
Wang, D., Zhang, Q., Yuan, C.-A., Qin, X., Huang, Z.-K., Shang, L.: Motif discovery via convolutional networks with K-mer embedding. In: Huang, D.-S., Jo, K.-H., Huang, Z.-K. (eds.) ICIC 2019. LNCS, vol. 11644, pp. 374–382. Springer, Cham (2019). https://doi.org/10.1007/978-3-030-26969-2_36
Zhu, L., Guo, W.-L., Huang, D.S., Lu, C.-Y.: Imputation of ChIP-seq datasets via low rank convex co-embedding. In: 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pp. 141–144 (2015)
Chen, Z.-H., et al.: Prediction of drug-target interactions from multi-molecular network based on deep walk embedding model. Front. Bioeng. Biotechnol. 8, 338 (2020)
Acknowledgements
This work was supported by the grant of National Key R&D Program of China (No. 2018AAA0100100 & 2018YFA0902600) and partly supported by National Natural Science Foundation of China (Grant nos. 61861146002, 61732012, 61772370, 62002266, 61932008, 61772357, and 62073231) and supported by “BAGUI Scholar” Program and the Scientific & Technological Base and Talent Special Program, GuiKe AD18126015 of the Guangxi Zhuang Autonomous Region of China.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Xu, Y., Zhang, Q., Chen, Z., Yuan, C., Qin, X., Wu, H. (2021). Using Deep Learning to Predict Transcription Factor Binding Sites Combining Raw DNA Sequence, Evolutionary Information and Epigenomic Data. In: Huang, DS., Jo, KH., Li, J., Gribova, V., Premaratne, P. (eds) Intelligent Computing Theories and Application. ICIC 2021. Lecture Notes in Computer Science(), vol 12838. Springer, Cham. https://doi.org/10.1007/978-3-030-84532-2_35
Download citation
DOI: https://doi.org/10.1007/978-3-030-84532-2_35
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-84531-5
Online ISBN: 978-3-030-84532-2
eBook Packages: Computer ScienceComputer Science (R0)