
Abstract
The current study aims to develop robust contextual knowledge of deep-learning methodology for DNA/RNA motif sequence identification and recognition of correct transcription factor-binding sites (TFBS) for gene regulatory mechanisms in humans. Knowledge of the exact sequence specificities of DNA- and RNA-binding to particular transcriptional factors (TF) seems to be an excellent strategy to develop unique deep-learning models for gene regulatory processes. But uncertainty in the sequence specificity of genomic sequences to a particular TFBS is a big issue. It may be possible to resolve this issue using deep-learning techniques, and thus, it will be helpful to gain generalizable domain knowledge of deep-learning architectures, which offers researchers to know better, their performance to select a unified computational approach for the discovery of a selective kind of motif pattern. This scoping review serves to synthesize evidence for DNA/RNA motif sequences binding with transcriptional factor sites using the PRISMA-ScR guidelines (Preferred Reporting Items for Systematic reviews and Meta-Analyses of Scoping Reviews) to better understand and further assessment of the scope of literature on DNA or RNA motif mining using deep-learning methods. A deep-learning architecture literature survey for DNA and RNA sequence specificity for human ChIP-seq (Chromatin Immuno-Precipitation sequence), DNase-seq (DNase hypersensitive site sequence), CLIP-seq (Cross Linking Immuno-Precipitation sequence), ATAC-seq (Assay for Transposase-Accessible Chromatin sequence), etc. datasets, common motif pattern, and their corresponding TF-DNA/RNA-binding site affinities are included in this study. Deep-learning (DL) models have been used to find selective motifs and have been demonstrated to be more reproducible than traditional methods. As per our literature survey, 33 DL models exist to detect DNA/RNA motifs that have varied framework designs and implementation styles. Through literature survey and PRISMA-ScR reporting guidelines, it is easy to analytically evaluate the performances of each DL model in terms of model size, automatic calibration ability, tool selection, and training set, and it has been found that the DESSO (DEep Sequence and Shape mOtif), DeepFinder, and DeepBind are the selective DL models that are appropriate to study the true biological relationship, especially concerning gene expression patterns and sequence analysis. This study concludes that the application of existing deep-learning methods in the field of motif discovery is the faster way to process complex data relevant to genomic sequences. Through the PRISMA-ScR reporting guidelines and literature survey analysis, more than 30 existing deep-learning models are compared, and it is concluded that complex DL models are preferred over simpler DL models in terms of performance and scalability evaluation. Selective selection of a DL model architecture can be made to understand the complex behavior of motifs and their associated regulatory mechanism at the gene level.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
DNA/RNA interactions with proteins play an important part in the gene expression regulatory mechanism, involving transcription, translation, alternative splicing, and degradation (Huang et al. 2014; Zhu et al. 2015). Both DNA/RNA have short regulatory sequences known as transcriptional factors (TFs). Interaction between the biomolecules in the presence of specific TFs is the basic and foremost criterion in gene regulation (Zhu et al. 2013). Usually, TFs are shorter sequences, typically ranging from a few to approximately 20 bp (base pairs), localized in regulatory regions of genes.
Separate proteins have a specific TF with a characteristic binding capacity to the complementary genomic sequence, which is due to the presence of a short guided and recurring pattern of sequences also regarded as motifs. The presence of such short conservative sequences in genomic sequences specifically indicates the binding sites for particular proteins such as nucleases and TFs.
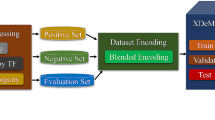
Nevertheless, RNA motifs are also involved in numerous significant RNA processes, including ribosomal binding and mRNA processing, and are typically useful in characterizing genomic regulatory pathways and decoding the regulatory code of different genes. Thereby, motif discovery acts as an important tool for computational biology in the post-genomic era (Dhaeseleer 2006). Similarly, motif discovery is imperative in providing insights into other primary problems like amyloid illnesses and has many applications in pharmaceutical and industrial purposes (Nair et al. 2012). However, the motif sequence specificity for the correct transcription factor-binding site (TFBS) identification is more accurately diagnosed using reliable and reproducible high-throughput sequencing technology with computational methods (Nutiu et al. 2011; Siggers and Gordan 2013). A review of the traditional techniques and algorithms employed for motif discovery can be found in Das and Dai (2007) and Hashim et al. (2019). The basic principles behind motif elicitation are threefold (Hashim et al. 2019), viz., data preprocessing, motif search, and motif evaluation (see Fig. 1). During the first phase, sequence data downloaded from motif databases, TFBS datasets, or other high-throughput experiment datasets are often clustered using state-of-the-art clustering algorithms to categorize the dataset based on some criteria. Then, data-cleaning procedures are performed on the clusters so that the effects of biases and noise are reduced satisfactorily. During the second and most important phase, the motif algorithms work on cleaned and clustered data to find conserved motifs. An encoding scheme for motif representation is applied to the data so that the chosen algorithm can work on the data efficiently through a scoring mechanism or scoring function to find statistically significant motif patterns. In the last stage, the elicited motifs are evaluated against known motif databases to determine the performance and accuracy of the motif discovery algorithm. Different flavors of motif search models use different motif discovery procedures in the second phase. Conventional methods such as the probabilistic approach and word enumeration techniques are now being replaced or augmented by neural network models in the second phase.

A basic flow diagram of the motif search process
In this aspect, the powerful machine learning concepts of “deep-learning” (DL) technology have been developed, which is typically built on the concept of convolutional neural networks (CNN). Their development is essential to capture the motif discovery relevant information which is used to define the selective transcriptional factor-binding sites accurately and the selection of appropriate computational biology (Alipanahi et al. 2015; Hassanzadeh and Wang 2016; Quang and Xie 2016; Zhou and Troyanskaya 2015). Conventional biological experimental techniques are less advantageous than modernized computational methods as they are simple to operate, cost-effective, and less tedious concerning motif research.
1.1 Motif mining with deep learning
The main task of motif finding is to interpret the complex behavior of motifs. This task can become difficult if one selects an inferior grade of experimental methodology randomly and therefore, being able to locate accurately, the binding specificity of TFs with variant motifs becomes problematic. Conversely, DL approaches can train on the various high-throughput datasets concerning biological research, especially to understand the regulatory changes that are directly concerned with human health and disease status. Furthermore, deep learning conventionally provides a framework to advance and communicate DL models for diverse genomic sequences (Avsec et al. 2019; Chen et al. 2019) and improve the interpretability of sequences via DL models (Shrikumar et al. 2017; Binder et al. 2021). It allows an automated optimization of network architecture (Zhang et al. 2021a) with improved power of accessibility. Thus, DL models are the most unprecedented technique, especially for elucidating several applications in the field of bioinformatics and computational biology (Eraslan et al. 2019), relying on the basic building blocks of CNNs (Krizhevsky et al. 2017).
The DeepBind application, which is the first of many models that employ DL methods, is used to predict the specificity of protein binding, and this application is based on a CNN (Alipanahi et al. 2015). In addition, some of the hybrid DL models are also used to find the function of particular DNA sequences, of which DanQ is one exemplar (Quang and Xie 2016). Furthermore, a variety of computational models are available that are based on novel convolutional architectures, its best example is a circular filter which is used efficiently to interpret data relevant to transcriptional factor specificity and binding to DNA/RNA (Blum et al. 2019). Many DL methods that came after the DeepBind method employ the CNN model and add some complex models on top of the CNN for gauging the long-term relationships between motif sequences. Some models use RNNs (Recurrent Neural Networks) or their improvements such as the Bi-LSTM (Bidirectional Long Short-Term Memory) networks, SAE (Stacked Autoencoders) instead of CNNs so that variable length input can be provided to the model, and long-term relationships are also captured. Some others use other regulatory elements such as DNA/RNA shape features, chromatin accessibility data, and histone modifications in addition to the convolutional kernels of the baseline CNN model to enhance the interpretability of the model. However, the basic motif discovery phase (Phase II of Fig. 1) consists of several convolution kernels that act like motif sequence finders. The kernel operations are performed across the input sequence such that the motif features are captured for each window of the sequence. DNA encoding as input to these kernels is achieved either via one-hot encoding or k-mer encoding. A general deep-learning framework for motif discovery that summarizes the broad steps involved is given in Fig. 2.

A generalized deep-learning framework for DNA/RNA motif elicitation. Any one or a combination of high-throughput datasets are pre-processed for noise, bias, etc., and encoded using either one-hot or k-mer encoding schemes before being used to train the deep neural network architecture of choice. Several deep neural networks may be combined for greater interpretability, performance, sensitivity, and specificity
A comprehensive meta-analysis of DL architectures via deepRAM can be used to locate the DNA and RNA-binding specificity and provide a valuable exhaustive investigation of the genome for the researcher (Trabelsi et al. 2019). A similar survey that reiterates the work done by Trabelsi et al. (2019) can be found in He et al. (2020). These DL methods are used to find motifs from human ChIP-seq (Chromatin Immuno-Precipitation sequence) data, which have common DNA sequence patterns and their corresponding TF-DNA-binding affinities (Yang et al. 2019). These features can be achieved by combining sequence and shape framework features of DNA (Sutskever et al. 2014) and making it possible to recognize the TFBS and sequence-specific motifs of DNA/RNA as well (Zhang et al. 2019a, b).
However, there are still some drawbacks existing in computational methods in discovering the task of genomic DNA/RNA motif mining. For example, the lack of big data profiles causes researchers to enhance their training datasets. Further, the more complex a DL model becomes, the interpretation of the model suffers. Even after prediction results present themselves to the researcher, it is often not fully understood how these results can be connected to the intricacies of our body’s regulatory networks. Moreover, choosing the correct network architecture along with the correctly tuned hyper-parameters is also very challenging. Thus, it is just as necessary to understand and work on these limitations as is necessary to understand the complex behavior of gene regulatory mechanisms concerning sequence-specific motifs and their respective transcriptional factors binding and affinities to genomic sequences (Das and Dai 2007).
1.2 Brief overview of our study
In this comprehensive review, more details about deep-learning predictive models are highlighted for DNA/RNA motif mining over the past few years, and the attributes of existing learning models are briefly described. The performance of existing DL models concerning predicting the transcriptional factor-binding interactions exactly to genomic DNA and RNA is also briefly explained. It also provides some promising evidence relevant to motif mining through PRISMA reporting and literature survey guidelines. It also includes other models that use datasets in addition to the ChIP-seq/CLIP-seq data, unlike the metanalysis reported by Trabelsi et al. (2019) such as DNase-seq, ATAC-seq, ChIP-exo, and ChIP-nexus. It also provides a methodology review of more than 30 models up to the year 2021 including benchmarking guidelines, whereas previous surveys (Trabelsi et al. 2019; Wang et al. 2020b; He et al. 2020) have included only 20 models. By including models that are scalable and flexible to work with more than one type of sequence dataset, this scoping methodology review intends to present a more comprehensive picture of the DNA/RNA motif mining problem that employs DL techniques. Furthermore, these facts and pieces of evidence of DL methods concerning DNA/RNA motifs are helpful to evaluate recent improvements in computational approaches. To the best of our knowledge, no past reviews have used PRISMA-ScR guidelines for systematic methodology review of human DNA/RNA motif discovery tools and algorithms that use deep-learning architectures with varied high-throughput datasets.
-
Review question: Is the prediction of DL models well suited for identifying the regulatory components and sequence structures that participate in the genomic rearrangement of DNA/RNA?
-
Inclusion criteria: The DL model accurately predicts the DNA/RNA protein sequence specificity pattern for gene regulatory mechanisms operating in a biological system.
-
Focus: The review will focus on short regulatory sequence elements or associated gene changes as a consequence of transcriptional factor binding.
-
Context: All surveyed literature reports are found to be original and peer-reviewed in all languages without date range limitations.
-
Types of sources: This scoping review will consider all full-text research papers, including experimental, case–control, quantitative studies and genome-wide studies, meta-analysis, and targeting the candidate gene studies. In addition, the research will not consider additional variants and non-variant sequences’ role in therapeutic approach development and disease diagnosis that may include an element of human cis-regulatory studies in gene expression.
2 Methods
We conducted a scoping review abiding by the reporting checklist of PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analyses of Scoping Reviews) guidelines (Moher et al. 2009; Tricco et al. 2018; Martin et al. 2020; Peters et al. 2021, 2022) (see Fig. 3). The goal of the bibliometric analysis is to explore motif information and provide an in-depth learning pathway to a biologist to understand the complex chemistry of DNA/RNA sequences. Although, this literature survey search was carried out on the scientific databases from Feb 2022 to June 2022. The strings used to explore the various scientific databases are as follows: (“motif mining” OR “motif discovery”) AND “data” AND “mining” AND (“deep learning techniques” OR “deep learning methods”) AND “load” AND “profil*”). This string is used to expose the items “article title, abstract, keyword, the content” of already existing reports in the literature database of 2012–2021. Meanwhile, following the positive and negative facts associated with motif mining, exploring specific TFBSs in a genomic sequence is a novel and growing field. Such a smart research initiative started around late 2004 (Häussler and Nicolas 2005). In addition, this report considers only research articles relevant to human DNA/RNA motif discovery and their data profiling.

PRISMA Flow Diagram for a scoping review of motif discovery using DL models for human DNA/RNA
2.1 PRISMA-ScR results: motif pattern discovery and transcription factor site binding
The total number (n = 13,801) of literature found in different databases (PubMed, BioMed Central, ScienceDirect, EBSCO, JASPAR, JSTOR, etc.) after the primary search is shown in Fig. 2 PRISMA flow chart. Automation tools help in speeding up systematic reviews and also aid in providing accuracy (Beller et al. 2018; Scott et al. 2021; Harrison et al. 2020), and tools such as LitSuggest (Allot et al. 2021), Abstrackr (Wallace et al. 2012), and Colandr (Cheng et al. 2018) were used for various automation tasks in this study for screening, extracting, eliminating duplicates, etc. After the preliminary identification phase, (n = 5420) are eliminated.
Of the remaining (n = 8381) records, these research articles are screened in PubMed (https://pubmed.ncbi.nlm.nih.gov/), JASPAR (Castro-Mondragon et al. 2021), Nature (https://www.nature.com/), EBSCO (https://www.ebsco.com/), ENCODE (Luo et al. 2019), PLOS (https://plos.org/), ScienceDirect (https://www.sciencedirect.com/), JSTOR (https://www.jstor.org/), UniPROBE (Universal Protein Binding Microarray Resource for Oligonucleotide Binding Evaluation, Hume et al. 2014), and BioMed Central (https://www.biomedcentral.com/). We carefully screened the literature relevant to the genomic datasets. Secondary search results from the ENCODE, UniPROBE, and JASPAR datasets include maximum papers (n = 4051) and it was first indexed to include short communication articles, books, book chapters, presented papers, and journals. On evaluating the results obtained from BioMed Central data which bears articles including research and book chapters and upon going through their titles and abstracts, around (n = 1042) related articles were selected. Analysis of titles and abstracts from all databases using manual screening and automation tools thereby reduces the number of articles to (n = 5316).
During the selection phase from the two search results, some studies excluded titles related to designing personalized therapeutic approaches and disease-associated risk factors as they do not come under the scope of this review. We considered only those articles that are related to genomic studies wherein, genetic and other risk factors with acquired biochemical activity along with genomic profiling are reflected. These factors have a substantial impact on motif profiling results. However, studies relevant to structural changes at the genomic level are also considered in this review, as these changes occur due to histone modifications, chromatin accessibility, or protein-to-protein binding. This is a broad subject area of research than the scope of review. In addition, many articles were concerned with motifs other than human DNA/RNA and corresponding TFs and TFBSs, and others were not relevant to genomic regulation pathways and these were thus excluded from the study. At this juncture, (n = 1209) records are eligible for retrieval. However, in the sorting of the google scholar database, additional (n = 1700) records were found which has a pivotal role in motif finding. These records are similarly screened in the screening phase and after removing duplicates and preprints, the results (n = 388) are merged to give (n = 1597) articles. Of these, (n = 303) studies are relevant to deep-learning frameworks, and (n = 58) are relevant to human DNA/RNA motif discovery from which (n = 33) quantitative studies that present novel DL models for the motif search problem from 2012 to 2021 are selected for inclusion in this review (Fig. 3).
3 Methods and deep-learning models
Several deep-learning model architectures are designed to improve the efficacy of DNA or RNA motif extraction. In this aspect, DL frameworks are designed to locate motifs that are based on low-cost variant CNN (Convolutional Neural Network) models; for example, it includes the Mobile Net family (Sandler et al. 2018), EfficientNet (Tan and Le 2019), CSPNet (Cross Stage Partial Network) (Wang et al. 2020a), and DenseNet (Huang et al. 2017). Furthermore, different deep-learning models have been used to explore the ChIP-seq data including recurrent neural networks (RNNs) (Kusupati et al. 2019), e.g., KEGRU (Gated Recurrent Unit with k-mer Embedding) used for RNA visual and textual motif mining (Shen et al. 2018; Xiong et al. 2016), Deep Belief Network (DBN) (Chen et al. 2015) and Graph Neural Networks (GNN) (Chiang et al. 2019; Zou et al. 2019) giving rise to over 30 specialized computational tools, e.g., DESSO (DEep Sequence and Shape mOtif, Yang et al. 2019), DeepBind (Alipanahi et al. 2015), and DeeperBind (Hassanzadeh and Wang 2016). These models are further modified to justify the problems found within the biological domain (Pouladi et al. 2015) and to reduce quadrant computational complexity and the memory cost of training. Moreover, not long ago, researchers designed deep-learning models such as DeepBind (Alipanahi et al. 2015), Basset (Kelley et al. 2016), and DeepSEA (Zhou and Troyanskaya 2015), which are methods based on CNN models for motif mining (Quang and Xie 2016). The motif discovery process involved in DeepBind is illustrated in Fig. 4. The convolution kernel filters detect low-level characteristics present in the one-hot encoded sequences which are shifted by a threshold in the rectified linear unit (ReLU). The average and maximum pooling account for the accumulative effects of shorter motifs and the detection of longer motifs, respectively.

Basic architecture of the DeepBind model. It uses a CNN architecture with several convolution kernels that extract low-level features from the input. The predictions are gradually improved via backpropagating the errors and updating model parameters
The neural network then trains on the feature vector generated and gives a final score which is improved via backpropagation until a desired performance is achieved. DeepSEA added single nucleotide sensitivity, and chromatin profiling, and increased the width of the kernel window to 1000 bp. While Basset (Kelley et al. 2016) used DNase I hypersensitive sites (DHS) to take account of DNA accessibility effects on TF binding, DeepHistone (Yin et al. 2019) used chromatin accessibility data with motif prediction. Dilated (Gupta and Rush 2017) further increased the spectrum of search by taking longer sequences to capture the long-range effects of DNA motifs. DeepSNR (Salekin et al. 2018) added a deconvolution layer after the CNN to increase specificity to single nucleotides using ChIP-exo datasets that remove noisy data and help to detect weak motifs as well. DESSO (Yang et al. 2019) added DNA shape features as well as a statistical analysis module to the baseline CNN model which is based on the binomial distribution for greater predictability. scFAN (Fu et al. 2020) added the feature of genome-wide TFBS prediction using a CNN with 3 layers for each cell. TFImpute (Qin and Feng 2017) and FactorNet (Quang and Xie 2019) impute the TFBSs for cell lines whose ChIP-seq data are not available by training the network on known cell line data. However, while TFImpute uses a CNN model, FactorNet uses a hybrid architecture composed of CNN and LSTM units in the RNN layer. FCNA (Zhang et al. 2021b) employs many fully connected CNNs to form an encoding and a decoding layer to do away with dataset disparity between positive and negative sets. RNA motif detectors such as iDeepE (Pan and Shen 2018b), iDeepV (Pan and Shen 2018a), and DeepRBP-Pred (Zheng et al. 2018) used CNNs to find the locations of RBPs (RNA Binding Proteins) from CLIP-seq datasets. While iDeepV used k-mer embedding and a one-dimensional CNN model, iDeepE used a local CNN and a global CNN to infer local and genome-wide features before merging the results. DeepVISP (Xu et al. 2021) used an attention mechanism after the CNN layer to identify virus integration sites (VISs) for cancer-causing viruses in humans.
Hybrid CNN–RNN-based models such as DanQ (Quang and Xie 2016), FactorNet (Quang and Xie 2019), DeepSite (Zhang et al. 2019b), iDeep (Pan and Shen 2017), iDeepS (Pan et al. 2018), and DeeperBind (Hassanzadeh and Wang 2016) are successfully prescribed to identify the sequence specificity of TF-DNA binding and RNA-binding proteins (Pan et al. 2018) with a good performance over existing motif-based statistical methods. TBiNet (Park et al. 2020), and DeepGRN (Chen et al. 2021) use an attention mechanism to discover long-range dependence in addition to the LSTM units in the RNN layer. Similarly, WSCNN LSTM (Zhang et al. 2019a), DeepSite (Zhang et al. 2019b), iDeepS (Pan et al. 2018), and DeepCLIP (Grønning et al. 2020) also employ Bidirectional LSTM units in the RNN layer to take into account the onward and reverse long-term dependencies among the motifs detected in the CNN layer. DeepCpG (Angermueller et al. 2017) and KEGRU (Shen et al. 2018) use gated units in the RNN layer to capture DNA methylation sites and TFBSs, respectively. AgentBind (Zheng et al. 2021) uses fine-tuned models after initial motif detection in the CNN layer which further enhances the specificity as each model is tuned for the target TF. iDeep (Pan and Shen 2017) uses Deep Belief Networks (DBN) for capturing different features such as motif information, structure information, region, and co-binding factors. iDeep also uses CNN filters to derive the motif locations for RBP sites directly from the sequence data. Then it merges all the results obtained from the various individual networks to classify RBP-binding sites. DeepFinder (Lee et al. 2018) uses a stacked autoencoder (SAE) for their ‘three-stage approach’ to detect motifs and TFBSs. DeepFinder tries to impute the other TFBSs from the small subset of training data.
The advantage of using the CNN-RNN hybrid model is that it is composed of multiple layers of data abstraction for accurate prediction of complex biological data relevant to functional biology e.g., phylogenetic inference, protein functions, and other aspects of computational biology (Krizhevsky et al. 2017). However, other most popular deep-learning architectures are applied to different areas of biological sciences such as CNN and ResNet (Residual Neural Networks) for phylogenetic inference, CNN, RNN, LSTM (Long Short-Term Memory), SAE (Stacked Auto Encoders), and VAE (Variable Auto Encoders) for system biology and data integration, MLP (Multi-Layer Perceptron) and CNN for genome engineering mainly for gRNA (guide RNA) sites on human genomes and CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) profile build-up, CNN, RNN, ResNet and GNN for protein function prediction, and lastly, CNN, ResNet, BLSTM (Bidirectional Long Short-Term Memory) and Transformers are preferred model architectures for protein structure prediction (Sapoval et al. 2022). In addition, all the advanced transcriptional DL models from 2012 to 2021 that deal with the problem of motif discovery are presented in Table 1 with a concise overview.
4 Deep-learning model selection benchmarks
Model selection benchmarks play a crucial role in the performance of deep-learning models for DNA/RNA motif discovery. To achieve the best performance, researchers need to carefully select the most suitable model for their specific dataset and research question. This requires extensive model benchmarking, which involves testing a range of models and selecting the best-performing one based on specific evaluation metrics. The adoption of the correct DL model for a specific purpose is relatively a confusing and challenging task for researchers without an assessment of model performance in terms of accuracy of motif finding, sequence classification, specificity, sensitivity, usability, and scalability. With a deeper understanding of DNA and RNA datasets, their comparative results were demonstrated using the deepRAM (Trabelsi et al. 2019) on human ChIP-seq/CLIP-seq data to reveal the performance of complex existing networks. Along with this, DL model selection is primarily based on the available volume of data, neural network type, and model outputs (Pouladi et al. 2015). Thereby, the deployment of new DL methodology from existing models and the origin of their variants is necessary to perform better when complex data and their size is sufficient (Pan and Shen 2017). Thus, recent research trends tend to move towards complex model construction despite choosing simpler models. Model selection is often difficult in motif mining also due to the many hyper-parameters that need to be carefully tweaked to attain the correct accuracy and acceleration. Training sample size also must be chosen to achieve the right representation of the datasets. While the generally accepted rule of thumb when it comes to training sample size is that training sample size should be larger than ten thousand samples at least; some researchers (Lee et al. 2018; Zia and Moses 2012; Hu et al. 2005) have observed that a smaller sample size of shorter sequences may suffice for the motif search problem and a larger number of sequences will not result in any further improvement in model performance. Thus, researchers must carefully select the most suitable model based on specific evaluation metrics, and model selection benchmarks such as validation set or cross-validation should be performed to achieve the best performance.
4.1 Performance evaluation of computational DL models
For computational biology applications, one approach for enhancing the efficacy of DL models is to exploit the inherent capacity to locate complex biological data sequences by focusing only on the small set of genomic sequences rather than the whole genome as discussed by Ke and Vikalo (2020). In this aspect, several researchers suggested transformer models for DNA/RNA sequence modeling (Zaheer et al. 2021). Nevertheless, Transformer models require higher training costs owing to the costly global attention procedure. Thereby, the practice of lightweight DL models with clustering methodology is recommended to reduce data pruning from the model and lower the neural network size, which has become a popular method in deployment.
Alternatively, a DL model known as deepBICS can compute the affinity of transcriptional factors to DNA target sites (Quan et al. 2022). This model applies to the human ChIP-seq datasets and differentiates disease-related variants and non-related variants. An improved version of deepBICS is also reframed (deepBICS4SNV) to improve accuracy and generalization capability to diagnose disease-related pathogenicity (Quan et al. 2022). In Trabelsi et al. (2019), Wang et al. (2020a, b) and He et al. (2020), some performance evaluation of DL models has been presented. The general consensus agrees upon CNN models as better at DNA/RNA motif discovery in terms of performance than others that are an amalgamation of various models. This is mainly due to the interpretability issue of the model in question which becomes more challenging as the models incorporate different types of DL sub-units to create a hybrid.
The performance of the DL models can be evaluated using various metrics, such as accuracy, sensitivity, specificity, and the area under the receiver-operating characteristic (ROC) curve (AUC). These metrics help to assess the quality of the model’s output and its ability to discriminate true motifs from false positives. The use of AUC is increasingly common in the evaluation of DNA/RNA motif discovery models as it provides a single numerical value that summarizes the overall model performance. In addition to these metrics, other state-of-the-art evaluation methods have emerged, such as precision–recall curves and F1 score. These evaluation methods can help researchers to identify the strengths and weaknesses of the DL models, which can be used to refine the models further. Furthermore, new metrics and evaluation techniques are constantly being developed, demonstrating the need for continuous improvement in DNA/RNA motif discovery applications.
Figure 5 provides a summary of the performance of various DL models for DNA and RNA motif discovery, in terms of the average AUC. Among the latest DNA motif discovery tools studied in this review, TBiNet, DeepSite, FactorNet, DeepGRN, AgentBind, and FCNA all outperform the advanced models such as DeepSEA, DeepBind, Basset, DanQ, and Zeng (Park et al. 2020; Zhang et al. 2019b, 2021b; Chen et al. 2021). These tools were tested for ChIP-seq datasets from the ENCODE database. However, DeepGRN was shown to outperform FactorNet for some DNase-seq (DNase hypersensitive sites sequencing) datasets that are considered to be superior to ChIP-seq datasets for TFs and TFBSs. DeepVISP was only tested on the traditional models and was found to outperform them with an average AUC (Area Under the receiver-operating characteristic Curve) of about 0.8 on several datasets (Xu et al. 2021). Overall, TBiNet and DeepFinder report the highest AUC for ChIP-seq datasets from ENCODE of greater than 0.9 and 0.95, respectively. However, among the RNA motif search models, RBPSuite was reported to be better than its counterparts like iDeepS, and other traditional methods with an approximate AUC of 0.85 (Pan et al. 2020). These models have demonstrated high performance in various benchmarks, and they are constantly being improved and refined to achieve better accuracy and generalization.

Performance of the various DL models in terms of average area under the receiver-operating characteristic (ROC) curve (AUC) for DNA/RNA motif mining problem
4.2 Scalability evaluation of computational DL tools
Researchers should know the appropriate deep-learning tools for assessing motif analysis studies and DNA/RNA sequence classification (Qin and Feng 2017). For this purpose, the performance of many DL tools was evaluated (Wang et al. 2020b), which is based on four matrix scores, namely the area of eight matrices radar (AEMR) score, motif prediction score, algorithm scalability, and tool usability. Based on eight metrics viz. sensitivity, specificity, precision, negative predictive value, accuracy, F1 score, Geometric-mean, and Matthews correlation coefficient (MCC), an overall score of AEMR and a score of motif prediction conclude the performance of developed DL tools, and then it was used, to rank every model from highest to lowest scores. The AEMR score provides a single summary metric that captures the overall performance of a deep-learning model across multiple metrics. This can be useful when comparing the performance of different models, as it provides a simple way to see which model is performing better overall. Out of recently developed DL tools, DESSO registers the maximum overall score for DNA sequence than any other DL tool while DeepBind is the perfect DL tool for RNA sequence-based analysis and is considered the next best DL tool for DNA sequences. Despite this, some researchers (Tang and Sun 2019) find the CNN network-based tools to be better than the CNN-RNN network tools for DNA sequences and inferior for micro-RNA sequences. It might be due to insufficient RNA motif data availability and a more variant nature of RNA CLIP-seq (Cross Linking Immuno-Precipitation with sequencing) data than DNA ChIP-seq data.
In addition, DeepHistone acquires the best AEMR score and DESSO was identified as the best tool to analyze the different motif patterns (LeCun et al. 2015). And for RNA sequences, iDeepV and iDeepS models are identified as the best tools that are based on CNN and BLSTM (Bidirectional Long Short-Term Memory) networks for RNA sequence cataloging and RNA motif mining, respectively.
Many of the models included in this review have been applied to the ENCODE-DREAM challenge datasets which consist of repositories of ChIP-seq, RNA-seq, and DNase-seq datasets for download (see https://www.synapse.org/#!Synapse:syn6131484/wiki/402028). Model scalability is thus often measured as how fast and accurately the model can be trained with the different datasets such as those available in the challenge. Models working on DNA motif discovery must be able to scale up well to ChIP-seq, ATAC-seq, DNase-seq, DNA shape features, etc. AgentBind, FactorNet, and DanQ scale up well to both ChIP-seq and DNase-seq datasets. DeepSEA and Dilated have only been tested on DNase-seq, whereas many state-of-the-art models such as DeepBind, DeeperBind, Zeng, TFImpute, DeepFinder, deepRAM, DESSO, DeFine, DeepSite, scFAN, FCNA, TBiNet, and AgentBind have been successfully trained and tested with ChIP-seq cell lines. Basset is one model that has scaled up well on many different types such as ChIP-seq, ATAC-seq, DNase1-seq, and CIS-BP (Catalog of Inferred Sequence Binding Proteins of RNA) datasets. DeepCpG has been tested and trained over two datasets CIS-BP and UniPROPE. RNA motif finders have scaled up well on CLIP-seq standard datasets such as iDeepS, iDeep, iDeepE, iDeepV, RBPSuite, and deepRAM. The authors of DeepVISP (Xu et al. 2021) on the other hand have created their own curated dataset called VISDB (Viral Integration Site Data Base) that they have used to train their model.
4.3 Research gaps identified
This study noted that the existence of numerous versions of motifs from several databases for a sole TF and the scarcity of a standardized evaluation system makes it problematic for biologists to select a suitable model and for algorithm designers to standardize, assess, and enhance their DL models. In addition, data scientists are not well versed with TFBSs which also hindered the capability to accurately find specific motif patterns and select appropriate algorithms to predict the true TF-binding sites. This is possible when there is a lack of interconnectivity between the researchers belonging to two different domains which affected the identification of unknown true TF-binding sites in genomic sequences. Such unknown information hinders the high-throughput screening of advanced techniques such as next-generation sequencing (NGS) and the identification of such specific sites may also be penalized. Until TF-binding sites are well annotated, sequencing techniques cannot be applied with confidence. In addition, inadequate knowledge of the entire gene expression dataset and an inappropriate tune setting of models, or performing the model selection before applying where it will be used in practice, can result in error-prone datasets. Thus, the incorporation of generalizable domain knowledge within DL architectures and adequate training of DL models that generate strong estimates on test data obtained from the data survey with comparison to previous studies concerned with the deeply learned mechanism can improve the performance of the model. Automatic calibration of complex datasets and training of biologists to keep themselves up to date can make it easy to predict the complex and variant nature of motifs which can help them to identify the complex chemistry behind the nucleic acid structure. More elaborately, the motif’s variant nature impacts the genomic sequences structurally and functionally and determines an exponential number of possible sequences of a given length. Deep-learning models can resolve the complex behavior of large genomic sequence datasets very well, especially for ChIP-seq data, and therefore, other techniques were discarded for computational reasons. However, these DL techniques come with their own set of limitations and challenges. Model interpretability is still an issue, especially with complex models that involve the use of many different types of DL concepts in a single DL framework. Further, training size and hyper-parameter selection along with other model selection benchmarks are also a challenge when designing a novel DL framework that is both scalable and efficient in eliciting motifs for TFs and TFBSs that have a low false-positive frequency. The representation of DNA/RNA sequence data in the DL model is also another area in which improvements and novelty are warranted.
5 Concluded comments
In this study, we have tried to present a comprehensive background of the deep-learning models that are state-of-the-art for human DNA/RNA motif mining that specifically uses ChIP-seq, DNase-seq, ATAC-seq, CLIP-seq, etc. This review concluded that the application of deep-learning methods in the field of motif discovery is decided in terms of the speed of complex data preprocessing, qualitative features of existing deep-learning architectures, and comparing the differences among the deep-learning models. Through the PRISMA-ScR reporting guidelines and literature survey, we have compared existing deep-learning models based on model size, automatic calibration ability, tool selection, and training set and have found that the DESSO, TBiNet, DeepSite, and DeepBind are the selective DL models in terms of performance and scalability of a true biological relationship especially concerning to gene expression pattern and sequence analysis. Other aspects of choosing the best DL models are when data are sufficient and briefly describe the characteristics of existing learning models. Therefore, it is necessary to conduct the literature survey on large datasets for motif mining and transcription factor recognition and an accurate choice selection of deep-learning methods. It will assist researchers to understand the current aspects of computational biology approaches and their concerned field of study.
Data availability
The data that support the findings of this scoping review are publicly available through the online bibliographic databases and search engines used, and are also mentioned in the Methods section, PRISMA-ScR flow diagram, and the included studies table. Details on the search strategy and inclusion criteria are provided in the Methods section of this article.
References
Alipanahi B, Delong A, Weirauch MT, Frey BJ (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 33(8):831–838. https://doi.org/10.1038/nbt.3300
Allot A, Lee K, Chen Q, Luo L, Lu Z (2021) Litsuggest: a web-based system for literature recommendation and curation using machine learning. Nucl Acids Res. https://doi.org/10.1093/nar/gkab326
Angermueller C, Lee HJ, Reik W, Stegle O (2017) DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. https://doi.org/10.1186/s13059-017-1189-z
Avsec Ž, Kreuzhuber R, Israeli J, Xu N, Cheng J, Shrikumar A, Banerjee A, Kim DS, Beier T, Urban L, Kundaje A, Stegle O, Gagneur J (2019) The kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat Biotechnol 37(6):592–600. https://doi.org/10.1038/s41587-019-0140-0
Beller E, Clark J, Tsafnat G, Adams C, Diehl H, Lund H, Ouzzani M, Thayer K, Thomas J, Turner T, Xia J, Robinson K, Glasziou P (2018) Making progress with the automation of systematic reviews: principles of the international collaboration for the automation of systematic reviews (ICASR). Syst Rev. https://doi.org/10.1186/s13643-018-0740-7
Binder A, Bockmayr M, Hägele M, Wienert S, Heim D, Hellweg K, Ishii M, Stenzinger A, Hocke A, Denkert C, Müller K-R, Klauschen F (2021) Morphological and molecular breast cancer profiling through explainable machine learning. Nat Mach Intell 3(4):355–366. https://doi.org/10.1038/s42256-021-00303-4
Blum CF, Kollmann M (2019) Neural networks with circular filters enable data efficient inference of sequence motifs. Bioinformatics 35(20):3937–3943. https://doi.org/10.1093/bioinformatics/btz194
Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Berhanu Lemma R, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, Fornes O, Leung TY, Aguirre A, Hammal F, Schmelter D, Baranasic D, Ballester B, Sandelin A, Lenhard B, Mathelier A (2021) Jaspar 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucl Acids Res. https://doi.org/10.1093/nar/gkab1113
Chen Y, Zhao X, Jia X (2015) Spectral-spatial classification of hyperspectral data based on deep belief network. IEEE J Sel Top Appl Earth Obser Remote Sens 8(6):2381–2392. https://doi.org/10.1109/jstars.2015.2388577
Chen KM, Cofer EM, Zhou J, Troyanskaya OG (2019) Selene: a PyTorch-based deep learning library for sequence data. Nat Methods 16(4):315–318. https://doi.org/10.1038/s41592-019-0360-8
Chen C, Hou J, Shi X, Yang H, Birchler JA, Cheng J (2021) DeepGRN: prediction of transcription factor binding site across cell types using attention-based deep neural networks. BMC Bioinform. https://doi.org/10.1186/s12859-020-03952-1
Cheng SH, Augustin C, Bethel A, Gill D, Anzaroot S, Brun J, DeWilde B, Minnich RC, Garside R, Masuda YJ, Miller DC, Wilkie D, Wongbusarakum S, McKinnon MC (2018) Using machine learning to advance synthesis and use of conservation and environmental evidence. Conserv Biol 32(4):762–764. https://doi.org/10.1111/cobi.13117
Chiang W-L, Liu X, Si S, Li Y, Bengio S, Hsieh C-J (2019) Cluster-GCN: an efficient algorithm for training deep and large graph convolutional networks. In: Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery and data mining. https://doi.org/10.1145/3292500.3330925
Das MK, Dai H-K (2007) A survey of DNA motif finding algorithms. BMC Bioinform. https://doi.org/10.1186/1471-2105-8-s7-s21
D'haeseleer P (2006) What are DNA sequence motifs? Nat Biotechnol 24(4):423–425. https://doi.org/10.1038/nbt0406-423
Eraslan G, Avsec Ž, Gagneur J, Theis FJ (2019) Deep learning: new computational modeling techniques for genomics. Nat Rev Genet 20(7):389–403. https://doi.org/10.1038/s41576-019-0122-6
Fu L, Zhang L, Dollinger E, Peng Q, Nie Q, Xie X (2020) Predicting transcription factor binding in single cells through deep learning. Sci Adv. https://doi.org/10.1126/sciadv.aba9031
Grønning AGB, Doktor TK, Larsen SJ, Petersen USS, Holm LL, Bruun GH, Hansen MB, Hartung A-M, Baumbach J, Andresen BS (2020) DeepCLIP: predicting the effect of mutations on protein–RNA binding with deep learning. Nucl Acids Res. https://doi.org/10.1093/nar/gkaa530
Gupta A, Rush AM (2017) Dilated convolutions for modeling long-distance genomic dependencies. https://doi.org/10.1101/200857
Harrison H, Griffin SJ, Kuhn I, Usher-Smith JA (2020) Software tools to support title and abstract screening for systematic reviews in Healthcare: an evaluation. BMC Med Res Methodol. https://doi.org/10.1186/s12874-020-0897-3
Hashim FA, Mabrouk MS, Al-Atabany W (2019) Review of different sequence motif finding algorithms. Avicenna J Med Biotechnol 11(2):130–148. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6490410/
Hassanzadeh HR, Wang MD (2016) DeeperBind: enhancing prediction of sequence specificities of DNA binding proteins. In: 2016 IEEE international conference on bioinformatics and biomedicine (BIBM). https://doi.org/10.1109/bibm.2016.7822515
Häussler M, Nicolas J (2005) Motif discovery on promotor sequences (Research Report). Inria. Retrieved September 24, 2022, from https://hal.inria.fr/inria-00070303
He Y, Shen Z, Zhang Q, Wang S, Huang D-S (2020) A survey on deep learning in DNA/RNA motif mining. Briefings Bioinform. https://doi.org/10.1093/bib/bbaa229
Hu J, Li B, Kihara D (2005) Limitations and potentials of current motif discovery algorithms. Nucl Acids Res 33(15):4899–4913. https://doi.org/10.1093/nar/gki791
Huang D-S, Zhang L, Han K, Deng S, Yang K, Zhang H (2014) Prediction of protein–protein interactions based on protein–protein correlation using least squares regression. Curr Protein Pept Sci 15(6):553–560. https://doi.org/10.2174/1389203715666140724084019
Huang G, Liu Z, Van Der Maaten L, Weinberger KQ (2017) Densely connected convolutional networks. In: 2017 IEEE conference on computer vision and pattern recognition (CVPR). https://doi.org/10.1109/cvpr.2017.243
Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML (2014) UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein–DNA interactions. Nucl Acids Res. https://doi.org/10.1093/nar/gku1045
Ke Z, Vikalo H (2020) A convolutional auto-encoder for haplotype assembly and viral quasispecies reconstruction. https://doi.org/10.1101/2020.09.29.318642
Kelley DR, Snoek J, Rinn JL (2016) Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 26(7):990–999. https://doi.org/10.1101/gr.200535.115
Krizhevsky A, Sutskever I, Hinton GE (2017) ImageNet classification with deep convolutional neural networks. Commun ACM 60(6):84–90. https://doi.org/10.1145/3065386
Kusupati A, Singh M, Bhatia K, Kumar A, Jain P, Varma M (2019) FastGRNN: a fast, accurate, stable, and tiny kilobyte-sized gated recurrent neural network. Retrieved March 12, 2022, from arXiv:1901.02358
Lan G, Zhou J, Xu R, Lu Q, Wang H (2019) Cross-cell-type prediction of TF-binding site by integrating convolutional neural network and adversarial network. Int J Mol Sci 20(14):3425. https://doi.org/10.3390/ijms20143425
LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521(7553):436–444. https://doi.org/10.1038/nature14539
Lee NK, Azizan FL, Wong YS, Omar N (2018) DeepFinder: an integration of feature-based and deep learning approach for DNA motif discovery. Biotechnol Biotechnol Equip 32(3):759–768. https://doi.org/10.1080/13102818.2018.1438209
Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, Baymuradov UK, Graham K, Litton C, Miyasato SR, Strattan JS, Jolanki O, Lee J-W, Tanaka FY, Adenekan P, Cherry JM (2019) New Developments on the encyclopedia of DNA elements (encode) data portal. Nucl Acids Res. https://doi.org/10.1093/nar/gkz1062
Martin GP, Jenkins DA, Bull L, Sisk R, Lin L, Hulme W, Wilson A, Wang W, Barrowman M, Sammut-Powell C, Pate A, Sperrin M, Peek N (2020) Toward a framework for the design, implementation, and reporting of methodology scoping reviews. J Clin Epidemiol 127:191–197. https://doi.org/10.1016/j.jclinepi.2020.07.014
Moher D, Liberati A, Tetzlaff J, Altman DG (2009) Preferred reporting items for systematic reviews and meta-analyses: the Prisma statement. PLoS Med. https://doi.org/10.1371/journal.pmed.1000097
Nair SS, Reddy NVS, Hareesha KS (2012) Motif mining: an assessment and perspective for amyloid fibril prediction tool. Bioinformation 8(2):70–74. https://doi.org/10.6026/97320630008070
Nutiu R, Friedman RC, Luo S, Khrebtukova I, Silva D, Li R, Zhang L, Schroth GP, Burge CB (2011) Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nat Biotechnol 29(7):659–664. https://doi.org/10.1038/nbt.1882
Pan X, Shen H-B (2017) RNA–protein binding motifs mining with a new hybrid deep learning-based cross-domain knowledge integration approach. BMC Bioinform. https://doi.org/10.1186/s12859-017-1561-8
Pan X, Rijnbeek P, Yan J, Shen H-B (2018) Prediction of RNA–protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genom. https://doi.org/10.1186/s12864-018-4889-1
Pan X, Fang Y, Li X, Yang Y, Shen H-B (2020) RBPsuite: RNA–protein binding sites prediction suite based on deep learning. BMC Genom. https://doi.org/10.1186/s12864-020-07291-6
Pan X, Shen H-B (2018a) Learning distributed representations of RNA sequences and its application for predicting RNA–protein binding sites with a convolutional neural network. Neurocomputing 305:51–58. https://doi.org/10.1016/j.neucom.2018.04.036
Pan X, Shen H-B (2018b) Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 34(20):3427–3436. https://doi.org/10.1093/bioinformatics/bty364
Park S, Koh Y, Jeon H, Kim H, Yeo Y, Kang J (2020) Enhancing the interpretability of transcription factor binding site prediction using an attention mechanism. Sci Rep. https://doi.org/10.1038/s41598-020-70218-4
Peters MDJ, Marnie C, Tricco AC, Pollock D, Munn Z, Alexander L, McInerney P, Godfrey CM, Khalil H (2021) Updated methodological guidance for the conduct of scoping reviews. JBI Evidence Implement 19(1):3–10. https://doi.org/10.1097/xeb.0000000000000277
Peters MDJ, Godfrey C, McInerney P, Khalil H, Larsen P, Marnie C, Pollock D, Tricco AC, Munn Z (2022) Best practice guidance and reporting items for the development of scoping review protocols. JBI Evidence Synth. https://doi.org/10.11124/jbies-21-00242
Pouladi F, Salehinejad H, Gilani AM (2015) Recurrent neural networks for sequential phenotype prediction in genomics. In: 2015 international conference on developments of E-systems engineering (DeSE). https://doi.org/10.1109/dese.2015.52
Qin Q, Feng J (2017) Imputation for transcription factor binding predictions based on deep learning. PLOS Comput Biol. https://doi.org/10.1371/journal.pcbi.1005403
Quan L, Chu X, Sun X, Wu T, Lyu Q (2022) How deepbics quantifies intensities of transcription factor-DNA binding and facilitates prediction of single nucleotide variant pathogenicity with a deep learning model trained on ChIP-seq data sets (Pre-Print). In: IEEE/ACM transactions on computational biology and bioinformatics. https://doi.org/10.1109/tcbb.2022.3170343
Quang D, Xie X (2016) DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucl Acids Res. https://doi.org/10.1093/nar/gkw226
Quang D, Xie X (2019) FactorNet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods 166:40–47. https://doi.org/10.1016/j.ymeth.2019.03.020
Salekin S, Zhang JM, Huang Y (2018) Base-pair resolution detection of transcription factor binding site by deep deconvolutional network. Bioinformatics 34(20):3446–3453. https://doi.org/10.1093/bioinformatics/bty383
Sandler M, Howard A, Zhu M, Zhmoginov A, Chen L-C (2018) MobileNetV2: inverted residuals and linear bottlenecks. In: 2018 IEEE/CVF conference on computer vision and pattern recognition. https://doi.org/10.1109/cvpr.2018.00474
Sapoval N, Aghazadeh A, Nute MG, Antunes DA, Balaji A, Baraniuk R, Barberan CJ, Dannenfelser R, Dun C, Edrisi M, Elworth RA, Kille B, Kyrillidis A, Nakhleh L, Wolfe CR, Yan Z, Yao V, Treangen TJ (2022) Current progress and open challenges for applying deep learning across the biosciences. Nat Commun. https://doi.org/10.1038/s41467-022-29268-7
Scott AM, Forbes C, Clark J, Carter M, Glasziou P, Munn Z (2021) Systematic review automation tool use by systematic reviewers, health technology assessors and clinical guideline developers: tools used, abandoned, and desired. https://doi.org/10.1101/2021.04.26.21255833
Shen Z, Bao W, Huang D-S (2018) Recurrent neural network for predicting transcription factor binding sites. Sci Rep. https://doi.org/10.1038/s41598-018-33321-1
Shrikumar A, Greenside P, Kundaje A (2017) Learning important features through propagating activation differences. PMLR. Retrieved September 24, 2022, from https://proceedings.mlr.press/v70/shrikumar17a.html
Siggers T, Gordân R (2013) Protein–DNA binding: complexities and multi-protein codes. Nucl Acids Res 42(4):2099–2111. https://doi.org/10.1093/nar/gkt1112
Sutskever I, Vinyals O, Le QV (2014) Sequence to sequence learning with neural networks. In: Proceedings of the 27th international conference on neural information processing systems. Conference proceedings. Retrieved September 24, 2022. https://doi.org/10.5555/2969033.2969173
Tan M, Le Q (2019) EfficientNet: rethinking model scaling for convolutional neural networks. PMLR. Retrieved March 12, 2022, from http://proceedings.mlr.press/v97/tan19a.html
Tang X, Sun Y (2019) Fast and accurate MicroRNA search using CNN. BMC Bioinform. https://doi.org/10.1186/s12859-019-3279-2
Trabelsi A, Chaabane M, Ben-Hur A (2019) Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities. Bioinformatics 35(14):i269–i277. https://doi.org/10.1093/bioinformatics/btz339
Tricco AC, Lillie E, Zarin W, O'Brien KK, Colquhoun H, Levac D, Moher D, Peters MDJ, Horsley T, Weeks L, Hempel S, Akl EA, Chang C, McGowan J, Stewart L, Hartling L, Aldcroft A, Wilson MG, Garritty C, Straus SE (2018) Prisma extension for scoping reviews (PRISMA-SCR): checklist and explanation. Ann Intern Med 169(7):467–473. https://doi.org/10.7326/m18-0850
Wallace BC, Small K, Brodley CE, Lau J, Trikalinos TA (2012) Deploying an interactive machine learning system in an evidence-based practice center. In: Proceedings of the 2nd ACM SIGHIT symposium on international health informatics—IHI’12. https://doi.org/10.1145/2110363.2110464
Wang M, Tai C, Weinan E, Wei L (2018) Define: deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucl Acids Res. https://doi.org/10.1093/nar/gky215
Wang C-Y, Mark Liao H-Y, Wu Y-H, Chen P-Y, Hsieh J-W, Yeh I-H (2020a) CSPNet: a new backbone that can enhance learning capability of CNN. In: 2020a IEEE/CVF conference on computer vision and pattern recognition workshops (CVPRW). https://doi.org/10.1109/cvprw50498.2020.00203
Wang Y, Zhang S, Ma A, Wang C, Wu Z, Xu D, Ma Q (2020b) Assessing deep learning algorithms in cis-regulatory motif finding based on genomic sequencing data. https://doi.org/10.1101/2020.11.30.403261
Xiong C, Merity S, Socher R (2016) Dynamic memory networks for visual and textual question answering. PMLR. Retrieved March 22, 2022, from https://proceedings.mlr.press/v48/xiong16.html
Xu H, Jia P, Zhao Z (2021) DeepVISP: deep learning for virus site integration prediction and motif discovery. Adv Sci 8(9):2004958. https://doi.org/10.1002/advs.202004958
Yang J, Ma A, Hoppe AD, Wang C, Li Y, Zhang C, Wang Y, Liu B, Ma Q (2019) Prediction of regulatory motifs from human ChIP-sequencing data using a deep learning framework. Nucl Acids Res 47(15):7809–7824. https://doi.org/10.1093/nar/gkz672
Yin Q, Wu M, Liu Q, Lv H, Jiang R (2019) Deephistone: a deep learning approach to predicting histone modifications. BMC Genom. https://doi.org/10.1186/s12864-019-5489-4
Zaheer M, Guruganesh G, Dubey A, Ainslie J, Alberti C, Ontanon S, Pham P, Ravula A, Wang Q, Yang L, Ahmed A (2021) Big bird: transformers for longer sequences. Retrieved April 24, 2022, from arXiv:2007.14062
Zeng H, Edwards MD, Liu G, Gifford DK (2016) Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 32(12):i121–i127. https://doi.org/10.1093/bioinformatics/btw255
Zhang Q, Shen Z, Huang D-S (2019a) Modeling in-vivo protein–DNA binding by combining multiple-instance learning with a hybrid deep neural network. Sci Rep. https://doi.org/10.1038/s41598-019-44966-x
Zhang Y, Qiao S, Ji S, Li Y (2019b) DeepSite: bidirectional LSTM and CNN models for predicting DNA–protein binding. Int J Mach Learn Cybern 11(4):841–851. https://doi.org/10.1007/s13042-019-00990-x
Zhang Q, Shen Z, Huang D-S (2021a) Predicting in-vitro transcription factor binding sites using DNA sequence + shape. IEEE/ACM Trans Comput Biol Bioinf 18(2):667–676. https://doi.org/10.1109/tcbb.2019.2947461
Zhang Q, Wang S, Chen Z, He Y, Liu Q, Huang D-S (2021b) Locating transcription factor binding sites by fully convolutional neural network. Briefings Bioinform. https://doi.org/10.1093/bib/bbaa435
Zheng J, Zhang X, Zhao X, Tong X, Hong X, Xie J, Liu S (2018) Deep-RBPPRED: Predicting RNA binding proteins in the proteome scale based on deep learning. Sci Rep. https://doi.org/10.1038/s41598-018-33654-x
Zheng A, Lamkin M, Zhao H, Wu C, Su H, Gymrek M (2021) Deep neural networks identify sequence context features predictive of transcription factor binding. Nat Mach Intell 3(2):172–180. https://doi.org/10.1038/s42256-020-00282-y
Zhou J, Troyanskaya OG (2015) Predicting effects of noncoding variants with deep learning—based sequence model. Nat Methods 12(10):931–934. https://doi.org/10.1038/nmeth.3547
Zhu L, Deng S-P, Huang D-S (2015) A two-stage geometric method for pruning unreliable links in protein–protein networks. IEEE Trans Nanobiosci 14(5):528–534. https://doi.org/10.1109/tnb.2015.2420754
Zhu L, You Z-H, Huang D-S, Wang B (2013) T-LSE: a novel robust geometric approach for modeling protein–protein interaction networks. PLoS ONE. https://doi.org/10.1371/journal.pone.0058368
Zia A, Moses AM (2012) Towards a theoretical understanding of false positives in DNA motif finding. BMC Bioinform. https://doi.org/10.1186/1471-2105-13-151
Zou D, Hu Z, Wang Y, Jiang S, Sun Y, Gu Q (2019) Layer-dependent importance sampling for training deep and large graph convolutional networks. Retrieved March 27, 2022, from arXiv:1911.07323
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
RC and UG conceptualized the review scope, RC carried out the literature survey and data analysis and wrote the manuscript and UG supervised and reviewed the manuscript preparation.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chaurasia, R., Ghose, U. Human DNA/RNA motif mining using deep-learning methods: a scoping review. Netw Model Anal Health Inform Bioinforma 12, 20 (2023). https://doi.org/10.1007/s13721-023-00414-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-023-00414-5