
Abstract
Knowing the transcription factor binding sites (TFBSs) is essential for modeling the underlying binding mechanisms and follow-up cellular functions. Convolutional neural networks (CNNs) have outperformed methods in predicting TFBSs from the primary DNA sequence. In addition to DNA sequences, histone modifications and chromatin accessibility are also important factors influencing their activity. They have been explored to predict TFBSs recently. However, current methods rarely take into account histone modifications and chromatin accessibility using CNN in an integrative framework. To this end, we developed a general CNN model to integrate these data for predicting TFBSs. We systematically benchmarked a series of architecture variants by changing network structure in terms of width and depth, and explored the effects of sample length at flanking regions. We evaluated the performance of the three types of data and their combinations using 256 ChIP-seq experiments and also compared it with competing machine learning methods. We find that contributions from these three types of data are complementary to each other. Moreover, the integrative CNN framework is superior to traditional machine learning methods with significant improvements.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
- Bioinformatics
- Machine learning
- Transcription factors binding sites
- Convolutional neural networks
- DNA accessibility
- Histone modification
1 Introduction
It has been well known that transcription factors (TFs) are key proteins decoding the information in the genome to express a precise and unique set of proteins and RNAs in each cell type in the cellular system [1]. How TFs bind to specific DNA-regulatory sequences (known as TF binding site, or TFBS for short) to cooperatively modulate the gene transcription and protein synthesis is an essential procedure, which plays key roles in many biological processes [2, 3]. Moreover, it has been reported that some genomic variants in such TFBSs are associated with serious diseases including cancer and so on [4]. In the past decade, large amount of immunoprecipitation followed by high throughput sequencing (ChIP-seq) data have been generated and profiled to study the mechanisms behind these regulatory processes [5]. However, the ChIP-seq experiment can only profile one TF binding map in a given cell type one time [6, 7]. Hence it is not possible to profile every TF binding maps in all cell types due to the large number of TF-cell combinations and the high experimental cost [6, 7]. Thus, accurate computational methods are desired to decode the underlying binding rules under different circumstances. Naturally, how to predict TFBSs in DNA sequences is a basic problem in bioinformatics.
In this background, using primary DNA sequences to predict the TFBSs has become a direct and promising paradigm. At first position weight matrices (PWMs) based methods achieved great success in modeling the DNA binding protein process [8]. Later, gkm-SVM (i.e., gapped k-mers along with support vector machine) shows great superiority over the PWM-based methods [9]. More recently, convolutional neural networks [10], coupled with the one-hot coding format of DNA sequences [11,12,13,14,15,16,17,18,19,20], attracted great interest in predicting TFBSs. However, prediction or imputation of TFBSs using solely primary DNA sequences lacks the ability of dealing with cell type-specific binding events.
As a result, more and more methods turn to using cell type-specific information for addressing this issue. In addition to primary DNA sequences, other local chromatin information such as chromatin accessibility and histone modifications also have great impact to the binding of TFs to their target sites [21]. Their analysis suggested models learned from one TF was transferable across diverse TFs. Xin and Rohs [22] built a L2-regularized multiple linear regression (MLR) model to analyze histone modification patterns associated with TFBSs and showed that histone modification patterns contribute to TF binding specificities. Their results suggested that adding histone modification or chromatin accessibility information could increase the prediction performance of a classifier. However, there still exist limitations to be addressed when integrating data from different sources.
In the last few years, the fast development of deep learning or deep neural networks such as the convolutional neural networks (CNNs) attracts great attentions for the predicting of TFBSs. First, the convolution filters fitting in well with the one-hot coding format of DNA sequence can mimic the characteristics of DNA motifs [12,13,14,15, 23, 24]. Meanwhile, the learning procedure of CNN automatically extract features, which may overcome the information loss of handcrafted features. Second, the deep learning framework is flexible enough to integrate different sources of data. In addition to DNA sequence data, other data sources can be put as inputs using a computational graph, which is a directed acyclic graph representing the arbitrary information flow [25]. Third, the use of graphics processing unit (GPU) makes the training process of deep learning and especially CNNs extremely faster than before. This enables the CNN models to be applicable to deal with large amount of biological samples. However, all the existing CNN based models use solely primary DNA sequence to predict TFBSs. Currently, it is not clear how to effectively integrate DNA sequence information with other local chromatin information (e.g., DNase and histone modification) using CNN.
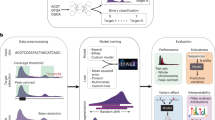
To this end, we disentangled the contributions of DNA sequence and DNase I hypersensitivity (DHS for short) and histone modifications (HMS for short) in distinguishing TFBSs from background based on a CNN model (Fig. 1). To explore how to use DHS and HMS to train the neural networks, we first benchmarked a series of architecture variants by changing network structure in terms of width and depth. We also explored the effects of sample length at flanking regions 5’ and 3’ of the motif binding sites ranging from 5 to 101 bp of DHS and HMS data. Based on detailed experimental setup, we evaluated the performance of the three types of data and their combinations using 256 ChIP-seq experiments [15]. We find that contributions from these three types of data are complementary to each other. Moreover, the results show distinct superiority of the integrative framework over traditional machine learning methods. We expect to see wide applications of integrating multiple types of data with deep learning methods not only for TFBSs prediction, but also for other genomic studies in near future.

Overview of the unified framework for predicting TFBSs using CNN.
2 Materials and Methods
2.1 Datasets
We downloaded 256 TF ChIP-seq experiments for 15 cell types from [15]. Each experiment includes training and testing datasets in fastq format. In the datasets, DNA sequences and its location in the reference genome (hg19) and labels are given. The positive and negative samples have matched GC-content and sequence length (101 bp). Then we downloaded normalized DNase-seq (DHS) and five core histone modifications (HMS) ChIP-seq data (H3K4me3, H3K4me1, H3K36me3, H3K9me3, H3K27me3) for the 15 cell types from the REMC database [26]. The DHS and HMS data are genome-wide –log10 (p-value) signal coverage tracks in bigwig format.
According to the location of the sample in the sequence datasets, we extracted the signal values of the corresponding positions from the DHS and HMS signal coverage tracks. The DNase-seq or each histone modification data was represented in a feature vector (where each nucleotide position has a value). Thus, TFBSs and non-TFBSs were described as three types of features: (1) a one-hot vector for a DNA sequence; (2) a vector for DHS at each nucleotide position; (3) a vector for each HMS at each nucleotide position. For each dataset, we used 70% samples for training, 10% samples for validating and 20% for testing.
2.2 Neural Network Setup
For a DNA sequence, TFBSs and non-TFBSs were described as one dimensional image with four channels. Each base pair (A, C, T, G) was denoted as a one-hot vector [1, 0, 0, 0], [0, 1, 0, 0], [0, 0, 1, 0] and [0, 0, 0, 1] respectively. For DNase-seq data and each histone modification data, TFBSs and non-TFBSs were described as one channel vector at each nucleotide position, For HMS, existing methods calculated the statistical values (such average reads number in each base pair) within the range of hundreds or thousands nucleotide. However, such a simplistic approach may not fully use the information in HMS data. So we used histone modification data of single base resolution in our study. HMS and DHS are contiguous attributes describing surrounding epigenetic marks and chromatin accessibility that may be related to the binding of specific TFs [27].
From the viewpoint of data, to examine how these models perform quantitatively in terms of the length of flanking regions used in calculating DHS and HMS, we tried different length scales ranging from 5 to 101 bp centered on the motif binding sites. For example, if we used DNase-seq data with 101 bp, the vector was of size 1 × 101 for a sample; if we used five histone modifications data with 71 bp, the dimension of a vector was of size 1 × 71, and they were combined as matrix with size of 5 × 71 for a sample.
For the purpose of combining DHS, HMS and sequence in the unified deep learning framework, after collecting DNA sequence, HMS, DHS, labels data and encoding features for each sample, we first implemented five different models: sequence CNN model, using DNA sequence as features; DHS CNN model, using DHS as features; DHS Deep Neural Networks (DNN) model, using DHS as features; HMS CNN model, using HMS as features; HMS DNN model, using HMS as features. We used CNN and DNN models to compare which one was more suitable for DHS and HMS data. The CNN consists of a convolutional layer, a max-pooling layer, a fully connected layer, a dropout layer [28] and an output layer. DNN consists of one or two full connection layers, a dropout layer after each full connection layer and an output layer. For CNN models, we vary the number of kernels, the size of kernel window, and the number of neurons in the full connection layer. For DNN models, we vary the number of layers, and the number of neural in each full connection layer.
After determining an appropriate model, hyper-parameters and sample length for each data, we then studied the combinations performances of two types of data implementing three different models: sequence + HMS model, using a combination of DNA sequence and HMS as features, sequence + DHS model, using a combination of DNA sequence and DHS as features, DHS + HMS model, using a combination of DHS and HMS as features. We suggest an integrative model combing all three types of data (sequence + HMS + DHS model) as features at last.
For training, we used the cross-entropy as the loss function. Given this loss function and different hyper-parameters (see below), the models were trained using the standard error back-propagation algorithm and AdaDetla method [29]. Passing all the training data through the model once is an epoch. We set each model for 100 epochs and 128 mini-batch size and validated the model after each epoch. Then the early-stop trick was used to stop training as the error on validation set is higher than the last four epochs. The best model was chosen according to the accuracy on the validation set.
2.3 Leave-One-Feature-Out of the HMS Model
To determine the importance of each histone modification feature in the classification models by combining five core histone modification features, we implemented CNN models where we left out one of the features at a time. We recorded the AUC for each model compared to the model that used all five histone modification features.
2.4 Comparison with Conventional Learning Methods with HMS and DHS Data
We evaluated whether conventional learning methods can get comparable predictions compared with CNN. We predicted the TFBSs using k-Nearest Neighbor (kNN), Logistic Regression (LR), Random Forest (RF) classifiers. For KNN, LR, RF, we implemented these baselines using the python based scikit-learn package.
For the kNN classifier implementation, this model was trained on varying hyper-parameter values of n_neighbors: 1, 3, or 5, weights: ‘uniform’, or ‘distance’, the algorithm was ‘auto’. The n_neighbors parameter defines the number of neighbors to be used for prediction. The weights define weight function used in prediction. Uniform means all points in each neighborhood are weighted equally. Distance means weight points by the inverse of their distance. In this case, closer neighbors of a query point will have a greater influence than neighbors which are a little far away.
For the LR classifier implementation, the model was trained on varying hyper-parameter values of penalty: ‘l1’ or ‘l2’, C: 0.1, 1, or 10. The penalty is used to specify the norm used in the penalization. C is the inverse of regularization strength, smaller values specify stronger regularization.
For the RF Classifier implementation, we varied the number of trees in the forest, n_estimators: 10, 20, 30, …, 100, 200, 300, used to train each model.
All the above models were trained on the training set, and evaluated on the corresponding testing set. For kNN, we selected n_neighbors = 5, weights = ‘distance’. For RF, we selected n_estimators = 100. For LR, we selected penalty = l2, C = 1.
2.5 Implementation
We used python and Keras framework to train neural networks. We used python and skcikit-learn to train conventional machine learning methods [30]. All the source codes are available at http://page.amss.ac.cn/shihua.zhang/.
3 Results
3.1 Long Sample Length and CNN Architecture Improve TFBSs Prediction Based on Histone Modification Profiles
For predicting TFBSs, we considered several practical aspects to make full use of HMS data. We first tested the effects of using different sample lengths. We used different sample lengths to train the CNN models and different hyper-parameters for each length. For each length, we selected the results of best hyper-parameters. As expected, the longer the sequence length was, the better the model performs (Fig. 2A). The improvement may come from the extra context information contained in the longer samples.

Performance evaluation of CNN with respect to sample length and model structure using HMS data in terms of the distribution of AUCs across 256 experiments. (A) The effect of sequence length. (B) The effect of kernel number. (C) The effect of neuron number. (D) The effect of kernel window size. (E) The effect of sample length and DNN model structure. (F) The performance comparison of DNN versus CNN.
In addition to different sequence lengths, proper model architecture was also needed. First, more convolutional kernels could also improve the prediction performance (Fig. 2B). This observation shows additional kernels add power in extracting features. However, when more than 64 kernels were used, the improvement seemed to be saturated for the 256 experiments (Fig. 2B). Second, more neurons in the full connection layer of CNN could improve the prediction performance (Fig. 2C). And adding more neurons could improve the results. We observe that small kernel window size achieves better performance than using large ones (Fig. 2D) while big kernel window size usually used in sequence-based CNN models. This suggests that HMS features is different from sequence, and big window size may lose some information. Since the small window size is good, we are wondering how DNN performs. For comparison, we trained DNN with HMS data. We find that deeper neural networks and longer sample length work better too for DNN (Fig. 2E). As model with more neurons and layers could represent more abstract features, this observation emphasizes sufficient neurons and layers are needed to extract abstract features. However, the performance of DNN is still slightly worse than that of CNN, indicating the importance of combining convolution operation with HMS data (Fig. 2F).
3.2 Different Histone Modification Features Contribute Diversely
How each individual histone modification feature contribute relative to all five features together? We conducted leave-one-feature-out feature selection experiments to train the CNN models by using merely four histone modifications data with the same hyper-parameters in previous section. Our results suggest that H3K4me3 mark is the most important mark and H3K4me1 is the second most important one (Fig. 3). We also known that H3K4me3 denotes a specific chemical modification of proteins used to package DNA in eukaryotic cells, which is commonly associated with active transcription of nearby genes [26]. While H3K4me1 has been shown distinct enrichment at active and primed enhancers, indicating its underlying strong connections with enhancer activity and function. However, the remaining three marks H3K27me3, H3K36me3, H3K9me3 play limited impacts on the prediction performance. This is very consistent with their well-known characteristics that H3K27me3, H3K36me3, H3K9me3 are found in facultatively repressed genes, actively transcribed gene bodies, and constitutively repressed genes respectively. Thus, this is reasonable that H3K9me3 shows the worst prediction ability to TFBSs. In summary, the histone modification importance observations are in consistent with their general functions and might provide further insights into the importance of different types of data in a similar way.

Performance comparison of different HMS combinations in terms of distribution of AUCs across 256 experiments. HMS means using all five histone modification marks. HMS-H3K4me3 means using other four histone modification marks except H3K4me3.
3.3 TFBSs Prediction Results Based on DNase-seq Profiles
Similar to HMS data, we also considered several practical aspects to make full use of DNase-seq data. We first tested the effects of using different sample lengths. As expected, the longer the sequence length is, the better the model performs (Fig. 4A). This indicates that the improvement may also come from the extra context information contained in the longer samples. For model architectures, more convolutional kernels could also improve the prediction performance (Fig. 4B). Thus, no matter what the data type is, the additional kernels are beneficial to enhance power in extracting features and improve model performance. By changing the number of neurons in the last dense layer of CNN, we can see that models with more hidden neurons achieve better performance (Fig. 4C). This observation was similar with that of HMS data. We also see that CNN models with small and large kernel window sizes (4 and 24) achieve almost the same performance for different sample lengths (Fig. 4D). This suggests that kernel window sizes (4 and 24) could not distinctly influence DHS data information. For comparison with CNN, we also trained DNN using different sequence lengths and hyper-parameters for the DHS data. Similarly, the deeper neural networks and longer sample length also work better based on DHS data (Fig. 4E). Moreover, the performance of DNN is slightly worse than that of CNN, indicating the importance of combining convolution operation with DHS data (Fig. 4F).

Performance evaluation of CNN with respect to sample length and model structure using DHS data in terms of the distribution of AUCs across 256 experiments. (A) The effect of sequence length. (B) The effect of kernel number. (C) The effect of neuron number. (D) The effect of kernel window size. (E) The effect of sample length and DNN model structure. (F) The performance comparison of DNN versus CNN.
3.4 Comparison of CNN with Conventional Learning Methods with HMS and DHS Data
We have shown that CNN models with HMS and DHS data could make very promising predictions for diverse TFs. In this section, we evaluated whether conventional learning methods can get such predictions compared to CNN. As we showed that for DHS and HMS, the longer the sequence length was, the better the model performed. Here all sample lengths used were set as 101 bp. We adopted the popular k-Nearest Neighbor (kNN), Logistic Regression (LR) and Random Forest (RF) for this task. The best hyper-parameters of these methods were also chosen according to the performance on testing set (Methods and Supplementary Information). In both HMS and DHS cases, CNN perform significantly better than conventional classifiers in term of the distribution of AUCs across 256 experiments (Fig. 5). This was not surprisingly, as deep learning models could automatically extract high-level features in the DHS or HMS data due to its elaborate architectures. We note that most conventional learning methods are shallow models, which limited their performance. Taken together, our study suggests that CNN model is a more reliable tool for predicting the TFBSs by integrating these three types of data.

Comparison of CNN with conventional learning methods in terms of the distribution of AUCs across 256 experiments. KNN: k-Nearest Neighbor; LR: Logistic Regression; RF: Random Forest.
4 Conclusion and Discussion
In this work, we systematically explored the effects of epigenomic information from the chromatin accessibility and histone modifications data on the basis of a series of CNN architectures. We suggest an integrative CNN framework to combine primary DNA sequence, DHS and HMS data to predict cell type-specific TFBSs. Thorough evaluation demonstrate that the integrative framework show much better performance than using primary DNA sequence data only.
Chromatin accessibility and histone modifications are critical factors enabling the binding of TFs to their target genes. Chromatin accessibility has been widely used in conventional methods. But conventional methods required a lot of time for large input data and they used low resolution canonical features. Thus, we expect to improve discrimination ability through deep learning approach by automatically extracting efficient features. Histone modifications data is less used in TFBSs prediction than chromatin accessibility. The reason is that DNase-seq can give base pair resolution whereas DNA sequence was nicked, histone modification ChIP-seq gives a region where protein interacting with DNA sequence, so it only gives low resolution information compared to DNase-seq data. Besides DNA sequence and DHS data, we suggest that the HMS data can also provide extra context information despite of the low experimental resolution. In short, our work suggests combining more data in deep learning model may be beneficial.
References
Mitchell, P.J., Tjian, R.: Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 245, 371–378 (1989)
Junion, G., Spivakov, M., Girardot, C., Braun, M., Gustafson, E.H., Birney, E., Furlong, E.E.: A transcription factor collective defines cardiac cell fate and reflects lineage history. Cell 148, 473–486 (2012)
Vaquerizas, J.M., Kummerfeld, S.K., Teichmann, S.A., Luscombe, N.M.: A census of human transcription factors: function, expression and evolution. Nature Rev. Genet. 10, 252–263 (2009)
Lee, T.I., Young, R.A.: Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 (2013)
Neph, S., Vierstra, J., Stergachis, A.B., Reynolds, A.P., Haugen, E., Vernot, B., Thurman, R.E., John, S., Sandstrom, R., Johnson, A.K.: An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90 (2012)
Gilfillan, G.D., Hughes, T., Sheng, Y., Hjorthaug, H.S., Straub, T., Gervin, K., Harris, J.R., Undlien, D.E., Lyle, R.: Limitations and possibilities of low cell number ChIP-seq. BMC Genom. 13, 645 (2012)
Park, P.J.: ChIP–seq: advantages and challenges of a maturing technology. Nature Rev. Genet. 10, 669–680 (2009)
Warner, J.B., Philippakis, A.A., Jaeger, S.A., He, F.S., Lin, J., Bulyk, M.L.: Systematic identification of mammalian regulatory motifs’ target genes and functions. Nat. Methods 5, 347–353 (2008)
Ghandi, M., Lee, D., Mohammad-Noori, M., Beer, M.A.: Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 10, e1003711 (2014)
LeCun, Y., Bengio, Y., Hinton, G.: Deep learning. Nature 521, 436–444 (2015)
Angermueller, C., Lee, H.J., Reik, W., Stegle, O.: DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 18, 67 (2017)
Qin, Q., Feng, J.: Imputation for transcription factor binding predictions based on deep learning. PLoS Comput. Biol. 13, e1005403 (2017)
Yang, B., Liu, F., Ren, C., Ouyang, Z., Xie, Z., Bo, X., Shu, W.: BiRen: predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics 33, 1930–1936 (2017)
Kelley, D.R., Snoek, J., Rinn, J.L.: Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26(7), 990–999 (2016)
Zeng, H., Edwards, M.D., Liu, G., Gifford, D.K.: Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 32, i121–i127 (2016)
Jurtz, V.I., Johansen, A.R., Nielsen, M., Almagro Armenteros, J.J., Nielsen, H., Sønderby, C.K., Winther, O., Sønderby, S.K.: An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics 33, 3685–3690 (2017)
Liu, Q., Xia, F., Yin, Q., Jiang, R.: Chromatin accessibility prediction via a hybrid deep convolutional neural network. Bioinformatics 34(5), 732–738 (2017). https://doi.org/10.1093/bioinformatics/btx679
Min, X., Zeng, W., Chen, N., Chen, T., Jiang, R.: Chromatin accessibility prediction via convolutional long short-term memory networks with k-mer embedding. Bioinformatics 33, i92–i101 (2017)
Bu, H., Gan, Y., Wang, Y., Zhou, S., Guan, J.: A new method for enhancer prediction based on deep belief network. BMC Bioinform. 18, 418 (2017)
Zhang, J., Peng, W., Wang, L.: LeNup: learning nucleosome positioning from DNA sequences with improved convolutional neural networks. Bioinformatics 34(10), 1705–1712 (2018). https://doi.org/10.1093/bioinformatics/bty003
Piqueregi, R., Degner, J.F., Pai, A.A., Gaffney, D.J., Gilad, Y., Pritchard, J.K.: Accurate inference of transcription factor binding from DNA sequence and chromatin accessibility data. Genome Res. 21, 447–455 (2011)
Xin, B., Rohs, R.: Relationship between histone modifications and transcription factor binding is protein family specific. Genome Res. (2018). https://doi.org/10.1101/gr.220079.116
Min, X., Zeng, W., Chen, S., Chen, N., Chen, T., Jiang, R.: Predicting enhancers with deep convolutional neural networks. BMC Bioinform. 18, 478 (2017)
Zhou, J., Troyanskaya, O.G.: Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12, 931–934 (2015)
Abadi, M., Barham, P., Chen, J., Chen, Z., Davis, A., Dean, J., Devin, M., Ghemawat, S., Irving, G., Isard, M.: TensorFlow: a system for large-scale machine learning. In: OSDI 2016, pp. 265–283 (2016)
Kundaje, A., Meuleman, W., Ernst, J., Bilenky, M., Yen, A., Kheradpour, P., Zhang, Z., Heravi-Moussavi, A., Liu, Y., Amin, V.: Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015)
Ziller, M.J., Edri, R., Yaffe, Y., Donaghey, J., Pop, R., Mallard, W., Issner, R., Gifford, C.A., Goren, A., Xing, J.: Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature 518, 355–359 (2015)
Srivastava, N., Hinton, G.E., Krizhevsky, A., Sutskever, I., Salakhutdinov, R.: Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 15, 1929–1958 (2014)
Zeiler, M.D.: ADADELTA: an adaptive learning rate method. arXiv preprint arXiv:1212.5701 (2012)
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., Blondel, M., Prettenhofer, P., Weiss, R., Dubourg, V.: Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011)
Acknowledgement
Fang Jing would like to thank the support of the National Center for Mathematics and Interdisciplinary Sciences, Academy of Mathematics and Systems Science, CAS, during his visit. The work was supported by the National Natural Science Foundation of China [No. 61473232 and 91430111 to SWZ; No. 61621003 and 11661141019 to SZ]; the Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) [No. XDB13040600], the Key Research Program of the Chinese Academy of Sciences, [No. KFZD-SW-219] and CAS Frontier Science Research Key Project for Top Young Scientist [No. QYZDB-SSW-SYS008].
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this paper

Cite this paper
Jing, F., Zhang, SW., Cao, Z., Zhang, S. (2018). Combining Sequence and Epigenomic Data to Predict Transcription Factor Binding Sites Using Deep Learning. In: Zhang, F., Cai, Z., Skums, P., Zhang, S. (eds) Bioinformatics Research and Applications. ISBRA 2018. Lecture Notes in Computer Science(), vol 10847. Springer, Cham. https://doi.org/10.1007/978-3-319-94968-0_23
Download citation
DOI: https://doi.org/10.1007/978-3-319-94968-0_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-94967-3
Online ISBN: 978-3-319-94968-0
eBook Packages: Computer ScienceComputer Science (R0)