
Abstract
Discovering DNA-protein binding sites, also known as motif discovery, is the foundation for further analyses of transcription factors (TFs). Deep learning algorithms such as convolutional neural networks (CNN) and recurrent neural networks (RNN) are introduced to motif discovery task and have achieved state-of–art performance. However, these methods still have limitations such as neglecting the context information in large-scale sequencing data. Thus, inspired by the similarity between DNA sequence and human language, in this paper we propose a hierarchical attention network for predicting DNA-protein binding sites which is based on a natural language processing method for document classification. The proposed method is tested on real ChIP-seq datasets and the experimental results show a considerable improvement compared with two well-tested deep learning-based sequence model, DeepBind and Deepsea.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Transcription factors (TFs) are proteins that directly interpret the genome, performing the first step in decoding the DNA sequence [1,2,3]. It recognizes specific DNA sequences to control chromatin and transcription, forming a complex system that guides expression of the genome [4, 5]. Such specific DNA sequences are called transcription factors binding sites (TFBSs) and play important roles in vital movement. It has proved that transcription factors binding sites are also associated with various human diseases and phenotypes. Mutation of TFs and TFBSs are often highly deleterious and may lead to diseases [6]. Thus, by discovering TFBSs, also called motif discovering, is helpful for further study in gene expression and therapy to diseases caused by gene mutation. Identification of DNA-binding motif provides a gateway to further analyses [7].
Data for study in transcription factors binding is pretty much nowadays due to the fast development of high-throughput sequencing technology which make obtaining DNA data a much easier work [8]. Traditionally, transcription factors binding sites are displayed by sequence logo which based on weight matrices (PWMs) [9, 10]. The PWM is computed by multiplying the scores for each base of a sequence and means a predicted relative affinity of the TF to that sequence. However, such model has an obvious defect that it is not able to process large-scale data from high-throughput sequencing technology [4, 11,12,13].
Thanks to the rapid development of deep learning in recent year, new computational methods such as convolutional neural network (CNN) and recurrent neural network (RNN) have been applied to many fields such as natural language processing and computer graphic and have made great achievement [14]. The recent applications of deep learning methods to processing biological data have also shown impressive improvement on performance [15,16,17,18]. Among these applications, DeepBind is one of the earliest attempts to apply deep learning to the motif discovery task and has proved to be an effective model. DeepBind’s success mainly based on the appropriate use of CNN, which is a variant of multilayer artificial neural network specialized in computer vision. By converting 1-D genomic sequence with four nucleotides {A, C, G, T} into 2-D image-like data through one-hot encoding method, CNN can be adapted to modeling DNA sequence protein-binding which is analogous to a two-class image classification problem [16]. Another impressive application of deep learning to bioinformatics field is Deepsea [19, 20]. Deepsea is an algorithmic framework for predicting the chromatin effects of sequence alterations with single nucleotide sensitivity that directly learns a regulatory sequence code from large-scale chromatin-profiling data. It also achieved a great performance by using CNN and related deep learning algorithm.
However, their performance results require much improvement, and in this study, we aim to enhance the solution to this issue with an innovative approach. Our idea is to transform the enhancer sequences into vectors using word embedding and then proceed to classify them with method of natural language processing (NLP). DNA sequence and human language are both sequence data which express certain meanings by the combination of a number of certain elements, which in human language is words or alphabet and in DNA is the four nucleic acids bases A, C, G, T. This idea has indeed been used in past experiments, where researches attempt to apply existing natural language processing algorithms to the study of biological sequences [21]. It was first presented by E. Asgari et al. and applied successfully in many latter biological applications [22]. Moreover, the word feature namely k-mer has also been applied in RNA sequence description and protein structure. In this paper, we proposed a hierarchical attention networks for motif discovery task [23], which based on a computational method for document classification proposed by Zichao Yang et al.
2 Proposed Approach
We propose a hierarchical attention network (HAN) for discovering DNA-protein binding sites. The model we used is based on the hierarchical attention network for document classification proposed by Zichao Yang et al. which includes two levels of attention mechanisms. The attention mechanism was used in two hierarchical level, word and sentence. By letting the model pay different attention on different part of sentence or document, the application of attention mechanism could generate a more reasonable representation of sentence or document. We believe the attention method is also suitable for bioinformatics data such as DNA sequence because different part of the DNA sequence may have different amount of influence to the expression of the whole sequence. DNA sequence also has the hierarchical structure like word and sentence in natural language, thus the hierarchy mechanism in the model could improve the performance of processing DNA data [23].
In order to transform DNA sequence into a form that could be read by our model based on natural language processing method while not losing information in the DNA sequence, we use k-mer and word embedding when processing the sequence data. The DNA sequence is first sliced by k-mer and word vectors is generated by word embedding according to the sliced result. The vectors are then fed into the hierarchical attention network for training. We also tried several different k-mer methods with different k lengths and gram lengths to figure out the best k length and gram length.
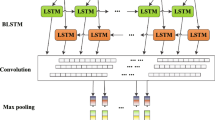
Figure 1 shows the structure of our proposed model. The result of word embeddding is fed into a hierarchical attention network. The network use a bidirectional LSTM based recurrent neural network as word encoder which encodes the word embedding into vectors containing information of context and the word itself. The followed attention layer extract words that are important to the meaning of the sentence and aggregate the representation of those informative words to form a sentence vector because not all words contribute equally to the representation of the sentence meaning. How attention mechanism work is specifically showed below.

A graphical illustration of HAN for motif discovery
Where hit is the output of word encoder which is generated according to the input word embedding. The word annotation hit is then fed into a one-layer MLP to get uit as a hidden representation of hit. After that, we measure the importance of the word as the similarity of uit with a word level context vector uw and get a normalized importance weight α it through a softmax function. Finally, the sentence vector si is computed as a weighted sum of the word annotations based on the weights. The context vector uw can be seen as a high-level representation of a fixed query “what is the informative word” over the words like that used in memory networks. The word context vector uw is randomly initialized and jointly learned during the training process.
The structure of sentence encoder and the following attention layer is as same as those for word except the sentence encoder take the sentence vector as input and finally output a vector representing the whole documentation. The documentation vector is fed into a softmax layer to get the final classification.
3 Experiments and Results
3.1 Data Sets
We collected 50 public ChIP-seq datasets from ENCODE to evaluate the performance of our proposed method. For each ChIP-seq dataset, 1000–5000 top ranking peaks were chosen as the foreground (positive) set in which each sequence consists of 200 bps. On the other hand, the way of generating background (negative) sequences is also crucial. It is widely recognized that the background sequences have to be selected to match the statistical properties of the foreground set, otherwise the elicited motifs could be biased [24, 25]. To satisfy such requirements, equal numbers of background sequences were generated by matching the length, GC content and repeat fraction of the foreground set following. According to this guideline, we selected the upstream and downstream DNA sequences of DNA-protein binding sites as negative sequences. Moreover, as mentioned before, the data sets are processed by several different k-mer methods with various k length and gram length to evaluate how value of k and gram influence the performance of the model [26, 27].
3.2 Evaluation Metrics
Area under the curve (AUC), a widely used evaluation metric in both machine learning and motif discovery, is one of the two evaluation metrics used in this paper [2, 28]. It is equal to the area under receiver operating characteristic curve (ROC curve), which is a graphical plot that illustrates the performance of a binary classifier, and indicates the probability that a classifier will rank a randomly chosen positive instance higher than a randomly chosen negative one [29, 30].
Another evaluation metrics applied in this paper is average precision (AP), which is also a commonly used metric to measure the ability of a proposed classifier [31]. Average precision is a measure that combines recall and precision for ranked retrieval results. For one information need, the average precision is the mean of the precision scores after each relevant document is retrieved [32].
3.3 Results
We compared the performance of HAN for predicting DNA-Protein binding sites with those of DeepBind and Deepsea. The AUCs and APs of proposed method HAN and competing methods DeepBind and Deepsea are calculated as the metrics of their performances. Each method is tested on the same 50 different datasets. The result of comparison is illustrated in Figs. 2 and 3.

The AUC and AP of our proposed HAN method across 50 experiments in the motif discovery task compared with DeepBind

The AUC and AP of our proposed HAN method across 50 experiments in the motif discovery task compared with Deepsea
From Figs. 2 and 3, which displays the AUC and AP comparisons between HAN and DeepBind, and between HAN and Deepsea, we observe that the AUCs and APs of MSC are much higher than those of DeepBind and Deepsea on most datasets, which means HAN could achieve a better performance than these two algorithms in most cases. In addition, APs of HAN are within a narrow range while these of DeepBind or Deepsea distribute in a quite larger area, which means the accuracy of our proposed HAN method are more stable than the other two compared methods across the various datasets.
The experiment results prove that with the help of hierarchical network structure and attention mechanism, our computation model extract critical information from DNA sequence more efficiently and process the data with a more reasonable method.
4 Conclusions and Future Work
In this paper, we propose a hierarchical attention network for predicting DNA-protein binding sites inspired by the model in natural language process area and has shown an impressive improvement on performance compared with DeepBind and DeepSea through a series of experiments. Our proposed HAN method obtains a higher accuracy than the two competing methods and shows a stable performance on various datasets. The results of experiments prove that the application of algorithms for NLP such as hierarchical attention network to motif discovery field are practicable and effective.
Although our proposed HAN method has achieved a relatively better performance, it still have some drawbacks such as that it shows poor performance on some datasets despite it perform well on most datasets, which means HAN for predicting DNA-protein binding sites still need to be improved.
References
Lambert, S.A., et al.: The human transcription factors. Cell 172, 650–665 (2018)
Huang, D.-S., Du, J.-X.: A constructive hybrid structure optimization methodology for radial basis probabilistic neural networks. IEEE Trans. Neural Netw. 19, 2099–2115 (2008)
Bao, W., Huang, Z., Yuan, C.-A., Huang, D.-S.: Pupylation sites prediction with ensemble classification model. Int. J. Data Min. Bioinform. 18, 91–104 (2017)
Deng, S.-P., Zhu, L., Huang, D.-S.: Predicting hub genes associated with cervical cancer through gene co-expression networks. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 13, 27–35 (2016)
Vaquerizas, J.M., Kummerfeld, S.K., Teichmann, S.A., Luscombe, N.M.J.N.R.G.: A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 10, 252 (2009)
Huang, D.-S., Zhang, L., Han, K., Deng, S., Yang, K., Zhang, H.: Prediction of protein-protein interactions based on protein-protein correlation using least squares regression. Curr. Protein Pept. Sci. 15, 553–560 (2014)
Elnitski, L., Jin, V.X., Farnham, P.J., Jones, S.J.J.G.R.: Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res. 16, 1455–1464 (2006)
Berger, M.F., Philippakis, A.A., Qureshi, A.M., He, F.S., Estep III, P.W., Bulyk, M.L.J.N.B.: Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24, 1429 (2006)
Stormo, G.D.J.B.: DNA binding sites: representation and discovery. Bioinformatics 16, 16–23 (2000)
Weirauch, M.T., et al.: Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 31, 126 (2013)
Furey, T.S.J.N.R.G.: ChIP–seq and beyond: new and improved methodologies to detect and characterize protein–DNA interactions. Nat. Rev. Genet. 13, 840 (2012)
Yu, H.-J., Huang, D.-S.: Normalized feature vectors: a novel alignment-free sequence comparison method based on the numbers of adjacent amino acids. IEEE/ACM Trans. Comput. Biol. Bioinform. (TCBB) 10, 457–467 (2013)
Zhu, L., Deng, S.-P., Huang, D.-S.: A two-stage geometric method for pruning unreliable links in protein-protein networks. IEEE Trans. Nanobiosci. 14, 528–534 (2015)
Bao, W., Jiang, Z., Huang, D.-S.: Novel human microbe-disease association prediction using network consistency projection. BMC Bioinform. 18, 543 (2017)
Liu, B., Li, K., Huang, D.-S., Chou, K.-C.: iEnhancer-EL: identifying enhancers and their strength with ensemble learning approach. Bioinformatics 34(22), 3835–3842 (2018)
Alipanahi, B., Delong, A., Weirauch, M.T., Frey, B.J.J.N.B.: Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831 (2015)
Shen, Z., Zhang, Y.-H., Han, K., Nandi, A.K., Honig, B., Huang, D.-S.: miRNA-disease association prediction with collaborative matrix factorization. Complexity 2017, 9 (2017)
Zhu, L., Guo, W.-L., Deng, S.-P., Huang, D.-S.: ChIP-PIT: enhancing the analysis of ChIP-Seq data using convex-relaxed pair-wise interaction tensor decomposition. IEEE/ACM Trans. Comput. Biol. Bioinform. 13, 55–63 (2016)
Zhou, J., Troyanskaya, O.G.J.N.M.: Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12, 931 (2015)
Huang, D.-S., Jiang, W.: A general CPL-AdS methodology for fixing dynamic parameters in dual environments. IEEE Trans. Syst. Man Cybern. B (Cybern.) 42, 1489–1500 (2012)
Le, N.Q.K., Yapp, E.K.Y., Ho, Q.-T., Nagasundaram, N., Ou, Y.-Y., Yeh, H.-Y.J.A.B.: iEnhancer-5Step: Identifying enhancers using hidden information of DNA sequences via Chou’s 5-step rule and word embedding. Anal. Biochem. 571, 53–61 (2019)
Asgari, E., Mofrad, M.R.J.P.O.: Continuous distributed representation of biological sequences for deep proteomics and genomics. PloS One 10, e0141287 (2015)
Yang, Z., Yang, D., Dyer, C., He, X., Smola, A., Hovy, E.: Hierarchical attention networks for document classification. In: Proceedings of the 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, pp. 1480–1489 (2016)
Fletez-Brant, C., Lee, D., McCallion, A.S., Beer, M.A.J.N.A.R.: kmer-SVM: a web server for identifying predictive regulatory sequence features in genomic data sets. Nucleic Acids Res. 41, W544–W556 (2013)
Orenstein, Y., Shamir, R.J.N.A.R.: A comparative analysis of transcription factor binding models learned from PBM, HT-SELEX and ChIP data. Nucleic Acids Res. 42, e63–e63 (2014)
Lee, D., et al.: A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 47, 955 (2015)
Yao, Z., MacQuarrie, K.L., Fong, A.P., Tapscott, S.J., Ruzzo, W.L., Gentleman, R.C.J.B.: Discriminative motif analysis of high-throughput dataset. Bioinformatics 30, 775–783 (2013)
Zeng, H., Edwards, M.D., Liu, G., Gifford, D.K.J.B.: Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 32, i121–i127 (2016)
Fawcett, T.J.P.R.L.: An introduction to ROC analysis. Pattern Recogn. Lett. 27, 861–874 (2006)
Zhu, L., Zhang, H.-B., Huang, D.-S.: Direct AUC optimization of regulatory motifs. Bioinformatics 33, i243–i251 (2017)
Aslam, J.A., Yilmaz, E., Pavlu, V.: A geometric interpretation of r-precision and its correlation with average precision. In: Proceedings of the 28th Annual International ACM SIGIR Conference on Research and Development in Information Retrieval, pp. 573–574. ACM
Davis, J., Goadrich, M.: The relationship between precision-recall and ROC curves. In: Proceedings of the 23rd International Conference on Machine Learning, pp. 233–240. ACM
Acknowledgements
This work was supported by the grants of the National Science Foundation of China, Nos. 61861146002, 61520106006, 61772370, 61873270, 61702371, 61672382, 61672203, 61572447, 61772357, and 61732012, China Post-doctoral Science Foundation Grant, No. 2017M611619, and supported by “BAGUI Scholar” Program and the Scientific & Technological Base and Talent Special Program, GuiKe AD18126015 of the Guangxi Zhuang Autonomous Region of China.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Yu, W., Yuan, CA., Qin, X., Huang, ZK., Shang, L. (2019). Hierarchical Attention Network for Predicting DNA-Protein Binding Sites. In: Huang, DS., Jo, KH., Huang, ZK. (eds) Intelligent Computing Theories and Application. ICIC 2019. Lecture Notes in Computer Science(), vol 11644. Springer, Cham. https://doi.org/10.1007/978-3-030-26969-2_35
Download citation
DOI: https://doi.org/10.1007/978-3-030-26969-2_35
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-26968-5
Online ISBN: 978-3-030-26969-2
eBook Packages: Computer ScienceComputer Science (R0)