
Abstract
This chapter describes the basic physical properties of wood and wood-based materials such as density, thermal properties, electrical properties, friction, optical properties, corrosion, and aging. In addition, selected new innovative testing methods are presented, which are still mainly used in research.
Together with the very high influence of (i) wood moisture on the properties of wood and wooden materials, (ii) the mechanical properties (elasticity and strength) as well as (iii) the heterogeneity and orthotropy of wood and wood-based materials, knowledge of the physical properties described in this chapter is very important.
This is necessary, for example, for processes such as the drying of wood, mechanical processing (e.g., machining, defibration, bonding), as well as the design and manufacture of products made of wood. Sound and thermal insulation of buildings, color change of wood during use, wood aging with changes in physical and mechanical properties are further examples.
Numerous processes from the area of nondestructive testing of wood are used to control the production. Examples are color measurement, the detection of the fiber angle by means of the tracheid effect, wood moisture measurement using electrical, dielectric, or spectrometric properties.
Ultrasound and eigenfrequency measurement already have a place in the industry for grading the quality of wood (strength) or blister detection with air-coupled ultrasound in particleboards or plywood.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Density
- Specific gravity
- Thermal properties
- Electrical and dielectric properties
- Acoustic properties
- Friction
- Optical properties
- Aging
- New innovative testing
1 Overview of Important Properties and Parameters
The physical–mechanical properties of wood and wood-based materials are described in Chaps. 6, 7, 8, and 9 in this handbook. In the specialist literature, these properties are often also summarized under the term ‘physics of wood and wood-based materials’ e.g., Niemz and Sonderegger [1] and Bosshard [2] (see also Fig. 6.1). Based on the classification of Kollmann and Côté [3], the above mentioned chapters are divided into:
-
Physical properties (Chap. 6)
-
Relationships between wood and water (Chap. 7)
-
Elasticity (Chap. 8)
-
Strength (Chap. 9)

Systematic of the properties of solid wood and wood-based materials
The knowledge of physical–mechanical properties of wood and wood-based materials, in combination with the biological properties (in particular the wood structure) and the chemical properties are an important basis for:
-
The working and processing of the wood (for example, cutting, joining, coating)
-
The development and optimization of new wood-based materials
-
The use of wood and wood-based materials (such as dimensioning for static properties, heat conduction, dimensional change in the case of moisture changes, utilization of constructions adapted to the material), and the development of products made of wood
Further literature for a deeper involvement to this topic are, for example, Niemz and Sonderegger, Kollmann and Côté, Bodig and Jayne, and the Wood Handbook [1, 3,4,5]. The biological and chemical wood properties are discussed in separate chapters of this book (Chaps. 3, 4, and 5) and also in several detailed items of specialist literature (e.g., Fengel and Wegener, Higuchi, Wagenführ, and Rowell [6,7,8,9]). As appropriate, a short recapitulation relating to these topics is appended to this chapter if it seems to be important for understanding.
All explanations refer to natural wood, i.e., solid wood, and also to wood-based materials as far as information is available.
By definition, solid wood is manufactured from green round timber through appropriate cuts (longitudinal, transverse) without altering the wood structure. In contrast, wood-based materials based on solid wood (laminated timber, sawn timber, cross-laminated timber), veneer (plywood, laminated veneer lumber), particles (particleboard, oriented strand board [OSB]), fibers (medium-density fiberboard [MDF], soft fiberboard) and their compounds are formed from structure elements that were previously produced from green timber. Normally, adhesives and additives (for sawn timber recently also mechanical joints such as dowels and conventional timber joints [tongue-and-groove, dovetail]) are inserted in order to join together the structure elements and to realize specific (predetermined) material properties. Wood-based materials are multipack products in which the structure of the natural wood is varied systematically for the purpose of property transformation.
All properties of wood and wood-based materials are subject to manifold influences that determine their level to a greater or lesser extent (Fig. 6.2). This has to be taken into account in material tests and test development in order to gain repeatable results. The most important influences are:
-
The structural wood composition (density, fiber-load angle, and cutting direction of solid wood; density and density profile, solid resin portion, additives and particle geometry of particle-based materials; layer thickness, layer orientation, and densification of laminated wood [laminated timber, plywood])
-
The surrounding conditions (e.g., relative humidity, temperature)
-
Wood history, e.g., aging (for wood-based materials, it is clearly influenced by the adhesive type, e.g. urea-formaldehyde resins are not moisture persistent), corrosion, heat treatment, fungal decay, and insect attack
-
The test specification (e.g., the span–thickness ratio at bending tests for the determination of modulus of elasticity (MOE), sample size at bending, tension and compression tests)

Systematic of the influencing factors of solid wood and wood-based materials
2 Density and Specific Gravity
Density is the ratio of mass to volume:
-
ρ – Density (kg m−3)
-
m – Mass (kg)
-
V – Volume (m3)
Wood is a capillary–porous system that can be filled with water, water vapor, air or an impregnating liquid (paint, adhesive). Wood swells when it absorbs moisture and shrinks when moisture is released.
Therefore, depending on the proportion of dry wood and water, both the mass (owing to the different density wood/water) and the volume of the wood change (see Chap. 7). This results in different wood densities, which can be distinguished as:
-
Density (density at a defined MC ω) ρω
-
Oven-dry density ρ0
-
Cell-wall density ρcw
-
Basic density R
Additional densities are used for particle-based materials:
-
Bulk density, spreading density, or mat density
-
Mass per unit area
-
Density profile
Instead of density, specific gravity is used in some countries, mainly the United States (see Kollmann and Côté, Stamm, and Kollmann [3, 10, 11]). Different specific gravities can be distinguished similar to the different densities [5]:
-
Specific gravity at MC (ω) Gω
-
Specific gravity of oven-dry wood G0
-
Basic specific gravity Gb
Further, the porosity can directly be derived from density.
2.1 Characteristics of Density and Specific Gravity
2.1.1 Density (Density at x% MC)
As mentioned above, the density of wood is moisture-dependent (Fig. 6.3 and ISO 13061-2). As the MC of the wood changes, the volume also changes below the fiber saturation point. Additionally, the density of bound water in wood increases exponentially from normal water density at fiber saturation point to 1300 kg m−3 under oven-dry conditions [3] (see also Chap. 7). The density ρω (or density at specified MC according to ISO 13061-2) is therefore moisture dependent and is defined as the quotient of the mass including moisture and the volume of the wood piece at the same MC.

Influence of moisture content (MC) on wood density [12]
-
mω – Mass of wood at MC ω (kg)
-
Vω – Volume of the wood at MC ω (m3)
-
ω – MC (%), which is defined as mass of water over mass of dry wood
The density in a normal climate (20 °C and 65% relative humidity (RH)) is referred to as normal density (often named ρ12). The equilibrium moisture content (EMC) in a normal climate is on average 12% for most European woods and 6–12% for particleboard and fiberboard. In scientific investigations, the MC of wood, which is tested in a defined climate (for example, in a normal climate), should be specified in each case.
2.1.2 Oven-Dry Density
The oven-dry density ρ0 is the quotient of the oven-dry mass and the oven-dry volume of wood:
-
m0 – Mass of oven-dry wood (without water; kg)
-
V0 – Volume of oven-dry wood (m3)
Since the oven-dry density is moisture independent, it seems most likely to be considered as a “material constant” (ignoring the growth-related variability). For the conversion of the density ρω into the density ρ0, the following applies for solid wood, taking into account the swelling and shrinkage behavior:
-
αVω – Maximum volumetric swelling (%) from oven-dry to MC ω
-
ω – MC (%)
At MCs of up to 25%, it is possible to approximate ρ0 using a linear dependence between moisture and swelling:
According to studies by the US Forest Products Research Laboratory, the following relationship between oven-dry density and maximum volumetric swelling αV (oven-dry to green) can be approximated:
2.1.3 Basic Density
The basic density Rg is the quotient of the mass of the oven-dry wood and the volume of the maximum swollen wood. The MC must therefore be above the fiber saturation range (green volume). The following applies:
-
m0 – Mass of oven-dry wood (without water; kg)
-
Vmax – Volume of maximum swollen wood (green volume; m3)
In the United States the following is also often used:
-
m0 – Mass of oven-dry wood (without water)
-
V12 – Volume of wood at 12% MC
The density Rg is often used in forestry, for example, for the calculation of the true wood mass of a timber stack. For the conversion of the density Rg into the density ρ0, the following results:
-
αV – Volumetric swelling (%)
-
βV – Volumetric shrinkage (%)
2.1.4 Cell-Wall Density
The cell-wall density ρcw is the quotient of the mass of the dry wood and the volume of the cell wall (without pores). It characterizes the density of the pure cell-wall substance. The following applies:
The cell-wall density is nearly the same for all wood species; it is on average 1500 kg m−3. According to Knigge and Schulz [13] the value varies between 1440 and 1600 kg m−3. These differences are caused by different test methods [10] and by the different lignin and cellulose content of the wood species. For lignin, a value of 1380–1460 kg m−3 and for cellulose a value of 1580 kg m−3 is indicated. For determination of the cell-wall density, e.g., the displacement method (liquids are often helium or gasoline but also water [10, 14]) and also mercury porosimetry are used [15].
2.1.5 Specific Gravity
The specific gravity Gω is the ratio between oven-dry mass and the mass of water displaced by the bulk specimen at a given MC [10] and is moisture dependent. The specific gravity is dimensionless. It is mainly used in the United States and not so often in Europe (see Kollmann and Côté, Stamm, and Kollmann [3, 10, 11]).
-
Vω – Moist volume (m3)
-
m0 – Oven-dry mass (kg)
-
ρw – Density of water (1000 kg m−3)
When the bound water content increases, it causes a swelling and the specific gravity decreases. The maximal value is reached under oven-dry conditions G0, the minimum (and a constant value) above the fiber saturation point. The latter is named basic specific gravity Gb.
We can also calculate a specific gravity for different MCs (e.g., in a normal climate: G12) and a specific cell-wall gravity [10]. Depending on the testing method, the dimensionless specific cell-wall gravity is in the range 1.53 (tested in water) to 1.44 (tested with mercury porosimetry).
2.1.6 Porosity Content (Void Fraction)
The porosity c is the volume of all the voids of the wood in the dry state, based on the volume of the wood. It results from the ratio of the oven-dry density to the cell-wall density of the wood.
-
for ρcw = 1500 kg m−3, it follows approximately:
-
ρ0 – Oven-dry density (kg m−3)
-
ρcw – Cell-wall density (kg m−3)
The pore volume changes at moisture absorption because the cell walls of the wood swell. Figure 6.4 shows the porosity of different wood species depending on the oven-dry density. On average, the porosity is around 50–60%. Porosity and pore size distribution are important for impregnability and seasoning of wood (strongly species dependent).

Pore content and cell-wall content of various wood species
2.1.7 Bulk Density, Spreading or Mat Density
The bulk density and spreading or mat density are parameters that are used for particle-based materials (particleboards and fiberboards).
The spreading density (or mat density) is the quotient of mass and volume of scattered falling wood particles randomly deposited in the plane.
Bulk density is the quotient of mass and volume of wood particles (in particular, wood chips and also particles) deposited as a heap. In this case, a largely three-dimensional, statistically random deposition of the particles takes place. The bulk density is used as a parameter for the dimensioning of storage bunkers for wood chips, fibers, or particles.
2.1.8 Board Weight Measurement (Area-Related Mass)
The area-related mass (kg m−2) is the quotient of the mass and the area of a board. It is an important characteristic in the production process of particle- and fiberboards. Together with the thickness and MC of the mat, it forms the basis for the calculation of the board density. The following applies:
-
ma – Basis weight (kg m−2)
-
m – Mass (kg)
-
A – Area (m2)
2.1.9 Density Profile Perpendicular to the Board Plane
The density profile is understood to be the distribution of the density over the cross section of a board (thickness). By changing the particle geometry, MC, and technological parameters (temperature, compression speed), the density profile can be varied within wide limits (Fig. 6.5). Most particleboards usually have the density profile, which is shown in Fig. 6.5b, with a density maximum in the edge zones and a lower density in the middle layer and at the edges (near the surface). By using a special pressing technique, the density profile can be largely homogenized (as desired, for example, in MDF and special particleboard types).

Typical density profiles of particle-based materials perpendicular to the plane: (a) Maximum density at the edge of the board. (b) Density drop at the board edge. (c) Homogeneous density over the entire board cross section
2.2 Influences on the Density and Density Distribution of Wood and Wood-Based Materials
2.2.1 Influence of the Wood Species
The ratio between cell wall and porosity (void fraction) varies considerably between the different wood species. The density is therefore dependent on wood species. Table 6.1 contains mean values and maximum deviation of oven-dry density and basic density of selected wood species.
In the case of balsa, which is the lightest industrially usable wood species, the average density is 130 kg m−3, in the case of lignum vitae it is a mean of 1230 kg m−3. Within a species of wood, significant differences in density are present owing to growth (soil) and location-related factors (climatic conditions). Figure 6.6 shows the frequency distribution of the density of different wood species. The density varies approximately in a ratio 2 to 3:1. It can be seen, that for most species, there is no Gaussian distribution, but there is a tendency to asymmetry. This means that the mean value is not identical to the peak value of the frequency distribution. Asymmetrical and uneven frequency curves are composed of symmetrical normal frequency curves [3].

Frequency distribution of the density of various wood species [13]
2.2.2 Influence of Growing Conditions and Location as well as the Sociological Position of the Tree in the Forest
There are different factors such as growing condition, location, and sociological position, that influence wood density. The differences can be distinguished into [16]:
-
Within-tree variation
-
Within-stand variation
-
Inter-regional variations
Soil and climate have a decisive effect on the growth of the wood. The density decreases under the same soil conditions from the climatic optimum to colder locations [17]. It is lower at higher altitudes than at lower levels (Fig. 6.7). In southern Sweden the density of spruce is higher than in northern Sweden [18]. Figure 6.8 shows the density distribution of Douglas fir in the USA and radiata pine in Chile. The geographical variability is recognizable.

Oven-dry density of Norway spruce and Scots pine from different regions [20]

Also, the growth-ring width changes significantly in softwood owing to the growth conditions. For example, it is higher in the Prealps than in the high altitudes of the Alps. Even within a forest there are differences in diameter, height, and stem shape of the trees. The density is lower in the predominant trees than in the suppressed trees. In the case of spruce, the basic density within a location increases with decreasing wood diameter.
In hardwood, the relationships have been studied less frequently. Schwappbach (cited in Kollmann [12]) found that the density of European beech in the northern hemisphere decreases evenly from south to north, and likewise from lower to higher altitudes.
Trendelenburg and Mayer-Wegelin [19, 20] compiled such trends and their causes very well. Likewise, Walker [16] and Butterfield [21] give a very good overview of the variation in density of plantation woods.
2.2.3 Influence of Structural Parameters
2.2.3.1 Earlywood and Latewood
Earlywood has a lower density than latewood. Figure 6.9a shows its frequency distribution for Douglas fir. Lanvermann [24] determined for Norway spruce minimum values of 200 kg m−3 in earlywood and maximum values of about 1000 kg m−3 in latewood. Yew as a softwood with very high density (590–670 kg m−3) has according to measurements by Keunecke, an earlywood density of 500–600 kg m−3 and a latewood density of about 1000 kg m−3 [25]. The maximum values in latewood are therefore about the same for spruce and yew. There is a strong correlation between latewood content and mean density for softwood (Fig. 6.9b) and between the proportion of strengthening tissue (sclerenchyma) and density for hardwood. In ring-porous hardwood, density depends on the percentage of latewood. For ash wood, the correlation is stronger at a low latewood percentage and flattens between 50 and 75%. Above 75%, nearly no influence is measured (Fig. 6.10).

(a) Frequency distribution of earlywood and latewood density in Douglas fir and (b) dependence of density on latewood content for Norway spruce [13]

Relationship between oven-dry density and latewood for ash [26]
2.2.3.2 Growth-Ring Width
The proportion of latewood and thus the density of softwood generally decreases as the growth rings become wider. Some softwoods have a pronounced maximum, so that the density only drops after a short rise (Fig. 6.11). These tendencies can be superimposed by location-related influences. For example, Norway spruce from the Alpine foothills usually have larger growth-ring widths, but also a higher density than those from high altitudes see [20]. Thinnings also have a significant impact.

Dependence of the oven-dry density of spruce, Douglas fir, pine and larch on the annual ring width [13]
Ring-porous hardwoods show a concordant increase in bulk density and growth-ring width [13]. The latewood percentage increases with increasing growth-ring width and thus also the bulk density. There is no clear tendency for diffuse porous trees.
2.2.3.3 Tree Age
Softwoods form denser wood with increasing age independently of the growth-ring width. For ring-porous hardwoods on the other hand, the density decreases with increasing tree age. Growth rings and/or early- and latewood width, in combination with density differences within the growth rings, are essential criteria for determining the age of wood (dendrochronology). Also, from the growth-ring course, conclusions can be drawn about the climatic conditions of bygone times (dendroclimatology). Special X-ray equipment and optical devices are used for this.
For age determination, a gapless annual growth-ring cycle is required up to the present. The longest European comparative chronology dates back to the year 5289 before our era. Dendrochronology and dendroclimatology have become increasingly important in recent decades (see, for example, Schweingruber and Günther [27, 28]). This research field has developed into an important field of climate research. Age determination based on growth-ring analysis is an established method today.
In the tropics, the trees do not grow in an annual rhythm, but in differently timed rainy seasons. Thus, there are no annual rings but some irregularities, which are difficult to detect even under the microscope [29].
Fast-growing wood from plantations (e.g., radiata pine, eucalyptus), which is often harvested after 10–20 years for use as sawn timber, has mainly juvenile wood; the density is lower, the microfibril angle (MFA) larger than of adult wood of the same species (see, for example, Butterfield [21]).
2.2.3.4 Heartwood/Sapwood
The formation of heartwood ends the physiologically active life phase of the young wood (sapwood). It causes a density increase by incorporating ingredients (extractives) into the cells. For many types of wood, this is evident in the significant differences in density between heartwood and sapwood. These are greater, the more they become visible as color differences.
2.2.3.5 Wood from Branches, Roots, and Reaction Wood (Tension and Compression Wood)
Wood of branches (knots) is heavier than that of logs; the wood of roots is lighter. The branch wood (knots) of spruce can reach twice the density of knotless wood. According to Mette [17], the density of spruce knots is around 900 kg m−3, that of clear wood around 450 kg m−3. But even in the vicinity of branches, wood has a higher density. At a greater distance from the branch, the density again reaches the density value of normal trunk wood. The branch density of pine, however, is only about 600 kg m−3. In European beech, the differences between branch and trunk wood are smaller (density of branches about 750 kg m−3).
The density of compression wood is up to 40% higher than that of normal wood. The density of root wood changes with the distance from the stump. At a greater distance, it has only about half the density of the stump wood.
2.2.3.6 Density Distribution in the Stem
Within the stem of a tree, there are regular density differences along and across the trunk axis. These differences depend on the wood species. Mette [17] shows the transverse density distribution for different types of wood (Fig. 6.12). The dependence on the tree height is shown in Fig. 6.13 and a cluster-like distribution over the stem with significant local density differences in Fig. 6.14.

Distribution of the oven-dry density over the cross section according to Mette [17]: (a) spruce type, (b) pine type, and (c) hardwood type

Changes in oven-dry density of earlywood and latewood within a stem of Douglas fir depending on (a) age and (b) height according to Knigge and Schultz [13]

Basic density distribution in a grand fir (Abies grandis Lindl.) log: (a) Density distribution. (b) Sampling scheme (according to Knigge and Schultz [13])
The density of the wood thus shows considerable differences, both within a log and between the logs. These must be taken into account when determining the required number of samples, but also when checking the quality online in order to obtain reproducible results.
2.2.4 Particleboard and Fiberboard
Particleboard and fiberboard (see also Chaps. 27 and 28) have significantly lower density variations in plane than a board made of solid wood for manufacturing reasons. The cause is the disintegration of the wood and the targeted mat formation. The density is controlled and regulated during mat forming. Figure 6.15 shows the relative frequency of the density of a particleboard. The coefficient of variation of the average density of wood-based materials is 3–5%; that of solid wood is 15–20%.

Relative frequency of density within a particleboard type
Particleboard and fiberboard have a significant density profile perpendicular to the board plane. The density profile can vary. Spread density and bulk density are influenced by the particle geometry and wood species (see Chap. 24).
2.3 Methods of Density Measurement
2.3.1 Conventional Methods
The density of wood and wood-based materials is usually determined on test specimens with constant dimensions in length, width, and thickness. The density then results from the quotient of mass and volume. Table 6.2 contains the usual dimensions of these test specimens.
For the density determination of wood, e.g., test specimens of dimensions 20 mm × 20 mm × 30 mm are used, whereas test specimens of wood-based materials usually have the dimensions 50 mm × 50 mm × board thickness. Often, the density test is also carried out on specimens (or parts of them) intended for other tests (e.g., on bending samples). This allows a direct correlation of the density with the respective tested property.
2.3.1.1 Mass Determination
The mass is usually determined by means of balance. Table 6.2 shows the measurement accuracy required.
2.3.1.2 Volume Determination
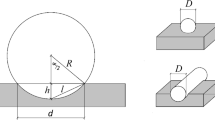
The volume of regularly shaped specimens is determined by measuring length, width, and thickness. For irregularly shaped specimens, such as chips, the volume can be easily determined by means of displacement methods. Overflow or immersion vessels with riser or a pycnometer are used (Fig. 6.16). In order to prevent water absorption by the wood, the test specimens are previously soaked with paraffin.

Volume determination of wood using displacement methods: (a) Overflow vessel. (b) Dip tank. (c) Pycnometer
When using a pycnometer, the test piece is first weighed and then the pycnometer is filled with distilled water and sealed. After determining the mass (m1) of the pycnometer (mpyc) and the water (mwatertotal), the test piece is placed in the pycnometer, whereby water is displaced according to the volume of the test piece. Thereafter, the total mass (m2) of the pycnometer (mpyc), residual water (mreswater), and test specimens (mwood) is determined. The following equations apply:
For the density of the wood (ρwood) the following then applies:
When using paraffin-sealed wood:
where:
-
\( {V}_{{\mathrm{water}}_{\mathrm{dis}}} \) – Volume of displaced water (m3)
-
\( {\rho}_{{\mathrm{water}}_{\mathrm{dis}}\left(\mathrm{T}\right)} \) – Density of displaced water at temperature T (kg m−3)
-
Vsample – Volume of the specimen (m3)
-
mwood – Mass of the wood (kg)
-
m′wood – Mass of paraffin-sealed wood (kg)
The volume of wet wood is often determined by immersing the wood in a vessel standing on a balance and filled with water. From the volume displacement and the density of water of 1000 kg m−3, the volume can be determined directly via the mass change. To determine oven-dry density, the specimen has to be dried at 103 °C (before volume determination) until mass constancy is reached (oven-dry mass). Afterward it is weighed and measured. To determine basic density, the specimens are stored in distilled water until maximum swelling. The change in dimensions must be checked by repeated measurements every 3 days.
2.3.2 Density Determination by Means of Electromagnetic Waves and Other Methods
For the continuous determination of the mass of the unit area or the bulk density, the absorption of electromagnetic waves is used to an increasing extent. For the most part X-rays are used (see also Chap. 20). The method is based on the effect that the electromagnetic waves are absorbed when passing through a material proportional to the area-related mass. The density is thus calculated as follows taking into account the radiated thickness of the material:
-
JO – Count rate in air
-
JN – Zero effect (count rates from background without radiation)
-
J – Count rate with absorber
-
ρ – Density (kg m−3)
-
μ/ρ – Mass attenuation coefficient (depending on material; m2 kg−1)
-
d – Thickness of the radiated sample (m)
Factors influencing the density measurement with X-rays are the wood MC and the mass attenuation coefficient. The mass attenuation coefficient for wood-based materials depends on the adhesive content and the type of adhesive. Increasingly, this method is used to determine local density differences or structural damage, e.g., bores from insect attack or fungal degradation (Fig. 6.17).

Computed tomography of spruce wood (a) in an air-dry state with fungal degradation (incipient rot near the pith) and (b) in a green (fresh) state. (Photos: A. Flisch, Empa)
Whereas the absorption of the radiation can be used to determine the average density, the diffraction of the X-radiation allows the density profile to be determined (see Sect. 6 of Chap. 20). X-ray scattering (e.g., with the device SilviScan) is further used to measure the microfibril angle in the S2-layer of the cell wall [21]. From these data (density profile, microfibril angle) the MOE can be derived [24].
Today, X-ray is used industrially to determine board weight and the density profile perpendicular to the board plane. Also, X-ray is used for grading logs and sawn timber (detection of board weight and knots; see also Chap. 20).
The industrial application of computed tomography (CT) (3D image of the structure) for quality assessment in logs for saw milling and veneer is currently also known in art, e.g., Microtec/I. Methodological principle is the creation of a spatial image of the examined body, which allows the localization of defects in the wood (for example, in works of art or in veneer wood). In research, X-ray microtomography and synchrotron light are used for tomography (Fig. 6.18) [30]. Today, the following resolution is reached:

Micro-CT and synchrotron images. (a) Micro-CT of glued wood (bond-line), photo T. Lüthi, Empa. (b) Synchrotron tomographies (high-resolution X-ray radiation) of spruce and fir samples during in situ compression [30]
Method | Resolution |
X-ray tomography | 50 μm |
X-ray microtomography | 1 μm |
Synchrotron tomography | 0.3 μm (1 mm sample diameter) |
2.3.3 Determination of the Density Profile of Wood-Based Materials
Owing to the importance of the density profile for the processing of wood-based materials, a number of determination methods have been developed. Figure 6.19 gives an overview.

Procedure for measuring the density profile of particle materials (particleboard and fiberboard) perpendicular to the plane
2.3.3.1 Milling Method
The principle of the milling method is shown schematically in Fig. 6.20. Grooves are milled in the samples according to the mass ratio of surface to middle layer. Then, the samples are separated into middle and surface layers. The density is determined by measuring the dimensions and weights of the single layers. Generally, a ratio of top to middle layer of 40:60 is used. The method allows only an approximate determination of the densities of the surface and middle layers.

Sample preparation for determining the density profile perpendicular to the plane according to the milling method
2.3.3.2 Planing Method
In the planing method, thin layers are removed from defined test specimens (50 mm × 200 mm, for example) by means of a planer (e.g., a long planer used in metalworking) until the middle of the specimen is reached and then the density from each layer is determined conventionally by its mass and volume. The thickness of one removed layer can be, for example, 0.5, 1.0, or 2 mm (lower in the outer zones than in the middle of the plate). The ablation takes place at 50% from the bottom of the test specimens and at 50% from the top to the middle of the board. Hence, this results in a complete density profile over the entire cross-section.
2.3.3.3 Drilling Method
In the drilling method, the torque or power consumption of a drill is determined as a function of density [31]. A thin needle with a specially shaped tip is drilled into the wood and the power consumption is measured. So far, the method has mostly been used for the detection of decay in living trees (see Fig. 6.78) or in wooden structures, but also for density profile measurements (in wood-based materials and for growth-ring analysis) and for density measurements of dry wood and even in living trees, for example, for forest genetics field tests and wood-quality survey programs [32,33,34,35].
2.3.3.4 Measurement of Density Profiles Using Electromagnetic Waves
A sharply focused X-ray or gamma ray penetrates the test specimen in layers parallel to the plane of the plate. With the aid of a detector, the count rate of the attenuated radiation is measured. The density is determined according to equation (6.20) by means of the material thickness (in the transmission direction), the mass attenuation coefficient, and the count rate of the attenuated radiation. By stepwise movement of the test specimen by 0.1 mm each time, its density profile can be determined (Fig. 6.21).

Typical density profile of a board perpendicular to the plane, determined by means of X-ray
2.3.4 Methods for Testing Bulk Density and Spreading or Mat Density
To determine the spreading density, the particles are sprinkled from the hand (from a height of about 500 mm) into a measuring vessel (content, for example, 2 dm3) by uniform, gradual dropping. From particle size and chip volume, the spreading density is determined according to equation (6.1).
To determine the bulk density, the chips are poured into a vessel (content 2 … 10 … 50 dm3). Particle mass and volume are used to determine the bulk density according to equation (6.1).
2.3.5 Determination of Pore Content and Pore Size Distribution in Wood-Based Materials
2.3.5.1 Mercury Intrusion Porosimetry
For the experimental determination of the pore content of wood and wooden materials as well as the pore size distribution, mercury intrusion porosimetry (ISO 15901-1) is often used.
This method takes advantage of the physical effect that a nonwetting liquid penetrates only under pressure in capillaries. The pressure to be applied is greater, the closer the capillaries. From the pressure, one can calculate the respective capillary radius.
From the recorded diagram, both the pore radius and the pore volume can be determined. The methodology uses the Washburn equation to calculate the pore radius from the applied pressure [36]. Pore radii are measurable within the range 58 μm to 1.8 nm. Extensive measurements have been made, for example, by Plötze and Niemz, Schneider, Schweitzer and Niemz, and Zauer et al. [15, 37,38,39,40].
2.3.5.2 Gas Adsorption
The pore size distribution can also be calculated from the sorption measurements [41]. According to the Kelvin equation, the equilibrium pressure in a capillary is less than the general saturation pressure as long as the contact angle is less than 90° [42]. The vapor therefore condenses under these conditions in all capillaries whose radius is smaller than that calculated from this equation for the pressure in question.
2.3.5.3 Other Methods
Other methods of pore size measurement are, for example, the pressure plate technique (often used in building physics [43]) and the thermoporosimetry (principle of dynamic differential calorimetry, based on heat flow measurements, [44]). A comprehensive compilation is included in the thesis by Zauer [45]. Zauer also gives an overview of the detectable pore size of the individual processes. Partial X-ray or synchrotron tomography is also used to detect larger pores [30, 46].
2.4 Influence of Density on Wood Properties
Density is one of the dominant characteristics. It influences almost all properties of wood. It is therefore often used as an indicator of strength, although it is not sufficient for reliable strength grading. Figure 6.22 shows schematically the effect of bulk density on selected properties. Density fluctuations of wood and wood-based materials therefore have a significant impact on strength. Data on the density of wood and wood-based materials are given in Tables 6.1, 6.2, and 6.3.

Schematic representation of the influence of bulk density on selected properties of wood and wood-based materials
3 Thermal Properties
Wood and wood-based materials have a specific thermal behavior, which is advantageous (e.g., insulation) or rather disadvantageous (burning) in comparison with other materials. Therefore, this chapter gives attention to different aspects of thermal influence on wood properties. Fundamental parameters are thermal conductivity and diffusivity, heat capacity, and thermal expansion. Also, the influence of heat on mechanical properties is essential. Additionally, thermal properties will be applied to some extent for quality control. As mentioned, a further important aspect of thermal behavior is thermal decomposition (pyrolysis), reaction to fire, combustion, and fire protection, which is implemented in Chap. 17.
3.1 Thermal Conductivity
Definition: Thermal conductivity (λ) is the quantity of heat that flows in 1 s through a cube of one cubic meter by a temperature difference of 1 Kelvin between the opposite sides. The unit of measurement is W m−1 K−1.
Wood is a poor heat conductor compared with other construction materials (Table 6.4). The thermal conductivity of wood and wood-based materials is substantially determined by their structure and is 1.5–2.75 as high in the direction of the fiber as it is perpendicular to the fiber orientation (see also Table 6.5). Values of the latter for many wood species are listed in the Wood Handbook [5]. Maku (cited in Kollmann and Malmquist [48]) calculated for the thermal conductivity of the net wood substance across the fiber 0.42 W m−1 K−1 and parallel to the fiber 0.65 W m−1 K−1, of which the latter is within the range of the thermal conductivity of water. Perpendicular to the fiber, there are only a few differences between the radial and the tangential direction. Generally, thermal conductivity is about 10% higher in the radial direction than in the tangential direction. This difference is highly influenced by the rays (increasing with increasing ray cell volume) in hardwoods and by the latewood (increasing with increasing latewood volume) in softwoods [49].
Thermal conductivity increases with increasing density (Fig. 6.23). It can be calculated for wood at 12% MC and room temperature (27 °C) perpendicular to the fiber orientation as follows [12]:

and parallel to the fiber orientation:
-
ρ – Density (kg m−3)
Thermal conductivity perpendicular to the grain increases with increasing MC owing to the higher thermal conductivity of water compared with oven-dry wood. Its influence is shown in Fig. 6.24. The values were deduced from the Wood Handbook [5] and were converted from values dependent on specific gravity to values dependent on density. The dependency from MC and specific gravity (>0.3) according to the Wood Handbook reads as follows:

Thermal conductivity of wood perpendicular to the grain depending on moisture content (MC) and density or specific gravity according to values in the Wood Handbook [5]
-
λ – Thermal conductivity (W m−1 K−1)
-
Gω – Specific gravity (−) at MC ω
-
ω – MC (%)
To calculate the thermal conductivity from a given value to a value under a different environmental condition, the influence of MC within the range 5 < ω < 35% is estimated according to Kollmann and Côté [3] as:
-
λ1,2 – Thermal conductivity (W m−1 K−1) at MC ω1 and ω2
-
ω1, ω2 – MC (%) at times/conditions 1 and 2
The effect of temperature on thermal conductivity is relatively minor and increases about 2–3% per 10 °C [5]. Kanter (cited in Steinhagen [49] also shows the influence of MC (Fig. 6.25). In the oven-dry state, thermal conductivity increases with increasing temperature within the range between −40 °C and 100 °C. In contrast, above fiber saturation, thermal conductivity also increases above zero degrees with increasing temperature but decreases below zero degrees with increasing temperature followed by a jump at zero degrees because ice has a much higher thermal conductivity than water, which increases with decreasing temperature (see Table 6.4).

Thermal conductivity of birch wood (specific gravity 0.515) depending on temperature and MC according to Kanter (cited in Steinhagen [49]) modified
3.2 Specific Heat
Definition: Specific heat (c) is defined as the heat quantity required to heat the temperature of 1 kg of a material by 1 Kelvin. The unit of measurement is J kg−1 K−1.
The specific heat of wood and wood-based materials is highly dependent on MC owing to the high specific heat of water and increases with increasing MC and also with increasing temperature. In contrast, it is not influenced by density. The dependency of temperature (according to Dunlap cited in Kollmann and Côté [3]) and MC [3] can be described as follows:
-
c0 – Heat capacity of oven-dry wood (J kg−1 K−1)
-
cω – Heat capacity of wood at MC ω (J kg−1 K−1)
-
cw – Heat capacity of water (J kg−1 K−1)
-
θ – Temperature ( °C)
-
ω – MC (−)
A mean heat capacity value within the temperature range 0–100 °C is suggested for drying technology [3], but for application in building physics, a value at 20 °C or even lower is more appropriate [51].
The specific heat of wood is about four times higher than that of iron or copper (Table 6.4). Therefore, wood is very appropriate for the grips of cooking utensils and heating devices owing to its high specific heat and low thermal conductivity. Table 6.6 shows the influence of MC on the specific heat of diverse wood species. In contrast to MC, the influence of wood species itself is low. Wood-based materials have similar specific heat to wood. It may be influenced by adhesives. Thus, according to Czajkowski et al. [55], specific heat of OSB at 6–7% MC is about 100 J kg−1 K−1 higher than that of low-density fiberboard or particleboard.
The high specific heat and the low thermal conductivity are advantageous for wood fiber insulation boards compared with other insulating materials. These properties induce a clearly higher damping of the temperature amplitude (decrement factor) and a much higher phase shift (decrement delay) from the outdoor to the indoor climate compared with, for example, rock and glass wool (Table 6.7). Thereby, Pavatex SA calculated the decrement delay with a high specific heat (according to the standard value). Determinations of specific heat of wood fiber insulation boards in Czajkowski et al. and Ghazi Wakili et al. [55, 56] result in clearly lower values between 1360 and 1630 J kg−1 K−1 at lower MC.
3.3 Thermal Diffusivity
Thermal diffusivity a is defined as the velocity of heat conduction in a material. It is influenced by thermal conductivity, specific heat, density, and inversely by MC. It is calculated as follows:
-
c – Specific heat (J kg−1 K−1)
-
ρ – Density (kg m−3)
-
λ – Thermal conductivity (W m−1 K−1)
Thermal diffusivity becomes important for wood in practice, i.e., for drying, bonding, compression (e.g., on production of wood-based materials as particle- and fiberboards, laminated veneer or solid wood), and for evaluation of low- and high-temperature performance. Table 6.8 shows the influence of wood density and MC on thermal conductivity, specific heat, and thermal diffusivity.
3.4 Thermal Expansion
Thermal expansion is characterized by the coefficient of thermal expansion, which is defined as the linear extension of a rod of 1 m at a temperature difference of 1 K and is calculated as follows:
-
αth – Coefficient of thermal expansion (m m−1 K−1) or (K−1)
-
Δl – Change in length (m)
-
l0 – Initial length (m)
-
Δt – Temperature difference (K)
A linear correlation exists between thermal expansion and temperature with a length increase by heating and a length decrease by cooling. The overall length l2 after a temperature change from t1 to t2 and an initial length l1 results in:
The coefficient of thermal expansion for wood is influenced by wood species, density, and cutting direction (Table 6.9).
According to Weatherwax and Stamm (cited in Kollmann [12]), thermal expansion depends on cutting direction and oven-dry density as follows:
-
αth.r – Linear radial thermal expansion (m m−1 K−1)
-
αth.t – Linear tangential thermal expansion (m m−1 K−1)
-
ρ0 – Oven-dry density (kg m−3)
-
αth⊥ − Linear thermal expansion perpendicular to the fiber (m m−1 K−1)
Thermal expansion has a clearly lower impact than moisture-related swelling and shrinkage. In general, change in temperature is combined with a MC change and therefore, swelling and shrinkage appear additionally. The swelling and shrinkage values are one order of magnitude higher than the values of thermal expansion. The wooden coefficient of thermal expansion in fiber direction account for 2.5 × 10−6 to 11 × 10−6 m m−1 K−1 within the temperature range −60 to +50 °C. It increases 2–3% in the fiber direction and 3–5% perpendicular to the fiber with decreasing MC at 1% MC change.
Thermal expansion of laminated wood is clearly influenced by the amount of adhesive and increases with increasing resin content [12]. In contrast, the influence of the degree of compression is low. Markings appear at the ends of laminated wood after temperature change owing to the different thermal expansion in the fiber direction and perpendicular to the fiber (Fig. 6.26). This phenomenon is called the washboard effect.

Schematic diagram of the washboard effect due to different linear thermal expansion of the separate layers on plywood. (a) Initial state. (b) After heating [12]
3.5 Thermal Influence on Wood Properties
The thermal influence on the properties of wood and wood-based materials is due to the fact that the volume of the material increases owing to the higher frequency of atom and molecule oscillation. Linked with that is a weakening of the intermolecular attraction and cohesion. Heating of the crystalline parts of structural substance results in an increased thermal vibration of the chain links against each other. Thereby, hydrogen bonds are reduced and softening of the lignin occurs additionally. This is similar to the influence of moisture. There, the strength reduction due to increased MC is attributed to a reduction in the partial valency forces and electrostatic bonds induced by water storage in the intermicellar and interfibrillar cavities (broadening of the cellulose chains).
In an oven-dry state, the cellulose chains lay close to each other so that dipole linkage, hydrogen bonds, and dispersibility operate. Thereby, structural substance and filler substance are stiff and brittle. The force of attraction is reduced with increasing heat and MC and the wood is more elastic. Simultaneous action of heat and increased wood moisture effects a clear property change and the ductility strongly increases, so that the wood can be permanently deformed. The plasticizing can be increased with additional vapor pressure (Fig. 6.27). These effects are used, for example, for wood bending (furniture from bentwood), for the thermo-mechanical pulping in a defibrator, or generally for the thermic plasticizing of wood.

Softening velocity of wood depending on vapor pressure, according to Lampert [58]
3.5.1 Thermal Influence of Short Duration
Temperature change clearly influences the properties of wood. The MOE decreases with increasing temperature. This behavior is strengthened with increasing wood moisture. Figure 6.28 shows the relative change in MOE with increasing temperature as mean values of several wood species related to 20 °C (= 100%). Analog tendencies exist for other elasto-mechanical properties as well as for strength (Figs. 6.29 and 6.30).

Relative change of the modulus of elasticity (MOE) of wood depending on temperature according to Sulzberger (cited in Kollmann [12])

Influence of temperature on strength (above) and modulus of elasticity (MOE) of spruce (below), according to Glos and Henrici [59]

Strong temperature variations also influence the processing characteristics of wood. For example, chipping of frozen wood results in an increase in the fines due to higher wood brittleness. Investigations on the freezing velocity of spruce wood show only a marginal influence on the strength properties at a fast cooling velocity of −10 °C/h. In contrast, the strength decrease was considerable at a cooling velocity of −1 °C/h [62]. Further, Geissen [63] researched fundamentals concerning the influence of temperature on the properties of wood.
The influence of temperature is of practical relevance at the burning of wooden constructions. The temperature behind the built charcoal layer is at most 100 °C according to investigations of Glos and Henrici [59] and therefore structural stability is ensured to some degree. However, a clear reduction of the elastic and strength properties is expected at this temperature (Table 6.10). Commonly, it is stated that tensile strength decreases least and compression strength is strongest. Also, a clear influence of the construction element and its size is stated (see also Chap. 17).
3.5.2 Long-Term Influence of Temperature
The duration of thermal exposure clearly influences the property change. Figure 6.31 show the relative change of density, compression strength, and impact bending strength of Sitka spruce depending on heating time. Also, color change takes place. The strength reduction with increasing duration is considerable and strongest for impact bending strength. Analog effects result at a thermal action of moist heat. Further, sorption is clearly influenced at long-term heating, which causes a decrease in the equilibrium MC [65].

Relative property change of Sitka spruce depending on the duration of thermal exposure (dry heat), according to Koehler and Pillow (cited in Kollmann [66])
Mönck and Erler [67] recommend for the long-term influence of heat (35–50 °C) on wood constructions a decrease in the allowable stress to 80% of the standard value (and at 50–80 °C to 60–70% of the standard value). This should especially be considered in buildings with specific conditions such as saunas and foundries.
3.6 Application of Thermal Properties for Quality Control
Thermal material properties were increasingly applied for quality control using thermographic methods . These methods take advantage of the effect that imperfections of the material such as branches, cracks, or delamination can clearly be distinguished from the temperature field of the surroundings owing to different heat emission or heating (Fig. 6.32). The methods are mainly applied in plastics and civil engineering (e.g., control of the heat transfer in construction), as well as in the metal industry, and can be subdivided into active and passive procedures. Three major classes for the main thermographic methods can be distinguished: pulse thermography, heating up thermography, and lock-in thermography [68].

Photos (left) and thermograms (right) of wood surfaces (spruce) with (a) compression failures and (b) a knot. (Images: P. Meinlschmidt, WKI/Braunschweig)
In the active method, a heat flux is induced in the test specimen using an external energy source. Thereby, the specimen is activated by means of a vibrator, ultrasonic equipment, or other excitation source (e.g., pulsed or transient thermography where the surface is pulse heated with flash lamps [69, 70]). In doing so, the specimen heats more in the field of defects (e.g., crack area, stress peaks at static strain) than in the remaining area, which is observed with an infrared camera and digitally stored and analyzed. On the basis of the evaluated differences in temperature, the quality of the material can be specified. With pulsed thermography and time-dependent analysis of the thermograms, the depth at which the defect is located can additionally be determined [71]. Further, the method can be combined with other nondestructive methods, e.g., for veneered wood diagnostics [70].
In the passive method, the test specimen is heated from an external source (e.g., during the production process) and the thermal radiation is then measured with an infrared camera or pyrometer. The method permits differences in density and MC (only high differences) to be distinguished, as well as knots and other surface defects to be localized [72, 73]. Applied to laminated wood-based panels, locally different surface temperatures (cold and hot spots) indicate defects either within the glued joint or in the carrier material [74]. Further, the method is widely implemented for the control of heat insulation in the building sector (Fig. 6.33).

Thermograms of a building (a) before and (b) after restoration. Relative scale with blue (low thermal radiation) and red (high thermal radiation). (Images: P. Meinlschmidt, WKI/Braunschweig)
4 Electrical and Dielectric Properties
The electrical properties of wood are, unlike the mechanical properties, not directly perceptible, but measurable.
The electrical properties result from the bonding of the electrons in the material structure. In the case of wood, the orthotropy of the material induces a directional dependence in the three main axes. Further, the electrical properties are influenced by the varying densities and chemical compositions (e.g., extractives) of the wood.
4.1 Electrical Resistance and Conductivity
Each material has its own electrical resistance, which depends on the atomic density and number of free electrons, and so does wood.
In the following, we introduce the most important parameters:
4.1.1 Electrical Resistance R
Electrical resistance is the resistance that wood puts up to the passage of an electrical current. It is based on the fact that the charge carriers must push between the atoms. In doing so, they trigger atoms and cause them to vibrate, which converts electrical energy into heat energy.
4.1.2 Electrical Resistivity ρ
The electrical resistivity (or specific electrical resistance) is the resistance of a material with a cross-sectional area A and a length l and is calculated as follows:
Further units used are (Ω cm) and (Ω mm2 m−1).
The following applies: 1 Ω m = 102 Ω cm = 106 Ω mm2 m−1.
4.1.3 Electrical Conductance G
The electrical conductance G is the ability of the substance to propagate electricity (i.e., electrons and/or ions) from the point of origin throughout the body. G is the reciprocal of the electrical resistance and is defined:
The unit is Siemens (S).
In the case of wood, the metrological primary effect is particularly based on the ionic conductivity.
4.1.4 Electrical Conductivity κ
Analogous to the specific resistance, the following applies to the electrical conductivity (also referred to as specific electrical conductance):
4.1.5 Test Methods
A standardized test procedure for wood does not currently exist in Germany. Riedel and Walter [75] apply circular plate electrodes with protective rings based on DIN 53482 (Fig. 6.34).

Experimental setup for determining the electrical resistance of wood [75]
The volume resistivity and surface resistivity are calculated as follows:
-
ρD – Volume resistivity (Ω)
-
ρS – Surface resistivity (Ω)
-
A – Surface of the electrode (m2)
-
h – Thickness of the sample (m)
-
dm – Mean electrode diameter (m)
-
RD – Volume resistance (Ω)
-
g – Gap width (m)
-
RS – Measured surface resistance (Ω)
4.1.6 Influences on the Electrical Resistance of Wood
4.1.6.1 Moisture Content ω
The MC significantly influences the conductivity or electrical resistance of wood. Very dry wood is a good insulator comparable with e.g., phenol-formaldehyde resin. If the MC increases, the conductivity increases and the electrical resistance decreases.
Up to the fiber saturation point, a linear relationship exists between the MC and the logarithm of the electrical conductivity or the electrical resistance. Above the fiber saturation point, the influence of moisture is significantly lower (Fig. 6.35).

Dependence of electrical conductivity on moisture content (MC) in redwood (according to Stamm (cited in Kollmann and Côté [3])
The strong influence of wood moisture on the conductivity or electrical resistance is used in particular for determining the MC of wood. Moisture meters operating on this principle are widely used.
4.1.6.2 Wood Species, Structure and Additives
Important influencing parameters are the species, the three main directions (longitudinal, radial, tangential) and the density (Tables 6.11 and 6.12). The influence of the wood species is significant, in particular owing to the variation in density and the structure. In the case of electrical moisture measurement, therefore, a species-specific characteristic curve is always used. Perpendicular to the fiber direction, the electrical resistance of the wood is about twice as large as in the fiber direction [12].
If the electrical resistance of particles or fibers is measured, it is necessary to keep the density constant. Particleboards for computer rooms and clean rooms are made by adding, for example, graphite, and have increased conductivity (antistatic particleboards). The electrical resistance of these particleboards is 105 to 109 Ω (solid wood about 1011 Ω). For these applications, special coating materials are also used (high-pressure laminate [HPL] conductive panels made using carbon fibers).
The bleeder resistance according to DIN 53482 is 1·105 to 9·106 (at 50% relative humidity and 500 V). Salt impregnation decreases the electrical resistance as well as the addition of adhesives and hardeners.
4.1.6.3 Temperature
The temperature of the wood significantly influences the electrical resistance and thus the conductivity. As the temperature increases, the electrical resistance drops sharply (Fig. 6.36).

Influence of moisture content (MC) and temperature on the electrical resistance of wood [77]
The temperature influence must be considered during the measurement. This is required for the electrical moisture measurement on wood and wooden particles or fibers.
The reduction of the electrical resistance at high temperatures makes it possible to measure particles within the range of very low wood MC, which is used in the wood industry for measuring the particle and fiber MC.
4.2 Dielectric Properties
Wood has dielectric properties. It can be considered to be a good insulator in its dry state and as a semiconductor in the air-dry state. When the fiber saturation region is reached (and above this region), no appreciable insulating properties are left.
The dielectric constant (or relative permittivity) indicates by how many times the dielectric conductivity of a material is greater than that of the vacuum (εr.vacuum = 1):
-
ε – Dielectric conductivity or permittivity in farads per meter (F m−1)
-
ε0 – Vacuum permittivity, also called electric constant = 8.854 ∙ 10−12 F m−1
The dielectric constant of water at 20 °C is about 81; that of dry wood is 2–3. A further characteristic is the loss tangent (loss angle) tan ϑ. The dielectric losses occur when applying an AC voltage to a capacitor charged with a medium being measured as a consequence of the continuous frequency-dependent charge transfer and the alignment of the water molecules with their dipole characteristics. Associated with this are molecular friction processes, so that heat energy is released. The loss tangent is determined as follows:
Where \( {\varepsilon}^{\prime } \) is the relative dielectric constant (real part) and ε″ the loss factor (imaginary part) of the complex dielectric constant ε*:
A detailed description with numerous values for wood and wood-based materials is given by Torgovnikov [78].
4.2.1 Influences on the Dielectric Constant of Wood
4.2.1.1 Moisture Content
The MC of wood has a significant influence on the dielectric constant. As the MC increases, the dielectric constant becomes larger. The reason for this is to be found in the already-mentioned differences in the dielectric constants of dry wood and water (Table 6.13, Fig. 6.37). The dielectric constant is also influenced by frequency and density (Fig. 6.37).

Influences on the dielectric constant of wood. (a) Influence of the moisture content (MC) and the measurement frequency of beech (cross section) according to Kröner (cited in Kollmann). (b) Influence of the density of wood perpendicular to the fiber direction, measuring frequency: 50 MHz [76]
4.2.1.2 Wood Structure and Wood Species
Further significant influencing parameters are the wood species, the cutting direction, and the grain angle (Table 6.14). Perpendicular to the fiber, the permittivity is lower than in the fiber direction. The differences between the radial and the tangential direction are small.
4.2.1.3 Temperature
The wood temperature also influences the dielectric properties. According to Ugolev [76], for example, the dielectric constant and the dielectric loss angle tan ϑ increases at low frequencies with increasing wood temperature (Fig. 6.38). For minus temperatures, Roig et al. [79] measured a peak at about −120 °C for the dielectric loss angle of wood and cellulose and at about −80 °C for that of lignin at a frequency of 1 Hz.

Influence of frequency and temperature on the dielectric loss angle tan ϑ and the relative permittivity εr of spruce perpendicular to the fiber direction [76]
4.2.2 Testing Methods
To determine the dielectric constant (relative permittivity), the wood is placed between the plates of a capacitor. The capacitance or the dielectric losses are measured. If the capacitance of a plate capacitor in the initial state is C0, it rises to the value C when a dielectric medium is placed between the plates.
The following equation applies:
The technique is mainly applied in the dielectric heating of wood for drying and curing of adhesive joints, especially on large cross sections (glued-laminated timber) but also for phytosanitation [80, 81]. Another application is the moisture measurement based on the strong influence of moisture on the dielectric properties of wood, in particular the dielectric constant. The method is used to determine the MC of sawn timber and wooden particles. A density correction is necessary.
In the temperature range below 0 °C, the method cannot be applied because the dielectric constant of the wood at these temperatures is approximately equal to the dielectric constant of frozen water (εr.wood ~ εr.ice).
4.3 Piezoelectric Properties
Piezoelectricity means the occurrence of electrical charges at the interfaces of crystals or materials, when elastic deformation causes changes in the electrical polarization of the structural elements, which cause an electric potential.
Wood cellulose contains crystalline elements. Therefore, piezoelectric effects are detectable in wood. They can be determined, for example, by the impact of a stress wave and by measuring the resulting charge (Fig. 6.39). The method can be used for following tasks, among others:
-
Localization of defects in wood
-
Grading of wood according to the strength
-
Measurement of stresses in the wood (e.g., during drying)

Experimental setup for the measurement of piezoelectric constants [85]
For corresponding investigations, see for example, Knuffel and Pizzi, and Fukada [82,83,84].
The piezoelectric modulus can be calculated as follows:
-
dQ – Differential separation of charge in Coulomb (C)
-
dP – Differential stress (N)
The value of the piezoelectric modulus of wood is about 5% of that of a quartz crystal [84].
4.3.1 Influences on the Piezoelectric Modulus of Wood
Significant influencing factors are the wood species, the crystallinity of the cellulose, the orthotropy of wood (three main directions, fiber angle), the density, the early and late wood content, the physical or chemical wood modification, the temperature, and especially the MC. The type and level of load (e.g., tension, compression, shear, bending) also have an influence. Under a compression load, the piezoelectric modulus has its maximum at a fiber angle of 45° [82] (Fig. 6.40). This effect may be caused by shear stresses in the wood. Tensile or compressive stresses (parallel and perpendicular to the fiber) do not produce electrical polarization. A change in the direction of the load always results in a change in the electrical polarization.

Influence of the fiber angle on the piezoelectric modulus [84]
As the temperature rises, the piezoelectric modulus rises too. The modulus is lower in earlywood than in latewood. Table 6.15 shows the piezoelectric modulus of wood determined by mechanical striking and charge measurement depending on wood moisture. Further investigations can be consulted, for example, in the proceedings on 50 years of nondestructive testing and evaluation of wood [86].
4.4 Magnetic Properties
Wood is diamagnetic, i.e., a test piece of wood is repelled in a magnetic field in the direction of the poles of lower field strength. The dimensionless physical characteristic of the magnetic properties is the susceptibility Sm. The following equation applies:
For wood, a value of −0.2 to −0.5 · 106 is given. Other materials have the following values:
-
Iron +720 · 106
-
Hard rubber +1.1 · 106
-
Paraffin −0.58 · 106
The magnetizability of the wood is very low.
4.5 Electrostatic Charge
The surface electrical potential differences between nonconductors are called electrostatic charges. Such charges can arise from relative movements (friction) by charge separation. The magnitude of the electrostatic charge depends, among other things, on the amount of relative movement, the insulation resistance of the surface material, the surface geometry, and the relative humidity. Notable electrical charges only occur above electrical resistances of 1010 Ω.
In the wood industry electrostatic charge affects, for example, the processing of resinated laminated boards (owing to adhesion of particles to boards in the processing of plastic-coated plates). Electrostatic charges may also arise in the pneumatic conveying of wood particles. The resulting field strengths are measured using a statometer [85, 87].
Also, for floor coverings and powder coating of surfaces, these properties are of practical importance. So, there must be a certain conductivity of the wood in the powder coating. This can be achieved via the wood moisture, additives, or primers of the surfaces. The adhesive type also has some influence on the conductivity of the boards. Phenolic resin and methylene diphenyl diisocyanate have higher conductivity than urea-formaldehyde (UF) or melamine formaldehyde (MF) resins [88]. The conductivity of wood-based boards can be increased by the addition of graphite admixtures, e.g., at particleboards for use in computer or clean rooms. Also, coating materials based on carbon fibers, e.g., HPLs were used if special reduced electrical resistivity is required. Riedel and Walter [75] assume that no electrostatic charges are expected for wood-based materials with a surface resistance ≤109 Ω under normal conditions (23 °C, 50% relative humidity; Fig. 6.41).

Electrostatic behavior of materials and recommended measuring field intensities for wood-based materials [89]
5 Acoustic Properties of Wood and Wood-Based Materials
Sound refers to mechanical vibrations or waves of an elastic medium. Depending on the type of medium in which the sound propagates, a distinction is made between airborne and structure-borne sound. Depending on frequency, sound is differentiated into infrasound (not audible, frequency < 16 Hz), audible sound (16 Hz to 20 kHz), and ultrasound (> 20 kHz). Important acoustic properties are:
-
The propagation velocity of sound waves in a material (sound velocity)
-
Acoustic emission (AE)
-
Sound absorption
-
Sound insulation
5.1 Physics of Sound
We distinguish two basic forms: longitudinal and transverse waves (Fig. 6.42). For the longitudinal waves (P-waves), the propagation direction and the vibration (oscillation) direction are identical; for the transverse waves (referred to as shear waves or S-waves ), the vibration direction is perpendicular to the propagation direction of the wave. In practice, both components usually occur, which often makes evaluation difficult (e.g., when measuring shear waves). The speed of transverse waves is significantly less than that of longitudinal waves. For example, Ozyhar [90] determined a sound velocity of 4682 m s−1 for longitudinal waves of beech in a normal climate (20 °C/65% RH), and 1485 m s−1 for transverse waves in an LR direction.

Wave types
Testing of wood usually involves modified forms such as Lamb waves, Rayleigh waves, bending waves, or plate waves. Bending waves occur in rod-shaped or plate-shaped parts.
Dispersion is a property of certain waves, which means that the propagation velocity of the waves depends not only on external conditions such as temperature and medium but also on the wavelength or on the frequency of the wave. This effect must be considered for wood when working at different frequencies. Additionally, wave propagation is influenced by the tested component dimensions (see, for example, Schubert and Baensch [91, 92]).
Further literature on the subject of ultrasound can be found, for example, in Krautkrämer and Krautkrämer [93] and Bucur [94].
5.2 Sound Velocity
The sound propagates in a multi-directional broad medium (seawater, air) as a longitudinal wave. For longitudinal waves, the two-way impeded transverse contraction must be taken into account as follows:
-
c – Sound velocity (m s−1)
-
ρ – Density (kg m−3)
-
E – MOE or Young’s modulus (Pa; usually indicated in MPa or N mm−2)
-
μ – Poisson’s ratio (−)
For a plate with unilaterally hindered transverse contraction, the following applies analogously:
In a bar whose width and thickness are small compared with the wavelength, the sound propagates only as a strain wave or quasi-longitudinal wave. Therefore:
Analogous to Eq. 6.46, the shear modulus G is determined from the velocity of the transverse waves cij (first index: oscillation direction, second index: direction of wave propagation) and the density as follows:
Often, (6.46) and (6.47) are also used at test specimen geometries other than long bars for calculating the MOE. This sometimes leads to significant inaccuracies. Eqs. (6.44, 6.45, 6.46, and 6.47) only apply to isotropic media. Additionally, the extended Hooke’s Law (see Chap. 9) applies to anisotropic materials. Thus, three elastic moduli, three shear moduli, and three Poisson’s ratios are required. The other three Poisson’s ratios can be derived with the help of the compliance matrix [S]. For their calculation, e.g., by means of ultrasound, the stiffness matrix [C] is first determined on the basis of sound velocity and density (6.48) and then converted into the compliance matrix [S] = [C−1]. For the conversion additionally, the elements of the sub-diagonal rigidities (e.g., at 45°) are required (Table 6.16, Fig. 6.43).

Sample geometry for the determination of strain and Poisson’s ratio on wood by means of ultrasound [95]
-
σ – normal stress, τ – shear stress, ε – strain, γ – shearing strain, C – stiffness
-
L – longitudinal, R – radial, T – tangential
The ultrasonic method can successfully be used for the determination of the MOE. Neglecting the transverse contraction, it applies approximately:
When using long lengths (e.g., boards) and relatively low frequencies of 20 kHz or below (as is usual with many devices for the measurement of wood), the deviations from the static MOE are low. In the case of plate-shaped or cubic samples, however, the values of MOE calculated from ultrasound without considering the transverse contraction are clearly too high. Therefore, for small samples within the range of about 10 mm edge length and frequencies within the range of 1–2 MHz, a correction has to be done. Otherwise, moduli that are much too high are calculated (see, for example, Ozyhar et al. and Bucur [90, 94]). The correction factors for the stiffnesses taking into account the transverse contraction (Poisson’s ratio) are given in (6.50). Table 6.17 shows the correction factors determined for cherry wood [95].
In contrast to the MOEs, the Poisson’s ratios (see Bucur [94]) could not be determined exactly enough from the ultrasound tests in extensive own measurements [90]. The differences between static shear modulus (determined in the Arcan test) and shear modulus (determined with transverse waves), however, are significantly lower. With ultrasound, about 20–30% higher values are determined than in the mechanical test by means of the Arcan test.
5.2.1 Further Acoustic Characteristics
The wavelength λ is defined as:
-
c – Sound velocity (m s−1)
-
f – Frequency (s−1)
The wavelength is for example, important for the dimensioning of transducers. It also provides information about the recognizability of failures. For example, at a usual frequency for wood of 20 kHz and a sound velocity in the fiber direction of 6000 m s−1, a wavelength of 0.3 m results. Therefore, small defects such as crushing but also branches or the early stages of rot in trees are not recognizable. In contrast, when examining trees for decay, large areas of rot can be well recognized owing to the resulting longer path of the sound wave and therefore lower sound velocity [91, 96]. The sound velocity reaches in wood in the direction of the grain values of 4000–6000 m s−1 and perpendicular to the grain values of 400–2000 m s−1. There is a strong dependence on the wood species (Table 6.18). Thereby, sound velocity correlates with the MOE (see Chap. 9.)
The sound waves are attenuated during propagation within wood or wood-based materials, whereby sound energy is transformed into heat. This effect is used to detect defects in wood or wood-based materials (see Chaps. 19 and 20).
For the detection of large defects in trees (large decay, hollows), the forced path change of the sound wave is used for defect detection (the wave propagates only in the solid state). According to the laws of sound propagation this results in significant path changes. The basics were described for example, by Schubert [91] and Sanabria [97].
5.2.2 Resonance Quotient
Tonal wood [87] is characterized by a high ratio of MOE to density. It is also usually assumed that sound wood has a high value of the resonance coefficient (QR), which is defined as follows:
-
QR – Resonance quotient (m4 kg−1 s−1 or m5 N−1 s−3)
-
c – Sound velocity (m s−1)
-
ρ – Density (kg m−3)
-
E – MOE or Young’s modulus (Pa)
Additionally, QR is used in combination with damping properties for the determination of the acoustic conversion efficiency (ACE) [98]. Further, for tonal wood, a high MOE in the fiber direction and in the radial direction as well as uniform growth-ring widths are required. Holz ([99, 100]) summarizes the requirements as follows:
-
The frequency distribution of the growth-ring width is unimodal
-
The standard deviation of the growth-ring width is less than 0.75 mm
-
The latewood content is between 20 and 33%
Recent work on resonant wood was described, for example, in Brémaud, Sonderegger et al., Sprossmann et al., and Krüger et al. [98, 101,102,103].
5.2.3 Influences on Sound Velocity
The sound velocity is significantly influenced by the wood structure. All properties that correlate with the MOE influence the speed of sound. Table 6.18 shows the sound velocity for different species and wood-based materials.
In solid wood, the sound velocity in the fiber direction is 3–4 times greater than perpendicular to the fiber and in the tangential direction, it is always slightly lower than in the radial direction. Particle-based materials have a significantly lower sound velocity than wood in the fiber direction owing to the largely random particle orientation. In OSB, sound velocity is significantly higher in the direction of particle orientation than perpendicular to it. According to investigations by Burmester [107], the following parameters are important for the speed of sound:
-
The fiber length
-
The density (Fig. 6.44)
-
The early and latewood relation
-
Grain angle (LR, LT) and ring angle (RT); reduction of the sound velocity with increasing grain angle, minimum of the speed of sound at 45° between the R and T directions (see also Bodig and Jayne [4] and Chaps. 8 and 9)
-
Wood defects (e.g., knots: reduction of sound velocity due to changes in the grain angle and density of the knots in relation to sound wood)
-
The MC (Fig. 6.45)

Influence of density on sound velocity in pine (Pinus sylvestris L.) [109]

Influence of moisture content (MC) on sound velocity perpendicular to the fiber of Roble (Nothofagus obliqua (Mirb.) Oerst.), according to Niemz [110]
The structural factors correlate with the MOE and thus with the sound velocity. The influence of fiber length has also been demonstrated in own measurements on various South American wood species [108].
Armstrong et al. [111] extensively researched the influence of fiber angle. They demonstrated that, as for elasto-mechanical properties (see Chaps. 8 and 9), the formula developed by Hankinson [112] applies:
-
c|| – Velocity in the fiber direction (m s−1)
-
c⊥ − Velocity perpendicular to the fiber direction (m s−1)
-
φ – Grain angle
-
n – Parameter (here: n = 1.7)
5.2.4 Selected Device Systems
For sound measurements on wood, the following systems are used:
-
Systems based on shock waves (also named stress wave techniques). The waves are generated by impact at frequencies of some 100 Hz (devices: for example, Fakopp, Picus Sonic Tomograph, both also used for tomography, Metrigard/USA; Fig. 6.46a)
-
Devices based on piezoelectrically generated waves within the range 20 kHz–50 kHz (measurements on boards, for example, BP-700 from UltraTest, Sylvatest). These devices usually show the transit time
-
Non-contact measuring devices (air-coupled ultrasound, frequency approximately 50 kHz (GreCon)). Principles of contactless coupled ultrasound and its use for delamination detection in glued-laminated timber are described in Sanabria [97]
-
Scientific studies for measuring elastic constants often use high-frequency devices of several MHz (e.g., Epoch XT from Olympus) and longitudinal and transverse waves [90] (see Fig. 6.46b). The waveform is used for the transit time measurement, where the time is measured at the top or the edge of the waves (Fig. 6.47; difference between outgoing and incoming waves). Thereby, transversal waves have much lower speeds than longitudinal waves

Ultrasonic measuring instruments. (a) Measuring instrument for determining sound velocity by means of the impulse hammer method (waves generated by impact, Metrigard). (b) Epoch XT ultrasonic measuring instrument (Olympus), waves generated piezoelectrically. Images: ETH Zurich

Waveforms: incoming waves for determination of sound velocity
A good coupling (constant contact pressure) is important; for example, special coupling gels or honey are used. In industrial applications, partially cone-shaped sensors are used, which are screwed or plugged into boreholes. Mostly, the transmission mode (one transmitter, one receiver) is used for the measurement of wood. Hasenstab [113] also successfully used the pulse-echo method. Thereby, transmitter and receiver are identical.
Mostly, the dynamic MOE is calculated from the measured sound velocity. Thereby, its correlation with the statically determined MOE and strength can be used, e.g., for the grading of strength classes (e.g., with the device Sylvatest). Figure 6.48 shows the correlation between sound velocity and MOE as well as the strength of particleboard. In practice, the correlation of the E-modulus with strength is used.

Correlation between sound propagation time and particleboard properties [114]
5.3 Sound Attenuation or Sound Absorption
The sound absorption is the ratio of the nonreflected to the incident sound power.
The following applies:
-
Ks – Nonreflected sound power (W)
-
Ka – Incident sound power (W)
The sound absorption is caused by dissipation of sound energy in the material (e.g., conversion into heat) and by transmission. It is frequency dependent and increases in particle materials with the porosity of the material. The sound absorption of a 12-mm thick wood fiber insulation board (density 250 kg m−3) is 20–30%; by punching or slitting, it can be increased to 60–80%. In the case of 5-mm thick hard fiberboards, on the other hand, only values of 5–8% are achieved, and in the case of particleboard, values of 10–15% result, depending on bulk density and surface finish [87] (Table 6.19).
5.4 Sound Insulation (Sound Reduction Index)
The sound reduction index R is used to measure the level of sound insulation provided by a structure.
The following applies:
-
R – Sound reduction index (dB)
-
P1 – Sound power striking the component (W m−2)
-
P2 – Sound power radiated on the back (W m−2)
Good sound insulation is achieved through reflection and dissipation. The sound reduction index increases with the frequency of the sound waves and the surface dimensions of the material. Lightweight wooden structures are therefore only slightly suitable for sound insulation. A double-shell design, on the other hand, effectively insulates the sound. For particleboard with a surface density of 15–20 kg m−2, the sound insulation is 24–26 dB. With each doubling of the surface density, the sound power is reduced by 5 dB [115].
5.5 Acoustic Emission
Acoustic emission is the emission of sound waves in the audible and ultrasonic range (frequency > 20 kHz), which is caused by microscopic fractures, friction of fracture surfaces, outflow of liquids, transport processes in capillaries, or other effects. Strictly speaking, the process is therefore not really nondestructive.
A characteristic of wood is that it emits clearly audible sound signals at pressures below the breaking load. This characteristic appearance (“crackling”) is called the warning ability of the wood and is used practically in mining. Long before the wood crackles, i.e., at a much lower load, sound waves are emitted from ultra-micro-cracks whose frequency spectrum is between 50 kHz and 1.5 MHz. This acoustic emission in the ultrasonic range forms the basis for a method used for the analysis of metal constructions (containers, machines) in particular. Practical applications such as the monitoring of drying processes or the condition monitoring of tools in woodworking have not yet been established.
Since the middle of the 1970s, acoustic emission analysis has been increasingly used in wood research where frequency signals within the range 50–150 kHz are evaluated. The bases are mostly crack and friction phenomena. The acoustic waves are released when the material is loaded and propagate at the surface. They are converted into electrical signals by means of piezoelectric transducers. Depending on the type of sound signals, a distinction is made between continuous emission and burst signals (Fig. 6.49). For wood and wood-based materials, burst signals are typical.

(a) Schematic of acoustic emission (AE) signal parameters in the time domain. (b) Parameters of AE signal in the frequency domain [92]
Important fundamentals of the sound emission are described in the following standards:
-
DIN EN 1330-9, Nondestructive testing – Terminology – Part 9: acoustic emission test
-
DIN EN 13554, Nondestructive testing – Acoustic emission – General principles
The following parameters are used (the signals are evaluated by means of a computer program with special software):
Arrival time, time of first threshold crossing | t0 |
Counts of the signal’s threshold crossings | C |
Frequency | f |
Peak amplitude | A |
Rise time | tR |
Signal duration | tD |
True signal energy | E |
An overview is available in Baensch [92] and Sause [116].
5.5.1 Influences on Acoustic Emission and the Practical Use of Acoustic Emission
Figure 6.50 gives an overview of the most important parameters; dominating are the structure (density, fiber length, particle geometry, type and percentage of adhesive, etc.), the MC, and the history (e.g., fungal infections or insect attack, mechanical or climatic pre-stress) of the wood or wood-based material.

Factors influencing the acoustic emission of wood
Applications of acoustic emission analysis in the wood industry are shown in Fig. 6.51. The main focus is the investigation of the relationships between structure and properties [92], the measurement of cracking in spruce under tension perpendicular to the grain [117], and measurements on living trees [118].

Applications of acoustic emission analysis in the wood industry
5.5.2 Analysis of Fracture Processes
Fracture phenomena at the microscopic and submicroscopic levels are characteristic of damage of the wood substance under load. They are already detectable well below the breaking load, but at a low load, they do not result in any reduced bearing capacity of the wood. The AE is largely determined by the structure of the material. It is initiated at 10–30% of the ultimate load for particleboards and at 50–60% for solid wood. A strong variation of the measurement results is characteristic between individual test specimens. At repeated exposure, the sound emission begins only above the pre-stress; this appearance is called the Kaiser effect (named after Kaiser, who carried out the first fundamental work on sound emission of metals in a PhD thesis in 1950).
The presence of wood defects (e.g., knots) and notches significantly influences the sound emission.
In particular, defects that lead to long cracks when broken cause a considerable increase in the sound signals emitted. Modern methods such as synchrotron radiation already allow online monitoring of failure processes. By volume correlation of the tomographic data, statements about relationships between structure and sound signals are possible [30]. Noguchi and Nishitoma [119] have shown that sound emission of fungus-infected wood already starts at 4–32% of ultimate load, but only at 60% in the case of sound wood. By means of AE analysis, the test material could be differentiated much more than by determination of mass loss. For particleboards, a clear influence of chip geometry was found. As the particle size decreases, the AE begins only at higher levels of load; the same effect applies with decreasing bulk density [108, 120]. The MC particularly influences the counts and the beginning of the AE. As the MC increases, fewer sound signals occur (Fig. 6.52).

Influence of moisture content on the relative sum of hits in bending tests of spruce wood and particleboard, according to Niemz [121]
As a result of the considerable damping of the sound waves in the wood, sound signals can only be detected at a limited distance from the source of generation. Our own measurements showed that sound waves at a frequency of 150 kHz could no longer be detected at a distance of 500 mm from the source of origin (in the fiber direction).
Characteristics of the fracture process in wood and wood-based materials are the classical macroscopic fracture and the subsequent slipping of the fracture surfaces. Fracture and friction (slipping) show distinct differences in energy distribution (Fig. 6.53).

Energy distribution as a function of time at bending tests of spruce wood, according to Niemz et al. [121]
Even climatic stresses (e.g., drying) can be acoustically detected. However, a direct correlation between the total counts of AE signals (hits) until breakage and the breaking force has not yet been confirmed.
With the acoustic emission analysis, fracture processes can be detected, which occur during creep or relaxation of wood and wood-based materials. There is a good agreement between deflection and counts as a function of time. The sound emission starts at a load factor of 30%. Under a dynamic load, a significant increase in emission could be determined with increasing load cycles.
5.5.3 Proof of Brittleness of Adhesive Joints
Comparative investigations on bonded elements and on wood-based materials showed that the acoustic emission analysis is well suited to demonstrating the brittleness of the adhesive joints. The relatively brittle urea resins emitted significantly more pulses than other adhesives such as PVAc or 1C-PUR. Recent work on glued wood has been carried out, for example, by Baensch [92].
5.5.4 Optimization of Wood Drying
Anisotropic shrinkage, moisture gradient, and orthotropy of wood may cause cracking during wood drying. With the help of acoustic emission, cracks can be detected in the formation phase (Fig. 6.54). This makes it possible to prevent macroscopic cracks by appropriate control engineering. Acoustic emission is influenced by:
-
The board geometry
-
The wood species
-
The drying parameters
-
The temperature
-
The MC

Acoustic emission and moisture content during drying of sawn timber, according to Schmidt [123]
Noise emission is greatest when the MC reaches or falls below the fiber saturation range [122].
5.5.5 Monitoring of Cutting Processes
According to studies by Lemaster and Dornfeld [124] as well as by Michailow and Niemz [125], there is a clear correlation between machining parameters (e.g., cut geometry, cut wear, cutting path), type of material, and acoustic emission by cutting of wood and wood-based materials. The number of counts increases with increasing feed rate and contact size, as well as increasing point of tool life. At the same time, there is a significant influence of the type of coating material. In principle, it is possible to deduce the edge quality from sound emission (e.g., direct correlation with the number and size of edge breakouts).
5.5.6 Measuring Systems for Acoustic Emission Analysis
Figure 6.55 shows the schematic structure of a tensile test assembly for the measurement of AE with simultaneous detection of the strain with digital image correlation. The most important elements are the transducer (frequency > 20 kHz, usually 100–150 kHz), the amplifier, the main amplifier, the signal filtering, and the signal processing.

Schematic representation of a measuring system for acoustic emission analysis in combination with digital image correlation (DIC) at tensile test [92]
Special systems with several channels are available for signal processing (e.g., Vallen GmbH). This permits a certain location of the signal source. However, this is hampered in wood and wood-based materials by its orthotropy and the influences of grain angle and ring angle on sound velocity. It should also be noted that there is a strong weakening (damping) of the high-frequency signals in porous wood. The signal amplitude decreases strongly with increasing distance from the acoustic source. Figure 6.56 shows a clustering of the signals under tensile load in the radial and tangential directions. There are two clusters separated by frequency, independent of the orientation of the layers. The combination of AE and digital image correlation allows strains and sound signals to be correlated. If the sample is tomographed at the same time (for example, in the synchrotron or with X-ray microtomography [92]), 3-D evaluations are possible and structural changes in the interior of the material can be compared via volume correlation with the sound signals. Figure 6.57 shows selected results. Modern evaluation algorithms are described, e.g., in Sause [116].

Clustering of acoustic emission signals from plywood (Cross and PLY 3, 4, 5, 6; → no influence of the layer structure), solid wood (Solid) and laminated wood (Unidirectional) made of spruce wood [92]

Acoustic emission signals and strains measured by means of digital image correlation on spruce wood [92]
5.6 Natural Frequency (Eigenfrequency) and Modal Analysis
Natural frequency (eigenfrequency) of a material excited to vibrate can be used as a criterion for determining the elastic constants. Rayleigh described its theoretical basis as early as 1929 [126]. The process is increasingly used industrially for quality control (grading of boards).
Longitudinal, bending or torsional vibrations are generated by impact and the elastic constants are then calculated from the determined natural frequency. The corresponding device systems are based on the basic principles described below. Thereby, a rod, which is excited to oscillate is borne on two supports [127].
5.6.1 Determination of Tensile/Compressive Modulus of Elasticity
In the case of the longitudinal (axial) vibration of a beam (activated, for example, by impact in the longitudinal direction of the beam), if the lateral contraction is neglected it applies as shown in Fig. 6.58.

Stress of a rod in longitudinal vibrations (schematic) according to Bodig and Jayne [4]
-
∂u – Displacement of the cross-section in time t (m s−1)
-
l – Rod length (m)
-
x – Location of the cross section (m)
-
ρ – Density (kg m−3)
-
E – MOE (Pa)
-
F – Force (N)
For the dynamic MOE, the following equation results after transformation:
-
l – Rod length (m)
-
f – Natural frequency (s−1)
-
n – Order of vibration
Equation (6.57) applies to all cross-sectional shapes when the rod length is multiple larger than the rod cross-sectional area. With known rod length and density, the tensile or compressive MOE can thus be determined from natural frequency.
5.6.2 Determination of the Elastic Modulus in Bending (Flexural Vibration)
Activation of flexural vibration yields according to Fig. 6.59 to:

Analysis of a rod in flexural vibration [4]
Out of it results according to Görlacher [128] for the calculation of the MOE taking into account the transverse contraction:
-
E – MOE (Pa)
-
Gxy – Shear modulus in the bending plane (Pa)
-
I – Moment of inertia (m4)
-
A – Cross section (m2)
-
ρ – Density (kg m−3)
-
l – Rod length (m)
-
i – Radius of inertia in the direction of the bending vibration (i2 = I/A); for rectangular cross sections: i2 = h2/12 (h = rod height (m))
-
f – Frequency (s−1)
-
K1, K2, mn – Constants (depending on the order of vibration)
-
s – Form factor (for isotropic rectangular cross sections 1.20, for wood 1.06)
K1, K2, and mn are constants, which depend on the order of vibration, s is a form factor that assumes a value of 1.20 for isotropic rectangular cross sections (wood is expected to have a shape factor of 1.06 as determined by Hearmon [129]). Görlacher [128] describes the method in detail and emphasizes for first order vibrations: K1 = 49.84; K2 = 12.3; mn4 = 500.6.
5.6.3 Determination of Torsion Modulus
The determination of the torsion modulus applies as follows:
The natural frequency measurement is often used to determine the elastic properties (see Chap. 8). Often, modal analysis is used, which makes it possible to determine all elastic constants of wood or wood-based materials (see, for example, for cross-laminated timber [130]).
The natural frequency measurement is today often used industrially for the strength grading of wood. Thereby, several measuring principles such as natural frequency measurement, X-ray for the detection of knots and the propagation of a laser beam in the wood (tracheid effect, see Sect. 6.7) are combined. Figure 6.60 shows the correlation of the MOE determined from eigenfrequency with that determined in the bending test for particleboard. Experiments to measure the stiffness of adhesive joints were also carried out using this method [46]. Thereby, to a certain extent, the penetration of the adhesive into the wood is integrated into the measurement result.

Correlation of moduli of elasticity measured by eigenfrequency (MOEEF) on particleboard with those determined in the static bending test (MOEST) [131]
6 Friction Properties of Wood and Wood-Based Materials
Friction is the resistance that counteracts the mutual contact of two bodies. With increasing speed, increasing smoothness, and hardness of the friction surfaces, the friction decreases. The parameter used is the coefficient of static (μs) or kinetic (dynamic, sliding) friction (μk). According to Fig. 6.61, the following balance of forces is required to overcome the static or sliding friction:

Relation FS and FN for determining the coefficient of static friction μs
Static friction Fs is the resistance of two touching body surfaces against their displacement. This resistance is greater the higher the MC and the softer and rougher the wood surfaces. With regard to the influence of the main cutting direction of the wood on friction, there is the following order of precedence (coefficient of static friction decreasing from top to bottom):
-
End-grain on end-grain
-
Radial section
-
Tangential section
The smaller the growth rings, the higher the amount of latewood, the smaller and more uniform the pores on the wood surface, the lower the static friction. For very smooth surfaces, it is often overlaid with adhesion phenomena [52].
Kinetic friction Fk is the resistance to movement that occurs when one body surface moves on another. The resistance to movement decreases with increasing smoothness and hardness of the surfaces. Kinetic friction is largest in wood perpendicular to the grain and in the dry state. Oil, water, or grease lubrication reduces sliding friction [52, 132].
Tables 6.20, 6.21, 6.22, and 6.23 contain coefficients of friction for selected types of wood, derived timber products, and combinations of materials.
The design values used (Table 6.21) are significantly lower than those listed in Table 6.20. Möhler and Maier [134] recommend a maximum value of 0.4 (rough sawn) or 0.25 (planed) for fiber-parallel contact surfaces. Meisel et al. [135] propose a design value of 0.18 (characteristic value: 0.25) for longitudinal cut on longitudinal cut and a value of 0.25 (characteristic value: 0.35) for end grain on longitudinal cut.
Wood friction on steel strongly depends on the MC and the surface condition [133, 136,137,138]. Price and Manwiller (cited in Koch [136]) investigated sliding friction of 22 hardwoods at 12–14% MC on smooth tool steel plates. They evaluated mean values between 0.097 and 0.127. Four selected species are listed in Table 6.22. Möhler and Herröder [133] determined much higher values of static and sliding (in brackets) friction coefficients for spruce wood at 20–25% MC on steel with different surface textures:
-
Rolled steel plates: 0.56 (0.44 … 0.49); skin surface primed with red lead: 0.81 … 1.03 (0.65 … 0.66)
-
Slightly rusty surface: 0.99 … 1.11 (0.74 … 0.75); in the case of a test repeat reduced by approximately 30%
Friction is of great importance in securing loads against slipping (e.g., transport of particleboards or furniture parts) but also when dealing with wood products, such as work plates for kitchens as well as when setting up wooden scaffolding (proof of stability), and also for wood joints, manufacturing of wood-based materials, and wood cutting.
7 Optical Properties of Wood and Wood-Based Materials
7.1 Color
Owing to its natural character, wood has significant color variations both within a wood species and between the different species. The color varies from almost white to yellowish (spruce) through dark red (Rauli from southern South America) to brown (walnut, teak) or even black (African ebony). The color differences are mainly based on the extractives (proportion, type). Heartwood and sapwood usually differ significantly in color. Although, for example, in yew, the heartwood is dark red, the sapwood is yellowish-white. Only a few dark wood species exist in Europe such as the hardwoods walnut, oak, and elm or the softwood yew. Woods from the tropics and also from the subtropics are often much darker than European or North American wood species [140]. Some tropical wood species, which provide natural pigments were previously used for coloring [29].
The color changes significantly during heat treatment, but also by boiling or steaming. This darkening of the color is the preferential criterion for some applications (furniture, parquet) previously to other treatment effects such as the reduction of sorption and swelling. In addition to the color, the texture (surface structure of color variations in the wood, e.g., between early wood and late wood) is also an important feature, e.g., for the choice of suitable wood for furniture construction.
The color characteristics are, for example, measured according to the CIELab system (color space according to ISO 11664-4; Fig. 6.62). Characteristic values are the lightness L* (white–black) as well as the color components green–red a* and blue–yellow b*. The system can be used to quantify the color characteristics or to determine age-related color changes. The characteristics L*, a*, b* are measured and from those, the color difference ΔE*ab based on the reference measurements is determined as follows:

CIELab color space
where:
-
ΔL* = L*T − L*R
-
Δa* = a*T − a*R
-
Δb* = b*T − b*R
-
ΔE*ab – Color difference
-
L* – Lightness
-
a*, b* – Color components green–red (a*) and blue–yellow (b*)
-
T – Test value (treated)
-
R – Reference value (untreated)
Colorimetric systems are widely used in industrial quality control (also on-line systems).
Wood-staining fungi (such as blue, white, and brown rot), the effects of temperature, chemicals, solar radiation, but also oxidation by oxygen, lead to changes in the color of wood. Color deviations are also caused by certain special features (e.g., knots, compression wood, red heart, foxiness). In softwood, the color differences between clear wood and knots are usually greater than in hardwood. Therefore, detection in hardwood is usually more difficult (Fig. 6.63).

Color change in Douglas fir wood, knot-free and with knots (L* value). (Measurements: Niemz)
Even in wood-based materials (particleboards), depending on the material used (wood species, bark content), significant color deviations may occur. Thus, for example, bark shows clear differences in color, which has an effect on the coating. In the case of particle-based materials, glue spots, chatter marks, or very large single particles can be well distinguished in color from the defect-free environment.
7.1.1 Color Change
7.1.1.1 Effect of Transparent Coatings
Surface treatments with paints, oils, or waxes significantly influence the visual color impression [141]. The surface treatments lead to a grain-raising of the surfaces. The color effect is significantly intensified. In many cases, this effect is already visible by slightly moistening wood with a sponge.
7.1.1.2 Aging Indoors
The color of the wood changes in the interior of the room over time owing to oxidation. The change in color is strongly dependent on the room interior conditions and the species. Many types of wood darken inside a room (Fig. 6.64), a few species become lighter. Especially noticeable is the color change when exposed to UV light (area of windows, balcony doors). This should be considered for parquet but also for furniture. A good overview of the color change in parquet woods is compiled in Pitt [142]. The color change due to UV light can be significantly reduced by coating with UV blockers [143].

ΔE* values after 120 and 600 h of simulated indoor solar irradiation [148]
In the wood lexicon [29] and other sources, the following tendencies regarding the natural color change are given:
-
Oak: greyish brown
-
Mahogany, cherry: deepening in color
-
Softwoods such as pine or larch: red–brown coloration in the heartwood, spruce turns to slightly yellowish brown [144,145,146] (Figs. 6.65 and 6.66)
-
Maple and birch: turn to yellow
-
Ammonia fumed oak and beech wood is more color stable than thermally treated wood; thermally treated wood (but also, for example, rosewood (Dalbergia latifolia)) becomes significantly brighter owing to UV radiation (e.g., by outdoor weathering or on parquet floors in the window area); see also Volkmer et al. [147]

(a) Naturally aged spruce with brownish color change over the entire surface area and (b) spruce wood before aging. (Photos: ETH Zurich)

Color change of spruce wood related to the age after first installation [146]
7.1.1.3 Color Change by Outdoor Weathering
Weathered wood (sunshine, rain) changes its color significantly. This is the result of a superposition of color change, chemical degradation of wood at the surface zones, and colonization by staining fungi [143, 149]. Figure 6.67 shows the color change of weathered, thermally treated, and untreated wood.

Comparison of weathering after 1 year . (a) Untreated beech wood. (b) Thermally treated ash wood. 1: reference before weathering, 2: vertical (façade cladding), protected by roof construction; 3: 45°, unprotected. (Photos: ETH Zurich)
7.2 Other Optical Properties (Tracheid Effect)
A special visual feature of wood is the tracheid effect . It can be visualized as follows. Owing to the anisotropic wood structure, a laser beam applied to the surface spreads out in the cell elements according to their structure orientation. Hence, deformations and grain flow in the area of branches influence the propagation. Figure 6.68 shows the effect schematically. It is especially detectable in coniferous trees. The major-axis/minor-axis ratio of the quasi-ellipses formed on the wood surface is lower for hardwoods than for softwood. Therefore, the effect is only restrictedly suitable for hardwoods [150].

Tracheid effect in wood [151]. (a) Schematic experimental setup. (b) Visible change in the scanned surface structure (image: Ridley-Ellis, 2000). (c) Wood surface as photograph (1) and recorded with scanner (2)
The tracheid effect is applied for defect detection in grading machines (e.g., detection of knots, grain deviation [151, 152]). Growth-related irregularities such as healthy branches, which are hardly recognizable in color, thus become visible.
Another nondestructive method for the determination and documentation of fiber direction in hardwood (e.g., European beech) is based on the analysis of the spindles formed by the rays with the charge-coupled device (CCD) technique [153]. Optical methods can also be used to measure the orientation of the particles as well as the particle geometry in wood-based materials [154]. Further optical properties that can be used for quality control, are gloss, luminescence, roughness, etc. [155].
Wood also has some fluorescent properties. These occur in certain types of wood, such as Pterocarpus indicus (commonly known as: Narra wood, amboyna wood (as valuable burls), Malay padauk, Papua New Guinea rosewood) and Eysenhardtia polystachya (Mexican kidneywood) [156]. Robinia also has these effects as well as fungus-infected wood. Often, fluorescent effects are only shown in interaction with water or alcohol. An overview is available in Gahle [157]. Further fluorescent woods are, for example, afzelia, merbau, okan, and mangium. Sometimes, these properties are also used for damage analysis [157].
7.3 Spectrometric Properties
Light of certain wavenumbers (or wavelengths, \( \mathrm{wavelength}=\frac{1}{\mathrm{wavenumber}} \)) is more strongly or less strongly absorbed depending on the chemical composition of the material. There is a correlation between the absorbed radiation and the proportion of essential substance components. Absorption can occur in both the infrared (IR) and near infrared (NIR) regions. Although the measurements within the IR range are largely bound to laboratory conditions (measurements by means of a spectrometer in the transmitted light method or by FTIR technique in the reflection method), the measurements within the NIR range can be done online (analogous to the MC testing of wooden particles). The NIR method is applied primarily for quantitative material analysis. Industrial applications so far are MC testing of particles and protein detection in animal feed [158].
Infrared investigations [159] showed (Table 6.24) that this technique amongst others applies to:
-
The determination of the mixing ratio of hardwood and softwood (using differences in lignin content; Fig. 6.69)
-
The qualitative and quantitative detection of adhesive content in glued wood particles (Fig. 6.70)
-
The detection of lignin or cellulose degradation by wood-destroying fungi

Correlation between the mixing ratio of hardwood/softwood particles and the extinction or band area (wavenumber range 1735 … 1744 cm−1) [160]

Correlation between solid resin content of pine wood shavings and extinction at a wavenumber of 1660 cm−1 [161]
The NIR method is primarily used for quantitative detection under industrial conditions. For example, the wavelengths of 1930 or 1430 nm are used to determine the MC of wooden particles. For the detection of urea resin, the wavelength of 2020 nm has proven to be suitable (Fig. 6.71a). By using wavelength combinations (e.g., 2020 nm, l900 nm, and 2120 nm), the determined regression equation and the coefficient of determination can be improved. However, a significant influence of the chemical structure of the resin and hardener used is present (Fig. 6.71b).

Near infrared spectra of (a) glued wooden particles with different solid resin content, (b) particles glued with different types of adhesive (own measurements, Niemz)
The NIR method can also be used for the detection of particular substances in a material, such as the detection of bark, sugarcane bagasse, or a certain type of adhesive. At wavelengths of 1300 and 1400 nm, it can further be used to determine the mixing ratios of hardwood and softwood chips or particles.
For about 15 years, spectroscopy, in particular NIR spectroscopy, has been increasingly used to control the wood quality (including mechanical properties), the adhesive content of particles, wood properties such as density and strength, and also fungal resistance.
For that purpose, a multitude of characteristics are determined from the spectra and correlated by means of multivariate statistics with the parameters determined in the standard experiment (e.g., bending, tension, pressure) such as strength, MOE (Fig. 6.72), density, microfibril angle but also concerning fungal resistance or wood aging. This requires a very high number of experiments in order to obtain a reliable correlation. Investigations on this topic were provided amongst others by Fujimoto et al., Leblon et al., Meder et al., Sandak et al., Tsuchikawa and Kobori, and Tsuchikawa and Schwanninger [162,163,164,165,166,167]. The method is based on the complex interaction between the chemical structure and the physico-mechanical properties of the wood.

Correlation between the modulus of elasticity (MOE) determined by near-infrared spectroscopy (NIRS) using multivariate statistics and by a static test [164]
8 Aging and Corrosion
Wood and wood-based materials have many factors in practical use that more or less influence their behavior. An overview is shown in Fig. 6.73. The main influencing factors are:
-
Climate
-
Mechanical pre-stress
-
Effect of aggressive media
-
Natural aging and wood preservation measures (see Chaps. 4 and 15)

Influences on the corrosion behavior and on the weather resistance of wood
In general, wood in the dry state is indefinitely stable, if the climate does not change and no infestation by fungi or insects takes place. This results, for example, in wood used in interiors. However, depending on the wood species, the color change is striking (see Sect. 6.7). The color may get darker (spruce, pine, larch) but also lighter (walnut, teak) or there is a yellowing (birch, maple).
8.1 Influence of Climate and Methods for Determination of Climatic Resistance
The ambient climate decisively influences the lifetime of wood and wood-based materials. Important parameters are MC, UV radiation and visible solar radiation, temperature or temperature changes, as well as rainfall and humidity changes, which in turn cause changes in MC and thus swelling or shrinkage of wood (basics of biodeterioration, see Chap. 4 and Sect. 6.7).
8.1.1 Wood
Ultraviolet (UV) radiation causes brownish discoloration in the near-surface zones, followed by a degradation and leaching of the lignin and thus a greying of the surface of the wood (the white cellulose is preserved). The degraded wood components are washed out by water, creating a shaky, rough surface. The impact of weathering on erosion depends to a large extent on the structural composition of the wood. The density and thickness of the cell walls as well as wood extractives have an influence on the weathering resistance of wood. With increasing density, the weathering resistance is improved (Fig. 6.74). With time, a colonization of the surface by fungi often occurs; this can lead to blackening. Therefore, depending on the weathering and microclimate, different discolorations are often visible on wood facades (gray, brown to black). Only in high mountain regions with a relatively dry climate and with good constructional wood preservation, the entire surface turns to largely even brown.

Influence of oven-dry density on the erosion of wood [168]
A strong change in MC leads to swelling and shrinkage of the wood and, owing to the associated tensions, to cracking. At low thicknesses of a few cm, as used for facades, this only occurs near the surface. At large cross-sections such as beams and piles, it can result in severe cracks, which cause moisture penetration into the interior and thus fungal attack.
The weathering resistance between the wood species varies very strongly owing to differences in the wood structure and of the constituents. The durability of the wood is regulated in the European standard EN 350, the corresponding humidity range of use in EN 335. Figure 6.75 shows the influence of fungal activity on the properties of ash wood. After several weeks of fungal activity, there is a considerable loss of strength. However, a suitable protective treatment can significantly increase the life time of the wood.

Influence of the duration of fungal activity of Polyporus hispidus on the properties of ash wood (after Cartwright and Campbell, cited in Vorreiter [52])
If wood is stored for a long time at an elevated temperature, the life of the wood is reduced. Mönck [169] recommends the following reduction factors Kt:
8.1.2 Wood-Based Materials
As for solid wood, the structure of wood-based materials is also affected when the climatic conditions change. Of important influence is the type of adhesive used. UF resin is relatively quickly destroyed by hydrolysis and by swelling and shrinkage of the glued elements. The strength (e.g., internal bond) is reduced and the thickness swelling increases. Phenol-resorcinol-formaldehyde, polyurethane (PUR) and urea-melamine resins are much more stable in relation to UF resin (Table 6.25). Further, the adhesives themselves are hygroscopic [170]. Thus, hide, bone, and fish glues, for example, have high MCs in the upper part of the sorption isotherms.
By using phenolic resin, melamine resin, isocyanate, PUR, and mixed condensates, a limited weather resistance of the wood-based materials can be achieved (particleboards, plywood and glued-laminated timber, cross-laminated timber). In general, wood particle materials are considered to be unsuitable for long-term use under increased humidity (for example, when used as facades), whereas solid wood, plywood, and cross-laminated timber are suitable. Surface coatings can cause a significant increase in weathering resistance. It should be noted, however, that if the surface coating is damaged, moisture can diffuse very quickly into the material, but cannot escape from the material to the same extent.
The testing of the climate resistance of wood and wood-based materials is carried out either by storing appropriate samples outdoors (e.g., at an angle of 45°, south oriented) or by artificial aging (e.g., xenon test: UV radiation and water exposure).
Both the xenon test and the BAM (Federal Institute for Materials Research and Testing) method have been proven in practice by Deppe [171]. Figure 6.76 shows the water absorption and thickness swelling of coated chipboard using various weathering methods.

(a) Water absorption and (b) thickness swelling of coated particleboards after comparative weathering tests [172]
8.2 Aging of Wood and Wood-Based Materials
Aging is the change in properties under natural or artificial conditions over a long period. It is known that many plastics partially age and embrittle owing to UV radiation or emigration of plasticizers. In solid wood, the effect of aging is comparatively rarely researched; there are different statements. An overview of the state of knowledge is given in Kránitz, Kránitz et al., Obataya, Yokoyama et al., and Cavalli et al. [144, 145, 173,174,175].
For wood-based materials, the influence of the adhesives and the material structure must be taken into account. It is caused by the different orientation of the particles or layers. We have a restrained swelling. In addition, the moisture resistance of the adhesives must be observed.
8.2.1 Solid Wood
The age of wood is nearly unlimited under dry conditions. This is the case not only indoors but also outdoors in dry desert areas such as the desert Atacama in northern Chile, where buildings such as in Humberstone survived many decades without major damage.
Aging of wood, stored under dry conditions inside, is described in different reports. According to the ‘Holz-Lexikon’ [29], it is state of the art that the coefficient of variation of the swelling and shrinkage values of aged wood is reduced. This is based on publications by Dietrich Holz [176]. A review paper on this topic shows strongly varying results [145]. Obataya [173] gives a very good overview on the change in chemical and mechanical properties due to aging. Our own measurements showed that the color changes (spruce becomes darker), but the strength properties change only slightly or not, and a reduction is most evident in impact bending strength (Fig. 6.77) [146].

Impact bending strength and modus of elasticity tested with ultrasound depending on density for recent and aged wood [146]
In contrast, the chemical properties change more clearly [144]. Differences compared with non-aged wood were found, particularly on oak wood relative to lignin and polysaccharide content as well as to the degree of cellulose crystallinity. The composition of wood extractives of aged and non-aged wood samples revealed degradation processes due to oxidation and slight hydrolysis. The investigation showed that the relative content of the structural wood constituents depends on the sample age as well as the conditions under which they were aged. Cellulose crystallinity values are affected by extractives content. A mild thermal treatment is often used today for artificial aging (for interior design but also for musical instruments) [177].
8.2.2 Wood-Based Materials
Extensive work has been carried out on wood-based panels, especially for particleboards and fiberboards [178,179,180]. There is less experience with glued-laminated wood. Moisture resistance is mostly tested by means of delamination tests (EN 302-2) or tensile shear tests (EN 302-1) after various storage series A1 (dry), A4 (storage in hot and cold water, wet), A5 (redried).
Glued-laminated timber constructions that are more than 100 years old are still in use. It turns out that even these do not show any reduction in load-bearing capacity. In contrast, works by Konnerth et al. [181] showed that urea resins used in the construction of wood-based aircraft age (aging of adhesives see also the Wood Handbook [5]). The aging effect of urea resins has been known for a long time. It is often attempted to simulate aging using methods such as climate changes (wet–dry), UV radiation, water storage (cooking, drying), or by simulated natural stresses such as the glass house test (Materials Testing Institute Stuttgart, Germany), or stress, e.g., of glulam under mechanical load and climate change (see, for example, Aicher [182]).
For plastic materials and metals, cyclic load tests on adhesive joints are common; also, there are first experiences of wood bonding. An effect of UV radiation on adhesive joints is generally non-existent for usual lamella thicknesses of 30–40 mm (except most likely at the edges). Particularly in the case of large wood cross sections, cracks and, in some cases, delamination, can occur both in solid wood and in bonded wood materials (glued-laminated timber, cross-laminated timber). Problems arise mostly in buildings under very dry conditions in the winter at very low humidity.
In the case of glued-laminated timber and cross-laminated timber, the properties of the elements, the growth ring orientation of the lamellae, the lamella thickness, and moisture differences between lamellae during bonding are important. For cross-laminated timber, the stresses are higher when changing the humidity than for parallel laminated wood (glulam).
8.3 Influence of Mechanical Pre-Stress, Load History
In practical use, wood is partly exposed to considerable mechanical stress. However, the load is often only 10–30% of the ultimate load. Therefore, damage caused by exceeding the permissible tensions occurs relatively rarely. This is usually the case in combinations with shrinkage cracks or when the tensile strength perpendicular to the grain of curved glulam beams is too high. Important for the selection of wood and wood-based materials is creep deformation, or the relaxation of stresses. In the case of particleboard, but also in solid wood, no permanent reduction in strength occurs under the usual stress levels. This happens only at extremely high loads [183,184,185,186].
The frequently mentioned influence of the lunar phase on the properties of wood [187] could not yet be proved with certainty [188,189,190].
Potential wood damage in trees or wood can be detected by among other things increment coring, drilling resistance (Fig. 6.78 [191]), and sound velocity measurement. Sound tomography is also partially used for defect detection in wood, especially in standing trees.

Drilling resistance of sound wood (a) and decayed wood (b). (Images: S. Herrmann, F. Bächle, ETH Zürich)
Mönck [169] recommends for the static proof of older wooden structures the following reduction factors:
-
For continuous use after more than 20 years: reduction of the load capacity to 0.6 up to 0.9 of the initial value
-
At existing seasoning cracks:
-
1.
At moderate weakening of the cross section:
Reduction of the load-bearing cross section to 0.95 of the modulus of resistance or 0.91 of the moment of inertia. Here, the determination of the critical stress for tear propagation using fracture toughness, the fracture energy (see Chap. 9), would be an alternative for more exact calculation. First works have been carried out on this topic of solid wood [192,193,194,195]
-
2.
Substantial weakening of the cross section:
Exact determination of the load-bearing cross section, which is relevant for the moment of inertia or the modulus of resistance and calculation with the determined values
-
1.
8.4 Influence of Aggressive Media
Wood corrosion is understood as the damage or destruction that migrates from the surface into the wood as a result of chemical and/or physico-chemical reactions by interaction with its environment [196]. Wood has a high resistance to corrosion compared with other materials. Because of this property, it is predestined for use, for example, as roof trusses in fertilizer halls, storage of de-icing salt, etc. [197]. It should be noted, however, that aggressive media attack primarily the hemicelluloses and lignin and attack the cellulose less. Therefore, softwood is generally more corrosion resistant than hardwood. A good overview of corrosion is given in Unger et al. [198].
8.4.1 Water
At normal temperatures, water does not cause any chemical change in the wood. However, storing wood in water for a very long time (e.g., during rafting) causes certain wood constituents to leach out, which increases the wood resistance to insects. If water acts on the wood at high temperatures and pressures, hydrolytic degradation can occur [12, 58]. This can be observed for example, in wood that has previously been used for construction of wooden cooling towers.
8.4.2 Chemical Substances
According to Erler [196], the aggressive effect of chemicals on wood, depending on the physical state of the medium, depends on the following factors:
-
For gaseous media: type, concentration, humidity
-
For liquid media: pH, concentration, degree of dissociation
-
For solid media: type, solubility in water, hygroscopicity, pH, humidity
Certain chemicals such as DMF (dimethylformamide) cause a strong swelling and probably also mechanical damage to the wood. When priming wood for bonding, damage of the surface zones adjacent to the bond line by the primer is often brought into discussion. Also, improved wetting is achieved by primers.
The corrosion phenomena are associated for most media with [196]:
-
A brown or yellowish-brown coloring of the wood, which penetrates from the surface zones inwards
-
An increased MC of the wood surface area
-
A storage of salt crystals or acid radicals in the wood
-
A fibrous structure at the surface, e.g., a wool-like surface after the influence of salt [199]
-
Delamination along the growth rings
-
Strength reduction of wood in the surface area
-
Flat breaks in the case of acid exposure
Figure 6.79 shows the increase in the corrosion layer thickness on wood as a function of the service life for different moisture classes. A reduction in strength occurs only within the relatively narrow margins of the wood, whereas the strength in 10–15 mm depth already corresponds mostly to the strength of the native wood. In load-bearing capacity calculations, it is therefore customary to reduce the load-bearing cross section of the timber.

Increase in the corrosion layer thickness of wood used in the fertilizer salt industry as a function of service life under different humidity conditions [196]
8.4.3 Metals
The contact of wood with metals partially causes corrosion. This depends on the type of wood and the wood moisture. In particular, iron induces discoloration in tannin-rich wood species (for example, oak). Even thermally treated wood causes increased corrosion owing to the lower pH value [200]. For fasteners, therefore, stainless steel is used.
Unger [201] gives the following specifications concerning the influences of metals:
-
Wood at a MC of <10% is less affected by metals. At a MC of 20–30%, the influence is more pronounced, it comes to interactions between metal and wood. The reason for this is that often, weakly acidic extracts are formed at these MCs, which promote corrosion
-
Chestnut and oak wood are highly corrosive, poplar and mahogany slightly corrosive
-
Iron stains oak and other wood that are rich in tannins (oak to black, linden to green)
-
Tensile strength of wood decreases after sufficiently long contact with iron (degradation of the polyoses by corrosion products of the iron); compression strength remains unaffected
9 Innovative Testing and Characterization Methods
Materials science necessitates a detailed analysis of the structure, mechanics, and chemistry of a given material. A comprehensive, scientific understanding of these three characteristics is crucial in grasping their influence on the materials’ properties, especially for materials characterized by a complex hierarchical structure, such as wood. An investigation of the properties at each structural level is highly relevant, as their interactions determine the entire macroscopic performance of the material [202, 203]. In wood science, a broad range of analytical tools is used for mechanical, chemical, and structural characterization, with scales of analysis ranging from several angstrom to entire macroscopic elements [204, 205]. Some of these techniques have been utilized for many decades, such as conventional optical microscopy, and continue to play an essential role in materials science laboratories. However, new technologies and methodologies are constantly emerging. These innovative analytical tools allow the spatially resolved characterization of a material’s structure, chemistry, composition, and mechanics to exist on very small length scales or provide a simultaneous recording of mechanics and structure or chemistry by in situ approaches [206,207,208,209].
This section provides a brief introduction to recent developments in select characterization and testing methods and assesses each method for its potential to analyze wood.
Selected Testing and Characterization Methods
Comparison of structural biological materials with man-made engineering materials reveals remarkable differences in design strategies. Engineering materials are based on a predesign of the material according to the targeted functionality and application. A wide variety of elements can be used and manufactured in a static process, which includes an acute knowledge of the material’s requirements prior to fabrication. In contrast, nature possesses a limited amount of useable elements, mainly light elements such as C, N, and O, that can be processed under physiological conditions (water based; low temperatures). In order to compensate for these limitations, nature developed various strategies to create materials that are stiff and long-living [202, 203].
The Scaling Effect in Wood Characterization
The hierarchical structure across multiple length scales of many biological materials, including wood, results in excellent mechanical properties in comparison with the light weight [202, 210]. To analyze the structure, chemistry, or mechanics of wood, it is crucial that the wood’s inherent hierarchical, multi-component structure is taken into consideration, as the results strongly depend on the length scale of the analysis.
The exact contribution of each hierarchical level to the bulk wood properties is still under debate. However, significant progress has been made within the last few years, such as in situ characterization approaches, which provide new insights into relevant structure–function relationships [211,212,213,214,215]. Usually, a decrease in the mechanical properties with increasing sample size is observed owing to the influence of knots and rays, irregularities in fiber direction, density variations, and changes in MFA [216,217,218,219].
Mechanical Characterization Techniques – From the Macro- to the Nanoscale
This sub-section gives a short introduction to recent developments in the field regarding innovative mechanical characterization tools, ranging from the testing of bulk wood samples to the analysis of mechanical properties on a sub-micron level. It briefly covers the theoretical background and measurement principle of the analysis technique, followed by certain application examples.
Advanced Macroscopic Mechanical Analysis by Digital Image Correlation and Electronic Speckle Pattern Interferometry
Measuring displacements and strains is one of the key tasks in mechanically characterizing solid wood materials. On the macroscopic scale, measurements are usually performed using mechanical extensometers, video extensometers, or machine cross-head displacements. However, their limits in detecting local deformations and strain concentrations with high spatial resolution have necessitated the development of alternatives, namely electronic speckle pattern interferometry (ESPI)) and digital image correlation (DIC) [220, 221].
The working principle of ESPI can be easily explained by a Michelson interferometer. Either out-of-plane or in-plane deformations are investigated, dependent upon the set up. For out-of-plane deformation measurements, a laser beam is split into two beams, with one beam reflected by a mirror and the other from the sample surface. Both beams are then imaged onto a detector (e.g., CCD detector). As a consequence of the phase interference between the two beams, a speckle pattern is formed, that is characteristic of the microstructure of the material. An out-of-plane material deformation causes a change in the phase difference between the two laser beams, and a new speckle pattern is created, which allows the mechanical loading of the sample to be followed. For in-plane measurements, the sample is illuminated by two laser beams. A speckle pattern is obtained owing to the interference of the two beams, which are reflected from the sample. In this case, sample deformation causes a new phase difference and speckle pattern. ESPI has been used intensively for various purposes, for example, the analysis of multiaxial deformations of wood samples, the detection of strain distributions in the vicinity of adhesive glue lines, the detection of elastic properties of adhesive polymers, and the analysis of interactions at the interface of wood–cement composites [222,223,224,225].
Digital image correlation is based on the observation of in-plane displacements. DIC is achieved, by tracking the deformation of a random pattern in an image using pattern recognition algorithms. Two patterns are used: either the natural surface structure of wood, or an artificial speckle pattern applied via an air-brush system. To obtain good correlation results, the speckle pattern should be isotropic, of high contrast, stochastic, and nonrepetitive. Moreover, a planar parallel orientation and a constant distance between the object and the camera must be upheld during the investigation. If the orientation and distance are not maintained, it will not be possible to distinguish between an isotropic deformation of the object and a change in the distance between object and camera. DIC has been used to measure the orthotropic properties of wood and wood-based materials. Moreover, this method can help to determine full-field strain distribution and can quantify two-dimensional strain behaviors, which provide the elastic properties of the sample [226,227,228,229,230,231,232,233].
In Situ Mechanical Characterization
In the field of mechanical characterization in situ tests have recently gained popularity that enable a simultaneous mechanical and structural (chemical) analysis. Mechanical testing equipment is placed within a microscopic, synchrotron, or spectroscopic device [215, 234,235,236,237,238]. In this regard, an in situ mechanical testing device was recently designed by Zauner et al., where the cellular response of wood to mechanical loading is followed by synchrotron-based tomography [215]. Computed tomography (CT) is defined as the generation of virtual cross sections of the inner structure of an object, created through the transmission of images obtained from different projections. Based on this consecutive stack of images, a 3D reconstruction is received. Depending on the sample size and required spatial resolution, different radiation sources are used for tomography, such as synchrotron, X-ray, and neutron radiation. X-ray tomography is applicable for many different materials. However, owing to the rather limited sensitivity of wood, only the macroscopic scale is accessible for wood samples. To conduct a mesoscale analysis neutron tomography is preferred, as it has the advantage of a high sensitivity for hydrogenous components such as adhesives, varnishes, or water. This high sensitivity limits the sample size to a few centimeters. If a micro-level analysis is desired (anatomical wood regions such as pits, cell wall, etc.) synchrotron radiation must be used [239,240,241]. In general, loading samples is usually stopped between the individual microscopic, X-ray, and synchrotron measurements, which may lead to a relaxation of the sample.
Micromechanical Testing – Single Fiber Tests
Testing plants micromechanically comprise mechanical analysis on a tissue and cell level, ranging from the characterization of entire organisms and small tissue samples to individual cells (fibers, tracheids). On the micromechanical level, uniaxial tests are mainly conducted to gain information on the stiffness, strength, and toughness of the material, as well as time-dependent properties such as relaxation and creep behavior [206, 207].
In this example, single wood fiber testing, was chosen for a more detailed discussion, as single wood fiber testing is one of the main examples of micromechanical testing. The first single wood fiber tests were reported by Jayne et al. in 1959, using tensile testing machines equipped with load cells based on strain gauges [242]. Since then, numerous studies have been dedicated to developing the technique. To review these pioneering studies, the reader is referred to the publication by Ehrnrooth and Kolseth [243]. Most developments have focused on the fixation of the fibers, as traditional mechanical clamping cannot be used for wood fibers because of their potential to slip and their short length. Currently, the standard approach for tensile tests is based on gluing fibers on support frames, which are then fixed in the testing device (e.g., foliar frames developed by Burgert et al.) [244]. Alternatively, a ball-and-socket setup may be used, where glue droplets are placed at the end of the fibers. After hardening, the epoxy spheres are placed behind two pillars, acting as a socket. The spheres can rotate freely and allow the fibers to align [245]. This approach was recently advanced by Yu et al., who developed a new method of gripping plant fibers with attached epoxy glue droplets at the ends of the fiber (Fig. 6.80) [246]. For all set-ups, it is crucial that the fibers are properly aligned, as even small deviations from the uniaxial loading conditions can significantly influence the results owing to potential shear stresses [206]. Prior to mounting, the fibers are isolated using one of two possible processes: mechanical approaches with tweezers, or chemical isolation. When analyzing the fibers in their natural state, the mechanical approach is preferred. If the influence of industrial processes on the mechanical properties is of interest, chemical isolation is the method of choice [207, 244]. In both cases, the fibers must retain a constant MC to avoid twisting [247].

Scheme of the improved ball and socket set-up for fiber fixation of the custom-built micro-tensile testing system presented in [246]
In early tensile tests of wood fibers, results were fairly inaccurate, as the strain was measured by simple cross head displacement. Today, video extensometry is typically used to measure the small strain values needed to rupture individual wood fibers [248]. To transform the obtained force displacement curves into stress–strain curves, the cross sections of the fibers must be measured using different microscopy techniques, such as confocal laser scanning microscopy [245, 246, 249] or environmental scanning electron microscopy (ESEM) [248]. Based on the stress–strain curves, various mechanical properties can be extracted. The stiffness is determined by the slope of the elastic region of the curve. After a yield point, the curve exhibits a nonlinear behavior characteristic for irreversible deformation. The total area below the curve represents the energy of deformation and is often considered an approximation of the toughness of the sample. The ultimate tensile strength is found at the highest point in the curve [234] (Fig. 6.81).

Schematic drawing of single fiber tests and schematic stress–strain curves recorded during tensile tests
In Fig. 6.82, a graph compiled by Eder et al. [207] depicts tensile stiffness values of dry and wet fibers isolated by mechanical and chemical processes depending on their MFA. It clearly reveals the well-known behavior of decreasing tensile moduli with increasing MFA. Interestingly, the influence of MC on the mechanical properties is more pronounced for fibers with high MFA, when the mechanical properties are significantly dependent on the matrix properties, which are more affected by hydration compared with cellulose [207]. Regarding tensile strength values, existing literature covers comparisons as a function of environmental conditions, fiber modifications, fiber isolation procedures, and fiber geometry [234, 243, 250, 251].

Tensile moduli plotted against microfibril angle: 2x2 mm wood samples (circles), dry chemically isolated wood fibres (squares), mechanically isoclated wood fibres in wet (open triangles) and dry (full triangles) condition [207]
It is relatively simple to combine single-fiber tests with simultaneous nano- and microstructural analysis tools, which provide a detailed observation of the respective deformation mechanisms. Mechanical tests were combined with polarized light microscopy [234, 252], scanning electron microscopy [234, 253], X-ray scattering [238, 254,255,256], and spectroscopy techniques. These tests were used to gain insight into deformation mechanisms of the individual polymeric cell-wall constituents, the load-bearing capacities of the components, or the cellulose fibril reorientation during loading [235, 257].
Nanoindentation
The measurement principle of nanoindentation (NI) is quite similar to traditional hardness tests (e.g., Brinell and Monnin hardness tests) where a stiff indenter of defined geometry is pushed into the surface of a sample with a determined load. The hardness is defined as the ratio of the maximum load and the indent area, which provides information on the resistance of the material to permanent deformation. The main differences in NI are the much smaller indenter size, the exact on-line monitoring of the displacement, and the loading of the indenter. These factors allow for mechanical characterization on a smaller-length scale (μm level), an accurate measurement of the hardness, and the possibility of extracting further mechanical properties from the recorded load-displacement curves, such as the local stiffness [258, 259].
Figure 6.84 displays a schematic of the experimental procedure of NI and a representative loading–unloading curve for viscoelastic materials. The curve can be divided into three main phases: a loading phase, followed by a holding and a de-loading phase. Based on this curve, hardness is calculated by converting the residual depth to the residual indent area by using a “shape function,” which depends on the indenter geometry that was used. Local stiffness is accessible through analysis of the unloading phase of the load-displacement curve. During the loading phase of a nanoindentation experiment the material deforms plastically and elastically below the indenter. In comparison, only the elastic deformation relaxes in the unloading phase. The slope of the elastic part of the unloading curve is then used to determine an indentation modulus (reduced modulus) by a method initially developed by Oliver and Phar in 1992 [260]. In combination with Poisson’s ratio of the material and the elastic properties of the indenter the indentation modulus is directly related to the materials elastic modulus. It should be noted that this relationship is only valid for elastically isotropic materials [207, 258, 260]. For a more detailed description of the theoretical background, nanoindentation instrumentation developments, measurement approaches, and data analysis, the reader is referred to the comprehensive book “Nanoindentation” edited by Anthony C. Fischer Cripps or the reviews by Warren C. Oliver and George M. Pharr, one of the main drivers of NI technology [258, 260,261,262].
On wood samples (cross-sectional wood fiber walls), NI was first applied by Wimmer et al. in 1997 [263, 264]. Since then, numerous studies have been published that advance the measurement procedure, sample preparation, and influence of MFA or chemical modifications [265,266,267,268,269,270,271]. NI was initially developed for the characterization of homogeneous, mechanically isotropic materials, but wood and many other materials are characterized by highly anisotropic behavior. This anisotropy leads to complex deformation behavior under the indenter tip, which means that the indentation modulus obtained should be regarded as a type of “geometric average” of the elastic tensor. An anisotropic indentation theory can be used to account for this circumstance, which was developed by Vlassak et al. and successfully adapted for wood by Jäger et al. [272, 273].
If NI is used for the mechanical characterization of wood, specific constraints and restrictions should be considered. The affected volume in NI is about three times larger than in the indent area, which problematizes an indentation close to an edge, e.g., a free space (lumen) or heterogeneous interface (cell wall/compound middle lamella). This was investigated in detail by Jakes et al. [274], who suggested implementing a correction factor to account for the compliance. To minimize edge effects, wood samples are usually embedded prior to nanoindentation, which additionally facilitates sample preparation in order to obtain flat surfaces. However, the embedding material may hinder cell-wall swelling and instigate stresses within the cell wall, which could potentially influence the mechanical properties. Furthermore, possible penetration of the embedding materials and its influence on the cell mechanics are still under debate [275, 276]. Another important limitation of NI is the dearth of knowledge on the MC of the tested volume, especially considering the substantial influence of MC on mechanical properties. Most of the NI experiments have been performed in noncontrolled environments, which could possibly explain the strong scattering in the values obtained [206, 207]. In this context, a new measurement setup that allows for NI experiments under controlled moisture conditions has been developed by Bertinetti et al. [277]. Despite these constraints and the relatively complex data analysis necessary to account for the mechanical anisotropy of wood, NI is a highly valuable tool for the mechanical characterization of wood on a cell-wall level.
Stiffness values of wood samples obtained by NI (Fig. 6.83) reveal a clear dependency on the MFA, especially in the range between 20° and 60°. These results show a stiffness decrease with increasing MFA. Thus, close attention must be paid to the alignment of the sample. The relatively high data scattering is attributed to the high sensitivity of NI measurement toward the cell-wall composition, as the data are based on experiments of different wood species (softwoods and hardwoods), reaction wood (compression wood, opposite wood), as well as earlywood and latewood [207]. The high sensitivity of NI makes the technique a valuable tool for the characterization of changes in the cell-wall composition or chemistry, as a result, for example, of synthetic modifications or natural lignification of the wood [278,279,280,281].

Reduced moduli obtained by nanoindentation plotted against microfibril angle [207]
In contrast to stiffness values, no significant dependency between hardness and MFA was found [264, 266, 282]. Even between the compound middle lamella (CML) region (mainly composed of lignin) and the secondary cell wall, composed of cellulose, hemicelluloses, and lignin, similar values were obtained [263, 274]. These results indicate that hardness mainly depends on the mechanical properties of the matrix, which was further supported by NI results of thermally treated wood, revealing a substantial decrease in the hardness values and a reduced modulus that was only minimally affected [269, 280]. The exact reason for this compositional independence is still under discussion, although some authors suggest that it could be attributed to the complex loading below the indenter and the strong plastic deformation, as a result of the structural reorganization due to damaged cellulose microfibrils and a high strain in the matrix. For a more in-depth understanding of the phenomena, further research on the relevant plastic deformation mechanisms on wood should be conducted. In addition to stiffness and hardness properties, the creep behavior of cell walls can be investigated by NI. Aside from the mechanical characterization of wood itself, NI has also been used to characterize the interactions of wood and adhesives, as well as the viscoelastic behavior of adhesives [206, 207, 269, 283].
Atomic Force Microscopy
To obtain the mechanical properties of individual wood cell-wall layers, the CML region, or microfibril aggregates the spatial resolution of NI is insufficient. This limitation is overcome by recent developments in atomic force microscopy, which provide mechanical information with sub-micron resolution [209, 284, 285]. The substantial progress in the nanomechanical characterization via AFM represents an important addition to the manifold examples of AFM in the field of wood cell-wall ultrastructural characterization [286, 287]. For structural analysis by AFM, a cantilever with a very sharp tip is raster-scanned across a sample surface. The deflection of the cantilever, as a result of the specific interaction of the tip with the sample surface, is recorded by a laser that is reflected from the cantilever to a photo diode (PSD). These interactions can be of different natures depending on the tip–sample distance, environment, or mode of utilization. The interactions include van der Waals, electrostatic, magnetic, capillary forces, or chemical bonding [288]. These interactions can be converted into a high-resolution topographical image (nm scale) of the surface structure. Comprehensive AFM studies have analyzed the molecular architecture of primary cell walls, the orientation and size of microfibrils within secondary cell walls, and the pore–matrix distribution in cell walls [286, 287, 289].
Aside from these structural imaging capabilities, suppliers have developed new AFM modes for mechanical characterization, mainly based on the mapping of the surface with a grid of force–distance (FD) curves [290]. The elimination of previous drawbacks of AFM–FD curve-based modes, including the amount of time it took to take measurements and analyze data, has significantly improved the applicability of these types of AFM modes. Similar to NI, the FD curve is separated into an approach part and a retract part. First, the cantilever is outside of the attractive regime of the sample with zero force acting on the cantilever. When the cantilever is approaching the sample, weak attractive forces between the tip and the sample (such as the van der Waals and capillary forces) begin to affect the cantilever, and it starts to bend toward the sample. As soon as the attractive forces surpass the stiffness of the cantilever, it jumps into contact with the surface. Until a pre-determined set-point, the cantilever will slightly indent the sample and the cantilever begins bending in the opposite direction. Once the set-point is reached, the retracting phase starts, and the cantilever is removed from the sample. At the pull-off force, the cantilever snaps back to its initial stage. Possible adhesion between the cantilever and the sample surface leads to a hysteresis [288, 291] (Fig. 6.84).

(a) Schematic experimental procedure of nanoindentation (NI) and (b) force–distance (FD) curve-based atomic force microscopy (AFM) and schematic experimental data obtained using the two techniques
To obtain the final FD curves, the deflection signal of the PSD is converted into an FD signal using Hooke’s law. From the slope of the FD curve (extend or retract part) an indentation modulus can then be calculated by applying a contact mechanics model (e.g., Hertz, Johnson–Kendall–Roberts, Derjaguin–Müller–Toporov models). The simplest model is the Hertz model, which assumes a hemispherical tip shape and does not account for adhesive forces, whereas more advanced contact mechanics models (Derjaguin–Müller–Toporov or Johnson–Kendall–Roberts) were developed for measurements where the adhesion force cannot be neglected [292]. In recent years, the potential of FD curve-based AFM modes for the mechanical characterization of different types of lignocellulosic samples has been demonstrated, which include natural fibers, and native and modified wood samples [285, 289, 293, 294]. These efforts helped to gain a better understanding of the ultrastructural arrangement, which aided in specifying current cell-wall models and understanding mechanical properties within the cell wall. FD curves for the characterization of the adhesion behavior of a sample with high sensitivity have been used to study nm-thick coatings of polyelectrolytes applied layer-by-layer to wood surfaces, the variability in the polarity of wood surfaces, and the working principle of primers used to promote the adhesion in 1C-PUR bond lines. This approach is further expanded by functionalization of the tips with specific functional groups to reveal interaction of the tip with specific anatomical regions and for following aging behavior of wood surfaces [295,296,297].
Nevertheless, it remains challenging in AFM to deliver quantitative measurements, as demonstrated by the large discrepancy between the values obtained in the different FD curve-based AFM publications, but also compared with NI measurements. A detailed comparison of the individual works on AFM is complicated, as different measurement setups are used, such as cantilever type, cantilever stiffness, fitting procedure, parameters, and measurement velocities. These factors, as well as unresolved questions, specifically regarding the influence of the tip wear during measurements and the contact situation between the probe and the sample surface must be addressed in future studies [293, 294, 296].
An alternative mode to FD curve-based AFM for the mechanical characterization is contact resonance AFM (CR-AFM), which belongs to the group of force acoustic microscopy. In CR-AFM local elasticity is measured by determining the resonance frequency of an AFM tip in contact with the material. Depending on the sample stiffness, the resonance frequency changes and the contact stiffness obtained is then converted into an elastic modulus [284].
Mechanical Characterization under Dynamic Load and as a Function of Temperature
Thermomechanical Analysis And dynamic Mechanical Analysis
Thermomechanical analysis (TMA) and dynamic mechanical analysis (DMA) represent powerful analytical techniques for studying the elastic and viscoelastic behavior of polymers under a static or dynamic force as a function of time or temperature. Both techniques provide valuable insight into the relationships between structure, morphology, and properties of a material. In Fig. 6.85, a scheme of a TMA apparatus is shown [298]. A constant load is applied to the sample via a probe, which is placed on a sample support located within a furnace. Every change in sample length due to temperature changes moves the probe either upward or downward, which is recorded by a highly sensitive linear variable differential transformer. Depending on the measurement setup, different types of loads can be applied. TMA is commonly used to determine the materials’ coefficients of thermal expansion (CTE), glass transition temperature (Tg), and heat deflection. Additionally, TMA allows stress–strain, creep/recovery, or stress relaxation tests. Both the anisotropic CTE of solid wood and the evolution of Young’s modulus in wood systems as a function of temperature can be measured by TMA, which offers an indication of stiffness and Tg for wooden structures. Both methods address the concerns of quality control and product development. In the field of wood-based materials, the interfacial adhesion of wood–plastic composites (WPCs) and the quality of particleboard or plywood with varied wood adhesives can be evaluated using TMA to ensure the optimal process parameters and chemical agents for manufacturing [299, 300].

Schematic diagram of thermomechanical analysis (TMA Q400EM; reprinted with permission from TA Instruments)
Dynamic mechanical analysis is a technique where sinusoidal force is applied to a sample, and the material’s response is analyzed afterward. Figure 6.86 shows a scheme of a DMA apparatus [301]. First, a sinusoidal force is applied as an input signal, where the resulting sinusoidal deflection or deformation is then detected as an output signal by an optical encoder. This result provides information on the stiffness (storage modulus E′) and damping (loss modulus E″, and damping factor tanδ) of the sample. Additional dynamic methods, including varying temperature, frequency, strain, or stress to static force tests (e.g., stress–strain, creep/recovery, and stress relaxation) are available for analysis and a wide range of fixtures or jigs allow samples to be tested in a variety of shapes and forms. The adjustable temperature and humidity conditions enable investigations at different temperature and humidity levels.

Schematic diagram of dynamic mechanical analysis (DMA Q800 apparatus; reprinted with permission from TA Instruments)
Using DMA, the variability of viscoelastic properties in normal wood–reaction wood, sapwood–heartwood, and earlywood–latewood were investigated. Their structural differences result in variations of (1) the linear viscoelastic response region, (2) of E′ with elevating temperature, (3) the E″ or tanδ peak temperature location and the E″ or tanδ peak value of transition processes, and (4) time-dependency of creep compliance or relaxation modulus. The differences in the viscoelastic properties result from differences in the chemical compositions of the samples. DMA is sensitive to the molecular responses of wood constituents in strained wood samples, providing the opportunity to study the response of individual wood components in situ. DMA also allows for improved understanding of the contributions made by individual wood polymers to the overall properties of the composites.
As the wood cell is composed of cellulose, hemicelluloses, and lignin, wood typically reveals multi-relaxation characteristics between −200 °C and 300 °C. Within this temperature range, three peaks for tanδ could be found (e.g., Fig. 6.87): (1) the low-temperature γ-peak (~−80 °C and below): assigned to the methylol groups’ motion; (2) β-peak (~−50 °C to 50 °C): related to the motion of adsorbed water and the hydrophilic cell-wall hemicelluloses; (3) α-peak (~75 °C to 300 °C): mostly attributed to the softening of lignin, and also the micro-Brownian motions of the cell-wall polymers in the noncrystalline region [302, 303]. The decreasing Tg, near the constant activation energy for the softening process, and a slight tendency toward a more pronounced E′ drop as a function of temperature after hemicellulose dissolution, indicate a more direct stress connection between lignin and cellulose, and lignin plays a more active role in the stress transfer after hemicellulose dissolution [304]. The Tg can be measured by the onset point of E′, or by the E″ peak, or tanδ peak. Stretching time and increasing temperature have equivalent effects on the E′ drop, i.e., time–temperature superposition principle (TTSP). Regarding the TTSP of wood or wood-based materials, DMA could conduct static measurements (e.g., creep or stress relaxation) or dynamic measurements using frequency-multiplexing experiments over a wide range of temperatures. Salmén et al. [305] originally demonstrated that wood follows TTSP for temperatures above the Tg of lignin. Salmén et al. additionally showed that TTSP can be used to identify wood/adhesive interactions and evaluate the distribution of relaxation times in both systems. A general approach to tracking the characterization of the interphase region in WPCs and the degree of adhesion is to calculate adhesion factor (A) using tanδ according to Kubat et al. [306]

Dynamic mechanical analysis (DMA) curve of dry Chinese fir (Cunninghamia lanceolata [Lamb.] Hook.) in radial direction. Tanδ is the ratio between the loss modulus E″ and the storage modulus E′
where, the subscripts “c” and “m” denote the composite and matrix (polymers), and Vf is the wood fiber volume fraction. A low value of A thus indicates a high degree of interaction or adhesion between the two phases. Of course, other wood composition and structure changes can also be evaluated by DMA. Ormondroyd et al. (cited in Siau [10]) found a correlation between observed E′ and the onset of decay as indicated by the fungal gDNA quantification technique.
Numerous works have sought to determine the wood–water interaction. Water acts as plasticizer, accelerating the dynamic and static relaxation process of wood. All three tanδ peaks shift to higher temperatures and decrease in magnitude with decreasing MC [302]. The response of viscoelastic behavior to MC variation is known as the mechano-sorptive (MS) effect [307]. Initially, the creep tests under a static load were used to explore the MS effect. It was also confirmed that the MS effect could be detected by the dynamic loading test. The MS effect is a result of an unstable state in wood cell walls under an external load when relative humidity varies, which is associated with the molecular packing mode, rearrangement of hydrogen bonds, and the shear slip between the crystalline and amorphous parts. In dynamic viscoelastic tests, the variation of MC may cause an increase in damping behavior, especially in the early stages of the adsorption/desorption process [308,309,310]. Under a set of ambient conditions, the evolution of parameters as a function of time monitored by DMA provides a simple and comprehensive way of studying the MS effect upon MC changes. DMA data can be plotted in a Cole–Cole graph or complex plane using the complex modulus (E″ with respect to E′) or compliance (loss compliance J″ with respect to storage compliance J′). The analysis provides a visual assessment of an unstable state during MC changes, especially for cyclic changes.
Chemical Imaging
Numerous analytical tools are available for the chemical analysis of lignocellulosic materials, including wood. Tremendous progress has been made in terms of the chemical and spatial resolution of such analytical tools. In general, analytical methods that require a sample disintegration prior to the analysis (e.g., wet chemical methods or nuclear magnetic resonance (NMR)) differ from techniques that allow for a 2D/3D mapping of the sample. A spatially resolved analysis of the constituents is necessary given that biological materials are characterized by a multi-hierarchical structure. Moreover, various spectroscopy methods in combination with microscopy can nondestructively follow the chemical composition in context with their microstructure. In the next section the main spectroscopy imaging methods for the analysis of wood are explained and selected applications of the techniques are highlighted.
Ultraviolet Spectroscopy Imaging
Ultraviolet spectroscopy is based on the sample’s absorption of light with a wavelength between 10 nm and 700 nm. These specific wavelengths are mainly absorbed by nonbonding n-electrons and π-electrons, which are characteristic of aromatic compounds and conjugated double bonds. In the case of wood, the analysis is limited to lignin and other aromatic constituents (e.g., extractives) respectively. The remaining major components of wood, namely cellulose and hemicelluloses, are not accessible by UV spectroscopy. UV spectroscopy measurements of wood samples are usually performed in transmission, which requires a defined sample thickness between 0.5 and 1 μm. In order to obtain thin cuts, the samples are typically embedded into resins (e.g., Spurr) and cut with an ultra-microtome equipped with a diamond knife [311] (Table 6.26). The main UV absorbance peak at ~280 nm is attributed to the lignin characteristic delocalized π-electron system of aromatic rings. Depending on the amount of lignin monomers (S, G, H units) in the sample, variations in the position and the intensity of the peak are observed. This was first analyzed by Musha and Goring by investigations of lignin model compounds, where the dependency of the lignin monomer unit on the absorbance maximum was clearly shown. The maximum absorbance for G units is between 280 and 285 nm, for S units between 270 and 275 nm, and for H units between 255 and 260 nm. Therefore, depending on the exact lignin composition of the wood samples, a slight adjustment of the wavelength may be conceptualized [312]. Owing to chromophoric groups, which are found in highly condensed phenolic structures, the UV spectra of wood samples can reveal an additional absorbance band at higher wavelengths. The high degree of conjugation leads to a stabilization of π – π* transitions, resulting in a shift toward higher wavenumbers [311]. In general, the UV absorbance bands of aromatic origin overlap and are relatively broad, which makes an exact assignment challenging [313].
Early works of UV micro-spectroscopy were mainly focused on topochemical investigations of different cell and tissue types, revealing a higher lignin content in the cell corner (CC) and middle lamella than in the secondary cell wall [314,315,316]. Detailed studies were performed following the xylem formation and lignification processes within the living tree [317,318,319,320]. Studies have also focused on the analysis of reaction wood of different wood species to understand the difference in lignin content and distribution [321,322,323]. UV spectroscopy measurements of the influence of wounding on the xylem were performed by Frankenstein et al., and fungal infections were studied by Baum et al. [324, 325]. Moreover, UV spectroscopy is also a valuable tool for the characterization of wood cell walls modified by furfurylation, as the modified cell walls reveal a significantly enhanced UV absorbance owing to the formation of condensed aromatic compounds [326].
Fluorescence Spectroscopy Imaging
In fluorescence spectroscopy, the sample is illuminated by a light source with a specific wavelength. The light is then absorbed by the sample’s inherent fluorophores, typically containing aromatic groups or π bonds, which is followed by re-emission of the light at longer wavelengths. The fluorescence signal obtained is significantly weaker than the illumination light and is separated by spectral emission filters. For fluorescence spectroscopy, different setups and configurations have been used to measure plants, including wood. In short, there are two main methods: endogenous and exogenous fluorescence measurements. In the former method, the auto-fluorescence of the sample is used to study the topochemistry. In the latter method, specific fluorophores are incorporated into the sample prior to the analysis [327].
Wood itself exhibits a distinct auto-fluorescence and can be excited with UV and visible light characterized by a broad emission in the blue, green, and red range. The multimodal auto-fluorescence of wood is attributed to different fluorescent structures within wood constituents such as extractives (phenolics, resins, tannins) and lignin. As lignin is rich in fluorescent aromatic units, constituting around 30% of the dry mass of wood, it is considered to be the main fluorophore. In contrast, the other two main wood components – hemicelluloses and cellulose – do not reveal fluorescence owing to their lack of fluorescent units. The fluorescence of lignin depends on various factors including lignin composition, type, and amount of inter-linkages between the monolignol units. The combination of these factors makes a spatially resolved analysis of the chemistry of wood/plant cells possible [328, 329]. In this regard, the lignification of different wood species, reaction wood, or genetically modified samples was analyzed [313, 328, 330,331,332]. Fluorescence spectroscopy has also provided a valuable insight into the penetration behavior of adhesives in pre-modified samples and in the distribution of wood-modifying agents with high resolution, as shown by Thygesen et al. in the case of furfurylated wood [333]. A specific type of fluorescence spectroscopy was recently used by Donaldson and Radotic, fluorescence lifetime imaging (FLM). Fluorescence lifetime is defined as the average time between the excitation of a fluorophore until the emission of a fluorescence photon. FLM makes it possible to distinguish between fluorophores of similar wavelength emission but different lifetimes and provides improved differentiation among the cell-wall layers and wood types compared with fluorescence intensity, which is commonly used [334].
In exogenous fluorescence spectroscopy, the signal of a probe is used for the measurement. The probe is either conjugated to the (bio)molecule that will be analyzed, or solely incorporated into the sample. The probe must be carefully chosen depending on the properties needed, for example, size, sample, and probe specificity. Historically, many different fluorescent molecules have been used to label the constituents of wood cells, including safranin, phloroglucinol, Congo red, or calcofluor [327]. A recent approach labels molecules, which are subsequently metabolized by the plant to provide a dynamic analysis of the lignification of wood cell walls [335].
Raman Spectroscopy-Infrared Spectroscopy (Imaging)
Chemical imaging of lignocellulosic materials (LMs) via Raman and infrared (IR) microscopy has gained tremendous interest in recent years, as they provide a nondestructive, spatially resolved visualization of the chemical composition. In contrast to UV and fluorescence spectroscopy all the main constituents can be investigated. Additionally, the analysis is not limited to lignin and other types of aromatic compounds [313, 336].
Both techniques (Raman and IR) monitor the molecular vibrations of the sample, but they differ in their underlying physical principle. In Raman spectroscopy, the vibrations of a molecular system due to inelastic scattering of photons (monochromatic light in the visible, UV or NIR region) are studied, whereas IR is based on the absorption of a photon from a polychromatic light source, which excites the molecule from the ground state to a higher vibrational energy level [336,337,338]. The theoretical prediction of the inelastic scattering (Raman effect) goes back to Adolf Smekal, which was experimentally proven by C.V. Raman in 1928 [339, 340]. Nevertheless, it took an additional 50 years for the technique to be used in routine analysis, as most of the light is elastically scattered (Rayleigh scattering). Therefore, the Raman signal is relatively weak, which results in a lower signal to-noise ratio than for IR spectroscopy [336].
The differences in the underlying physical principles of the two techniques also lead to both divergent selection rules and sensitivity toward different molecular structures. IR requires a change in the dipole moment of a bond, whereas for Raman, a change in the polarizability is necessary. Therefore, IR is highly sensitive for polar functional groups and Raman is mainly used for the carbon backbone or molecular structures with high symmetry. For all imaging approaches, the spatial resolution is highly relevant, as it defines the minimum distance between two points of a sample that can still be separated. In microscopy, this distance is determined by Abbe’s diffraction limit, which is given by the following formula: r = 0.61λ/NA, where λ is the wavelength of the light and NA the numerical aperture of the objective. As a consequence, the highest spatial resolution is achieved by the UV-visible excitation used for Raman spectroscopy, and the lowest by the IR excitation utilized in IR spectroscopy. The resolution in IR spectroscopy is further reduced by the necessity of using so-called Cassegrain objectives, possessing a maximum NA of 0.6. In comparison, Raman microscopy can use immersion objectives with a NA of up to 1.4. In short, in Raman spectroscopy, the maximum resolution of 0.3–2 μm and in IR of 5–20 μm is achievable [336, 337, 341] (Table 6.26).
In the 1980s, the first Raman spectra of lignocellulosic samples were published. Since then, significant improvements in the Raman instrumentation (laser, filter, spectrometer, CCD) have resulted in a tremendous spread of the technique, especially for mapping and imaging approaches [336, 337, 342]. A particular problem of early Raman spectroscopy was the hindrance of laser-induced fluorescence of the LMs, which masks the Raman signal. The acquisition of the spectra in a wet state and the usage of NIR lasers and Kerr gated Raman spectroscopy instruments were especially important steps toward diminishing the influence of fluorescence [336, 343,344,345,346]. In general, the spectra obtained by Raman and IR spectroscopy are rather complex, as all the constituents of the sample are probed at the same time. Simultaneous probing results in a multitude of partially overlapping bands. Therefore, many studies focused on the analysis of isolated plant cell-wall polymers, as well as model compounds. The goal of this analysis was to develop the basis for interpretation of plant cell-wall spectra in terms of band assignment, band variations, or band intensities. Detailed band assignments for Raman-and IR spectroscopy can be found in the literature [347, 348].
Infrared and Raman spectroscopy have been used intensively for the analysis of the composition and the structure of plant cell-wall constituents. Studies have covered the analysis of cellulose types and degree of crystallinity [349,350,351,352]. Recently, Raman spectroscopy demonstrated that native plant cell walls lack crystalline cellulose parts, and that crystalline cellulose parts are formed upon drying [353]. Vibrational spectroscopy can also be used to study the molecular orientation of components, such as the changes of component-specific bands upon changes in the polarization of the excitation source [336, 337]. Therefore, Raman and IR were used to show that lignin follows the orientation of the cellulose microfibrils. Similar results were reported for the hemicelluloses [354,355,356,357].
A major strength of both techniques is the ability to record spatial position-resolved spectral images, which are based on up to many thousands of spectra. IR and Raman spectroscopy were applied to map the distribution of the main constituents of wood (e.g., cellulose, xylan, pectin, lignin), providing an insight into the chemical build up of the lignocellulosic material [358,359,360,361]. Further works covered the analysis of lignification and delignification processes, heartwood formation, UV degradation, and natural decay of waterlogged spruce or mineral deposits in lignocellulosic materials [341, 362,363,364,365,366,367,368]. In recent years, Raman spectroscopy has been also extensively used for analyzing genetically engineered wood and functionalized/modified wood, as it allows for detailed analysis of the distribution of various modifying agents (minerals, polymers, nanoparticles) [369,370,371,372]. Overall, more imaging approaches using Raman spectroscopy are reported than for IR. This can be explained by the spatial resolution achievable by Raman microscopy, which is significantly higher and makes analysis of distinct anatomical regions such as the CC, CML, pits, or individual secondary cell-wall layers possible. Nevertheless, IR spectroscopy represents a valuable tool for the analysis on a tissue and cell level.
Mappings in Raman spectroscopy, as well as IR, can be easily composed of thousands of individual spectra with partially overlapping signals from all the components. In order to fully exploit the available data, it is important to develop smart analysis strategies. One can distinguish between univariate and multivariate analysis strategies. In univariate approaches, the analysis is limited to a specific band (intensity, band position, full width at half maximum, etc.) attributed to a specific functional group of a component, followed by visualization of these parameters in false color images (Fig. 6.88) [373]. However, for lignocellulosic materials, the characteristic, overlapping bands and the large number of spectra within one mapping sometimes limit the application of univariate methods, necessitating the use of multivariate methods. Over the years, many different multivariate methods have been used, including principal component analysis, cluster analysis (hierarchical, k-means clustering), vertex component analysis, non-negative matrix factorization, and multivariate curve resolution alternating least squares [337, 374,375,376]. For a detailed description of these methods and their application in wood science, the reader is referred to the Prats-Mateu et al. [377].

Raman images of tension wood based on (a) integrations from 1545–1698 cm−1 (lignin-specific band); (b) from 2774–3026 cm−1 (cellulose + lignin); (c) from 1067–1106 cm−1 (orientation-sensitive band), and (d) the band width at 50% height of the Carbon-Hydrogen band between 2823 and 2917 cm−1 [373]
References
Niemz, P., Sonderegger, W.: Holzphysik – Physik des Holzes und der Holzwerkstoffe. Carl Hanser Verlag, Munich (2017)
Bosshard, H.H.: Holzkunde 1–3. Birkhäuser, Basel (1982–1984)
Kollmann, F., Côté Jr., W.A.: Principles of Wood Science and Technology, vol. 1. Springer, Berlin/Heidelberg (1968)
Bodig, J., Jayne, B.A.: Mechanics of Wood and Wood Composites. Krieger Publishing Company, Malabar (1993)
Wood Handbook: Wood as an Engineering Material. Forest Products Laboratory, Madison (2010)
Fengel, D., Wegener, G.: Wood: Chemistry, Ultrastructure. Reactions/De Gruyter, Berlin/New York (1984)
Higuchi, T.: Biochemistry and Molecular Biology of Wood. Springer, Berlin/Heidelberg (1997)
Wagenführ, R.: Anatomie Des Holzes. DRW-Verlag, Leinfelden-Echterdingen (1999)
Rowell, R.: Handbook of Wood and Wood Composites. CRC Press/Taylor and Francis Group, Boka Ratan/London/New York (2005)
Siau, J.F.: Wood: Influence of Moisture on Physical Properties. Department of Wood Science and Forest Products, Virginia Polytechnic Institute and State University, Keene, NY (1995)
Stamm, A.J.: Wood and Cellulose Science. Ronald Press, New York (1964)
Kollmann, F.: Technologie des Holzes und der Holzwerkstoffe, vol. 1. Springer, Berlin/Göttingen/Heidelberg (1951)
Knigge, W., Schulz, H.: Grundriss der Forstbenutzung. Parey, Hamburg (1966)
Zauer, M., Pfriem, A., Wagenführ, A.: Wood Sci. Technol. 47, 1197–1211 (2013)
Plötze, M., Niemz, P.: Eur. J. Wood Prod. 69, 649–657 (2011)
Walker, J.C.F.: Primary Wood Processing: Principles and Practice. Springer, Dordrecht (2006)
Mette, H.J.: Holzkundliche Grundlagen Der Forstnutzung. Dt. Landwirtschaftsverlag, Berlin (1984)
Grönlund, A., Grönlund, U., Hagmann, O.: Nordkalottfura. TH Lulea, Lulea (1992)
Trendelenburg, R.: Das Holz Als Rohstoff. J. F. Lehmanns Verlag, Munich (1939)
Trendelenburg, R., Mayer-Wegelin, H.: Das Holz Als Rohstoff. Carl Hanser Verlag, Munich (1955)
Butterfield, B.G.: Proceedings of IAWA/IUFRO International Workshop of Significance of the Microfibril Angle to Wood Quality, Westport/New Zealand (1997)
Western Wood Density Survey: Report No. 1. USDA, Forest service, Forest Products Laboratory, Research Paper FPL 27 (1965)
Delmastro, R., Diaz-Vaz, J.E., Schlatter, J.: Variabilidad De Las Características Tecnolóficas Hereditables Del Pinus Radiata (D. Don). CONAF/PNUD/FAO-CHI/76/003, Documento de trabajo No. 43 (1982)
Lanvermann, C.: Sorption and Swelling within Growth Rings of Norway Spruce and Implications on the Macroscopic Scale, PhD Thesis. ETH Zurich, Zurich (2014)
Keunecke, D.: Elasto-Mechanical Characterisation of Yew and Spruce Wood with Regard to Structure-Property Relationships, PhD Thesis. ETH Zurich, Zurich (2008)
Kollmann, F.: Die Esche und Ihr Holz. Julius Springer, Berlin (1941)
Schweingruber, F.H.: Wood Structure and Enviroment. Springer, Berlin/Heidelberg/New York (2007)
Günther B.: Erarbeitung einer Methode zur Erfassung von dendrochronologisch relevanten Jahrringmerkmalen der Trauben-Eiche (Quercus Petraea [Matt.] Liebl.) auf Grundlage der Röntgendensitometrie, PhD Thesis. TU Dresden, Dresden (2013)
Holz-Lexikon. DRW-Verlag, Leinfelden-Echterdingen (2003)
Zauner, M.: In-Situ Synchrotron Based Tomographic Microscopy of Uniaxially Loaded Wood: In-Situ Testing Device, Procedures and Experimental Investigations, PhD Thesis. ETH Zurich, Zurich (2014)
Helms, D., Niemz, P.: HOB, 78–80 (1993)
Görlacher, R., Hättich, R.: Die Bohrwiderstandsmessung, Bauen mit Holz. 92, 455–459 (1990)
Winistorfer, P.M., Xu, W., Wimmer, R.: Forest Prod. J. 45, 90–93 (1995)
Rinn, F., Schweingruber, F.-H., Schär, E.: Holzforschung. 50, 303–311 (1996)
Gao, S., Wang, X., Wiemann, M.C., Brashaw, B.K., Ross, R.J., Wang, L.: Ann. For. Sci. 74, 1–13 (2017)
Washburn, E.W.: Phys. Rev. 17, 273–283 (1921)
Schneider, A.: Holz Roh Werkst. 37, 295–302 (1979)
Schneider, A.: Holz Roh Werkst. 40, 415–420 (1982)
Schweitzer, F., Niemz, P.: Holz Roh Werkst. 49, 27–29 (1991)
Zauer, M., Hempel, S., Pfriem, A., Mechtcherine, V., Wagenführ, A.: Wood Sci. Technol. 48, 1229–1240 (2014)
Popper, R., Bariska, M.: Holz Roh Werkst. 30, 289–294 (1972)
Popper, R.: Das Holz/Sorbat-System mit Rücksicht auf die submikroskopische Betrachtungsweise. In: Kucera, L.J. (ed.) Xylorimba: Trends in Wood Research, pp. 155–163. Birkhäuser Verlag, Basel (1985)
Zauer, M., Meissner, F., Plagge, R., Wagenführ, A.: Holzforschung. 70, 137–143 (2016)
Zauer, M., Kretzschmar, J., Großmann, L., Pfriem, A., Wagenführ, A.: Wood Sci. Technol. 48, 177–193 (2014)
Zauer, M.: Untersuchungen zur Porenstruktur und zur kapillaren Wasserleitung in Holz und deren Änderung infolge einer thermischen Behandlung, PhD Thesis. TU Dresden, Dresden (2011)
Hass, P.F.S.: Penetration Behavior of Adhesives into Solid Wood and Micromechanics of the Bondline, PhD Thesis. ETH Zurich, Zurich (2012)
Marutzky, R., Schwab, H.: Span- und Faserplatten, Osb. Schriftenreihe: Informationsdienst Holz Spezial. Fraunhofer-Institut für Holzforschung (WKI), Verband der Deutschen Holzwerkstoffindustrie e. V. (VHI) (2009)
Kollmann, F., Malmquist, L.: Holz Roh Werkst. 14, 201–204 (1956)
Steinhagen, H.P.: Thermal Conductive Properties of Wood, Green or Dry, from –40° to +100 °C: A Literature Review. Tech. Rep. Forest Products Laboratory, U.S. Department of Agriculture, Forest Service, Madison (1977)
Schneider, A., Engelhardt, F.: Holz Roh Werkst. 35, 273–278 (1977)
Sonderegger, W., Hering, S., Niemz, P.: Holzforschung. 65, 369–375 (2011)
Vorreiter, L.: Holztechnologisches Handbuch, vol. 1. Fromme, Vienna (1949)
Sonderegger, W., Niemz, P.: Eur. J. Wood Prod. 67, 313–321 (2009)
Sonderegger, W., Niemz, P.: Eur. J. Wood Prod. 70, 25–35 (2012)
Czajkowski, L., Olek, W., Weres, J., Guzenda, R.: Wood Sci. Technol. 50, 537–545 (2016)
Ghazi Wakili, K., Binder, B., Vonbank, R.: Energ. Buildings. 35, 413–415 (2003)
Sonderegger, W., Niemz, P.: Holz Roh Werkst. 64, 11–20 (2006)
Lampert, H.: Faserplatten. Fachbuch, Leipzig (1966)
Glos, P., Henrici, D.: Festigkeit von Bauholz bei hohen Temperaturen. Forschungsbericht, Institut für Holzforschung der Universität München, Munich (1990)
Niemz, P., Hug, S., Schnider, T.: Forstarchiv. 85, 163–168 (2014)
Sonderegger, W., Niemz, P.: Holz Roh Werkst. 64, 385–391 (2006)
Szmutku, M.B., Campean, M., Porojan, M.: Eur. J. Wood Prod. 71, 205–210 (2013)
Geissen, A.: Über den Einfluß der Temperatur und Feuchtigkeit auf die Elastizitäts- und Festigkeitseigenschaften des Holzes im Gefrierbereich, PhD Thesis. Universität Hamburg, Hamburg (1976)
Holz-Brandschutz-Handbuch: Deutsche Gesellschaft für Holzforschung, Munich (1983)
Hill, C.A.S.: Wood Modification: Chemical, Thermal and Other Processes. Wiley, Chichester (2006)
Kollmann, F.: Technologie des Holzes und der Holzwerkstoffe, vol. 2. Springer, Berlin (1955)
Mönck, W., Erler, K.: Schäden an Holzkonstruktionen: Analyse und Behebung. Huss-Medien Verlag Bauwesen, Berlin (2004)
Bucur, V.: Nondestructive Characterization and Imaging of Wood. Springer, Berlin (2003)
Shepard, S.M.: Anti-Corros. Method. M. 44, 236–239 (1997)
Sfarra, S., Theodorakeas, P., Avdelidis, N.P., Koui, M.: Russ. J. Nondestruct. 49, 234–250 (2013)
López, G., Basterra, L.-A., Ramón-Cueto, G., de Diego, A.: Int. J. Archit. Herit. 8, 517 (2013)
Conde, M.J., Liñán, C.R., De Hita, P.R., Gálvez, F.P.: Res. Nondestruct. Eval. 23, 32–45 (2012)
López, G., Basterra, L.-A., Acuña, L.: Infrared Phys. Techn. 89, 242–246 (2018)
Meinlschmidt, P.: Thermographic Detection of Defects in Wood and Wood-Based Materials. Proceedings of the 14th International Symposium on Nondestructive Testing of Wood, pp. 41–45. Shaker Verlag (2005)
Riedel, B., Walter, T.: HK Holz- und Kunststoffverarbeitung. 25, 775–777 (1989)
Ugolev, B.N.: Holzkunde und Grundlagen der Holzwarenkunde. Lesn. Prom, Moscow (1986)
Keylwerth, R., Noack, D.: Holz Roh Werkst. 14, 162–172 (1956)
Torgovnikov, G.I.: Dielectric Properties of Wood and Wood-Based Materials. Springer, Berlin/Heidelberg/New York (1993)
Roig, F., Ramanantsizehena, G., Lahatra Razafindramisa, F., Dantras, E., Dandurand, J., Hoyet, H., Bernés, A., Lacabanne, C.: Wood Sci. Technol. 51, 1389–1404 (2017)
Koubaa, A., Perré, P., Hutcheon, R.M., Lessard, J.: Dry. Technol. 26, 568–578 (2008)
Avramidis, S.: Dry. Technol. 34, 753–760 (2016)
Knuffel, W., Pizzi, A.: Holzforschung. 40, 157–162 (1986)
Fukada, E.: Holzforschung. 21, 186–187 (1967)
Fukada, E.: Wood Sci. Technol. 2, 299–307 (1968)
Niemz, P., Lühmann, A., Wagner, J.: Holz Roh Werkst. 50, 484 (1992)
Nondestructive Testing and Evaluation of Wood – 50 Years of Research: International Nondestructive Testing and Evaluation of Wood Symposiums Series, FPL-GTR-213. U.S. Department of Agriculture, Forest Service, Forest Product Laboratory, Madison (2012)
Lexikon Der Holztechnik. Fachbuchverlag, Leipzig (1990)
Prieto, J., Kiene, J.: Holzbeschichtung. Chemie Und Praxis. Vincentz Verlag, Hannover (2007)
Lüttgens, G.: Elektrostatische Aufladungen. Expert-Verlag, Ehringen (1979)
Ozyhar, T., Hering, S., Sanabria, S.J., Niemz, P.: Wood Sci. Technol. 47, 329–341 (2013)
Schubert, S.: Acousto-Ultrasound Assessment of Inner Wood-Decay in Standing Trees: Possibilities and Limitations, PhD Thesis. ETH Zurich, Zurich (2007)
Baensch, F.: Damage Evolution in Wood and Layered Wood Composites Monitored in Situ by Acoustic Emission, Digital Image Correlation and Synchrotron Based Tomographic Microscopy, PhD Thesis. ETH Zurich, Zurich (2015)
Krautkrämer, J., Krautkrämer, H.: Werkstoffprüfung mit Ultraschall. Springer, Berlin (1986)
Bucur, V.: Acoustics of Wood. Springer, Berlin (2006)
Bachtiar, E.V., Sanabria, S.J., Niemz, P.: Elastic characteristics of wood by means of ultrasonic waves and mechanical test, Proc. ISCHP 2015: 5th International Scientific Conference on Hardwood Processing, pp. 12–19 (2015)
Niemz, P., Bächle, F.: Stadt und Grün. 51, 52–55 (2002)
Sanabria, S.J.: Air-Coupled Ultrasound Propagation and Novel Non-Destructive Bonding Quality Assessment of Timber Composites, PhD Thesis. ETH Zurich, Zurich (2012)
Brémaud, I.: J. Acoust. Soc. Am. 131, 807–818 (2012)
Holz, D.: Holztechnologie. 8, 221–224 (1967)
Holz, D.: Holztechnologie. 14, 195–202 (1973)
Sonderegger, W., Alter, P., Niemz, P.: Holz Roh Werkst. 66, 345–354 (2008)
Sprossmann, R., Zauer, M., Wagenführ, A.: Results Phys. 7, 1737–1742 (2017)
Krüger, R., Zauer, M., Wagenführ, A.: Eur. J. Wood Prod. 76, 1663–1668 (2018)
Bekhta, P., Niemz, P., Kucera, L.J.: Holz Roh Werkst. 60, 41–45 (2002)
Keunecke, D., Sonderegger, W., Pereteanu, K., Lüthi, T., Niemz, P.: Wood Sci. Technol. 41, 309–327 (2007)
Keunecke, D., Merz, T., Sonderegger, W., Schnider, T., Niemz, P.: Wood Mater. Sci. Eng. 6, 91–94 (2011)
Burmester, A.: Holz Roh Werkst. 26, 113–117 (1968)
Niemz, P.: Holz Roh Werkst. 53, 100 (1995)
Burmester, A.: Holz Roh Werkst. 23, 227–236 (1965)
Niemz, P.: Holz Roh Werkst. 54, 60 (1996)
Armstrong, J.P., Patterson, D.W., Sneckenberger, J.E.: Wood Fiber Sci. 23, 32–43 (1991)
Hankinson, R.: Investigation of Crushing Strength of Spruce at Varying Angles of Grain, Air Force Information Circular No. 259, U. S. Air Service (1921)
Hasenstab, A.G.M.: Integritätsprüfung von Holz mit dem zerstörungsfreien Ultraschallechoverfahren, PhD Thesis. TU Berlin (2006)
Hoyle, R.J., Pellerin, R.: Stress Wave Inspection of Wood Structure, Proceedings of fourth nondestructive testing symposium of wood (1978)
Autorenkollektiv: Werkstoffe aus Holz und andere Werkstoffe der Holzindustrie. Fachbuchverlag, Leipzig (1975)
Sause, M.G.R.: Identification of Failure Mechanisms in Hybrid Materials Utilizing Pattern Recognition Techniques Applied to Acoustic Emission Signals, PhD Thesis. Augsburg University, mbv-Verlag, Berlin (2010)
Aicher, S., Höfflin, L., Dill-Langer, G.: Holz Roh Werkst. 59, 104–116 (2001)
Rosner, S.: Bioresources. 7, 1253–1263 (2012)
Noguchi, M.N., Nishitoma, K.: Tagung Schallemission. (1985)
Niemz, P., Plotnikov, S.: Holztechnologie. 29, 207–210 (1988)
Niemz, P., Hänsel, A., Schweitzer, F.: Holztechnologie. 30, 44–47 (1989)
Skaar, C., Simpson, W.T., Honeycutt, R.M.: Forest Prod. J. 30, 21–22 (1980)
Schmidt, K.: Internationaler Holzmarkt. 76, 1–3 (1985)
Lemaster, R., Dornfeld, D.A.: Tagung Schallemission. (1985)
Michailow, W., Niemz, P.: HK Holz- und Kunststoffverarbeitung. 25, 1129–1131 (1990)
Rayleigh, S.J.H.: Theory of Sound. Macmillan, London (1929)
Görlacher, R.: Klassifizierung von Brettschichtholzlamellen durch Messung von Longitudinalschwingungen, PhD Thesis. Universität Karlsruhe, Karlsruhe (1990)
Görlacher, R.: Holz Roh Werkst. 42, 219–221 (1984)
Hearmon, R.F.S.: Forest Prod. J. 16, 29–40 (1966)
Gülzow, A.: Zerstörungsfreie Bestimmung der Biegesteifigkeiten von Brettsperrholzplatten, PhD Thesis. ETH Zurich, Zurich (2008)
Grundström, F., Kucera, L.J., Niemz, P.: Holz-Zentralblatt. 125, 1734, 1736 (1999)
Xu, M., Li, L., Wang, M., Luo, B.: Tribol. T. 57, 871–878 (2014)
Möhler, K., Herröder, W.: Holz Roh Werkst. 37, 27–32 (1979)
Möhler, K., Maier, G.: Holz Roh Werkst. 27, 303–307 (1969)
Meisel, A., Wallner, B., Schickhofer, G.: Bautechnik. 92, 412–423 (2015)
Koch, P.: Utilization of Hardwoods Growing on Southern Pine Sites, vol. 1. USDA Forest Service, Agriculture Handbook 605, Washington, DC (1985)
Koubek, R., Dedicova, K., Dorn, M., Serrano, E., Vessby, J.: Friction of Wood on Steel, Tech. Rep. Linnaeus University, Faculty of Technology (2014)
Svensson, B.A., Nyström, S., Gradin, P.A., Höglund, H.: Tribol. Int. 42, 190–196 (2009)
Gressel, P., Redecker, P.: Reibbeiwerte von Holzwerkstoffen. Forschungsbericht. FH Rosenheim, Rosenheim (1991)
Wagenführ, R.: Holzatlas. Fachbuchverlag Leipzig im Carl Hanser Verlag, Munich (2007)
Teischinger, A., Zukal, M.L., Meints, T., Hansmann, C., Stingl, L.: Colour characterization of various hardwoods. In: Nemeth, R., Teischinger, A. (eds.) Hardwood Science and Technology. The 5th Conference of Hardwood Research and Utilization in Europe, pp. 180–188. University of West Hungary Press, Sopron (2012)
Pitt, W.: 33 Farbtafeln Parkett. Holzmann Buchverlag, Bobingen (2010)
Lukowsky, D.: Schadensanalyse Holz Und Holzwerkstoffe. Fraunhofer IRB Verlag, Stuttgart (2013)
Kránitz, K.: Effect of Natural Aging on Wood, PhD Thesis. ETH Zurich, Zurich (2014)
Kránitz, K., Sonderegger, W., Bues, C.-T., Niemz, P.: Wood Sci. Technol. 50, 7–22 (2016)
Sonderegger, W., Kránitz, K., Bues, C.-T., Niemz, P.: J. Cult. Herit. 16, 883–889 (2015)
Volkmer, T., Lorenz, T., Hass, P., Niemz, P.: Eur. J. Wood Prod. 72, 249–259 (2014)
Oltean, L., Teischinger, A., Hansmann, C.: Holz Roh Werkst. 66, 51–56 (2008)
Willeitner, H., Schwab, E.: Holz-Aussenverwendung im Hochbau. Verlagsanstalt Alexander Koch GmbH, Stuttgart (1981)
Schlotzauer, P., Wilhelms, F., Lux, C., Bollmus, S.: Eur. J. Wood Prod. 40, 118 (2018)
Wendland, G.: Beitrag zur automatischen Oberflächeninspektion von Holz anhand optischer Verfahren, PhD Thesis. TU Dresden, Dresden (1999)
Hu, M., Briggert, A., Olsson, A., Johansson, M., Oscarsson, J., Säll, H.: Wood Sci. Technol., 7–27 (2018)
Ehrhart, T., Steiger, R., Frangi, A.: Eur. J. Wood Prod. 76, 925–935 (2018)
Plinke, B.: Größenanalyse an nicht separierten Holzpartikeln mit regionenbildenden Algorithmen sm Beispiel bon OSB-Strands, PhD Thesis. WKI Braunschweig, TU Dresden, Dresden (2012)
Böhme, P.: Industrielle Oberflächenbehandlung von plattenförmigen Werkstoffen aus Holz. Fachbuchverlag, Leipzig (1980)
Weiss, D., Brandl, H.: Chem. Unserer Zeit. 47, 122–131 (2013)
Gahle, C.: Holz-Zentralblatt. 142, 603–604 (2016)
Stark, E., Luchter, K., Margoshes, M.: Appl. Spectrosc. Rev. 22, 335–399 (1986)
Wienhaus, O., Niemz, P., Fabian, S.: Holzforsch. Holzverw. 32, 120–125 (1988)
Niemz, P., Wienhaus, O., Schaarschmidt, K., Ramin, R.: Holzforsch. Holzverw. 41, 22–26 (1989)
Niemz, P., Wienhaus, O., Körner, S., Szaniawski, M.: Holzforsch. Holzverw. 42, 25–28 (1990)
Fujimoto, T., Kobori, H., Tsuchikawa, S.: J. Near Infrared Spec. 20, 353–359 (2012)
Leblon, B., Adedipe, O., Hans, G., Haddadi, A., LaRocque, A., Tsuchikawa, S., Burger, J., Stirling, R., Pirouz, Z., Groves, K., Nader, J.: Forest. Chron. 89, 595–606 (2013)
Meder, R., Thumm, A., Bier, H.: Holz Roh Werkst. 62, 159–164 (2002)
Sandak, A., Sandak, J., Negri, M.: Wood Sci. Technol. 45, 35–48 (2011)
Tsuchikawa, S., Kobori, H.: J. Wood Sci. 61, 213–220 (2015)
Tsuchikawa, S., Schwanninger, M.: Appl. Spectrosc. Rev. 48, 560–587 (2013)
Sell, J., Feist, W.C.: Forest Prod. J. 36, 57–60 (1986)
Mönck, W.: Schäden an Holzkonstruktionen: Analyse und Behebung. Bauwesen, Berlin (1999)
Wimmer, R., Kläusler, O., Niemz, P.: Wood Sci. Technol. 47, 763–775 (2013)
Deppe, H.-J.: Beurteilung von Holzwerkstoffen im Kurzzeit- (Xeno-Test) und Langzeittest (Bewitterungsversuch). In: Sammelband Bundesanstalt Für Materialforschung und Prüfung (BAM), Berlin (1987)
Deppe, H.-J., Stolzenburg, R., Schmidt, K.: Holz Roh Werkst. 34, 379–384 (1976)
Obataya, E.: Characteristics of Aged Wood and Japanese Traditional Coating Technology for Wood Protection, Proc. Cité de la Musique – Conserver aujourd’hui: les “vieillissements” du bois, journée d’étude du 2 février 2007, pp. 26–43, Philharmonie de Paris (2007)
Yokoyama, M., Gril, J., Matsuo, M., Yano, H., Suigiyama, J., Ozaki, H., Imamura, M., Kawai, S.: C. R. Phys. 10, 601–611 (2009)
Cavalli, A., Cibecchini, D., Togni, M., Sousa, H.S.: Constr. Build. Mater. 114, 681–687 (2016)
Holz, D.: Holztechnologie. 22, 80–85 (1981)
Montero, C., Gril, J., Legeas, C., Hunt, D.G., Clair, B.: Holzforschung. 66, 757–764 (2012)
Deppe, H.-J., Ernst, K.: Taschenbuch der Spanplattentechnik. DRW-Verlag, Leinfelden-Echterdingen (2000)
Dunky, M., Niemz, P.: Holzwerkstoffe und Leime: Technologie und Einflussfaktoren. Springer, Berlin (2002)
Paulitsch, M., Barbu, M.C.: Holzwerkstoffe der Moderne. DRW-Verlag, Leinfelden-Echterdingen (2015)
Konnerth, J., Müller, U., Gindl, W., Buksnowitz, C.: Eur. J. Wood Prod. 70, 381–384 (2012)
Aicher, S.: Bestandsaufnahme: Schadensfälle, Dauerstandfestigkeit, Prüfmethoden. MPA Stuttgart, Stuttgart (2006)
Gressel, P.: Untersuchungen über das Zeitstandbiegeverhalten von Holzwerkstoffen in Abhängigkeit von Klima und Belastung, PhD Thesis. Universität Hamburg, Hamburg (1971)
Kjucukow, G., Nikolajewa, R.: Derevoobr. i Mebelna Prom. 23, 264–271 (1980)
Niemz, P.: Untersuchungen zum Kriechverhalten von Spanplatten unter besonderer Berücksichtigung des Einflusses der Werkstoffstruktur, PhD Thesis. TU Dresden, Dresden (1982)
Ehlbeck, J., Görlacher, R.: Bauen mit Holz. 92, 117–121 (1990)
Zürcher, E., Schlaepfer, R., Conedera, M., Giudici, F.: Trees. 24, 31–41 (2010)
Fellner, J., Teischinger, A.: Alte Holzregeln. Von Mythen und brauchbarem über Fehlinterpretationen zu neuen Erkenntnissen. Österreichischer Kunst- u. Kulturverlag, Vienna (2001)
Niemz, P., Kucera, L.J.: Schweiz. Z. Forstwes. 151, 444–450 (2000)
Bariska, M., Rösch, P.: Schweiz. Z. Forstwes. 151, 439–443 (2000)
Niemz, P., Bues, C.-T., Herrmann, S.: Schweiz. Z. Forstwes. 153, 201–209 (2002)
Gustafsson, P.J., Hoffmeyer, P., Valentin, G.: Holz Roh Werkst. 56, 307–317 (1998)
Dill-Langer, G.: Schädigung von Brettschichtholz bei Zugbeanspruchung rechtwinklig zur Faserrichtung. Universität Stuttgart, Stuttgart, Diss (2004)
Hassani, M.M.: Adhesive Bonding of Structural Hardwood Elements, PhD Thesis. ETH Zurich, Zurich (2015)
Ammann, S.D.: Mechanical Performance of Glue Joints in Structural Hardwood Elements, PhD Thesis. ETH Zurich, Zurich (2015)
Erler, K.: Korrosion von Vollholz und Brettschichtholz (1998)
Titner, J., Smid, E., Tieben, J., Reschreiter, H., Kovarik, K., Grabner, M.: Wood Sci. Technol., 953–961 (2016)
Unger, A., Schniewind, A.P., Unger, W.: Conservation of Wood Artifacts. Springer, Berlin/Heidelberg (2001)
Rug, W., Lissner, A.: Bautechnik. 88, 177–188 (2011)
Niemz, P., Wetzig, M.: Holz-Zentralblatt. 137, 24–26 (2011)
Unger, A.: Holzkonservierung. Fachbuchverlag, Leipzig (1988)
Fratzl, P., Weinkamer, R.: Prog. Mater. Sci. 52, 1263–1334 (2007)
Lakes, R.: Nature. 361, 511–515 (1993)
Fernandes, A.N., Thomas, L.H., Altaner, C.M., Callow, P., Forsyth, V.T., Apperley, D.C., Kennedy, C.J., Jarvis, M.C.: P. Natl. Acad. Sci. USA. 108, E1195–E1203 (2011)
Steiger, R., Feltrin, G., Weber, F., Nerbano, S., Motavalli, M.: B. Earthq. Eng. 15, 3265–3291 (2017)
Burgert, I., Keplinger, T.: J. Exp. Bot. 64, 4635–4649 (2013)
Eder, M., Arnould, O., Dunlop, J.W.C., Hornatowska, J., Salmen, L.: Wood Sci. Technol. 47, 163–182 (2013)
Milani, P., Braybrook, S.A., Boudaoud, A.: J. Exp. Bot. 64, 4651–4662 (2013)
Zlotnikov, I., Zolotoyabko, E., Fratzl, P.: Prog. Mater. Sci. 87, 292–320 (2017)
Gibson, L.J.: J. Roy. Soc. Interface. 9, 2749–2766 (2012)
Burgert, I.: Am. J. Bot. 93, 1391–1401 (2006)
de Borst, K., Jenkel, C., Montero, C., Colmars, J., Gril, J., Kaliske, M., Eberhardsteiner, J.: Comput. Struct. 127, 53–67 (2013)
Salmen, L., Burgert, I.: Holzforschung. 63, 121–129 (2009)
Baensch, F., Zauner, M., Sanabria, S.J., Sause, M.G.R., Pinzer, B.R., Brunner, A.J., Stampanoni, M., Niemz, P.: Holzforschung. 69, 1015–1025 (2015)
Zauner, M., Keunecke, D., Mokso, R., Stampanoni, M., Niemz, P.: Holzforschung. 66, 973–979 (2012)
Reiterer, A., Burgert, I., Sinn, G., Tschegg, S.: J. Mater. Sci. 37, 935–940 (2002)
Lanvermann, C., Hass, P., Wittel, F.K., Niemz, P.: Bioresources. 9, 105–119 (2014)
Lanvermann, C., Sanabria, S.J., Mannes, D., Niemz, P.: Holzforschung. 68, 113–122 (2014)
Reiterer, A., Lichtenegger, H., Tschegg, S., Fratzl, P.: Philos. Mag. A. 79, 2173–2184 (1999)
McCormick, N., Lord, J.: Mater. Today. 13, 52–54 (2010)
Sharp, B.: Opt. Laser. Eng. 11, 241–255 (1989)
Frybort, S., Mauritz, R., Teischinger, A., Muller, U.: Bioresources. 7, 2483–2495 (2012)
Gindl, W., Sretenovic, A., Vincenti, A., Muller, U.: Holzforschung. 59, 307–310 (2005)
Konnerth, J., Gindl, W., Muller, U.: J. Appl. Polym. Sci. 103, 3936–3939 (2007)
Muller, U., Sretenovic, A., Vincenti, A., Gindl, W.: Holzforschung. 59, 300–306 (2005)
Jeong, G.Y., Park, M.J.: Constr. Build. Mater. 113, 864–869 (2016)
Jeong, G.Y., Hindman, D.P., Zink-Sharp, A.: J. Mater. Sci. 45, 5820–5830 (2010)
Serrano, E., Enquist, B.: Holzforschung. 59, 641–646 (2005)
Ljungdahl, J., Berglund, L.A., Burman, M.: Holzforschung. 60, 190–195 (2006)
Konnerth, J., Valla, A., Gindl, W., Muller, U.: Wood Sci. Technol. 40, 631–636 (2006)
Jeong, G.Y., Zink-Sharp, A.E., Hindman, D.P.: Wood Fiber Sci. 41, 51–63 (2009)
Jeong, G.Y., Zink-Sharp, A., Hindman, D.P.: Holzforschung. 64, 729–734 (2010)
Valla, A., Konnerth, J., Keunecke, D., Niemz, P., Muller, U., Gindl, W.: Wood Sci. Technol. 45, 755–765 (2011)
Eder, M., Stanzl-Tschegg, S., Burgert, I.: Wood Sci. Technol. 42, 679–689 (2008)
Gierlinger, N., Schwanninger, M., Reinecke, A., Burgert, I.: Biomacromolecules. 7, 2077–2081 (2006)
Kolln, K., Grotkopp, I., Burghammer, M., Roth, S.V., Funari, S.S., Dommach, M., Muller, M.: J. Synchrotron Radiat. 12, 739–744 (2005)
Peura, M., Grotkopp, I., Lemke, H., Vikkula, A., Laine, J., Muller, M., Serimaa, R.: Biomacromolecules. 7, 1521–1528 (2006)
Peura, M., Muller, M., Serimaa, R., Vainio, U., Saren, M.P., Saranpaa, P., Burghammer, M.: Nucl. Instrum. Meth. B. 238, 16–20 (2005)
Mannes, D., Lehmann, E., Cherubini, P., Niemz, P.: Trees-Struct. Funct. 21, 605–612 (2007)
Mannes, D., Marone, F., Lehmann, E., Stampanoni, M., Niemz, P.: Wood Sci. Technol. 44, 67–84 (2010)
Mannes, D., Sonderegger, W., Hering, S., Lehmann, E., Niemz, P.: Holzforschung. 63, 589–596 (2009)
Jayne, B.A.: TAPPI J. 42, 461–467 (1959)
Ehrnrooth, E.M.L., Kolseth, P.: Wood Fiber Sci. 16, 549–566 (1984)
Burgert, I., Keckes, J., Fruhmann, K., Fratzl, P., Tschegg, S.E.: Plant Biol. 4, 9–12 (2002)
Groom, L., Mott, L., Shaler, S.: Wood Fiber Sci. 34, 14–27 (2002)
Yu, Y., Jiang, Z.H., Fei, B.H., Wang, G., Wang, H.K.: J. Mater. Sci. 46, 739–746 (2011)
Kolseth, P.: Torsional Properties of Single Wood Pulp Fibers, PhD Thesis. The Royal Institute of Technology Stockholm, Stockholm (1983)
Burgert, I., Fruhmann, K., Keckes, J., Fratzl, P., Stanzl-Tschegg, S.E.: Holzforschung. 57, 661–664 (2003)
Sedighi-Gilani, M., Sunderland, H., Navi, P.: Wood Sci. Technol. 39, 419–430 (2005)
Burgert, I., Eder, M., Fruhmann, K., Keckes, J., Fratzl, P., Stanzl-Tschegg, S.: Holzforschung. 59, 354–357 (2005)
Goswami, L., Eder, M., Gierlinger, N., Burgert, I.: J. Mater. Sci. 43, 1286–1291 (2008)
Thygesen, L.G., Eder, M., Burgert, I.: J. Mater. Sci. 42, 558–564 (2007)
Mott, L., Shaler, S.M., Groom, L.H., Liang, B.H.: TAPPI J. 78, 143–148 (1995)
Keckes, J., Burgert, I., Fruhmann, K., Muller, M., Kolln, K., Hamilton, M., Burghammer, M., Roth, S.V., Stanzl-Tschegg, S., Fratzl, P.: Nat. Mater. 2, 810–814 (2003)
Montero, C., Clair, B., Almeras, T., van der Lee, A., Grila, J.: Compos. Sci. Technol. 72, 175–181 (2012)
Zabler, S., Paris, O., Burgert, I., Fratzl, P.: J. Struct. Biol. 171, 133–141 (2010)
Eichhorn, S.J., Sirichaisit, J., Young, R.J.: J. Mater. Sci. 36, 3129–3135 (2001)
Fischer-Cripps, A. (ed.): Nanoindentation. Springer, New York (2011)
Oyen, M. (ed.): Handbook of Nanoindentation with Biological Applications. Pan Stanford Publishing, Singapore (2011)
Oliver, W.C., Pharr, G.M.: J. Mater. Res. 7, 1564–1583 (1992)
Oliver, W.C., Pharr, G.M.: MRS Bull. 35, 897–907 (2010)
Oliver, W.C., Pharr, G.M.: J. Mater. Res. 19, 3–20 (2004)
Wimmer, R., Lucas, B.N.: IAWA J. 18, 77–88 (1997)
Wimmer, R., Lucas, B.N., Tsui, T.Y., Oliver, W.C.: Wood Sci. Technol. 31, 131–141 (1997)
de Borst, K., Bader, T.K., Wikete, C.: J. Struct. Biol. 177, 532–542 (2012)
Gindl, W., Gupta, H.S., Schoberl, T., Lichtenegger, H.C., Fratzl, P.: Appl. Phys. A-Mater. 79, 2069–2073 (2004)
Gindl, W., Schoberl, T.: Compos. Part A-Appl. S. 35, 1345–1349 (2004)
Konnerth, J., Gierlinger, N., Keckes, J., Gindl, W.: J. Mater. Sci. 44, 4399–4406 (2009)
Stanzl-Tschegg, S., Beikircher, W., Loidl, D.: Holzforschung. 63, 443–448 (2009)
Wagner, L., Bader, T.K., de Borst, K.: J. Mater. Sci. 49, 94–102 (2014)
Wang, X.Z., Zhao, L.G., Deng, Y.H., Li, Y.J., Wang, S.Q.: Holzforschung. 72, 301–309 (2018)
Jäger, A., Bader, T., Hofstetter, K., Eberhardsteiner, J.: Compos. Part A-Appl. S. 42, 677–685 (2011)
Vlassak, J.J., Ciavarella, M., Barber, J.R., Wang, X.: J. Mech. Phys. Solids. 51, 1701–1721 (2003)
Jakes, J.E., Frihart, C.R., Beecher, J.F., Moon, R.J., Resto, P.J., Melgarejo, Z.H., Suarez, O.M., Baumgart, H., Elmustafa, A.A., Stone, D.S.: J. Mater. Res. 24, 1016–1031 (2009)
Konnerth, J., Harper, D., Lee, S.H., Rials, T.G., Gindl, W.: Holzforschung. 62, 91–98 (2008)
Kamke, F.A., Lee, J.N.: Wood Fiber Sci. 39, 205–220 (2007)
Bertinetti, L., Hangen, U.D., Eder, M., Leibner, P., Fratzl, P., Zlotnikov, I.: Philos. Mag. 95, 1992–1998 (2015)
Gindl, W., Gupta, H.S.: Compos. Part A-Appl. S. 33, 1141–1145. Pii s1359-835x(02)00080-5 (2002)
Zickler, G.A., Schoberl, T., Paris, O.: Philos. Mag. 86, 1373–1386 (2006)
Yin, Y.F., Berglund, L., Salmen, L.: Biomacromolecules. 12, 194–202 (2011)
Konnerth, J., Eiser, M., Jager, A., Bader, T.K., Hofstetter, K., Follrich, J., Ters, T., Hansmann, C., Wimmer, R.: Holzforschung. 64, 447–453 (2010)
Tze, W.T.Y., Wang, S., Rials, T.G., Pharr, G.M., Kelley, S.S.: Compos. Part A-Appl. S. 38, 945–953 (2007)
Ammann, S., Obersriebnig, M., Konnerth, J., Gindl-Altmutter, W., Niemz, P.: Int. J. Adhes. Adhes. 50, 45–49 (2014)
Arnould, O., Arinero, R.: Compos. Part A-Appl. S. 74, 69–76 (2015)
Casdorff, K., Keplinger, T., Burgert, I.: Plant Methods. 13, 60 (2017)
Fahlen, J., Salmen, L.: J. Mater. Sci. 38, 119–126 (2003)
Fahlen, J., Salmen, L.: Biomacromolecules. 6, 433–438 (2005)
Butt, H.J., Cappella, B., Kappl, M.: Surf. Sci. Rep. 59, 1–152 (2005)
Casdorff, K., Keplinger, T., Ruggeberg, M., Burgert, I.: Planta. 247, 1123–1132 (2018)
Chopinet, L., Formosa, C., Rols, M.P., Duval, R.E., Dague, E.: Micron. 48, 26–33 (2013)
Bhushan, B.: J. Vac. Sci. Technol. B. 21, 2262–2296 (2003)
Kopycinska-Muller, M., Geiss, R.H., Hurley, D.C.: Ultramicroscopy. 106, 466–474 (2006)
Arnould, O., Siniscalco, D., Bourmaud, A., Le Duigou, A., Baley, C.: Ind. Crop. Prod. 97, 224–228 (2017)
Muraille, L., Aguie-Beghin, V., Chabbert, B., Molinari, M.: Sci. Rep.-UK. 7, 44065 (2017)
Casdorff, K., Keplinger, T., Bellanger, H., Michen, B., Schon, S., Burgert, I.: ACS Appl. Mater Inter. 9, 13793–13800 (2017)
Casdorff, K., Klausler, O., Gabriel, J., Amen, C., Lehringer, C., Burgert, I., Keplinger, T.: Int. J. Adhes. Adhes. 80, 52–59 (2018)
Frybort, S., Obersriebnig, M., Muller, U., Gindl-Altmutter, W., Konnerth, J.: Colloid. Surface. A. 457, 82–87 (2014)
Thermomechanical Analyzer TMA Q400. Online accessible brochure. TA Instruments. http://www.tainstruments.com/wp-content/uploads/BROCH-TMA-2014-EN.pdf (2018)
Osman, Z.: J. Therm. Anal. Calorim. 107, 709–716 (2012)
Zanetti, M., Marini, D., Pasqualini, E., Masetto, E., Cavalli, R.: Eur. J. Wood Prod. 74, 127–130 (2016)
Dynamic Mechanical Analyzer DMA Q800. Online accessible brochure. TA Instruments. http://www.tainstruments.com/wp-content/uploads/BROCH-DMA-2014-EN.pdf (2018)
Havimo, M.: Wood Sci. Technol. 43, 627–642 (2009)
Jiang, J.L., Lu, J.X., Huang, R., Li, A.X.: Dry. Technol. 27, 1229–1234 (2009)
Salmen, L., Stevanic, J.S., Olsson, A.M.: Holzforschung. 70, 1155–1163 (2016)
Salmen, L.: J. Mater. Sci. 19, 3090–3096 (1984)
Kubat, J., Rigdahl, M., Welander, M.: J. Appl. Polym. Sci. 39, 1527–1539 (1990)
Hunt, D.G.: Present knowledge of mechano-sorptive creep of wood. In: Morlier, P. (ed.) Creep in Timber Structures. CRC Press, London (1992)
Zhan, T.Y., Jiang, J.L., Peng, H., Lu, J.X.: Holzforschung. 70, 547–555 (2016)
Zhan, T.Y., Jiang, J.L., Peng, H., Lu, J.X.: Thermochim. Acta. 633, 91–97 (2016)
Zhan, T.Y., Lu, J.X., Jiang, J.L., Peng, H., Li, A.X., Chang, J.M.: Materials. 9, 1020 (2016)
Koch, G., Kleist, G.: Holzforschung. 55, 563–567 (2001)
Musha, Y., Goring, D.A.I.: Wood Sci. Technol. 9, 45–58 (1975)
Fackler, K., Thygesen, L.G.: Wood Sci. Technol. 47, 203–222 (2013)
D. E. Bland, W. E. Hillis: Appita 23, 204–210 (1969)
Fergus, B.J., Goring, D.A.I.: Pulp Paper Mag. Can. 70, T314–T322 (1969)
B. J. Fergus, D. A. I. Goring: Holzforschung 24, 118–124 (1970)
Dunisch, O., Fladung, M., Nakaba, S., Watanabe, Y., Funada, R.: Holzforschung. 60, 608–617 (2006)
Grunwald, C., Ruel, K., Joseleau, J.P., Fladung, M.: Trees-Struct. Funct. 15, 503–517 (2001)
Grunwald, C., Ruel, K., Kim, Y.S., Schmitt, U.: Plant Biol. 4, 13–21 (2002)
Prislan, P., Koch, G., Cufar, K., Gricar, J., Schmitt, U.: Holzforschung. 63, 482–490 (2009)
Lehringer, C., Gierlinger, N., Koch, G.: Holzforschung. 62, 255–263 (2008)
Ruelle, J., Yoshida, M., Clair, B., Thibaut, B.: Trees-Struct. Funct. 21, 345–355 (2007)
Yoshida, M., Ohta, H., Okuyama, T.: J. Wood Sci. 48, 99–105 (2002)
Baum, S., Schwarze, F., Fink, S.: New Phytol. 147, 347–355 (2000)
Frankenstein, C., Schmitt, U., Koch, G.: Ann. Bot. 97, 195–204 (2006)
Ehmcke, G., Pilgard, A., Koch, G., Richter, K.: Holzforschung. 71, 821–831 (2017)
Paes, G.: Molecules. 19, 9380–9402 (2014)
Donaldson, L., Radotic, K., Kalauzi, A., Djikanovic, D., Jeremic, M.: J. Struct. Biol. 169, 106–115 (2010)
Radotic, K., Kalauzi, A., Djikanovic, D., Jeremic, M., Leblanc, R.M., Cerovic, Z.G.: J. Photoch. Photobio. B. 83, 1–10 (2006)
Donaldson, L., Hague, J., Snell, R.: Holzforschung. 55, 379–385 (2001)
Donaldson, L.A., Knox, J.P.: Plant Physiol. 158, 642–653 (2012)
Wagner, A., Donaldson, L., Kim, H., Phillips, L., Flint, H., Steward, D., Torr, K., Koch, G., Schmitt, U., Ralph, J.: Plant Physiol. 149, 370–383 (2009)
Thygesen, L.G., Barsberg, S., Venas, T.M.: Wood Sci. Technol. 44, 51–65 (2010)
Donaldson, L.A., Radotic, K.: J. Microsc.-Oxford. 251, 178–187 (2013)
Tobimatsu, Y., Wagner, A., Donaldson, L., Mitra, P., Niculaes, C., Dima, O., Kim, J.I., Anderson, N., Loque, D., Boerjan, W., Chapple, C., Ralph, J.: Plant J. 76, 357–366 (2013)
Gierlinger, N.: Appl. Spectrosc. Rev. (2017)
Gierlinger, N., Keplinger, T., Harrington, M.: Nat. Protoc. 7, 1694–1708 (2012)
Wiercigroch, E., Szafraniec, E., Czamara, K., Pacia, M.Z., Majzner, K., Kochan, K., Kaczor, A., Baranska, M., Malek, K.: Spectrochim. Acta A. 185, 317–335 (2017)
Smekal, A.: Z. Phys. 32, 241–244 (1925)
Raman, C.V., Krishnan, K.S.: Nature. 121, 501–502 (1928)
Richter, S., Mussig, J., Gierlinger, N.: Planta. 233, 763–772 (2011)
Gierlinger, N., Schwanninger, M.: Spectrosc.-Int. J. 21, 69–89 (2007)
Agarwal, U.P., Atalla, R.H.: Planta. 169, 325–332 (1986)
Atalla, R.H., Agarwal, U.P.: J. Raman Spectrosc. 17, 229–231 (1986)
Barsberg, S., Matousek, P., Towrie, M., Jorgensen, H., Felby, C.: Biophys. J. 90, 2978–2986 (2006)
Yakovlev, V.V.: Spectroscopy. 22, 34–41 (2007)
Agarwal, U.P., Ralph, S.A.: Appl. Spectrosc. 51, 1648–1655 (1997)
Schwanninger, M., Rodrigues, J.C., Pereira, H., Hinterstoisser, B.: Vib. Spectrosc. 36, 23–40 (2004)
Agarwal, U.P., Reiner, R.S., Ralph, S.A.: Cellulose. 17, 721–733 (2010)
Kataoka, Y., Kondo, T.: Macromolecules. 29, 6356–6358 (1996)
Kataoka, Y., Kondo, T.: Macromolecules. 31, 760–764 (1998)
Szymanska-Chargot, M., Cybulska, J., Zdunek, A.: Sensors. 11, 5543–5560 (2011)
Agarwal, U.P., Ralph, S.A., Reiner, R.S., Baez, C.: Cellulose. 23, 125–144 (2016)
Atalla, R.H., Agarwal, U.P.: Science. 227, 636–638 (1985)
Atalla, R.H., Whitmore, R.E., Heimbach, C.J.: Macromolecules. 13, 1717–1719 (1980)
Salmen, L., Olsson, A.M., Stevanic, J.S., Simonovic, J., Radotic, K.: Bioresources. 7, 521–532 (2012)
Olsson, A.M., Bjurhager, I., Gerber, L., Sundberg, B., Salmen, L.: Planta. 233, 1277–1286 (2011)
Naumann, A., Polle, A.: New Zeal. J. For. Sci. 36, 54–59 (2006)
Labbe, N., Rials, T.G., Kelley, S.S., Cheng, Z.M., Kim, J.Y., Li, Y.: Wood Sci. Technol. 39, 61–U19 (2005)
Horvath, L., Peszlen, I., Gierlinger, N., Peralta, P., Kelley, S., Csoka, L.: Holzforschung. 66, 717–725 (2012)
Ma, J.F., Zhang, Z.H., Yang, G.H., Mao, J.Z., Xu, F.: Bioresources. 6, 3944–3959 (2011)
Schmidt, M., Schwartzberg, A.M., Perera, P.N., Weber-Bargioni, A., Carroll, A., Sarkar, P., Bosneaga, E., Urban, J.J., Song, J., Balakshin, M.Y., Capanema, E.A., Auer, M., Adams, P.D., Chiang, V.L., Schuck, P.J.: Planta. 230, 589–597 (2009)
Segmehl, J.S., Studer, V., Keplinger, T., Burgert, I.: Materials. 11, 517 (2018)
Belt, T., Keplinger, T., Hanninen, T., Rautkari, L.: Ind. Crop. Prod. 108, 327–335 (2017)
Felhofer, M., Prats-Mateu, B., Bock, P., Gierlinger, N.: Tree Physiol. 38, 1526–1537 (2018)
Pedersen, N.B., Gierlinger, N., Thygesen, L.G.: Holzforschung. 69, 103–112 (2015)
Gierlinger, N., Sapei, L., Paris, O.: Planta. 227, 969–980 (2008)
Kataoka, Y., Kiguchi, M., Williams, R.S., Evans, P.D.: Holzforschung. 61, 23–27 (2007)
Keplinger, T., Cabane, E., Chanana, M., Hass, P., Merk, V., Gierlinger, N., Burgert, I.: Acta Biomater. 11, 256–263 (2015)
Merk, V., Chanana, M., Keplinger, T., Gaan, S., Burgert, I.: Green Chem. 17, 1423–1428 (2015)
Segmehl, J.S., Lauria, A., Keplinger, T., Berg, J.K., Burgert, I.: Front. Chem. 6, 28 (2018)
Ozparpucu, M., Ruggeberg, M., Gierlinger, N., Cesarino, I., Vanholme, R., Boerjan, W., Burgert, I.: Plant J. 91, 480–490 (2017)
Dieing, T., Hollrichter, O., Toporski, J. (eds.): Confocal Raman Microscopy, Optical Sciences. Springer, Heidelberg (2010)
Zhang, X., Ji, Z., Zhou, X., Ma, J.F., Hu, Y.H., Xu, F.: Anal. Chem. 87, 1344–1350 (2015)
Gierlinger, N.: Front. Plant Sci. 5, 306 (2014)
Felten, J., Hall, H., Jaumot, J., Tauler, R., de Juan, A., Gorzsas, A.: Nat. Protoc. 10, 217–240 (2015)
Prats-Mateu, B., Felhofer, M., de Juan, A., Gierlinger, N.: Plant Methods. 14, 52 (2018)
Acknowledgment
Sections 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, and 6.8 of this chapter include several parts that have been previously published in the following book: Peter Niemz and Walter Sonderegger: Holzphysik, Physik des Holzes und der Holzwerkstoffe (Wood physics. Physics of wood and wood-based materials). Fachbuchverlag Leipzig im Carl Hanser Verlag, Munich 2017.
The selected paragraphs from the original publication have been translated into English, and the content has been expanded and adapted to the structure of the Springer Handbook of Wood Science and Technology. The authors and Springer are grateful to Carl Hanser Verlag Munich for kind permission.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Niemz, P., Sonderegger, W., Keplinger, T., Jiang, J., Lu, J. (2023). Physical Properties of Wood and Wood-Based Materials. In: Niemz, P., Teischinger, A., Sandberg, D. (eds) Springer Handbook of Wood Science and Technology. Springer Handbooks. Springer, Cham. https://doi.org/10.1007/978-3-030-81315-4_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-81315-4_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-81314-7
Online ISBN: 978-3-030-81315-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)