
Abstract
Liver cancer is a major cause of cancer-related death, and its incidence keeps rising. The liver is exposed to gut microbial products and metabolites via portal blood and influenced by the gut microbiome. Alteration of the gut microbiome is commonly observed in high-risk factors for liver cancer such as obesity, nonalcoholic fatty liver disease, and cirrhosis. The association between dysbiosis and liver cancer has been suggested. Importantly, animal studies provide direct evidence that the gut microbiome promotes liver cancer. The current knowledge of the gut microbiome’s contribution to liver cancer and the reported mechanisms will be reviewed in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Chronic viral hepatitis
- Hepatocellular carcinoma
- Helicobacter pylori
- Gut-liver axis
- Liver fluke infection
- Virus
- Parasites
- Obesity
- Nonalcoholic fatty liver disease
- Microbiome
7.1 Liver Cancer Types and Risk Factors
7.1.1 Hepatocellular Carcinoma
Liver cancer is a leading cause of cancer-related death worldwide, and its incidence is rising (Tanaka et al. 2010; Mokdad et al. 2017; Forner et al. 2018; Lauby-Secretan et al. 2016; Villanueva 2019). Hepatocellular carcinoma (HCC), the most frequent (~80%) primary liver cancer, ranks as the sixth most common malignancy and mainly occurs in men. HCC is closely related to chronic liver diseases, arises frequently in patients with cirrhosis, and is considered a typical inflammation-linked cancer (Capece et al. 2013; Colotta et al. 2009). Although early HCC can be potentially cured by surgical resection or liver transplantation, most HCC patients are diagnosed with unresectable disease. Despite the great improvement of modern cancer treatments and increased survival in many cancers, the mortality rate of HCC is still rising (Tanaka et al. 2010; Mokdad et al. 2017; Forner et al. 2018; Lauby-Secretan et al. 2016; Villanueva 2019).
Currently, hepatitis C infection and excessive alcohol consumption are the main risk factors for HCC. In recent years, excessive body weight or obesity has been linked to higher risk of cancer at several organ sites including the liver (Lauby-Secretan et al. 2016; Tyson and El-Serag 2011; Calle et al. 2003). Significantly higher incidence and mortality from liver cancer is observed in people with high baseline body mass index (Calle et al. 2003; Campbell et al. 2016). Besides a chronic low-grade inflammation with insulin resistance and metabolic abnormalities, roughly one third of obese patients present an accumulation of large amounts of lipids inside the liver, a disease condition called nonalcoholic fatty liver disease (NAFLD). NAFLD is a spectrum of liver diseases characterized by excessive accumulation of triglycerides in hepatocytes without heavy alcohol consumption, which range from simple steatosis to hepatic triglyceride accumulation with inflammation and liver damage (nonalcoholic steatohepatitis [NASH]) and finally hepatic fibrosis and cirrhosis (Brunt 2010; Michelotti et al. 2013; Wree et al. 2013). NAFLD has been established as an important risk factor for HCC (Brunt 2010; Michelotti et al. 2013; White et al. 2017; Kanwal et al. 2018; Anstee et al. 2019). In NAFLD patients, retrospective assessments support the association between metabolic syndrome, diabetes, and HCC (Siegel and Zhu 2009). In contrast to the success in preventing and controlling of viral hepatitis with hepatitis B vaccination and curable treatments for hepatitis C, the worldwide prevalence of obesity is continuing to rise. Accompanying the increasing prevalence of obesity, NAFLD has become the most common cause of liver dysfunction globally (Li et al. 2018). The attribution of metabolic syndrome and NAFLD to HCC is expected to increase in the future (Streba et al. 2015).
7.1.2 Intrahepatic Cholangiocarcinoma
Intrahepatic cholangiocarcinoma (ICC) is the second most common (~10%–20%) primary hepatic malignancy and arises from the bile ducts within the liver parenchyma (Tanaka et al. 2010; Tyson and El-Serag 2011; Razumilava and Gores 2014; Banales et al. 2016). Established risk factors for ICC are primary sclerosing cholangitis, choledochal cysts, fibropolycystic liver disease, hepatolithiasis, parasitic infection, and toxic exposure such as the radiologic contrast agent thorotrast (Tanaka et al. 2010; Tyson and El-Serag 2011). However, the majority of ICC patients do not present any of these risk factors. Many other less-established risk factors have been suggested such as inflammatory bowel disease, hepatitis C, hepatitis B, cirrhosis, obesity, diabetes, alcohol consumption, and tobacco use (Tanaka et al. 2010; Tyson and El-Serag 2011; Welzel et al. 2007). Surgery remains the only curative treatment option, but most ICC patients present with unresectable disease at the time of diagnosis with a median survival of less than 3 years.
7.1.3 Metastatic Liver Malignancies
Metastases are responsible for majority of cancer-related death (Lambert et al. 2017). The liver is a common site to form metastatic spread especially from cancers of the gastrointestinal tract, breast, and lung. It is often overlooked that secondary hepatic malignancies (liver metastases) account for the majority (95%) of all hepatic cancers (Disibio and French 2008). Treatment for liver metastasis is often difficult, and patients have a poor prognosis.
7.2 Carcinogenesis of Liver Cancer
7.2.1 Oncogenic Pathways in HCC
HCC tumors are highly complex and heterogenous with multiple signaling pathways contributing to hepatocarcinogenesis. Epidermal growth factor (EGF) signaling is one of the most thoroughly evaluated proliferation cascades in human HCC. EGF upregulation is an important signature for predicting late HCC recurrence after surgical resection (Hoshida et al. 2008). Insulin-like growth factor (IGF) signaling is an essential regulator of liver growth and development. Overexpression of IGF2 is frequently observed in HCC and even found in preneoplastic lesions (Breuhahn et al. 2006). Hepatocyte growth factor (HGF), the ligand for the MET receptor, is a potent mitogen for hepatocytes. Upon EGFR, IGFR, or MET activation, extracellular signals can be transduced through AKT or MAPK pathways (Johnson and Lapadat 2002). Molecules belonging to both signaling cascades (such as KRAS or AKT) have been identified as oncogenes in human cancer. Although RAS mutations are infrequent, overexpression of RAS is often found in human HCC (Calvisi et al. 2006). AKT phosphorylation has been described as a predictor of tumor recurrence after surgical resection (Nakanishi et al. 2005). Inhibition of MTOR, one of the most important molecules downstream of AKT, demonstrates antitumor function in experimental HCC models (Treiber 2009), which further increases the relevance of this pathway. The WNT/β-catenin pathway is not only involved in liver development and differentiation but also implicated in cell proliferation and metabolism (Thompson and Monga 2007). β-catenin is encoded by the CTNNB1 gene. Mutations in CTNNB1 and AXIN1 (important for β-catenin ubiquitination and subsequent degradation) are frequently found in HCC (Villanueva et al. 2007). Nuclear accumulation of β-catenin induces upregulation of genes for cell differentiation and proliferation (Boyault et al. 2007). The hedgehog pathway is involved in the embryonic liver development, and its reactivation plays a substantial role in sustaining cancer cell growth and progression in HCC (Tada et al. 2008). Chronic inflammation is closely associated with HCC. Inflammatory interleukin 6 (IL-6) signaling has been suggested to be responsible for the gender disparities of HCC incidence (Naugler et al. 2007). IL-6/STAT3 has been identified as a major pathway in maintaining stem-cell-like features in HCC (Lin et al. 2009). Substantial evidence supports the important role of NF-κB signaling in inflammation-related HCC (Elsharkawy and Mann 2007). Vascular endothelial growth factor (VEGF) and fibroblast growth factors (FGFs) have been revealed as the major drivers for angiogenesis in HCC (Imura et al. 2004). Mutation of tumor suppressor P53 has also been associated with HCC progression (Lowe et al. 2004). The oncogene MYC encodes a protein which is involved in nucleic acid metabolism. The activation of the MYC oncogene is considered to be an important mechanism of tumor evolution in HCC. Amplification of MYC can be detected in all the HCC stages and is considered an important driver for HCC disease progression (Kaposi-Novak et al. 2009). Nuclear receptors are ligand-modulated transcription factors that play diverse roles in cell differentiation, development, proliferation, and metabolism. Nuclear receptors such as farnesoid X receptor (FXR) are associated with liver cancer (Huang et al. 2015). FXR is considered to be a multifunctional tumor suppressor and tightly controls bile acids synthesis (Claudel et al. 2005). Significant reduction of FXR expression was found in human HCC. FXR−/− mice develop spontaneous HCC with disrupted bile acid metabolism as the major defect (Yang et al. 2007). Overload of bile acids due to the depletion of the FXR gene is the causative factor for induction of chronic liver inflammation, enhancement of hepatocyte proliferation, and development of liver tumors. In FXR−/− SHP−/− double knockout mice, the sharply elevated bile acid levels lead to the activation of YAP protein (Anakk et al. 2013), which is a core component of the Hippo pathway and considered as a crucial promoter of hepatocarcinogenesis (Lu et al. 2010). In addition, FXR shows anti-inflammation function. Activation of FXR inhibits NF-κB transcriptional activity through decreased DNA binding of NF-κB (Wang et al. 2008).
7.2.2 Oncogenic Pathways in Cholangiocarcinoma
Chronic inflammation and cholestasis contribute to cholangiocarcinoma through a complex process involving multiple genomic alterations and signaling pathway deregulations. KRAS and P53 mutations are commonly found in cholangiocarcinoma with relative low mutations in BRAF and EGFR (Andersen et al. 2012; Borad et al. 2014; Simbolo et al. 2014). Overlapping molecular profile between subclasses of cholangiocarcinoma and HCC has been found, indicating that these two cancer types may share a common ancestor such as hepatic progenitor cells (HPCs) (Hoshida et al. 2009; Roskams 2006; Woo et al. 2010). Alterations in the Hippo pathway components in the liver (such as YAP, SAV1, MST1/2) expand progenitor-like cells and lead to the development of both HCC and cholangiocarcinoma in animal models (Lu et al. 2010). Gain-of-function IDH mutations are often reported in cholangiocarcinoma (Saha et al. 2014; Wang et al. 2013). The expression of these IDH mutations inhibited hepatocyte differentiation and expanded HPCs in mice. Furthermore, the concurrence of IDH and KRAS mutations in mice shows pronounced oncogenic cooperation and led to the development of premalignant biliary lesions and subsequent progression to cholangiocarcinoma (Saha et al. 2014). Recent studies have suggested the emerging roles for NOTCH and WNT signaling in cholangiocarcinoma pathogenesis. The NOTCH signaling pathway plays an important role during embryonic development and is essential for liver regeneration and repair (Zender et al. 2013). NOTCH pathway deregulation has been implicated in the induction of inflammation and the development and progression of cholangiocarcinoma. In human cholangiocarcinoma, the upregulation of NOTCH1 and NOTCH4 has been commonly observed (Wu et al. 2014). Liver-specific expression of NOTCH1 intracellular domain in mice resulted in the formation of cholangiocarcinoma (Zender et al. 2013). The WNT pathway is highly activated in the tumor epithelium of human cholangiocarcinoma (Boulter et al. 2015). Tumor surrounding macrophages have been demonstrated to be responsible for this highly activated WNT signaling status (Loilome et al. 2014). Mimicking human cholangiocarcinoma, the progressive activation of WNT pathway during the course of cholangiocarcinoma has been demonstrated in animal models (Boulter et al. 2015). Furthermore, WNT singling inhibition successfully controlled tumor growth in the tumor-bearing animals (Boulter et al. 2015).
7.3 Infectious Disease and Liver Cancer
7.3.1 Chronic Viral Hepatitis and HCC
Currently chronic HBV and HCV infections still represent the leading cause for HCC. The majority of viral hepatitis-related HCC arise from cirrhosis and are closely associate with liver inflammation and tissue damage. However, a significant proportion of HBV-related HCC arise in the absence of liver inflammation, indicating that the virus directly contributes to hepatocarcinogenesis (Kew 1998). The HBV genome can randomly integrate into the host genome. Although random integration rarely leads to direct oncogene activation or inactivation of tumor suppressor genes, HBV integration contributes to the genetic instability (Robinson 1994). In addition, several HBV viral proteins have been identified to promote hepatocyte transformation including the HBV envelope and HBx protein (Twu and Schloemer 1987; Paterlini et al. 1995; Sunami et al. 2016). HBV envelope proteins induce endoplasmic reticulum stress and cause liver cancer when expressed in mice (Xu et al. 1997). HBV-DNA sequences coding for a C-terminally truncated envelope protein are frequently found integrated in HCC. This truncated envelope protein induces the activation of c-Raf-1/Erk2, Ap-1, and NF-κB pathways and increases hepatocyte proliferation (Hildt et al. 2002). The HBx protein, which is essential for initiating and maintaining HBV virus replication, has been linked to chromatin modulation. With these intrinsic carcinogenic characters, up to 20% of HBV-related HCC cases occur in the absence of cirrhosis (Chayanupatkul et al. 2017).
HCV is classified into seven genotypes. Genotypes 1b and 3 are associated with an increased risk of developing HCC (Kanwal et al. 2014; Raimondi et al. 2009). Even after virus elimination by antiviral treatment, the history of infection with HCV genotype 3 confers an increased HCC risk in patients with advanced fibrosis or cirrhosis (El-Serag et al. 2016). The reported DNA methylation of enhancers in HCV-associated HCC tumors highlights a role for HCV to influence host transcription (Okamoto et al. 2014). HCV stabilizes hypoxia-inducible factor-1α to induce de-differentiation through regulating the epithelial-to-mesenchymal transition (Wilson et al. 2012). HCV core transgenic mice show an imbalance in the oxidant/antioxidant state and develop HCC. HCV-encoded polymerase NS5B has been reported to bind the tumor suppressor protein Rb and induce its degradation through host ubiquitin (Munakata et al. 2005). Despite the many potentially oncogenic features of HCV infection, unlike HBV, the HCV-related HCC almost exclusively occurs in cirrhosis. An altered gut microbiome has been discovered in chronic HBV infection (Zhu et al. 2019; Wang et al. 2019). However, whether the gut microbiome affects disease progression of viral hepatitis and its impact on HCC development is currently unknown.
7.3.2 Liver Fluke and Cholangiocarcinoma
Liver fluke infection is a well-known risk factor for cholangiocarcinoma. Liver flukes are parasitic worms that live in the bile ducts and the liver of the infected host. Currently, liver fluke infection remains a major public health problem in East Asia and Eastern Europe. The prevalence of Opisthorchis viverrini (Southeast Asian liver fluke) infection has a strong positive correlation with the incidence of cholangiocarcinoma in Thailand and Laos (Sripa et al. 2011; Sriamporn et al. 2004), whereas such relationship cannot be found in HCC. Liver fluke-related cholangiocarcinoma is generally considered to be caused by chronic inflammation (Holzinger et al. 1999). Fluke feeding activity and migration contribute to biliary damage (Bhamarapravati et al. 1978). Fluke eggs entrapped in the periductal tissue induce granulomatous inflammation. The liver fluke can secrete or excrete metabolic products, some of which are highly immunogenic (Wongratanacheewin et al. 1988). Oxygen radicals such as nitric oxide (NO) released from activated immune cells contribute to biliary cell damage (Pinlaor et al. 2004). Furthermore, an in vitro co-culture study found that Opisthorchis viverrini induces murine fibroblasts to produce growth-promoting proteins such as transforming growth factor, which contributes to cell proliferation and tumor development (Thuwajit et al. 2006). It has been reported that liver fluke infection changes the gut and biliary microbiome (Xu et al. 2018). However, its contribution to cholangiocarcinoma development is unknown.
7.4 Overview of Gut Microbiome and Cancer
The human gastrointestinal (GI) tract is colonized with a large and immensely complex community of commensal microbials termed the gut microbiome (Donaldson et al. 2016; Gilbert et al. 2018). While the gut microbiome mostly consists of bacteria, it also contains fungi, protozoa, archaea, and viruses. It is estimated that in total there is about 1014 bacteria present in an adult’s intestine, but that number varies greatly among individuals (Sender et al. 2016). The number and type of microbes also vary dramatically from site to site within the GI tract (Donaldson et al. 2016; Human Microbiome Project 2012; Hillman et al. 2017). Largely due to the bactericidal activity of gastric acid, microbial density increases from the stomach to the distal aspect of the GI tract (Hunt et al. 2015). In healthy individuals, stomach and proximal small intestine contain only a very limited amount of microbes with a density around 101 to 103 cells per gram of lumen content, most of which are aerobes and facultative anaerobes. In sharp contrast, the microbial density in the colon can reach up to 1012 cells per gram content and predominantly present as strict anaerobes. Bacterial diversity also increase along the GI tract from proximal to distal (Hillman et al. 2017).
It is estimated that a typical person harbors 500–1000 bacterial species within the GI system (Lloyd-Price et al. 2016). Surprisingly, all the bacteria belong to only a few phyla, with the majority classified under Firmicutes and Bacteroidetes. Minor representation includes Proteobacteria, Verrucomicrobia, Actinobacteria, Fusobacteria, and Cyanobacteria (Donaldson et al. 2016; Human Microbiome Project 2012; Huse et al. 2008; Rajilic-Stojanovic and de Vos 2014). Due to the large variations in both taxonomic composition and abundance of shared taxa among healthy individuals, using a universally “core” set of microbial taxa to define a “healthy” gut microbiome is believed to be unpractical (Lloyd-Price et al. 2016). In contrast, the abundance of metabolic pathways or the “functional core” of gut microbiome seems considerably consistent across people and remains stable over time after establishment in early life (Human Microbiome Project 2012; Lloyd-Price et al. 2016; Turnbaugh and Gordon 2009; Abubucker et al. 2012). The combined gut microbiome genome contains more than five million genes and has a large capacity to provide diverse metabolic activities, some of which are essential for host biology such as production of essential vitamins and fermentation of polysaccharides indigestible by the host (Rowland et al. 2018). A healthy gut microbiome covers a core set of functions and is likely to be ecologically diverse. Conversely, decreased diversity within the gut microbiome is associated with diseases such as obesity, inflammatory bowel disease, and diabetes.
The gut microbiome has a profound influence on maintaining structure, function, and tissue homeostasis of the GI tract, development of the intestinal immune system, and defense against opportunistic pathogens (Gilbert et al. 2018; Thaiss et al. 2016; Gopalakrishnan et al. 2018a). The association between the gut microbiome and cancer development was discovered a while ago (Gopalakrishnan et al. 2018a). In recent years, various cancer-associated bacteria have been identified, and both pro- and antitumor functions of these bacteria have been alluded to. So far, the most clear and direct evidence for the contribution of intestinal bacteria to carcinogenesis was the discovery of Helicobacter pylori as the strongest known risk factor for gastric cancer (Boland et al. 2005; Wroblewski et al. 2010; Sepulveda 2013). Helicobacter pylori produces a protein called cytotoxin-associated gene A, a class I carcinogen, which can induce proteasome-mediated p53 degradation in gastric epithelial cells and promote gastric cancer formation (Hatakeyama 2014). GI cancers also have a strong link with chronic inflammatory diseases that demonstrate changes in the gut microbiome. Inflammatory bowel disease, in particular Crohn’s disease, is associated with the development of colorectal cancer (CRC) (Axelrad et al. 2016; Cipe et al. 2015; Jahani-Sherafat et al. 2018). Inflammatory bowel diseases present with an imbalance of gut microbial community or dysbiosis. Independent of inflammatory bowel disease, many bacteria such as Bacteroides fragilis, Clostridium septicum, Enterococcus faecalis, Fusobacterium spp., and Streptococcus bovis have been suggested to contribute to CRC (Gagniere et al. 2016; Purcell et al. 2017; Mirza et al. 2009; Shang and Liu 2018; Abdulamir et al. 2011). Potential carcinogenic functions of many bacterial products have been discovered such as Fusobacterium nucleatum effector adhesin A and Bacteroides fragilis metalloproteinase toxin. Both of these toxins are capable of interacting with host’s epithelial E-cadherin, disrupt the intercellular junctions, and activate β-catenin signaling which triggers cell proliferation and potentially malignant transformation (Rubinstein et al. 2013; Boleij et al. 2015).
Besides their pro-tumor functions, a number of microbial-derived products show antitumor activity (Zitvogel et al. 2016). The microbial-derived short-chain fatty acids (SCFAs) such as butyrate and propionate have been found to inhibit tumor histone deacetylases and suppress CRC and lymphoma (Scheppach et al. 1995; Hinnebusch et al. 2002; Gorres et al. 2014). Monophosphoryl lipid A (MPL) from Salmonella enterica has been used as adjuvant in the vaccine formulation for cervical carcinoma (Monie et al. 2008). Bacillus Calmette-Guerin (BCG) vaccine, a weakened form of Mycobacterium bovis, has been used as immunotherapy in patients with bladder cancer (Kawai et al. 2013).
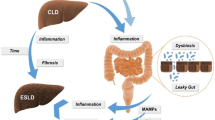
The GI tract and the liver have a close anatomical and functional relationship which is termed the “gut-liver axis.” It is important to note that the communication in the gut-liver axis is bidirectional (Fig. 7.1) (Tripathi et al. 2018; Wiest et al. 2017). The liver produces and secretes bile into the intestine which contains bile acids, immunoglobulin A, and antimicrobial molecules. The bile not only helps fat digestion and absorption but also maintains intestinal hemostasis and regulates microbial number and composition (Wiest et al. 2017; Urdaneta and Casadesus 2017). On the other hand, the intestine also influences liver function by providing nutrient-rich blood via the portal vein. The single thin layer of intestinal epithelium not only facilitates nutrient absorption but also makes it easy for small microbial components to cross and enter the blood stream. The blood supply to the liver carries both nutrients from digestion and also a large number of microbial components, metabolites, and even intact bacteria. Many of the intestinal metabolites function as signaling messengers regulating metabolic processes in the liver (Levy et al. 2016). In addition, the liver prevents harmful microbial products from entering the systemic circulation and thus serves as a critical “filter” to clear and detox microbial toxins such as lipopolysaccharide (LPS) (Tripathi et al. 2018; Wiest et al. 2017). Due to this close relationship, the liver is under great influence from the gut microbiome. Many important risk factors for liver cancer such as NAFLD, ALD, and cirrhosis commonly present with dysbiosis (Leung et al. 2016; Mokhtari et al. 2017; Da Silva et al. 2018; Sharpton et al. 2019; Rao 2009). It has been suggested that there is an association between alterations within gut microbiome and liver cancer (Zitvogel et al. 2016; Llorente and Schnabl 2015; Yu and Schwabe 2017). Importantly, preclinical animal models demonstrate that commensal intestinal bacteria play a critical role in the regulation of liver cancer development (Shalapour et al. 2017; Dapito et al. 2012; Yoshimoto et al. 2013; Singh et al. 2018; Ma et al. 2018). The relevant basic knowledge of the liver and its interaction with the gut microbiome, current findings, and proposed mechanisms of intestinal bacteria in liver tumor development will be discussed below.

The interaction between liver and the gut microbiome. The liver secretes bile into the intestine which not only helps fat digestion but also modulates the microbiome composition. In the intestine, the gut microbiome mediates many metabolic processes such as primary to secondary bile acid conversion and SCFA production. Bile acids, SCFA, and many microbial products such as LPS are absorbed and travel to the liver through portal blood circulation. Portal blood mixes with arterial blood and passes through liver sinusoids; during the process, nutrients are taken up, and microbial products are detoxicated by hepatocytes. The liver is heavily populated by immune cells, including Kupffer cells, NKT, T cells, and myeloid cells. Immune cells and other nonhepatic cells such as LSEC and HSC make up ~30% of the total cell population in normal liver
7.5 Relevant Liver and GI Features for the Gut-Liver Axis
7.5.1 Intrahepatic Circulation
The liver has a characteristic blood flow system (Fig. 7.1). About 75% of the blood supply to the liver is from the intestine venous system via the portal vein, which contains a significant amount of intestinal microbial products and metabolites (Abdel-Misih and Bloomston 2010). Arterial and portal vein blood mixes and passes through the thin-walled sinusoids which are lined by a single layer of liver sinusoidal endothelial cells (LSECs). Due to the small diameter, sinusoidal blood flow rate is low and often static which helps nutrient extraction and detoxification of harmful substances (Vollmar and Menger 2009).
7.5.2 Liver as an Immunological Organ
The liver is heavily populated by immune cells, and non-hepatocytes make up ~30% of the total cell population in normal livers (Fig. 7.1) (Racanelli and Rehermann 2006; Bogdanos et al. 2013; Heymann and Tacke 2016; Robinson et al. 2016). Macrophages are phagocytic innate immune cells and play an essential role in host defense. The liver harbors the largest population of tissue-resident macrophages, known as Kupffer cells, in the body. Kupffer cells comprise ~20% of the non-hepatocytes population and have multiple functions within the liver (Dixon et al. 2013; Toth and Thomas 1992; Bilzer et al. 2006). For example, they play an important role in tissue homeostasis, liver inflammation, and liver tumor progression. LSECs make up the lining of sinusoids and are in directly contact with mixed portal and arterial blood. LSECs act as efficient antigen presenting cells (APCs) and express MHC class I and II, CD1, MR1, and the co-stimulatory molecules CD40, CD80, and CD86 (Bogdanos et al. 2013; Crispe 2011; Knolle and Wohlleber 2016). The slow blood flow rate inside sinusoids lengthens the contact between immune cells and APCs, including LSECs, which promotes leukocyte extravasation. In mice, hepatic natural killer T (NKT) cells make up ~30% of the total lymphocyte population in the liver (Gao et al. 2009; Bandyopadhyay et al. 2016; Crosby and Kronenberg 2018). NKT cells are innate-like lymphocytes which recognize lipid antigens presented on CD1 molecules, which is expressed on various APCs including Kupffer cells and LSECs. Although the endogenous lipid ligand for NKTs are still elusive, lipid components of intestinal bacteria have been suggested to be able to activate NKT cells (Brennan et al. 2014; Wolf et al. 2015; Zajonc and Girardi 2015; An et al. 2014). Upon stimulation, NKT cells rapidly release cytokines to initiate diverse immune responses and act as a bridge between innate and adaptive immunity (Terabe and Berzofsky 2008; Nishimura et al. 2000). The role of NKT cells in acute liver inflammation, alcoholic steatohepatitis, NASH, fibrosis, liver regeneration, and tumor growth has been reported (Gao et al. 2009; Bandyopadhyay et al. 2016). MAITs are MR1 molecule-restricted lymphocytes that share several characters with NKT cells (Le Bourhis et al. 2011; Toubal et al. 2019). Interestingly, MAITs recognize and are activated by metabolites derived from bacterial vitamin B2 (riboflavin) biosynthesis and are thus affected by intestinal bacteria. MAITs are specifically enriched in the intestinal system and can make up ~50% of the hepatic lymphocyte population in humans. The functional study of MAITs has just began, and the knowledge is very limited. In the lung, the potential role of MAITs in controlling Mycobacterium tuberculosis infection has been suggested (Gold et al. 2015).
Facing the continuous exposure of microbial products and the potential challenge of microbial infection from the GI tract, the liver local immune system is skewed toward a unique tolerance stage in order to avoid reacting with non-harmful antigens but is still able to recognize pathogens (Crispe 2003; Tiegs and Lohse 2010; Horst et al. 2016). Accumulating evidence shows that alternation of the gut microbiome has profound influences on hepatic immune cells. The role of immune cells in the gut microbiome-regulated liver tumor developed will be discussed below.
7.5.3 Pattern Recognition Receptors
One method by which the host sense the presence of microbes is through pattern recognition receptors (PRRs) which recognize pathogen-associated molecular patterns (PAMPs). PAMPs are various microbial-specific molecules including bacterial carbohydrates (such as LPS), bacterial or viral nucleic acids, bacterial peptides (such as flagellin), peptidoglycans, lipoteichoic acid, and fungal glucans (Zitvogel et al. 2016; Chu and Mazmanian 2013; Takeuchi and Akira 2010; Mogensen 2009). Based on localization, PRRs can be grouped into membrane PRRs including Toll-like receptors (TLRs) and C-type lectin receptors and cytoplasmic PRRs including NOD-like receptors and RIG-I-like receptors. PRRs recognize specific PAMPs and trigger anti-pathogenic responses through different signaling pathways. TLRs, the most well-studied PRRs, contain ten members. TLR1, 2, 4, 5, 6, and 10 are expressed on the cell membrane, while TLR3, 7, 8, and 9 are found in the endosomal compartment (Janssens and Beyaert 2003). TLR4 is a major component of the receptor complex recognizing LPS (Park and Lee 2013). TLR5 recognizes flagellin (Andersen-Nissen et al. 2007). TLR2 forms homodimers or heterodimers with TLR1, 6, and 10 to recognize protozoa, bacteria, fungi, and viruses (Oliveira-Nascimento et al. 2012). The intracellular TLRs (TLR3, 7, 8, and 9) sense nucleic acids. TLR3 recognizes double-stranded RNA and the synthetic analog polyriboinosinic polyribocytidylic acid (poly(I:C)) (Matsumoto and Seya 2008). TLR9 recognizes unmethylated CpG motifs of DNA (Ramirez-Ortiz et al. 2008). After stimulation most TLRs induces MyD88-dependent downstream signaling often involving the NF-κb pathway to trigger various cytokine production (such as interferons) and co-stimulatory molecule expression (Kawasaki and Kawai 2014; Bagchi et al. 2007; Kawai and Akira 2007). TLRs are widely expressed in liver cells (Mencin et al. 2009; Seki and Brenner 2008). Hepatocytes and biliary epithelium express mRNA for all TLRs. LSECs constitutively express TLR4. Kupffer and hepatic stellate cells express functional TLR2 and TLR4 and produce proinflammatory cytokines upon stimulation with TLR2/4 ligands. Intrahepatic T cells and NK cells are rich in TLR1, 2, 4, 5, and 9. Although the expression and the functional role of TLRs in the liver has not been fully delineated, TLRs have been found to play a critical role in liver tissue homeostasis and various pathologic conditions including acute liver failure, ischemia-reperfusion injury, viral hepatitis, ALD, liver regeneration, fibrosis, and liver cancer (Chen and Sun 2011; Schwabe et al. 2006; Zhang and Lu 2015; Petrasek et al. 2010; Yang and Seki 2012).
7.5.4 Intestinal Barrier
The intestinal lumen microbes, especially the large amount of colon bacteria, pose a continuous threat to the host. In order to deal with this threat, a multilayer barrier has been developed to retain intestinal microbes inside the lumen (Bischoff et al. 2014; Groschwitz and Hogan 2009). The mucus layer is the first defense line separating the gut microbiome from the host, which primarily composes of a thick gel-like polysaccharide called mucin secreted from goblet cells. The colon has two layers of mucus of which the inner layer is impermeable to the luminal bacteria and has protective function. Below the mucous layer lies the intestinal epithelial layer, which is organized in crypt and villus structure to increase surface area. The intestinal epithelial cells form an extremely close lining through intercellular connections with tight and adherens junctions (Peterson and Artis 2014). Besides the physical barrier, an immune barrier exists in the lamina propria which contains gut-associated lymphoid tissue, IgA-producing plasma cells, resident macrophages, neutrophils, dendritic cells, effector cells, and regulatory T cells (Tregs) (Turner 2009). Macrophages in the intestinal lamina propria are highly phagocytic and responsible for clearing the “leaked” bacteria (Smith et al. 2011). Intestinal dendritic cells (DCs) sample lumen microbes by extending projections beyond the epithelial layer or via specialized microfold cells (Lelouard et al. 2012; Chieppa et al. 2006). Instead of immediate killing, DCs can hold living bacteria and transport them to mesenteric lymph nodes (MLN) and subsequently present microbial antigens to the immune system (Macpherson and Smith 2006). The primed local immune system can elicit quick responses against a microbial invasion if ever there is barrier dysfunction. The lamina propria also contains regulatory T cells whose development is under great influence from the intestinal bacteria (Zeng and Chi 2015; Smith et al. 2013). The presence of regulatory T cells is critical to limit unwanted inflammatory responses and avoid tissue damage.
7.5.5 Bacterial Translocation
The impairment of barrier function increases intestinal permeabilization which promotes paracellular transportation of microbial products such as LPS and even intact bacteria under severe conditions. The movement of bacteria across the intestinal barrier is termed bacterial translocation. Bacterial translocation is influenced by several factors including intestinal bacterial overgrowth, physical barrier impairment, and immune system functional status (Brenchley and Douek 2012). Rodent studies show that intestinal epithelia cells can uptake and transport latex particles similar in size to E. coli, suggesting that there is a continuous low-level trafficking of bacteria across the intestinal epithelial layer (Howe et al. 2014; Hodges et al. 1995; LeFevre et al. 1978). Most of the crossed microbes will be immediately destroyed by the intestinal phagocytes such as macrophages. Other bacteria will be taken up by DCs and transported to MLNs for training of the adaptive immune system. Clinical meaningful bacterial translocation often requires intestinal bacterial overgrowth and rarely occurs in its absence. Under bacteria overgrowth conditions and physical barrier damage, more bacteria enter the intestinal tissue and are subsequently carried to MLNs by immune cells. MLNs are often the first site to detect live translocated bacteria (Berg 1995). If there is a sufficient functional immune system, the translocated bacteria will be localized and controlled. In an immunocompromised state, uncontrolled bacteria will spread via the blood or lymphoid circulation (Berg 1995). Inside the intestinal blood stream, the translocated bacteria can move to the liver via the portal vein and even progress further to the systemic circulation. The uncontrolled bacteria inside MLNs can pass through the lymphoid vessels and enter the systemic circulation via the thoracic duct. Opposite to the low oxygen tension in the intestinal lumen, especially inside the colon, tissue and the blood stream contain relative high oxygen levels which decrease the survival of anaerobic bacteria.
Increased intestinal permeability and bacteria translocation are commonly observed in chronic liver diseases and contribute to hepatic inflammation. Consumption of high fat-containing diets are associated with increased intestinal permeabilization and elevated LPS levels in portal blood (Yoshimoto et al. 2013; Moreira et al. 2012). The critical role of elevated LPS in low-grade systemic inflammation, insulin resistance, and metabolic syndrome has been proposed. Importantly, the elevated LPS has been connected to liver carcinogenesis in animal studies (Dapito et al. 2012). The significance of bacterial translocation in liver tumors is still not clear. However, recent reports show that bacterial 16s rRNA can be detected from pancreatic cancer tumor tissue and metastatic liver tissue (Pushalkar et al. 2018; Sethi et al. 2018). Importantly, bacterial taxa composition of metastatic liver tissue can also be influenced by oral antibiotic treatment. This finding suggests that translocated bacteria can directly interact with the liver tumor environment. Its role in liver cancer development needs to be investigated further.
7.6 Gut Microbiome and Liver Cancer-Associated Conditions
Intestinal bacterial overgrowth and dysbiosis are commonly seen in risk factors for liver cancer such as obesity, NAFLD, ALD, and cirrhosis. The current knowledge of the gut microbiome in these conditions will be discussed below.
7.6.1 Obesity
Intestinal microbiomes play a critical role in regulation of energy extraction from food and in part affect obesity (Krajmalnik-Brown et al. 2012). The link between the gut microbiome and obesity was initially suggested from studies using germ-free mice, which are raised in sterile conditions and free of microorganisms. Compared to regular specific pathogen-free (SPF) mice, germ-free mice have less body fat content even though they consume more food (Backhed et al. 2004). Transferring fecal bacteria from SPF mice to germ-free mice causes a quick increase in body fat content without any change in food consumption (Ridaura et al. 2013). Importantly, germ-free condition provides a protective function against diet-induced obesity (Ley et al. 2005; Turnbaugh et al. 2006, 2008). The gut microbiome can promote energy intake through several mechanisms such as breakdown of plant polysaccharides and complex carbohydrates which normally cannot be digested by the host (Flint et al. 2012). As expected, lower caloric release from dietary plant polysaccharides was observed in germ-free mice (Turnbaugh et al. 2008). Interestingly, host metabolic processes such as energy deposition in adipocytes, hepatic fatty acid oxidation, de novo fatty acid biosynthesis, and glycogen utilization are affected by germ-free conditions and favor catabolism (Backhed et al. 2007). These studies demonstrate that the gut microbiome not only affects energy uptake from food but also influences energy expenditure and storage.
The composition of the gut microbiome has been suggested to be important in obesity development. Increase of Firmicutes especially some Clostridium clusters is involved in harvesting energy from diet (Clarke et al. 2012). Genetically obese (ob/ob) mice have been reported to contain higher proportion of intestinal Firmicutes and parallel enrichment of microbial genes for polysaccharide degradation compared to their lean siblings (Ley et al. 2005). Fecal transplantation studies demonstrate that germ-free mice who receive microbiota from obese humans develop higher adiposity compared to controls (Ridaura et al. 2013). Antibiotics, especially those with broad spectrum, affect intestinal microbiome composition. Early-life usage of penicillin causes a long-lasting effect on mouse body composition including increased fat mass and hepatic expression of adipogenesis genes (Cho et al. 2012). This supports the hypothesis that antibiotic use maybe contributing to the obesity epidemic. In humans, low fecal bacterial diversity has been found to associate with high adiposity and dyslipidemia (Le Chatelier et al. 2013). High Firmicutes and low Bacteroidetes have been reported in obese people (Koliada et al. 2017). Interestingly, in obese volunteers who lost body weight by consuming a low fat and carbohydrate diet for a year showed a reversal of the Firmicutes and Bacteroidetes populations in the colon (Wu et al. 2011). Of note the reduction of Firmicutes/Bacteroidetes ratio is not always present in obese people likely due to large interpersonal variations and the large influence diets have on the intestinal bacterial community (Singh et al. 2017). Probiotics are live microorganisms and can confer a health benefit to the host. Animal research suggests that administration of various lactobacillus may reduce weight gain in response to a high-fat diet (Kobyliak et al. 2016). However, in humans the data is less consistent.
7.6.2 Nonalcoholic Fatty Liver Disease
NAFLD is an import high-risk factor for HCC. Excessive ROS production, inflammatory cytokines, endoplasmic reticulum (ER) stress, circadian dysregulation, and immune cells have been suggested to contribute to the NAFLD-promoted hepatocarcinogenesis. In a lipid-rich environment, excessive ROS causes lipid peroxidation and generation of highly reactive aldehydic derivatives including 4-HNE and malondialdehyde (MDA), which subsequently causes DNA damage and promotes hepatocyte malignant transformation. Increased ROS production in NAFLD liver and its contribution to disease progression from NASH to HCC have been described in animal models (Kathirvel et al. 2010; Sutti et al. 2014; Gandhi et al. 2015). NAFLD presents an increase of inflammatory cytokines including TNF-α and IL-6 (Dowman et al. 2010). Both TNF-α and IL-6 have been demonstrated to play a critical role in obesity-/NAFLD-enhanced HCC through enhancing cell proliferation and preventing apoptosis of hepatocytes in mice (Park et al. 2010). Hepatic ER stress is common in NAFLD and can be observed in NAFLD animal models and NASH patients (Puri et al. 2008). Its critical role in promoting NAFLD to HCC has been demonstrated in HFD-fed MUP-uPA mice through increasing macrophage TNF production and the subsequent activation of TNFR1-IKKβ-NF-κB pathway in the HCC progenitor cells (Nakagawa et al. 2014). Importantly, the HFD-fed MUP-uPA mice develop spontaneous HCC even without carcinogen treatment, which mimics the clinical disease progression from NASH to HCC. Circadian dysregulation has been demonstrated to cause dysfunction of hepatic metabolic pathways such as in mice with jet lag (Adamovich et al. 2014). Its contribution to NASH and HCC has been suggested (Kettner et al. 2016). The liver of jet-lag mice shows a genome-wide deregulation of gene expression, and a global metabolic disruption with cholesterol, bile acid, and xenobiotic metabolism are the most affected pathways. Disrupting the hepatic metabolic pathways has been shown to promote HCC in the context of NAFLD; however, the underlying mechanisms are complex. Ablation of farnesoid X receptor (FXR), one of the key regulators of bile acid metabolic pathway, increased liver bile acids and enhanced the tumor-promoting effect of jet lag in NAFLD-HCC, while the opposite effect was found after deletion of constitutive androstane receptor (CAR), a critical modulator of xenobiotic and endobiotic metabolism (Kettner et al. 2016). The liver is rich in various immune cells. The contribution of different immune cells in NAFLD-HCC progression has been reported. An increase of hepatic NKT cells has been reported to promote steatosis through secreting LIGHT (TFNSF14), a ligand for lymphotoxin β receptor (LTβR), which acts LTβR on hepatocyte and causes enhanced lipid uptake (Wolf et al. 2014). In addition, LIGHT also activates NF-κB signaling in hepatocytes and promotes malignant transformation. The increase of IL-17-producing Th17 cells has been found in both NAFLD animal models and NASH patients (Paquissi 2016). Increased IL-17 acts on IL17RA-expressing myeloid cells and leads to release of FFAs from white adipose tissues, which promotes NASH progression and HCC formation (Gomes et al. 2016). Blocking Th17 cells decreases NASH and delays HCC (Gomes et al. 2016).
Emerging evidence suggests that the gut microbiome is an important environmental factor that contributes to NAFLD development (Leung et al. 2016; Mokhtari et al. 2017; Da Silva et al. 2018; Sharpton et al. 2019). Germ-free mice fed with high-fat diet are resistant to hepatic steatosis and dyslipidemia (Rabot et al. 2010; Cani et al. 2008). NAFLD is transmissible to germ-free mice by fecal microbial transplantation, and two bacterial strains Barnesiella intestinihominis and Lachnospiraceae have been positively associated with the development of metabolic features (Le Roy et al. 2013). NASH patients display frequent intestinal bacterial overgrowth (Augustyn et al. 2019). In humans, the association between dysbiotic environment and NAFLD has been discovered (Schnabl and Brenner 2014), and many NAFLD disease-associated bacteria have been reported. Children with NAFLD have been found to display higher presentations of Gammaproteobacteria and Epsilonproteobacteria than heathy lean and obese children (Michail et al. 2015). Increased Proteobacteria has also been observed in NASH patients compared to obese individuals (Zhu et al. 2013). However, so far, no single bacterial species has been identified to mechanistically associate with the development of fatty liver. In addition, some of the correlation studies yield controversial results. Lower percentage of Bacteroidetes was found in NASH patients compared to healthy controls (Mouzaki et al. 2013). In contrast, higher Bacteroidetes has also been linked with NASH patients (Boursier et al. 2016). In a different report, no change in Bacteroidetes was found when comparing NASH with healthy controls (Wong et al. 2013). The discrepancy is likely due to cofounding factors such as diet which plays a more important role in shaping the microbiome than genetic factors.
Several mechanisms have been suggested for the effects the gut microbiome has on NAFLD progression including regulation of intestinal protein expression, intestinal barrier breakdown, inflammatory responses, and changes in metabolites. Dysbiosis is linked with reduced synthesis and secretion of fasting-induced adipose factor (FIAF) in enterocytes, which leads to increased uptake of fatty acids in the liver and adipose tissue and ultimately favors hepatic steatosis and expansion of adipose tissue (Backhed et al. 2004; Mandard et al. 2006). NAFLD has been suggested to be associated with increased intestinal permeability. A meta-analysis comprehensively assessing the association between intestinal permeability and risk of developing NAFLD has been performed (Luther et al. 2015). Indeed, NAFLD patients had enhanced intestinal permeability (Luther et al. 2015; Miele et al. 2009). The underlying mechanism is still not clear, but bacteria involvement has been suggested. Bacterial toxic metabolites such as acetaldehyde and ethanol are associated with gut permeability. With the increased intestinal permeability and the presence of dysbiosis, the liver is exposed to more bacterial products via portal blood, which will be recognized by liver PRRs such as TLRs and leads to the production of inflammatory cytokines. A positive correlation between plasma inflammatory cytokines and blood LPS has been found (Ceccarelli et al. 2015). In addition, plasma inflammatory cytokines have been reported to be negatively correlated with intestinal Bifidobacteria count (Okada et al. 2009; Cani et al. 2007). High-fructose diet is considered to be a significant risk factor for NAFLD (Lim et al. 2010). Chronic intake of fructose is associated with bacterial overgrowth and an increase in blood LPS (Vos and McClain 2009). Fat consumption promotes production of chylomicrons, which facilitate the translocation of LPS toward other organs. All these findings suggest a link between dysbiosis, microbial products, and the inflammatory component of NAFLD.
Choline deficiency is related to NAFLD pathogenesis (Corbin and Zeisel 2012). As a methyl group donor, choline contributes to the synthesis of phosphatidylcholine which is required for the synthesis and secretion of very-low-density lipoprotein (VLDL) which transports lipids from the liver to the peripheral organs (Yao and Vance 1988). Although it can be synthesized in the body, choline is considered an essential nutrient, and dietary intake is required. Reduction of choline bioavailability is associated with increased ROS, hepatic lipid accumulation, and reduced hepatic VLDL (Zhu et al. 2014). In humans, rigorously controlled choline deficiency diet leads to hepatic lipid accumulation (Spencer et al. 2011). Recently, it was discovered that intestinal bacteria can convert dietary choline to a variety of metabolites such as trimethylamine, thus reducing choline bioavailability (Romano et al. 2015). Several choline-metabolizing bacteria have been identified, and a low level of colonization of trimethylamine-producing bacteria species can induce significant reduction of host choline levels (Romano et al. 2015). In mice, high-fat diet increases choline-metabolizing microbes and leads to the development of hepatic steatosis (Boutagy et al. 2015). Interestingly, in the liver, trimethylamine can be converted to trimethylamine-N-oxide which is associated with atherosclerosis and cardiovascular disease (Janeiro et al. 2018). The association between trimethylamine-N-oxide and hepatic lipid metabolism in addition with the microbe-host interactions has been suggested, and its role in NALD needs further investigation (Chen et al. 2016a).
7.6.3 Alcoholic Liver Disease
Alcoholic liver disease is the most prevalent chronic liver disease worldwide and accounts for ~30% of HCC cases and HCC-specific death (Ganne-Carrie and Nahon 2019). Alcohol consumption is an independent risk factor for HCC and has been associated with a high risk of serval other malignancies starting at a dose as low as 10 g/1 unit/day (Testino et al. 2014; European Association for the Study of the Liver 2018). Alcohol is classified as a group 1 carcinogen. Although not fully understood, several mechanisms have been found to contribute to the alcohol-induced hepatocarcinogenesis. Alcohol is mainly metabolized in hepatocyte cytoplasm to acetaldehyde, which is subsequently oxidized to acetate in the mitochondria (Lieber 2005). After consumption of a large amount of ethanol, cytochrome P450 2E1 (CYP2E1) also contributes to the metabolization of alcohol to acetaldehyde (Lieber and DeCarli 1968). Acetaldehyde has been shown to be a carcinogen in animal studies (Seitz and Homann 2007). Acetaldehyde interacts with DNA and proteins to form adducts which plays an important role in carcinogenesis. The formation of adducts with O6-methylguanine methyltransferase causes DNA repair system dysfunction (Collier et al. 1996). The CYP2E1-dpendent alcohol metabolization process generates various ROS, such as hydroxyethyl, superoxide anion, and hydroxyl radicals (Haorah et al. 2008). The increased ROS leads to the generation of lipid peroxidation products, such as malondialdehyde and 4-hydroxy-2-nonenal, and causes DNA damage. 4-hydroxy-2-nonenal has been found to cause a mutation at codon 249 of the p53 gene which is commonly found in HCC (Hu et al. 2002). In addition to the mutagenic effects on DNA, ROS can act as an important mediator of tumor angiogenesis and metastasis. It has been shown that alcohol-induced ROS can activate NF-kB signaling and upregulate VEGF and MCP-1 (Liu et al. 2016). Aberrant DNA methylation and protein methylation are involved in HCC development. It has been reported that alcohol inhibits the synthesis of S-adenosyl-L-methionine (SAMe), which is a universal methyl group donor. The generation of SAMe is mediated by the enzyme methionine adenosyltransferase (MAT). MAT1A knockout mice develop SAMe deficiency, fatty liver, and HCC (Santamaria et al. 2006). Furthermore, decreased hepatic MAT activity and MAT1A gene expression have been found in ALD patients (Tsukamoto and Lu 2001). ALD presents hepatic activation of innate immunity and increased proinflammatory cytokines (Kasztelan-Szczerbinska et al. 2015; Kawaratani et al. 2017). The involvement of the altered immune system in ALD-induced HCC needs further investigation.
ALD is mainly due to the accumulation of acetaldehyde, a toxic metabolite of ethanol, in hepatocytes which causes liver inflammation and fibrosis (Setshedi et al. 2010). Recently, the gut microbiome has been found to contribute to ALD development, and a growing body of evidences suggest that LPS is closely associated with ALD (Rao 2009). Chronic ethanol consumption leads to bacterial overgrowth and dramatic changes within intestinal bacterial composition (Malaguarnera et al. 2014). Specifically, there is an increase in gram-negative bacteria such as Proteobacteria which are the main source of LPS (Purohit et al. 2008). ALD patients display increased intestinal permeability (Zhou and Zhong 2017). Although ethanol can directly disrupt the intestinal barrier, its concentration is relatively low inside the intestine. Intestinal bacteria can metabolize ethanol to produce acetaldehyde, and a significant amount of evidences indicates that microbial-derived acetaldehyde plays a crucial role in the disruption of the intestinal barrier function in ALD (Ferrier et al. 2006). Acetaldehyde-induced disruption of tight and adherens junctions was validated in human colonic mucosa (Basuroy et al. 2005). The increased intestinal permeability and more abundant gram-negative bacteria contribute to increased absorption of LPS. Numerous studies demonstrate that ALD patients have elevated plasma LPS compared to healthy individuals (Fujimoto et al. 2000; Bala et al. 2014; Fukui et al. 1991). LPS itself fails to mimic ethanol-induced steatosis or hepatitis. However, LPS shows a synergistic effect with ethanol to exacerbate liver damage (Pennington et al. 1997). The mechanism involves multiple factors inducing downregulation of IL-10-mediated protection, ROS production, and adrenergic stimulation (Hill et al. 2000). TLR4 knockout studies suggest that TLR4 plays a critical role in LPS-promoted live damage (Soares et al. 2010). LPS-/TLR4-mediated stimulation of different liver cells including Kupffer cells, LSECs, stellate cells, neutrophils, and hepatocytes induces secretion of proinflammatory cytokines, chemokines, and ROS which subsequently leads to liver damage and inflammation (Duryee et al. 2004; Quiroz et al. 2001; Adachi et al. 1994). In a more recent report, cytolysin, an exotoxin secreted by Enterococcus faecalis, has been shown to contribute to hepatocyte death and liver injury in ALD (Duan et al. 2019). ALD patients have increased level of Enterococcus faecalis. Importantly, the presence of cytolysin-positive Enterococcus faecalis correlates with the severity of liver disease. Using humanized mice colonized with fecal bacteria from ALD patients, it has been demonstrated that targeting cytolysin-positive Enterococcus faecalis correlates with bacteriophages and attenuates ethanol-induced liver disease. This study offers a novel therapeutic approach for ALD through precisely editing the gut microbiome.
7.6.4 Cirrhosis
Cirrhosis represents the final stage of liver fibrosis and is characterized by distortion of liver parenchyma associated with fibrous septa and nodule formation as well as alterations in blood flow. Bacterial translocation is often observed in cirrhosis, and cirrhotic patients have increased susceptibility to bacterial infections, most commonly spontaneous bacterial peritonitis (Alexopoulou et al. 2017; Bonnel et al. 2011; Jalan et al. 2014). About 10% of cirrhotic patients without selective intestinal decontamination show MLN bacterial translocation (Cirera et al. 2001). In addition, a positive correlation between cirrhosis disease severity and bacterial translocation has been reported (Alexopoulou et al. 2017; Cirera et al. 2001). Consistent with the correlation results, experimental cirrhotic animals show an increase in intestinal permeability, and ~40% of cirrhotic rats with ascites have MLN bacteria translocation (Giannelli et al. 2014). Bacteria strains isolated from MLN have been shown to be genetically identical to strains causing spontaneous bacterial peritonitis in the same animal supporting the process of bacterial translocation to infection (Bert et al. 2010).
Several pathologic changes in cirrhosis promote the bacterial translocation including bacterial overgrowth, intestinal barrier dysfunction, and impaired immune function (Pijls et al. 2013). Bacterial overgrowth and dysbiosis are often present in cirrhotic patients (Fukui 2017). Bacterial overgrowth can even be found in the proximal small intestine likely due to a shift toward to alkaline gastric secretions (Chen et al. 2016b). In cirrhosis, a marked decrease in intestinal luminal concentration of bile acids and increased deconjugation by bacteria have been observed (Ridlon et al. 2013). In addition to its role in digestion, bile acids limit microflora proliferation and contribute to maintaining the integrity of the small intestine. Obstruction of bile flow in humans or rodents causes bacterial overgrowth and mucosal injury followed by bacterial translocation (Inagaki et al. 2006). Oral administration of conjugated bile acids in cirrhotic rats results in a reduction of intestinal bacterial overgrowth, bacteria translocation, and endotoxemia (Lorenzo-Zuniga et al. 2003). The impaired intestinal barrier function in cirrhosis is often associated with portal hypertension-related structural and functional alterations including vascular congestion, edema, widened intracellular spaces in the intestinal wall, and functional abnormalities such as reduced small bowel motility (Kalaitzakis 2014). Advanced liver diseases often display impaired chemotaxis, phagocytosis, and intracellular killing by polymorphonuclear leukocytes and monocytes (Andrews and Sullivan 2003). Cirrhosis is accompanied by impaired reticuloendothelial system (RES) which is the main defensive system against bacteremia. Most of the RES activity is located in the liver where Kupffer cells are the major component. Portosystemic shunting that bypasses the liver (escaping the action of RES) and impaired Kupffer cell phagocytic activity leads to not only the failure to clear bacteria but also failure to clear bacterial products such as endotoxins and cytokines (Moller et al. 2014; Pinzone et al. 2012).
7.6.5 Autoimmune Hepatitis
Autoimmune hepatitis (AIH) is an immunologic mediated, chronic, and progressive inflammatory liver disease of uncertain cause. Without appropriate treatment, AIH can lead to cirrhosis and HCC (Teufel et al. 2009). Genetics have been implicated as susceptibility factors for AIH. Recent evidence suggests that there is an association between the gut microbiome and AIH (Lin et al. 2015). AIH patients are reported to have increased intestinal permeability, dysbiosis, and bacterial translocation, which correlates with disease severity (Lin et al. 2015; Cai et al. 2017; Czaja 2016).
The intravenous injection of the plant lectin concanavalin A (Con A) is a well-established hepatitis model for investigating T cells and macrophage-dependent liver injury in mice (Wang et al. 2012). The model mimics pathological features of AIH patients and is considered the best experimental model for AIH research so far. Recently, several studies have shown that the gut microbiome has a profound impact on Con A liver injury (Celaj et al. 2014; Chen et al. 2014). Severity of Con A liver injury varies greatly among genetically identical mice raised in different environments harboring distinct microbiota. BALB/c mice from TAC, NCI, and JAX show clear vendor-specific Con A liver damage. Germ-free and co-housing studies show that manipulating the intestinal flora alters susceptibility to Con A liver injury (Celaj et al. 2014). In addition, administration of pathogenic bacteria such as Salmonella and Streptococcus exacerbates Con A liver injury (Chen et al. 2014). In contrast, depletion of gut gram-negative bacteria alleviated Con A liver injury. Several mechanisms have been proposed, such as the ability of the gut microbiome to regulate the sensitivity to Fas-induced liver injury from TLR/MyD88 signaling (Celaj et al. 2014). Other proposed mechanism suggests that the presence of pathogenic bacteria stimulates dendritic cells, enhances NKT cell cytotoxicity, and exacerbates liver damage (Chen et al. 2014).
7.7 Gut Microbiome Regulates Liver Cancer
Emerging evidences suggest that there is an association between altered intestinal bacteria and the presence of HCC following advances in bacterial sequencing and profiling techniques (Yu and Schwabe 2017; Mima and Baba 2019; Gupta et al. 2019). Enriched Actinobacteria was observed in early HCC compared to cirrhosis (Ren et al. 2019). Increased Bacteroides and decreased Bifidobacterium were also found in HCC patients (Ponziani et al. 2019). Compared to non-HCC cirrhotic patients, HCC patients presented with high levels of fecal E. coli (Grat et al. 2016). Building on this evidence and the close association between dysbiosis and HCC risk factors, the gut microbiome has been suggested to play a critical role in liver cancer development.
Recently, direct evidence showing that intestinal bacteria contributes to liver cancer has emerged using preclinical animal models mainly focusing on HCC (Shalapour et al. 2017; Dapito et al. 2012; Yoshimoto et al. 2013; Singh et al. 2018; Ma et al. 2018; Sethi et al. 2018; Loo et al. 2017). A profound influence of the gut microbiome on liver cancer has been demonstrated in gut bacteria-depleted mice using antibiotic cocktails or germ-free mice. Although through different mechanisms, a consistent liver tumor-promoting effect of the gut microbiome has been observed from several research groups using various liver tumor models (Fig. 7.2 and Table 7.1). The current findings are discussed below.

Mechanisms by which the gut microbiome regulates liver cancer. (1) Lipopolysaccharides (LPS) produced by gram-negative bacteria activate TLR4 signaling in hepatocytes and hepatic stellate cells (HSC), which promotes cell proliferation, survival, and tumor formation. (2) Gut bacterial-metabolized secondary bile acid deoxycholic acid (DCA) induces senescence in HSC, and the senescence-associated secretory phenotype (SASP) contributes to HCC. (3) Secondary to primary bile acids ratio, which is controlled by gut bacteria, regulates chemokine CXCL16 expression on liver sinusoidal endothelial cells (LSEC) and thus modulates the level of hepatic NKT cells which express CXCR6. NKT cells control liver tumor development. (4) Short-chain fatty acids (SCFA), which are produced by bacteria through fermentation of dietary fibers, together with bile acids induce cholestasis, hepatocyte death, fibrosis, and liver inflammation which leads to HCC formation
7.7.1 Lipopolysaccharides
Lipopolysaccharides, also known as endotoxins or lipoglycans, are the main components of the outer membrane of gram-negative bacteria (Alexander and Rietschel 2001). LPS are a group of large molecules consisting of three parts: O polysaccharide, core oligosaccharide, and lipid A. Intestinal epithelial cells internalize LPS and transport them to the Golgi complex where LPS bind and form complex with newly generated chylomicrons (Ghoshal et al. 2009). LPS have a high affinity for chylomicrons, and chylomicron formation promotes LPS transportation. Under healthy condition, most intestinal-absorbed LPS are present on chylomicron remnants within the blood. Intestinal barrier damage enhances unbound LPS absorption through paracellular movement. LPS is recognized by pattern recognition receptor TLR4 in association with protein partners such as MD2 and CD14, which signal through two major pathways: MyD88/ NF-κB and TRIF/IRF3 (Park and Lee 2013). LPS can induce a massive production of proinflammatory cytokines and even septic shock in extreme conditions (Yamamoto et al. 2011). TLR4 is expressed by hepatocytes and various cell types in the liver (Mencin et al. 2009; Chen and Sun 2011). Indeed, the liver is a critical organ responsible for clearing LPS from blood circulation. Due to its continuous exposure, the liver immune system is tolerant to low levels of LPS. The exact mechanism of LPS tolerance is still not fully understood but the immunomodulatory cytokine IL-10 and other molecules such as SHIP-1, A20, and IRAK-M are believed to be critical (Bagchi et al. 2007; Xiong and Medvedev 2011).
Elevated blood LPS is present in many conditions considered to be high risk for liver cancer. Increased serum LPS has been found in NAFLD patients (Miele et al. 2009; Harte et al. 2010). Excessive alcohol consumption is correlated with gram-negative bacteria overgrowth and high LPS in the circulation (Fujimoto et al. 2000; Bala et al. 2014; Fukui et al. 1991). Consumption of high fat-containing diets is associated with increased intestinal permeabilization with elevated LPS level in the portal blood (Pendyala et al. 2012). Importantly, the TLR4/MyD88 pathway, a major pathway downstream of LPS signaling, has been linked to carcinogenesis in colorectal cancer (Wang et al. 2010, 2018). In addition, LPS promotes liver metastasis of colorectal cancer by directly acting on TLR4 expressed in tumor cells (Hsu et al. 2011). All these findings suggest LPS as a potential link between the gut microbiome and liver cancer.
The research group led by Robert Schwabe was the first to demonstrate the crucial role of LPS in liver carcinogenesis using animal models (Fig. 7.2) (Dapito et al. 2012). In their studies, spontaneous mouse HCC was induced by early-life exposure to the chemical carcinogen diethylnitrosamine (DEN) followed by chronic treatment with the hepatotoxin, carbon tetrachloride (CCL4) (Dapito et al. 2012). The model demonstrates a pattern of chronic liver inflammation, fibrogenesis, and increased blood LPS level which mimics features of the microenvironment from which the majority of human HCC tumors arise. To test the role of the gut microbiome in DEN-CCL4-induced HCC, a TLR4 mouse strain carrying a nonfunctional mutant TLR4 was chosen. Interestingly, a TLR4 functional deficiency causes a robust reduction in both HCC tumor number and size. The finding was recaptured in gut-sterilized mice treated with an oral antibiotic cocktail and in germ-free mice, which proves that the commensal gut microbiome is responsible. Furthermore, prolonged low nontoxic LPS exposure increases HCC size and number which directly confirms that LPS promotes HCC.
The liver is rich in immune cells and is considered a lymphoid organ (Racanelli and Rehermann 2006). TLR4 is expressed on different cell types within the liver (Mencin et al. 2009; Chen and Sun 2011; Soares et al. 2010). Besides hepatocytes, liver non-parenchymal cells such as hepatic stellate cells and various immune cells, particularly Kupffer cells, express TLR4 and can respond to LPS. The contribution of liver immune cells or parenchyma cells to gut microbiome-enhanced HCC was investigated using bone marrow chimeric study. Notably, the chimeric study was combined with Kupffer cell depletion to exclude the potential contribution of liver resident macrophages. Using this chimeric protocol, the study clearly demonstrated that the non-Kupffer resident liver cells mediate the gut microbiome-enhanced HCC.
Another interesting finding of the Dapito et al. paper was that the gut microbiome does not affect the initiation of carcinogenesis in the DEN-CCL4 HCC model (Dapito et al. 2012). Microarray analysis showed no difference in expression of cancer stem cell markers, suggesting the regulation of hepatocarcinogenesis is not likely from progenitor cells. Unexpectedly, the time frame seems to be critical for the gut microbiome’s influence on hepatocarcinogenesis. Early sterilization followed by antibiotic treatment withdrawal does not affect hepatocarcinogenesis. In contrast, gut sterilization at the time when tumors start to appear greatly reduces tumor number and size. Cell proliferation was investigated, and it was demonstrated that TLR4 functional deficiency strongly reduced cell proliferation. Consistent with the chimeric results, NF-κB activation, a surrogate for LPS response, was mainly observed in the hepatic stellate cells and a large percentage of hepatocytes in DEN-CCL4 mice. Furthermore, epiregulin, a mitogen which can stimulate cell proliferation, was found to be significantly affected by TLR4 status and gut sterilization. In vivo LPS challenge significantly upregulates epiregulin in the hepatic stellate cells. As expected, epiregulin-deficiency inhibits DEN-CCl4-induced hepatocarcinogenesis. Besides the altered cell proliferation, a significant increase in cleaved-caspase3 positive cells within nontumor hepatocytes was observed in both gut-sterilized and TLR4 mutant mice. Importantly, hepatocyte apoptosis is inversely correlated with tumor number and size. Together, the data supports the mechanism that LPS act on hepatic stellate cells to produce epiregulin which enhances proliferation of malignant hepatocytes and LPS directly act on hepatocytes and promote their survival during malignant transformation.
The work described above is the first to provide direct evidence that the gut microbiome controls liver cancer. Using LPS, a common structural component of gram-negative bacteria, the gut microbiome modulates proliferation and survival of hepatocytes through targeting liver-resident cells, thus promoting HCC. It is well accepted that chronic inflammation contributes to cancer, but the underlying mechanism is still not clear (Colotta et al. 2009; Boland et al. 2005). Robert Schwabe’s work demonstrates that gut bacteria-derived LPS is an important factor in malignancy and helps shed light on the contributions of chronic liver inflammation’s role in hepatocarcinogenesis.
7.7.2 Bile Acids
The liver metabolizes cholesterol to produce primary bile acids which are conjugated with glycine or taurine in hepatocytes (Chiang 2013). Bile acids are secreted into the small intestinal lumen to help digestion and absorption of fat and lipid-soluble vitamins. In the presence of bacteria, the primary bile acids are converted into secondary bile acids with the main transformation steps being deconjugation and dehydroxylation (Ridlon et al. 2006, 2016; Dawson and Karpen 2015).The deconjugation step is the hydrolysis of the glycine or taurine from the steroid nucleus of primary bile acids which is catalyzed by bacterial bile salt hydrolases (BSH). BSH are mainly expressed in gram-positive bacteria but have also been found in some gram-negative bacteria such as Bacteroides spp. (Urdaneta and Casadesus 2017). Following deconjugation, free primary bile acids are converted into secondary bile acids through 7α/β-dehydroxylation. Unlike BSH, only a small number of bacterial species belonging to Clostridia have the 7α/β-dehydroxylation activity, and the 7α/β-dehydroxylation reaction mainly occurs in the colon (Ridlon et al. 2006, 2016). The body will then reuse the bacteria-modified bile acids with close to 95% of colonic bile acids being reabsorbed and shuttled back to the liver through enterohepatic circulation (Hofmann 2009).
Bile acids can activate signaling cascades and transcriptional networks and significantly influence liver function by binding to bile acid receptors such as farnesoid X receptor (FXR), pregnane X receptor (PXR), vitamin D3 receptor (VDR), constitutive androstane receptor (CAR), and membrane-bound G protein-coupled bile acid receptor1 (GPBAR1, also known as TGR5) (Copple and Li 2016; Schaap et al. 2014). Many target genes in the transcriptional networks are involved in metabolism of bile acids, cholesterol, lipid, and carbohydrates as well as inflammation, fibrosis, and carcinogenesis (Li and Chiang 2014). Each bile acid receptor shows different affinity for individual bile acids, thus changes in bile acid composition fine tune bile acid receptor signaling. Through controlling primary-to-secondary bile acid conversion, intestinal bacteria can influence liver function. On the other hand, bile acids also regulate the size and composition of the bacterial community. Bile acids have antibacterial function through multiple mechanisms including disruption of bacterial membranes, denaturing proteins, chelation of iron and calcium, and causing oxidative damage to DNA (Urdaneta and Casadesus 2017). Overgrowth of enteric bacteria is presented in cirrhotic patients with low levels of fecal bile acids. In addition, bile acids in part control growth of pathogenic bacteria such as Clostridium difficile (Allegretti et al. 2016; Sorg and Sonenshein 2008).
The contribution of bile acids to carcinogenesis has long been suggested (Phelan et al. 2017). As early as the 1930s, injection of deoxycholic acid (DCA), a secondary bile acid, has been found to cause malignant tumors at injection sites in mice. High level of bile acid exposure induces the generation of reactive oxygen species in cells which leads to disruption of the cell membrane, mitochondrial dysfunction, and DNA damage (Perez and Briz 2009). In addition, by binding to bile acid receptors, bile acids can regulate the expression of a lot of genes, many of which are involved in inflammation and carcinogenesis (Schaap et al. 2014; Li and Chiang 2014). Substantial evidences suggest that bile acids, especially secondary bile acids, promote colon cancer (Ajouz et al. 2014).
Naoko Ohtani’s group was the first to demonstrate that the secondary bile acid DCA acts as a critical messenger linking obesity-associated dysbiosis with liver cancer (Fig. 7.2) (Yoshimoto et al. 2013). High-fat diet (HFD)-fed mice with chemical carcinogen exposure recapitulates the liver tumor-promoting effect of obesity in humans (Park et al. 2010). In a study from Naoko Ohtani’s group, mice received a single injection of the chemical carcinogen DMBA (7,12-dimethylbenz[a]anthracene), which causes oncogenic Ras mutation, and were fed HFD (Yoshimoto et al. 2013). In line with other reports, HFD-fed obese mice develop marked increase of HCC. Interestingly, depleting intestinal bacteria with an oral antibiotic cocktail greatly reduced obesity-enhanced HCC, suggesting that the gut microbiome is critical in this process. In contrast to the findings of Robert Schwabe’s group (Dapito et al. 2012), TLR4 deficiency does not influence HCC in HFD-fed DMBA mice, suggesting that LPS is not involved in this setting. Obesity was associated with dysbiosis and a dramatic increase in gram-positive bacteria. Oral vancomycin treatment, which preferentially targets gram-positive bacteria, is sufficient to block HCC development. Serum metabolites were analyzed, and the secondary bile acid DCA was substantially increased in HFD mice. Lowering DCA reduces whereas DCA-feeding increases obesity-enhanced HCC. Intriguingly, DCA feeding alone is sufficient to enhance HCC in lean mice. As expected, HFD feeding causes expansion of cluster XI Clostridium which is a gram-positive bacteria and contains the majority of 7α/β-dehydroxylation required for primary-to-secondary bile acid conversion (Ridlon et al. 2006, 2016).
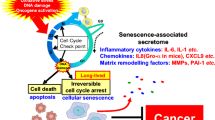
Cellular senescence is a process occurring in normal cells in response to telomere erosion or oncogene activation (Kuilman et al. 2010). This process is a barrier to tumorigenesis by acting through checkpoint activation and cell-cycle arrest. The important role of senescence in hepatocarcinogenesis has been reported (Kang et al. 2011; Lujambio et al. 2013). The livers of HFD-fed DMBA mice show a strong increase of senescence mainly in hepatic stellate cells (HSCs), which can be blocked by either an oral antibiotic cocktail or vancomycin treatment. In recent years, senescent cells have been found to present a secretory profile composed mainly of inflammatory cytokines chemokines and proteases (Acosta et al. 2013; Kuilman et al. 2008). Some of the senescence-associated secretory phenotype (SASP) have cell-autonomous activities which reinforce cell cycle arrest and promote clearance of senescent cells, whereas other SASP factors are associated with inflammation and tumorigenesis promotion. Depending on the stage of tumor development, SASP can be tumor-inhibiting or tumor-promoting (Eggert et al. 2016). The HFD-DMBA mouse study shows that DCA provokes SASP phenotype in hepatic stellate cells with increased IL-6, Gro-a, and CXCL9. Next the role of SASP in HCC was tested. Inflammasome activation and subsequent IL1β can act as an upstream regulator of SASP. IL1β knockout greatly reduces SASP expression of HSCs and subsequent liver tumor development. In addition, depleting senescent HSC significantly reduces HCC. Importantly, the study demonstrates that it is SASP but not cell-cycle arrest that regulates obesity-associated HCC. Together, the study suggests that the DCA-SASP axis in stellate cells are a key regulator in obesity-associated HCC.
The HFD-DMBA mouse study suggests that DCA is a potential target to treat liver cancer. Lowering DCA by decreasing 7α-dehydroxylation activity with difructose anhydride III or stimulating bile acid secretion with ursodeoxycholic acid (UDCA) inhibits HCC in HFD-fed DMBA mice. Cholestyramine, a bile acid sequestering resin that promotes bile acid excretion, has been reported to inhibit HCC in a different mouse model (Singh et al. 2018). The findings have clinical implication since UDCA is commonly used to treat patients with primary sclerosing cholangitis and cholestatic diseases (Lindor et al. 2009). UDCA usage has been reported to be associated with reduced mortality in colorectal cancer patients (Pardi et al. 2003). A possible association between UDCA usage and lower HCC incidence has been reported in patients with hepatitis C-associated cirrhosis (Tarao et al. 2005). The potential benefit of bile acid-targeting therapy approaches for liver cancer treatment should be investigated.
7.7.3 Short-Chain Fatty Acids
Short-chain fatty acids (SCFAs) are a group of saturated fatty acids containing less than six carbon molecules and include acetate, propionate, butyrate, pentanoic acid, and hexanoic acid. Intestinal SCFAs are produced by bacteria through fermentation of dietary fibers such as non-starch polysaccharides and oligosaccharides that cannot be digested by host enzymes (den Besten et al. 2013). Lacking a gut microbiome causes the significant reduction of SCFAs in germ-free mice (Hoverstad and Midtvedt 1986). The proximal colon has the highest concentration of SCFAs (70–140 mM) with acetate, propionate, and butyrate being the major components (Topping and Clifton 2001; Cook and Sellin 1998). A wide range of bacteria can generate acetate, whereas the production of propionate and butyrate seems more specific. Akkermansia muciniphila is believed to be the major propionate-producing bacteria (Louis and Flint 2017). In the colon, the molar ratio of acetate, propionate, and butyrate is roughly 60:25:15, respectively. This ratio, however, can be affected by many factors such as diet and bacterial composition. SCFAs are readily absorbed by the host. Butyrate serves as an energy source for colon epithelial cells (Hamer et al. 2008), and propionate is metabolized by hepatocytes. Acetate can pass through the liver in portal blood and reach the systemic circulation (Bloemen et al. 2009).
SCFAs influence host systems both at the cellular and molecular levels through two major mechanisms. First, SCFAs can directly inhibit histone deacetylases (HDACs) to regulate gene expression (Waldecker et al. 2008). Second, SCFAs regulate signaling through G-protein-coupled receptors (GPCRs) (Husted et al. 2017). The major GPCRs activated by SCFAs includes GPR41, GFPR43, and GPR109A. Extensive evidence shows that SCFAs are important for host health and affect the pathogenesis of a wide range of diseases including allergies, metabolic disorders, neurological diseases, and cancer (Koh et al. 2016). SCFAs promote the differentiation of anti-inflammatory regulatory T cells (Smith et al. 2013). GPR43 activation by SCFAs is necessary for the normal resolution of certain inflammatory responses (Maslowski et al. 2009). Butyrate and propionate at low amounts exert beneficial effects, including prevention of oxidative stress, inflammation, and lipid oxidation in hepatocytes (McNabney and Henagan 2017).
In contrast to the numerous reported beneficial effects of SCFAs, recently Matam Vijay-Kumar’s team demonstrates that SCFAs can exert a detrimental function and promote HCC (Fig. 7.2) (Singh et al. 2018). Initially, the team investigated the influence of dietary fiber fermentation and SCFA production on metabolic changes in TLR5 knockout mice which are prone to develop metabolic syndrome. Inulin, a soluble fiber, was supplemented to the mice in the diet. Consistent with other reports that inulin ameliorates low-grade inflammation, insulin resistance, and obesity (Zou et al. 2018), inulin feeding lowered obesity incidence and improved the indices of metabolic syndrome in the TLR5 knockout mice. Unexpectedly, some inulin-fed mice had hyperbilirubinemia, hepatic injury and inflammation, and impaired liver detoxification functions. Perhaps most interesting, prolonged inulin feeding caused primary HCC in the TLR5 knockout mice. Dietary fiber can be broadly categorized as insoluble or soluble. By comparing different types of dietary fibers, the study concluded that soluble fibers, but not fermentable insoluble fibers, induce HCC. Importantly, soluble fiber-induced HCC only occurs in mice with dysbiosis, not in TLR5 mice with balanced gut microbiome. Cohousing and cross-fostering studies demonstrate that the soluble fiber-induced HCC is transmissible between mice exchanging intestinal microbes. Germ-free TLR5 KO mice fed with irradiated soluble fiber diet failed to develop hyperbilirubinemia and HCC, confirming that it is gut microbiome dependent. A high-fat diet causes dysbiosis in mice (Murphy et al. 2015). Consistently, combining soluble fiber with HFD induced HCC in a small portion of wild-type mice, suggesting the phenomena is generalizable. Unlike insoluble fibers, soluble fibers can be fermented by the gut bacteria to produce SCFAs. HCC-prone TLR5 KO mice display gut dysbiosis characterized by an increase in fiber-fermenting bacteria and Proteobacteria. As expected, blocking microbial fermentation by depleting SCFA-consuming bacteria or plant-derived β-acids protects mice from soluble fiber-induced HCC. Although it causes hyperbilirubinemia, liver inflammation, enhanced hepatocyte proliferation, and fibrosis, single SCFA butyrate feeding fails to induce HCC, suggesting other factors are required.
In this model HCC is closely associated with hyperbilirubinemia caused by chronic liver inflammation. Mice with hyperbilirubinemia show higher liver expression of pattern recognition receptor NLRC4, the important inducer of inflammasome (Duncan and Canna 2018), and TLR4 signaling. However, knockout of TLR4 or NLRC4 has no influence on HCC incidence, suggesting that LPS and the inflammasome pathway are not important in this setting. Higher serum levels of bile acids were found in mice that developed HCC following soluble fiber feeding. Furthermore, cholestasis is closely associated with dietary fiber-induced HCC, suggesting the contribution of bile acids in HCC development. Clostridia members, particularly Clostridium cluster XIVa, are the main producers of butyrate and secondary bile acids (Van den Abbeele et al. 2013; Riviere et al. 2016). Supporting these findings, bacterial taxa analysis shows that Clostridia predominantly distinguishes hyperbilirubinemia mice, which develop HCC, from other groups. Indeed, lowering bile acid levels using cholestyramine suppresses HCC formation from dietary fiber feeding. The study is in line with the finding from Naoko Ohtani’s group that the gut bacteria-controlled secondary bile acid DCA promotes HCC (Yoshimoto et al. 2013).
The finding is surprising since dietary fibers and SCFAs are known to have antitumor function. It has been well recognized that dietary fiber consumption is associated with reduced risk of colon cancer (Hinnebusch et al. 2002). Importantly, two clinical studies reported that high intake of fiber has a protective effect against HCC (Fedirko et al. 2013; Yang et al. 2019). In the EPIC cohort of Western Europeans, dietary fiber from cereals and cereal derivatives has been found to have a significantly inverse association with HCC risk (Fedirko et al. 2013). In a separate study using large NHS and HPFS cohorts of US adults, increased intake of whole grains and possibly cereal fiber has been suggested to be associated with a reduced risk of HCC (Yang et al. 2019). However, this study showed that in the presence of a dysregulated intestinal microbiome, soluble fibers and SCFAs have opposite effects and promote HCC development, suggesting that the benefit of soluble fibers and SCFA is context dependent. The finding has important implications. Half of US adults consume dietary supplements to improve health. However, adverse effects caused by dietary supplement consumption have been reported (Ronis et al. 2018). More studies are needed to improve the safety and appropriate usage of dietary supplements.
7.7.4 Immune Cells
The liver contains a large number of immune cells with particular concentrations of Kupffer cells, NKT, and MAIT cells (Racanelli and Rehermann 2006). HCC is a typical inflammation-related cancer, and HCC patients often have underlying chronic liver inflammation (Michelotti et al. 2013; Anstee et al. 2019; Ganne-Carrie and Nahon 2019; Ponziani et al. 2019). The important role of immune surveillance in liver cancer has been recognized. In mice, removing the adaptive immune system promotes carcinogen in DEN-induced HCC (Schneider et al. 2012; Mossanen et al. 2019). Together with macrophages, CD4 T cells remove senescent hepatocytes to prevent malignant transformation (Kang et al. 2011). In addition, the liver immune cells play a critical role in obesity-/NAFLD-promoted HCC. Several mechanisms have been identified including hepatic activation of NKT, CD8 T cells, IgA-producing B cells, Th17 cells, and loss of liver CD4 T cells (Shalapour et al. 2017; Wolf et al. 2014; Gomes et al. 2016; Ma et al. 2016). In humans, adoptive T-cell therapy using tumor-specific CD4 T cells induced a strong antitumor response and complete eradication of tumor lesions in a late-stage cholangiocarcinoma patient (Tran et al. 2014). The patient is still alive 10 years after diagnosis. Based on the promising results, immune checkpoint inhibitors nivolumab and pembrolizumab have recently been approved by FDA to treat HCC patients (Okusaka and Ikeda 2018).
The gut microbiome has been established as a modulator for antitumor immunity (Gopalakrishnan et al. 2018a). Emerging evidences suggest that the gut microbiome plays a critical role in regulating liver local antitumor immune responses. In a follow-up study by Naoko Ohtani’s group, lipoteichoic acid (LTA), a gram-positive bacterial component, has been shown to contribute to obesity-enhanced HCC in accordance with the bile acid DCA (Loo et al. 2017). Unlike DCA, LTA is a major ligand for TLR2. Indeed, TLR2-deficient mice have reduced HCC in the HFD-MDTA model. Interestingly, the binding of LTA to TLR2 leads to increased COX2 activity, which subsequently enhances the production of prostaglandin E2 (PGE2). Tumor tissues upregulate PTGER4, the receptor for PGE2 (Sugimoto and Narumiya 2007), which is predominately expressed on immune cells particularly on T cells, but not on hepatocytes or HSCs. In vitro culture experiments show that PGE2 attenuates the proinflammatory cytokines IFNγ and TNFα but increases anti-inflammatory cytokines IL10 and IL6. High production of COX2 and PGE2 can be detected in noncirrhotic NASH-associated HCC patients. To our knowledge, it is the first publication which links the gut microbiome and liver tumor development through inhibition of the immune system.
A recent mouse study from Vikas Dudeja’s group investigated the effects of the gut microbiome on tumor growth (Sethi et al. 2018). An oral antibiotic cocktail was applied to remove intestinal bacteria, and the subsequent influence on growth of both subcutaneous tumors and liver metastases was tested. Enhanced tumor development in both compartments were found after removing the gut microbiome. Interestingly, the tumor-promoting function of the gut microbiome depends on adaptive immunity and disappears in Rag1 knockout mice which lack mature T, B, and NKT cells. Removing intestinal bacteria induces higher T-cell activation and production of proinflammatory cytokines such as IFNγ in the tumor microenvironment, which correlates with improved survival. This report provides direct evidence that the adaptive immune system in involved in the gut microbiome-modulated HCC.
Our group also studies the influence of the gut microbiome on liver cancer (Fig. 7.2) (Ma et al. 2018). Consistent with previous observations, removing gut bacteria using oral antibiotic cocktail reduces liver tumor formation (Shalapour et al. 2017; Dapito et al. 2012; Sethi et al. 2018). The effect is not only observed in primary HCC but also seen in liver metastatic models with nonhepatic tumors. Interestingly, removing the gut bacteria causes a seemingly location-dependent opposite effect on the growth of metastatic lesion from the same tumor line in the liver compared to the lung. This observation suggests that the gut microbiome-influenced liver environment is crucial for controlling tumor growth. We focused on the changes within the liver immune system to understand the mechanism. A prominent increase in liver CXCR6+ NKT cells was found in the oral antibiotics-treated mice or germ-free mice. Importantly, either NKT cell deficiency or disrupting liver accumulation of NKT cells by knocking out CXCR6 eliminated the influence of the gut bacteria on liver tumor growth. In addition, hepatic NKT cells from antibiotic-treated mice are more active and produce higher levels of IFNγ, an important cytokine to induce antitumor immunity, after in vivo stimulation. Together these results suggest that there is a critical role of NKT cells in gut microbiome-controlled liver tumor growth.
NKT cells are a group of innate-like lymphocytes and serve as a bridge between the innate and adaptive immune system (Gao et al. 2009; Bandyopadhyay et al. 2016; Terabe and Berzofsky 2008; Nishimura et al. 2000). With limited TCR repertoire, NKT cells recognize lipids presented on CD1 molecules, which leads to a quick release of a larger amount and various cytokines that help initiate a variety of immune responses. NKT cells are enriched in the liver and make up ~30% of intrahepatic lymphocytes in mice (Bandyopadhyay et al. 2016; Terabe and Berzofsky 2008). The endogenous lipid ligands for NKT are still elusive, but studies have shown that the lipid components of gut bacteria can activate NKT cells (Brennan et al. 2014; Wolf et al. 2015; Zajonc and Girardi 2015), which suggest a potential role for gut bacteria regulation of liver NKT cells. It is well-known that NKT cells have antitumor functions (Terabe and Berzofsky 2008). NKT cells are reported to be important in controlling liver metastasis (Cullen et al. 2009). A recent publication demonstrates that NKT cells play an important role in preventing DEN-induced HCC (Mossanen et al. 2019). NKT cells can exert antitumor function by either directly killing tumor cells or an indirect function through secreting cytokines. The exact mechanism of how liver NKT cells mediate gut microbiome-controlled liver tumor formation is not clear and still under investigation.
Next, we focused on how the gut microbiome regulates the intrahepatic NKT cell population. Removing gram-positive bacteria is sufficient to increase NKT cells. Liver accumulation of NKT cells is mediated by CXCR6 receptor which recognizes its ligand CXCL16 (highly expressed on LSECs) (Geissmann et al. 2005) in the lining of liver sinusoids. Removing gut bacteria or selectively targeting gram-positive bacteria increases CXCL16 expression in LSECs. Bacteria responsible for primary-to-secondary conversion of bile acids are gram-positive. For this reason, we tested the possible link between bile acids and NKT cells. Interestingly, secondary bile acids decreased whereas primary bile acid increased CXCL16 expression. Feeding secondary bile acids to mice inhibits liver CXCL16 expression and reduces NKT cells in the liver. Colonization of mice with Clostridium scindens, a bacteria species well-known for primary-to-secondary bile acid conversion (Studer et al. 2016), accelerates the dropping of liver NKT cells following antibiotic withdrawal. Importantly, Clostridium scindens colonization increases liver tumor burden. Next, we checked whether the finding in mice can also be replicated in humans. Indeed, in nontumor liver tissues of HCC patients, a positive association between primary bile acid CDCA and CXCL16 mRNA level was found. In contrast, secondary bile acid DCA is negatively associated with CXCL16 mRNA levels, suggesting the bile acids/CXCL16 axis also exists in humans. Our study clearly demonstrated that the gut bacteria use bile acids as a messenger to regulate liver NKT cells thus influencing liver antitumor immunity.
Our understating of the gut microbiome’s effects on liver cancer is still in its infancy. Most of the studies still focus on HCC, the major type of primary liver cancer. The influence of the gut microbiome on cholangiocarcinoma has not been reported, although there are reports of the association between intrahepatic cholangiocarcinoma and chronic inflammatory bowel diseases which display dysbiosis (Tyson and El-Serag 2011). In addition, the majority of patients with primary sclerosing cholangitis, a well-established risk factor for cholangiocarcinoma, have a concomitant inflammatory bowel disease (Gulamhusein et al. 2016). Our study with liver metastatic models suggests that the gut microbiome regulates liver antitumor immunity and the tumor regulatory function is not limited to HCC but also apply to other liver cancer types including liver metastasis.
7.8 Gut Microbiome and Immunotherapy
Immunotherapies, particularly immune checkpoint inhibitors, have shown promising effects in treating patients with solid tumors (Khalil et al. 2016). The gut microbiome plays a fundamental role in the development and functional regulation of the host immune system. Importantly, alternation of the gut microbiome has been found to influence the efficacy of many types of immunotherapies for cancer including immune checkpoint inhibitors (Routy et al. 2018a). In patients with metastatic melanoma, non-small cell lung cancer, or renal cancer, the presence of intestinal bacteria such as Faecalibacterium prausnitzii, Bifidobacterium longum, or Akkermansia muciniphila has been linked to better anti-PD-1 responses (Routy et al. 2018b; Matson et al. 2018; Gopalakrishnan et al. 2018b; Frankel et al. 2017; Pinato et al. 2019). As expected, usage of antibiotics, especially the ones with broad spectrum, has been connected with impaired efficacy of checkpoint blockade in these studies. The timing of antibiotic application seems to be important. Prior, but not concurrent, administration of broad-spectrum antibiotic with anti-PD1 therapy is associated with worse overall survival and a higher risk of tumor refractory (Pinato et al. 2019). Of note, no close taxonomic relationship has been associated with improved efficacy of anti-PD1/PD-L1 therapy, suggesting that multiple bacteria-controlled pathways are involved or these bacteria are functionally related. Besides immunotherapy, proper immune responses are required for many chemotherapies to reach full-treatment potency such as cyclophosphamide- and platinum-based therapies. In mouse studies, the optimal antitumor efficacy of cyclophosphamide and oxaliplatin has been found to be dependent on intact gut commensal bacteria and is significantly dampened in germ-free mice or mice received antibiotic cocktails (Iida et al. 2013). Although the crucial role of intestinal bacteria in tumor immunotherapy has been established, the underlying mechanisms are still largely unknown.
Following the advancements of immunotherapy for solid tumors in the past few years, there is a global interest in immunotherapies for HCC (Hou et al. 2019; Johnston and Khakoo 2019; Floudas et al. 2019; Xie et al. 2018). Most HCC patients diagnosed with advanced disease are not eligible for curative approaches such as surgical resection, liver transplantation, or local percutaneous tumor ablation. Systemic treatments for HCC constitute mostly of multitargeted tyrosine kinase inhibitors (TKIs) with sorafenib being the only FDA-approved drug until 2017 (Johnston and Khakoo 2019; Floudas et al. 2019). Although additional TKIs have been approved for HCC treatment recently, its overall survival benefit is limited. Recently, nivolumab and pembrolizumab, two immune checkpoint inhibitors, have been approved by the FDA to treat HCC patients. Besides immune checkpoint inhibitors, other immune-based approaches for HCC are under investigation including DC vaccines, oncolytic viruses, adoptive cell therapy, antigen-targeting antibodies, and blocking inhibitory cytokines (Floudas et al. 2019; Xie et al. 2018). Currently, the influence of the gut microbiome on immunotherapy for liver cancer is unknown. A recent mouse study reported the impact of the gut microbiome on anti-PD1 therapy for pancreatic adenocarcinoma (PDA) which is a deadly GI malignancy (Pushalkar et al. 2018). Similar to liver cancer, the gut microbiome promotes PDA development. Interestingly, removing the gut bacteria sensitizes PDA tumors to anti-PD1 therapy accompanying an enhanced adaptive T-cell response. The observation is contradictory to many reports that the gut microbiome is needed for the success of anti-PD1 therapy (Routy et al. 2018b; Matson et al. 2018; Gopalakrishnan et al. 2018b; Pinato et al. 2019). Further investigations are needed to clarify the potential influences the gut microbiome has on immunotherapy against GI cancers.
7.9 Summary
Liver cancer is a deadly disease, and its incidence is rising. Following the success of controlling viral hepatitis and global pandemic of obesity, the major risk factors for HCC are shifting toward obesity and chronic metabolic dysregulation conditions such as NAFLD. Obesity and NAFLD are closely associated with the alteration of the gut microbiome. Together with the finding of liver cancer-associated bacteria, it has been suggested that the gut microbiome contributes to liver cancer formation. Importantly, animal studies prove direct evidence that the gut bacteria promote HCC development. The potential strategies targeting the gut microbiome to treat liver cancer, and the influence of the gut microbiome on available liver cancer treatment, especially immunotherapies, should be further investigated.
Acknowledgement
C.M. and T.F.G. were supported by the Intramural Research Program of the NIH, NCI.
References
Abdel-Misih SR, Bloomston M (2010) Liver anatomy. Surg Clin North Am 90(4):643–653. https://doi.org/10.1016/j.suc.2010.04.017
Abdulamir AS, Hafidh RR, Abu Bakar F (2011) The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J Exp Clin Cancer Res 30:11. https://doi.org/10.1186/1756-9966-30-11
Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, White O, Kelley ST, Methe B, Schloss PD, Gevers D, Mitreva M, Huttenhower C (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8(6):e1002358. https://doi.org/10.1371/journal.pcbi.1002358
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15(8):978–990. https://doi.org/10.1038/ncb2784
Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG (1994) Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 20(2):453–460
Adamovich Y, Rousso-Noori L, Zwighaft Z, Neufeld-Cohen A, Golik M, Kraut-Cohen J, Wang M, Han X, Asher G (2014) Circadian clocks and feeding time regulate the oscillations and levels of hepatic triglycerides. Cell Metab 19(2):319–330. https://doi.org/10.1016/j.cmet.2013.12.016
Ajouz H, Mukherji D, Shamseddine A (2014) Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol 12:164. https://doi.org/10.1186/1477-7819-12-164
Alexander C, Rietschel ET (2001) Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res 7(3):167–202
Alexopoulou A, Agiasotelli D, Vasilieva LE, Dourakis SP (2017) Bacterial translocation markers in liver cirrhosis. Ann Gastroenterol 30(5):486–497. https://doi.org/10.20524/aog.2017.0178
Allegretti JR, Kearney S, Li N, Bogart E, Bullock K, Gerber GK, Bry L, Clish CB, Alm E, Korzenik JR (2016) Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment Pharmacol Ther 43(11):1142–1153. https://doi.org/10.1111/apt.13616
An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, Lu X, Zeissig S, Blumberg RS, Kasper DL (2014) Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell 156(1–2):123–133. https://doi.org/10.1016/j.cell.2013.11.042
Anakk S, Bhosale M, Schmidt VA, Johnson RL, Finegold MJ, Moore DD (2013) Bile acids activate YAP to promote liver carcinogenesis. Cell Rep 5(4):1060–1069. https://doi.org/10.1016/j.celrep.2013.10.030
Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, Conner EA, Gillen MC, Roskams T, Roberts LR, Factor VM, Thorgeirsson SS (2012) Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 142(4):1021–1031. e1015. https://doi.org/10.1053/j.gastro.2011.12.005
Andersen-Nissen E, Smith KD, Bonneau R, Strong RK, Aderem A (2007) A conserved surface on toll-like receptor 5 recognizes bacterial flagellin. J Exp Med 204(2):393–403. https://doi.org/10.1084/jem.20061400
Andrews T, Sullivan KE (2003) Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev 16(4):597–621. https://doi.org/10.1128/cmr.16.4.597-621.2003
Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M (2019) From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 16(7):411–428. https://doi.org/10.1038/s41575-019-0145-7
Augustyn M, Grys I, Kukla M (2019) Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease. Clin Exp Hepatol 5(1):1–10. https://doi.org/10.5114/ceh.2019.83151
Axelrad JE, Lichtiger S, Yajnik V (2016) Inflammatory bowel disease and cancer: the role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol 22(20):4794–4801. https://doi.org/10.3748/wjg.v22.i20.4794
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101(44):15718–15723. https://doi.org/10.1073/pnas.0407076101
Backhed F, Manchester JK, Semenkovich CF, Gordon JI (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 104(3):979–984. https://doi.org/10.1073/pnas.0605374104
Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, Hellman J (2007) MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol 178(2):1164–1171. https://doi.org/10.4049/jimmunol.178.2.1164
Bala S, Marcos M, Gattu A, Catalano D, Szabo G (2014) Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS One 9(5):e96864. https://doi.org/10.1371/journal.pone.0096864
Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, Lind GE, Folseraas T, Forbes SJ, Fouassier L, Geier A, Calvisi DF, Mertens JC, Trauner M, Benedetti A, Maroni L, Vaquero J, Macias RI, Raggi C, Perugorria MJ, Gaudio E, Boberg KM, Marin JJ, Alvaro D (2016) Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European network for the study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 13(5):261–280. https://doi.org/10.1038/nrgastro.2016.51
Bandyopadhyay K, Marrero I, Kumar V (2016) NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol 13(3):337–346. https://doi.org/10.1038/cmi.2015.115
Basuroy S, Sheth P, Mansbach CM, Rao RK (2005) Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine. Am J Physiol Gastrointest Liver Physiol 289(2):G367–G375. https://doi.org/10.1152/ajpgi.00464.2004
Berg RD (1995) Bacterial translocation from the gastrointestinal tract. Trends Microbiol 3(4):149–154. https://doi.org/10.1016/s0966-842x(00)88906-4
Bert F, Johnson JR, Ouattara B, Leflon-Guibout V, Johnston B, Marcon E, Valla D, Moreau R, Nicolas-Chanoine MH (2010) Genetic diversity and virulence profiles of Escherichia coli isolates causing spontaneous bacterial peritonitis and bacteremia in patients with cirrhosis. J Clin Microbiol 48(8):2709–2714. https://doi.org/10.1128/JCM.00516-10
Bhamarapravati N, Thammavit W, Vajrasthira S (1978) Liver changes in hamsters infected with a liver fluke of man, Opisthorchis viverrini. Am J Trop Med Hyg 27(4):787–794. https://doi.org/10.4269/ajtmh.1978.27.787
Bilzer M, Roggel F, Gerbes AL (2006) Role of Kupffer cells in host defense and liver disease. Liver Int 26(10):1175–1186. https://doi.org/10.1111/j.1478-3231.2006.01342.x
Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, Wells JM (2014) Intestinal permeability—a new target for disease prevention and therapy. BMC Gastroenterol 14:189. https://doi.org/10.1186/s12876-014-0189-7
Bloemen JG, Venema K, van de Poll MC, Olde Damink SW, Buurman WA, Dejong CH (2009) Short chain fatty acids exchange across the gut and liver in humans measured at surgery. Clin Nutr 28(6):657–661. https://doi.org/10.1016/j.clnu.2009.05.011
Bogdanos DP, Gao B, Gershwin ME (2013) Liver immunology. Compr Physiol 3(2):567–598. https://doi.org/10.1002/cphy.c120011
Boland CR, Luciani MG, Gasche C, Goel A (2005) Infection, inflammation, and gastrointestinal cancer. Gut 54(9):1321–1331. https://doi.org/10.1136/gut.2004.060079
Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, Ellis B, Carroll KC, Albesiano E, Wick EC, Platz EA, Pardoll DM, Sears CL (2015) The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis 60(2):208–215. https://doi.org/10.1093/cid/ciu787
Bonnel AR, Bunchorntavakul C, Reddy KR (2011) Immune dysfunction and infections in patients with cirrhosis. Clin Gastroenterol Hepatol 9(9):727–738. https://doi.org/10.1016/j.cgh.2011.02.031
Borad MJ, Champion MD, Egan JB, Liang WS, Fonseca R, Bryce AH, McCullough AE, Barrett MT, Hunt K, Patel MD, Young SW, Collins JM, Silva AC, Condjella RM, Block M, McWilliams RR, Lazaridis KN, Klee EW, Bible KC, Harris P, Oliver GR, Bhavsar JD, Nair AA, Middha S, Asmann Y, Kocher JP, Schahl K, Kipp BR, Barr Fritcher EG, Baker A, Aldrich J, Kurdoglu A, Izatt T, Christoforides A, Cherni I, Nasser S, Reiman R, Phillips L, McDonald J, Adkins J, Mastrian SD, Placek P, Watanabe AT, Lobello J, Han H, Von Hoff D, Craig DW, Stewart AK, Carpten JD (2014) Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 10(2):e1004135. https://doi.org/10.1371/journal.pgen.1004135
Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, Ridgway RA, Samuel K, Van Rooijen N, Barry ST, Wigmore SJ, Sansom OJ, Forbes SJ (2015) WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest 125(3):1269–1285. https://doi.org/10.1172/JCI76452
Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, Hunault G, Oberti F, Cales P, Diehl AM (2016) The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63(3):764–775. https://doi.org/10.1002/hep.28356
Boutagy NE, Neilson AP, Osterberg KL, Smithson AT, Englund TR, Davy BM, Hulver MW, Davy KP (2015) Short-term high-fat diet increases postprandial trimethylamine-N-oxide in humans. Nutr Res 35(10):858–864. https://doi.org/10.1016/j.nutres.2015.07.002
Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, Herault A, Saric J, Belghiti J, Franco D, Bioulac-Sage P, Laurent-Puig P, Zucman-Rossi J (2007) Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 45(1):42–52. https://doi.org/10.1002/hep.21467
Brenchley JM, Douek DC (2012) Microbial translocation across the GI tract. Annu Rev Immunol 30:149–173. https://doi.org/10.1146/annurev-immunol-020711-075001
Brennan PJ, Tatituri RV, Heiss C, Watts GF, Hsu FF, Veerapen N, Cox LR, Azadi P, Besra GS, Brenner MB (2014) Activation of iNKT cells by a distinct constituent of the endogenous glucosylceramide fraction. Proc Natl Acad Sci USA 111(37):13433–13438. https://doi.org/10.1073/pnas.1415357111
Breuhahn K, Longerich T, Schirmacher P (2006) Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 25(27):3787–3800. https://doi.org/10.1038/sj.onc.1209556
Brunt EM (2010) Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 7(4):195–203. https://doi.org/10.1038/nrgastro.2010.21
Cai W, Ran Y, Li Y, Wang B, Zhou L (2017) Intestinal microbiome and permeability in patients with autoimmune hepatitis. Best Pract Res Clin Gastroenterol 31(6):669–673. https://doi.org/10.1016/j.bpg.2017.09.013
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348(17):1625–1638. https://doi.org/10.1056/NEJMoa021423
Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, Factor VM, Thorgeirsson SS (2006) Ubiquitous activation of Ras and Jak/stat pathways in human HCC. Gastroenterology 130(4):1117–1128. https://doi.org/10.1053/j.gastro.2006.01.006
Campbell PT, Newton CC, Freedman ND, Koshiol J, Alavanja MC, Beane Freeman LE, Buring JE, Chan AT, Chong DQ, Datta M, Gaudet MM, Gaziano JM, Giovannucci EL, Graubard BI, Hollenbeck AR, King L, Lee IM, Linet MS, Palmer JR, Petrick JL, Poynter JN, Purdue MP, Robien K, Rosenberg L, Sahasrabuddhe VV, Schairer C, Sesso HD, Sigurdson AJ, Stevens VL, Wactawski-Wende J, Zeleniuch-Jacquotte A, Renehan AG, McGlynn KA (2016) Body mass index, waist circumference, diabetes, and risk of liver Cancer for U.S. adults. Cancer Res 76(20):6076–6083. https://doi.org/10.1158/0008-5472.CAN-16-0787
Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM (2007) Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50(11):2374–2383. https://doi.org/10.1007/s00125-007-0791-0
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57(6):1470–1481. https://doi.org/10.2337/db07-1403
Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, Zazzeroni F, Alesse E (2013) The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int 2013:187204. https://doi.org/10.1155/2013/187204
Ceccarelli S, Panera N, Mina M, Gnani D, De Stefanis C, Crudele A, Rychlicki C, Petrini S, Bruscalupi G, Agostinelli L, Stronati L, Cucchiara S, Musso G, Furlanello C, Svegliati-Baroni G, Nobili V, Alisi A (2015) LPS-induced TNF-alpha factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 6(39):41434–41452. https://doi.org/10.18632/oncotarget.5163
Celaj S, Gleeson MW, Deng J, O’Toole GA, Hampton TH, Toft MF, Morrison HG, Sogin ML, Putra J, Suriawinata AA, Gorham JD (2014) The microbiota regulates susceptibility to Fas-mediated acute hepatic injury. Lab Investig 94(9):938–949. https://doi.org/10.1038/labinvest.2014.93
Chayanupatkul M, Omino R, Mittal S, Kramer JR, Richardson P, Thrift AP, El-Serag HB, Kanwal F (2017) Hepatocellular carcinoma in the absence of cirrhosis in patients with chronic hepatitis B virus infection. J Hepatol 66(2):355–362. https://doi.org/10.1016/j.jhep.2016.09.013
Chen Y, Sun R (2011) Toll-like receptors in acute liver injury and regeneration. Int Immunopharmacol 11(10):1433–1441. https://doi.org/10.1016/j.intimp.2011.04.023
Chen J, Wei Y, He J, Cui G, Zhu Y, Lu C, Ding Y, Xue R, Bai L, Uede T, Li L, Diao H (2014) Natural killer T cells play a necessary role in modulating of immune-mediated liver injury by gut microbiota. Sci Rep 4:7259. https://doi.org/10.1038/srep07259
Chen YM, Liu Y, Zhou RF, Chen XL, Wang C, Tan XY, Wang LJ, Zheng RD, Zhang HW, Ling WH, Zhu HL (2016a) Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci Rep 6:19076. https://doi.org/10.1038/srep19076
Chen Y, Ji F, Guo J, Shi D, Fang D, Li L (2016b) Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci Rep 6:34055. https://doi.org/10.1038/srep34055
Chiang JY (2013) Bile acid metabolism and signaling. Compr Physiol 3(3):1191–1212. https://doi.org/10.1002/cphy.c120023
Chieppa M, Rescigno M, Huang AY, Germain RN (2006) Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med 203(13):2841–2852. https://doi.org/10.1084/jem.20061884
Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, Li H, Alekseyenko AV, Blaser MJ (2012) Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488(7413):621–626. https://doi.org/10.1038/nature11400
Chu H, Mazmanian SK (2013) Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol 14(7):668–675. https://doi.org/10.1038/ni.2635
Cipe G, Idiz UO, Firat D, Bektasoglu H (2015) Relationship between intestinal microbiota and colorectal cancer. World J Gastrointest Oncol 7(10):233–240. https://doi.org/10.4251/wjgo.v7.i10.233
Cirera I, Bauer TM, Navasa M, Vila J, Grande L, Taura P, Fuster J, Garcia-Valdecasas JC, Lacy A, Suarez MJ, Rimola A, Rodes J (2001) Bacterial translocation of enteric organisms in patients with cirrhosis. J Hepatol 34(1):32–37. https://doi.org/10.1016/s0168-8278(00)00013-1
Clarke SF, Murphy EF, Nilaweera K, Ross PR, Shanahan F, O’Toole PW, Cotter PD (2012) The gut microbiota and its relationship to diet and obesity: new insights. Gut Microbes 3(3):186–202. https://doi.org/10.4161/gmic.20168
Claudel T, Staels B, Kuipers F (2005) The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol 25(10):2020–2030. https://doi.org/10.1161/01.ATV.0000178994.21828.a7
Collier JD, Bassendine MF, Burt AD, Major GN (1996) Characterisation of the DNA repair enzyme for O(6)-methylguanine in cirrhosis. J Hepatol 25(2):158–165. https://doi.org/10.1016/s0168-8278(96)80068-7
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30(7):1073–1081. https://doi.org/10.1093/carcin/bgp127
Cook SI, Sellin JH (1998) Review article: short chain fatty acids in health and disease. Aliment Pharmacol Ther 12(6):499–507. https://doi.org/10.1046/j.1365-2036.1998.00337.x
Copple BL, Li T (2016) Pharmacology of bile acid receptors: evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res 104:9–21. https://doi.org/10.1016/j.phrs.2015.12.007
Corbin KD, Zeisel SH (2012) Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol 28(2):159–165. https://doi.org/10.1097/MOG.0b013e32834e7b4b
Crispe IN (2003) Hepatic T cells and liver tolerance. Nat Rev Immunol 3(1):51–62. https://doi.org/10.1038/nri981
Crispe IN (2011) Liver antigen-presenting cells. J Hepatol 54(2):357–365. https://doi.org/10.1016/j.jhep.2010.10.005
Crosby CM, Kronenberg M (2018) Tissue-specific functions of invariant natural killer T cells. Nat Rev Immunol 18(9):559–574. https://doi.org/10.1038/s41577-018-0034-2
Cullen R, Germanov E, Shimaoka T, Johnston B (2009) Enhanced tumor metastasis in response to blockade of the chemokine receptor CXCR6 is overcome by NKT cell activation. J Immunol 183(9):5807–5815. https://doi.org/10.4049/jimmunol.0803520
Czaja AJ (2016) Factoring the intestinal microbiome into the pathogenesis of autoimmune hepatitis. World J Gastroenterol 22(42):9257–9278. https://doi.org/10.3748/wjg.v22.i42.9257
Da Silva HE, Teterina A, Comelli EM, Taibi A, Arendt BM, Fischer SE, Lou W, Allard JP (2018) Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci Rep 8(1):1466. https://doi.org/10.1038/s41598-018-19753-9
Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, Lefkowitch JH, Bower M, Friedman R, Sartor RB, Rabadan R, Schwabe RF (2012) Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21(4):504–516. https://doi.org/10.1016/j.ccr.2012.02.007
Dawson PA, Karpen SJ (2015) Intestinal transport and metabolism of bile acids. J Lipid Res 56(6):1085–1099. https://doi.org/10.1194/jlr.R054114
den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54(9):2325–2340. https://doi.org/10.1194/jlr.R036012
Disibio G, French SW (2008) Metastatic patterns of cancers: results from a large autopsy study. Arch Pathol Lab Med 132(6):931–939. https://doi.org/10.1043/1543-2165(2008)132[931:MPOCRF]2.0.CO;2
Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE (2013) Kupffer cells in the liver. Compr Physiol 3(2):785–797. https://doi.org/10.1002/cphy.c120026
Donaldson GP, Lee SM, Mazmanian SK (2016) Gut biogeography of the bacterial microbiota. Nat Rev Microbiol 14(1):20–32. https://doi.org/10.1038/nrmicro3552
Dowman JK, Tomlinson JW, Newsome PN (2010) Pathogenesis of non-alcoholic fatty liver disease. QJM 103(2):71–83. https://doi.org/10.1093/qjmed/hcp158
Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, White RC, Clarke TH, Nguyen K, Torralba M, Shao Y, Liu J, Hernandez-Morales A, Lessor L, Rahman IR, Miyamoto Y, Ly M, Gao B, Sun W, Kiesel R, Hutmacher F, Lee S, Ventura-Cots M, Bosques-Padilla F, Verna EC, Abraldes JG, Brown RS Jr, Vargas V, Altamirano J, Caballeria J, Shawcross DL, Ho SB, Louvet A, Lucey MR, Mathurin P, Garcia-Tsao G, Bataller R, Tu XM, Eckmann L, van der Donk WA, Young R, Lawley TD, Starkel P, Pride D, Fouts DE, Schnabl B (2019) Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575(7783):505–511. https://doi.org/10.1038/s41586-019-1742-x
Duncan JA, Canna SW (2018) The NLRC4 inflammasome. Immunol Rev 281(1):115–123. https://doi.org/10.1111/imr.12607
Duryee MJ, Klassen LW, Freeman TL, Willis MS, Tuma DJ, Thiele GM (2004) Lipopolysaccharide is a cofactor for malondialdehyde-acetaldehyde adduct-mediated cytokine/chemokine release by rat sinusoidal liver endothelial and Kupffer cells. Alcohol Clin Exp Res 28(12):1931–1938. https://doi.org/10.1097/01.alc.0000148115.90045.c5
Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, Heikenwalder M, Wang XW, Zender L, Greten TF (2016) Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell 30(4):533–547. https://doi.org/10.1016/j.ccell.2016.09.003
El-Serag HB, Kanwal F, Richardson P, Kramer J (2016) Risk of hepatocellular carcinoma after sustained virological response in veterans with hepatitis C virus infection. Hepatology 64(1):130–137. https://doi.org/10.1002/hep.28535
Elsharkawy AM, Mann DA (2007) Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 46(2):590–597. https://doi.org/10.1002/hep.21802
European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L (2018) EASL clinical practice guidelines: management of alcohol-related liver disease. J Hepatol 69(1):154–181. https://doi.org/10.1016/j.jhep.2018.03.018
Fedirko V, Lukanova A, Bamia C, Trichopolou A, Trepo E, Nothlings U, Schlesinger S, Aleksandrova K, Boffetta P, Tjonneland A, Johnsen NF, Overvad K, Fagherazzi G, Racine A, Boutron-Ruault MC, Grote V, Kaaks R, Boeing H, Naska A, Adarakis G, Valanou E, Palli D, Sieri S, Tumino R, Vineis P, Panico S, Bueno-de-Mesquita HB, Siersema PD, Peeters PH, Weiderpass E, Skeie G, Engeset D, Quiros JR, Zamora-Ros R, Sanchez MJ, Amiano P, Huerta JM, Barricarte A, Johansen D, Lindkvist B, Sund M, Werner M, Crowe F, Khaw KT, Ferrari P, Romieu I, Chuang SC, Riboli E, Jenab M (2013) Glycemic index, glycemic load, dietary carbohydrate, and dietary fiber intake and risk of liver and biliary tract cancers in Western Europeans. Ann Oncol 24(2):543–553. https://doi.org/10.1093/annonc/mds434
Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J (2006) Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 168(4):1148–1154. https://doi.org/10.2353/ajpath.2006.050617
Flint HJ, Scott KP, Duncan SH, Louis P, Forano E (2012) Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3(4):289–306. https://doi.org/10.4161/gmic.19897
Floudas CS, Brar G, Greten TF (2019) Immunotherapy: current status and future perspectives. Dig Dis Sci 64(4):1030–1040. https://doi.org/10.1007/s10620-019-05516-7
Forner A, Reig M, Bruix J (2018) Hepatocellular carcinoma. Lancet 391(10127):1301–1314. https://doi.org/10.1016/S0140-6736(18)30010-2
Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, Koh AY (2017) Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific Human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 19(10):848–855. https://doi.org/10.1016/j.neo.2017.08.004
Fujimoto M, Uemura M, Nakatani Y, Tsujita S, Hoppo K, Tamagawa T, Kitano H, Kikukawa M, Ann T, Ishii Y, Kojima H, Sakurai S, Tanaka R, Namisaki T, Noguchi R, Higashino T, Kikuchi E, Nishimura K, Takaya A, Fukui H (2000) Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: relation to severity of liver disturbance. Alcohol Clin Exp Res 24(Suppl 4):48S–54S
Fukui H (2017) Gut Microbiome-based therapeutics in liver cirrhosis: basic consideration for the next step. J Clin Transl Hepatol 5(3):249–260. https://doi.org/10.14218/JCTH.2017.00008
Fukui H, Brauner B, Bode JC, Bode C (1991) Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol 12(2):162–169. https://doi.org/10.1016/0168-8278(91)90933-3
Gagniere J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, Bringer MA, Pezet D, Bonnet M (2016) Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 22(2):501–518. https://doi.org/10.3748/wjg.v22.i2.501
Gandhi CR, Chaillet JR, Nalesnik MA, Kumar S, Dangi A, Demetris AJ, Ferrell R, Wu T, Divanovic S, Stankeiwicz T, Shaffer B, Stolz DB, Harvey SA, Wang J, Starzl TE (2015) Liver-specific deletion of augmenter of liver regeneration accelerates development of steatohepatitis and hepatocellular carcinoma in mice. Gastroenterology 148(2):379–391. e374. https://doi.org/10.1053/j.gastro.2014.10.008
Ganne-Carrie N, Nahon P (2019) Hepatocellular carcinoma in the setting of alcohol-related liver disease. J Hepatol 70(2):284–293. https://doi.org/10.1016/j.jhep.2018.10.008
Gao B, Radaeva S, Park O (2009) Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol 86(3):513–528. https://doi.org/10.1189/JLB.0309135
Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, Littman DR (2005) Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol 3(4):e113. https://doi.org/10.1371/journal.pbio.0030113
Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E (2009) Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 50(1):90–97. https://doi.org/10.1194/jlr.M800156-JLR200
Giannelli V, Di Gregorio V, Iebba V, Giusto M, Schippa S, Merli M, Thalheimer U (2014) Microbiota and the gut-liver axis: bacterial translocation, inflammation and infection in cirrhosis. World J Gastroenterol 20(45):16795–16810. https://doi.org/10.3748/wjg.v20.i45.16795
Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R (2018) Current understanding of the human microbiome. Nat Med 24(4):392–400. https://doi.org/10.1038/nm.4517
Gold MC, Napier RJ, Lewinsohn DM (2015) MR1-restricted mucosal associated invariant T (MAIT) cells in the immune response to Mycobacterium tuberculosis. Immunol Rev 264(1):154–166. https://doi.org/10.1111/imr.12271
Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, Waisman A, Theurillat JP, Perna C, Djouder N (2016) Metabolic inflammation-associated IL-17A causes non-alcoholic Steatohepatitis and hepatocellular carcinoma. Cancer Cell 30(1):161–175. https://doi.org/10.1016/j.ccell.2016.05.020
Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA (2018a) The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33(4):570–580. https://doi.org/10.1016/j.ccell.2018.03.015
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, Cogdill AP, Zhao L, Hudgens CW, Hutchinson DS, Manzo T, Petaccia de Macedo M, Cotechini T, Kumar T, Chen WS, Reddy SM, Szczepaniak Sloane R, Galloway-Pena J, Jiang H, Chen PL, Shpall EJ, Rezvani K, Alousi AM, Chemaly RF, Shelburne S, Vence LM, Okhuysen PC, Jensen VB, Swennes AG, McAllister F, Marcelo Riquelme Sanchez E, Zhang Y, Le Chatelier E, Zitvogel L, Pons N, Austin-Breneman JL, Haydu LE, Burton EM, Gardner JM, Sirmans E, Hu J, Lazar AJ, Tsujikawa T, Diab A, Tawbi H, Glitza IC, Hwu WJ, Patel SP, Woodman SE, Amaria RN, Davies MA, Gershenwald JE, Hwu P, Lee JE, Zhang J, Coussens LM, Cooper ZA, Futreal PA, Daniel CR, Ajami NJ, Petrosino JF, Tetzlaff MT, Sharma P, Allison JP, Jenq RR, Wargo JA (2018b) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359(6371):97–103. https://doi.org/10.1126/science.aan4236
Gorres KL, Daigle D, Mohanram S, Miller G (2014) Activation and repression of Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus lytic cycles by short- and medium-chain fatty acids. J Virol 88(14):8028–8044. https://doi.org/10.1128/JVI.00722-14
Grat M, Wronka KM, Krasnodebski M, Masior L, Lewandowski Z, Kosinska I, Grat K, Stypulkowski J, Rejowski S, Wasilewicz M, Galecka M, Szachta P, Krawczyk M (2016) Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis. Transplant Proc 48(5):1687–1691. https://doi.org/10.1016/j.transproceed.2016.01.077
Groschwitz KR, Hogan SP (2009) Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol 124(1):3–20; quiz 21-22. https://doi.org/10.1016/j.jaci.2009.05.038
Gulamhusein AF, Eaton JE, Tabibian JH, Atkinson EJ, Juran BD, Lazaridis KN (2016) Duration of inflammatory bowel disease is associated with increased risk of Cholangiocarcinoma in patients with primary Sclerosing cholangitis and IBD. Am J Gastroenterol 111(5):705–711. https://doi.org/10.1038/ajg.2016.55
Gupta H, Youn GS, Shin MJ, Suk KT (2019) Role of gut microbiota in Hepatocarcinogenesis. Microorganisms 7(5). https://doi.org/10.3390/microorganisms7050121
Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ (2008) Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27(2):104–119. https://doi.org/10.1111/j.1365-2036.2007.03562.x
Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y (2008) Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med 45(11):1542–1550. https://doi.org/10.1016/j.freeradbiomed.2008.08.030
Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, Tripathi G, Ashour E, Abdalla MS, Sharada HM, Amin AI, Burt AD, Kumar S, Day CP, McTernan PG (2010) Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond) 7:15. https://doi.org/10.1186/1476-9255-7-15
Hatakeyama M (2014) Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 15(3):306–316. https://doi.org/10.1016/j.chom.2014.02.008
Heymann F, Tacke F (2016) Immunology in the liver—from homeostasis to disease. Nat Rev Gastroenterol Hepatol 13(2):88–110. https://doi.org/10.1038/nrgastro.2015.200
Hildt E, Munz B, Saher G, Reifenberg K, Hofschneider PH (2002) The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J 21(4):525–535. https://doi.org/10.1093/emboj/21.4.525
Hill DB, Barve S, Joshi-Barve S, McClain C (2000) Increased monocyte nuclear factor-kappaB activation and tumor necrosis factor production in alcoholic hepatitis. J Lab Clin Med 135(5):387–395. https://doi.org/10.1067/mlc.2000.106451
Hillman ET, Lu H, Yao T, Nakatsu CH (2017) Microbial ecology along the gastrointestinal tract. Microbes Environ 32(4):300–313. https://doi.org/10.1264/jsme2.ME17017
Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA (2002) The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr 132(5):1012–1017. https://doi.org/10.1093/jn/132.5.1012
Hodges GM, Carr EA, Hazzard RA, Carr KE (1995) Uptake and translocation of microparticles in small intestine. Morphology and quantification of particle distribution. Dig Dis Sci 40(5):967–975. https://doi.org/10.1007/bf02064184
Hofmann AF (2009) The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci (Landmark Ed) 14:2584–2598. https://doi.org/10.2741/3399
Holzinger F, Z’Graggen K, Buchler MW (1999) Mechanisms of biliary carcinogenesis: a pathogenetic multi-stage cascade towards cholangiocarcinoma. Ann Oncol 10(Suppl 4):122–126
Horst AK, Neumann K, Diehl L, Tiegs G (2016) Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol Immunol 13(3):277–292. https://doi.org/10.1038/cmi.2015.112
Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, Gupta S, Moore J, Wrobel MJ, Lerner J, Reich M, Chan JA, Glickman JN, Ikeda K, Hashimoto M, Watanabe G, Daidone MG, Roayaie S, Schwartz M, Thung S, Salvesen HB, Gabriel S, Mazzaferro V, Bruix J, Friedman SL, Kumada H, Llovet JM, Golub TR (2008) Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med 359(19):1995–2004. https://doi.org/10.1056/NEJMoa0804525
Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, Watanabe G, Gabriel S, Friedman SL, Kumada H, Llovet JM, Golub TR (2009) Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 69(18):7385–7392. https://doi.org/10.1158/0008-5472.CAN-09-1089
Hou J, Zhang H, Sun B, Karin M (2019) The immunobiology of hepatocellular carcinoma in humans and mice: basic concepts and therapeutic implications. J Hepatol 72:167. https://doi.org/10.1016/j.jhep.2019.08.014
Hoverstad T, Midtvedt T (1986) Short-chain fatty acids in germfree mice and rats. J Nutr 116(9):1772–1776. https://doi.org/10.1093/jn/116.9.1772
Howe SE, Lickteig DJ, Plunkett KN, Ryerse JS, Konjufca V (2014) The uptake of soluble and particulate antigens by epithelial cells in the mouse small intestine. PLoS One 9(1):e86656. https://doi.org/10.1371/journal.pone.0086656
Hsu RY, Chan CH, Spicer JD, Rousseau MC, Giannias B, Rousseau S, Ferri LE (2011) LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res 71(5):1989–1998. https://doi.org/10.1158/0008-5472.CAN-10-2833
Hu W, Feng Z, Eveleigh J, Iyer G, Pan J, Amin S, Chung FL, Tang MS (2002) The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 23(11):1781–1789. https://doi.org/10.1093/carcin/23.11.1781
Huang XF, Zhao WY, Huang WD (2015) FXR and liver carcinogenesis. Acta Pharmacol Sin 36(1):37–43. https://doi.org/10.1038/aps.2014.117
Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature 486(7402):207–214. https://doi.org/10.1038/nature11234
Hunt RH, Camilleri M, Crowe SE, El-Omar EM, Fox JG, Kuipers EJ, Malfertheiner P, McColl KE, Pritchard DM, Rugge M, Sonnenberg A, Sugano K, Tack J (2015) The stomach in health and disease. Gut 64(10):1650–1668. https://doi.org/10.1136/gutjnl-2014-307595
Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML (2008) Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet 4(11):e1000255. https://doi.org/10.1371/journal.pgen.1000255
Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW (2017) GPCR-mediated signaling of metabolites. Cell Metab 25(4):777–796. https://doi.org/10.1016/j.cmet.2017.03.008
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, Molina DA, Salcedo R, Back T, Cramer S, Dai RM, Kiu H, Cardone M, Naik S, Patri AK, Wang E, Marincola FM, Frank KM, Belkaid Y, Trinchieri G, Goldszmid RS (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342(6161):967–970. https://doi.org/10.1126/science.1240527
Imura S, Miyake H, Izumi K, Tashiro S, Uehara H (2004) Correlation of vascular endothelial cell proliferation with microvessel density and expression of vascular endothelial growth factor and basic fibroblast growth factor in hepatocellular carcinoma. J Med Investig 51(3–4):202–209. https://doi.org/10.2152/jmi.51.202
Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA (2006) Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA 103(10):3920–3925. https://doi.org/10.1073/pnas.0509592103
Jahani-Sherafat S, Alebouyeh M, Moghim S, Ahmadi Amoli H, Ghasemian-Safaei H (2018) Role of gut microbiota in the pathogenesis of colorectal cancer; a review article. Gastroenterol Hepatol Bed Bench 11(2):101–109
Jalan R, Fernandez J, Wiest R, Schnabl B, Moreau R, Angeli P, Stadlbauer V, Gustot T, Bernardi M, Canton R, Albillos A, Lammert F, Wilmer A, Mookerjee R, Vila J, Garcia-Martinez R, Wendon J, Such J, Cordoba J, Sanyal A, Garcia-Tsao G, Arroyo V, Burroughs A, Gines P (2014) Bacterial infections in cirrhosis: a position statement based on the EASL special conference 2013. J Hepatol 60(6):1310–1324. https://doi.org/10.1016/j.jhep.2014.01.024
Janeiro MH, Ramirez MJ, Milagro FI, Martinez JA, Solas M (2018) Implication of Trimethylamine N-oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients 10(10). https://doi.org/10.3390/nu10101398
Janssens S, Beyaert R (2003) Role of toll-like receptors in pathogen recognition. Clin Microbiol Rev 16(4):637–646. https://doi.org/10.1128/cmr.16.4.637-646.2003
Johnson GL, Lapadat R (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298(5600):1911–1912. https://doi.org/10.1126/science.1072682
Johnston MP, Khakoo SI (2019) Immunotherapy for hepatocellular carcinoma: current and future. World J Gastroenterol 25(24):2977–2989. https://doi.org/10.3748/wjg.v25.i24.2977
Kalaitzakis E (2014) Gastrointestinal dysfunction in liver cirrhosis. World J Gastroenterol 20(40):14686–14695. https://doi.org/10.3748/wjg.v20.i40.14686
Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L (2011) Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479(7374):547–551. https://doi.org/10.1038/nature10599
Kanwal F, Kramer JR, Ilyas J, Duan Z, El-Serag HB (2014) HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. veterans with HCV. Hepatology 60(1):98–105. https://doi.org/10.1002/hep.27095
Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, Li L, Desiderio R, Thrift AP, Asch SM, Chu J, El-Serag HB (2018) Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 155(6):1828–1837. e1822. https://doi.org/10.1053/j.gastro.2018.08.024
Kaposi-Novak P, Libbrecht L, Woo HG, Lee YH, Sears NC, Coulouarn C, Conner EA, Factor VM, Roskams T, Thorgeirsson SS (2009) Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res 69(7):2775–2782. https://doi.org/10.1158/0008-5472.CAN-08-3357
Kasztelan-Szczerbinska B, Surdacka A, Celinski K, Rolinski J, Zwolak A, Miacz S, Szczerbinski M (2015) Prognostic significance of the systemic inflammatory and immune balance in alcoholic liver disease with a focus on gender-related differences. PLoS One 10(6):e0128347. https://doi.org/10.1371/journal.pone.0128347
Kathirvel E, Chen P, Morgan K, French SW, Morgan TR (2010) Oxidative stress and regulation of anti-oxidant enzymes in cytochrome P4502E1 transgenic mouse model of non-alcoholic fatty liver. J Gastroenterol Hepatol 25(6):1136–1143. https://doi.org/10.1111/j.1440-1746.2009.06196.x
Kawai T, Akira S (2007) Signaling to NF-kappaB by toll-like receptors. Trends Mol Med 13(11):460–469. https://doi.org/10.1016/j.molmed.2007.09.002
Kawai K, Miyazaki J, Joraku A, Nishiyama H, Akaza H (2013) Bacillus Calmette-Guerin (BCG) immunotherapy for bladder cancer: current understanding and perspectives on engineered BCG vaccine. Cancer Sci 104(1):22–27. https://doi.org/10.1111/cas.12075
Kawaratani H, Moriya K, Namisaki T, Uejima M, Kitade M, Takeda K, Okura Y, Kaji K, Takaya H, Nishimura N, Sato S, Sawada Y, Seki K, Kubo T, Mitoro A, Yamao J, Yoshiji H (2017) Therapeutic strategies for alcoholic liver disease: focusing on inflammation and fibrosis (review). Int J Mol Med 40(2):263–270. https://doi.org/10.3892/ijmm.2017.3015
Kawasaki T, Kawai T (2014) Toll-like receptor signaling pathways. Front Immunol 5:461. https://doi.org/10.3389/fimmu.2014.00461
Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, Katchy CA, Lee C, Moore DD, Fu L (2016) Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell 30(6):909–924. https://doi.org/10.1016/j.ccell.2016.10.007
Kew MC (1998) Hepatitis viruses and hepatocellular carcinoma. Res Virol 149(5):257–262. https://doi.org/10.1016/s0923-2516(99)89003-7
Khalil DN, Smith EL, Brentjens RJ, Wolchok JD (2016) The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 13(5):273–290. https://doi.org/10.1038/nrclinonc.2016.25
Knolle PA, Wohlleber D (2016) Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol 13(3):347–353. https://doi.org/10.1038/cmi.2016.5
Kobyliak N, Conte C, Cammarota G, Haley AP, Styriak I, Gaspar L, Fusek J, Rodrigo L, Kruzliak P (2016) Probiotics in prevention and treatment of obesity: a critical view. Nutr Metab (Lond) 13:14. https://doi.org/10.1186/s12986-016-0067-0
Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F (2016) From dietary Fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165(6):1332–1345. https://doi.org/10.1016/j.cell.2016.05.041
Koliada A, Syzenko G, Moseiko V, Budovska L, Puchkov K, Perederiy V, Gavalko Y, Dorofeyev A, Romanenko M, Tkach S, Sineok L, Lushchak O, Vaiserman A (2017) Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol 17(1):120. https://doi.org/10.1186/s12866-017-1027-1
Krajmalnik-Brown R, Ilhan ZE, Kang DW, DiBaise JK (2012) Effects of gut microbes on nutrient absorption and energy regulation. Nutr Clin Pract 27(2):201–214. https://doi.org/10.1177/0884533611436116
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133(6):1019–1031. https://doi.org/10.1016/j.cell.2008.03.039
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev 24(22):2463–2479. https://doi.org/10.1101/gad.1971610
Lambert AW, Pattabiraman DR, Weinberg RA (2017) Emerging biological principles of metastasis. Cell 168(4):670–691. https://doi.org/10.1016/j.cell.2016.11.037
Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, International Agency for Research on Cancer Handbook Working G (2016) Body fatness and cancer—viewpoint of the IARC working group. N Engl J Med 375(8):794–798. https://doi.org/10.1056/NEJMsr1606602
Le Bourhis L, Guerri L, Dusseaux M, Martin E, Soudais C, Lantz O (2011) Mucosal-associated invariant T cells: unconventional development and function. Trends Immunol 32(5):212–218. https://doi.org/10.1016/j.it.2011.02.005
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jorgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clement K, Dore J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T, HITc M, Bork P, Wang J, Ehrlich SD, Pedersen O (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500(7464):541–546. https://doi.org/10.1038/nature12506
Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, Perlemuter G, Cassard-Doulcier AM, Gerard P (2013) Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62(12):1787–1794. https://doi.org/10.1136/gutjnl-2012-303816
LeFevre ME, Olivo R, Vanderhoff JW, Joel DD (1978) Accumulation of latex in Peyer’s patches and its subsequent appearance in villi and mesenteric lymph nodes. Proc Soc Exp Biol Med 159(2):298–302. https://doi.org/10.3181/00379727-159-40336
Lelouard H, Fallet M, de Bovis B, Meresse S, Gorvel JP (2012) Peyer’s patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology 142(3):592–601. e593. https://doi.org/10.1053/j.gastro.2011.11.039
Leung C, Rivera L, Furness JB, Angus PW (2016) The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 13(7):412–425. https://doi.org/10.1038/nrgastro.2016.85
Levy M, Thaiss CA, Elinav E (2016) Metabolites: messengers between the microbiota and the immune system. Genes Dev 30(14):1589–1597. https://doi.org/10.1101/gad.284091.116
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102(31):11070–11075. https://doi.org/10.1073/pnas.0504978102
Li T, Chiang JY (2014) Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 66(4):948–983. https://doi.org/10.1124/pr.113.008201
Li B, Zhang C, Zhan YT (2018) Nonalcoholic fatty liver disease cirrhosis: a review of its epidemiology, risk factors, clinical presentation, diagnosis, management, and prognosis. Can J Gastroenterol Hepatol 2018:2784537. https://doi.org/10.1155/2018/2784537
Lieber CS (2005) Metabolism of alcohol. Clin Liver Dis 9(1):1–35. https://doi.org/10.1016/j.cld.2004.10.005
Lieber CS, DeCarli LM (1968) Ethanol oxidation by hepatic microsomes: adaptive increase after ethanol feeding. Science 162(3856):917–918. https://doi.org/10.1126/science.162.3856.917
Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH (2010) The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol 7(5):251–264. https://doi.org/10.1038/nrgastro.2010.41
Lin L, Amin R, Gallicano GI, Glasgow E, Jogunoori W, Jessup JM, Zasloff M, Marshall JL, Shetty K, Johnson L, Mishra L, He AR (2009) The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene 28(7):961–972. https://doi.org/10.1038/onc.2008.448
Lin R, Zhou L, Zhang J, Wang B (2015) Abnormal intestinal permeability and microbiota in patients with autoimmune hepatitis. Int J Clin Exp Pathol 8(5):5153–5160
Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Keach J, Mooney J, Sargeant C, Braaten J, Bernard T, King D, Miceli E, Schmoll J, Hoskin T, Thapa P, Enders F (2009) High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 50(3):808–814. https://doi.org/10.1002/hep.23082
Liu C, Gao H, Cao L, Gui S, Liu Q, Li C, Li D, Gong L, Zhang Y (2016) The role of FSCN1 in migration and invasion of pituitary adenomas. Mol Cell Endocrinol 419:217–224. https://doi.org/10.1016/j.mce.2015.10.021
Llorente C, Schnabl B (2015) The gut microbiota and liver disease. Cell Mol Gastroenterol Hepatol 1(3):275–284. https://doi.org/10.1016/j.jcmgh.2015.04.003
Lloyd-Price J, Abu-Ali G, Huttenhower C (2016) The healthy human microbiome. Genome Med 8(1):51. https://doi.org/10.1186/s13073-016-0307-y
Loilome W, Bungkanjana P, Techasen A, Namwat N, Yongvanit P, Puapairoj A, Khuntikeo N, Riggins GJ (2014) Activated macrophages promote Wnt/beta-catenin signaling in cholangiocarcinoma cells. Tumour Biol 35(6):5357–5367. https://doi.org/10.1007/s13277-014-1698-2
Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, Nakajima-Takagi Y, Iwama A, Koga T, Sugimoto Y, Ozawa T, Nakamura M, Kumagai M, Watashi K, Taketo MM, Aoki T, Narumiya S, Oshima M, Arita M, Hara E, Ohtani N (2017) Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov 7(5):522–538. https://doi.org/10.1158/2159-8290.CD-16-0932
Lorenzo-Zuniga V, Bartoli R, Planas R, Hofmann AF, Vinado B, Hagey LR, Hernandez JM, Mane J, Alvarez MA, Ausina V, Gassull MA (2003) Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 37(3):551–557. https://doi.org/10.1053/jhep.2003.50116
Louis P, Flint HJ (2017) Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol 19(1):29–41. https://doi.org/10.1111/1462-2920.13589
Lowe SW, Cepero E, Evan G (2004) Intrinsic tumour suppression. Nature 432(7015):307–315. https://doi.org/10.1038/nature03098
Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, Johnson RL (2010) Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci USA 107(4):1437–1442. https://doi.org/10.1073/pnas.0911427107
Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, Lowe SW (2013) Non-cell-autonomous tumor suppression by p53. Cell 153(2):449–460. https://doi.org/10.1016/j.cell.2013.03.020
Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, Motola DL, Luther S, Bohr S, Jeoung SW, Deshpande V, Singh G, Turner JR, Yarmush ML, Chung RT, Patel SJ (2015) Hepatic injury in nonalcoholic Steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol 1(2):222–232. https://doi.org/10.1016/j.jcmgh.2015.01.001
Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, Han M, Thornton AM, Zhang H, Egger M, Luo J, Felsher DW, McVicar DW, Weber A, Heikenwalder M, Greten TF (2016) NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531(7593):253–257. https://doi.org/10.1038/nature16969
Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, Ritz T, Longerich T, Theriot CM, McCulloch JA, Roy S, Yuan W, Thovarai V, Sen SK, Ruchirawat M, Korangy F, Wang XW, Trinchieri G, Greten TF (2018) Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360(6391):eaan5931. https://doi.org/10.1126/science.aan5931
Macpherson AJ, Smith K (2006) Mesenteric lymph nodes at the center of immune anatomy. J Exp Med 203(3):497–500. https://doi.org/10.1084/jem.20060227
Malaguarnera G, Giordano M, Nunnari G, Bertino G, Malaguarnera M (2014) Gut microbiota in alcoholic liver disease: pathogenetic role and therapeutic perspectives. World J Gastroenterol 20(44):16639–16648. https://doi.org/10.3748/wjg.v20.i44.16639
Mandard S, Zandbergen F, van Straten E, Wahli W, Kuipers F, Muller M, Kersten S (2006) The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J Biol Chem 281(2):934–944. https://doi.org/10.1074/jbc.M506519200
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461(7268):1282–1286. https://doi.org/10.1038/nature08530
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359(6371):104–108. https://doi.org/10.1126/science.aao3290
Matsumoto M, Seya T (2008) TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv Drug Deliv Rev 60(7):805–812. https://doi.org/10.1016/j.addr.2007.11.005
McNabney SM, Henagan TM (2017) Short chain fatty acids in the colon and peripheral tissues: a focus on butyrate, colon cancer, obesity and insulin resistance. Nutrients 9(12). https://doi.org/10.3390/nu9121348
Mencin A, Kluwe J, Schwabe RF (2009) Toll-like receptors as targets in chronic liver diseases. Gut 58(5):704–720. https://doi.org/10.1136/gut.2008.156307
Michail S, Lin M, Frey MR, Fanter R, Paliy O, Hilbush B, Reo NV (2015) Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol Ecol 91(2):1–9. https://doi.org/10.1093/femsec/fiu002
Michelotti GA, Machado MV, Diehl AM (2013) NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 10(11):656–665. https://doi.org/10.1038/nrgastro.2013.183
Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Masciana R, Forgione A, Gabrieli ML, Perotti G, Vecchio FM, Rapaccini G, Gasbarrini G, Day CP, Grieco A (2009) Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49(6):1877–1887. https://doi.org/10.1002/hep.22848
Mima K, Baba H (2019) The gut microbiome, antitumor immunity, and liver cancer. Hepatobiliary Surg Nutr 8(1):67–68. https://doi.org/10.21037/hbsn.2018.11.09
Mirza NN, McCloud JM, Cheetham MJ (2009) Clostridium septicum sepsis and colorectal cancer – a reminder. World J Surg Oncol 7:73. https://doi.org/10.1186/1477-7819-7-73
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22(2):240–273, Table of Contents. https://doi.org/10.1128/CMR.00046-08
Mokdad AH, Dwyer-Lindgren L, Fitzmaurice C, Stubbs RW, Bertozzi-Villa A, Morozoff C, Charara R, Allen C, Naghavi M, Murray CJ (2017) Trends and patterns of disparities in cancer mortality among US counties, 1980-2014. JAMA 317(4):388–406. https://doi.org/10.1001/jama.2016.20324
Mokhtari Z, Gibson DL, Hekmatdoost A (2017) Nonalcoholic fatty liver disease, the gut microbiome, and diet. Adv Nutr 8(2):240–252. https://doi.org/10.3945/an.116.013151
Moller S, Henriksen JH, Bendtsen F (2014) Extrahepatic complications to cirrhosis and portal hypertension: haemodynamic and homeostatic aspects. World J Gastroenterol 20(42):15499–15517. https://doi.org/10.3748/wjg.v20.i42.15499
Monie A, Hung CF, Roden R, Wu TC (2008) Cervarix: a vaccine for the prevention of HPV 16, 18-associated cervical cancer. Biologics 2(1):97–105
Moreira AP, Texeira TF, Ferreira AB, Peluzio Mdo C, Alfenas Rde C (2012) Influence of a high-fat diet on gut microbiota, intestinal permeability and metabolic endotoxaemia. Br J Nutr 108(5):801–809. https://doi.org/10.1017/S0007114512001213
Mossanen JC, Kohlhepp M, Wehr A, Krenkel O, Liepelt A, Roeth AA, Mockel D, Heymann F, Lammers T, Gassler N, Hermann J, Jankowski J, Neumann UP, Luedde T, Trautwein C, Tacke F (2019) CXCR6 inhibits Hepatocarcinogenesis by promoting natural killer T- and CD4(+) T-cell-dependent control of senescence. Gastroenterology 156(6):1877–1889. e1874. https://doi.org/10.1053/j.gastro.2019.01.247
Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, McGilvray ID, Allard JP (2013) Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 58(1):120–127. https://doi.org/10.1002/hep.26319
Munakata T, Nakamura M, Liang Y, Li K, Lemon SM (2005) Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc Natl Acad Sci USA 102(50):18159–18164. https://doi.org/10.1073/pnas.0505605102
Murphy EA, Velazquez KT, Herbert KM (2015) Influence of high-fat diet on gut microbiota: a driving force for chronic disease risk. Curr Opin Clin Nutr Metab Care 18(5):515–520. https://doi.org/10.1097/MCO.0000000000000209
Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, Koike K, Kaufman RJ, Karin M (2014) ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26(3):331–343. https://doi.org/10.1016/j.ccr.2014.07.001
Nakanishi K, Sakamoto M, Yamasaki S, Todo S, Hirohashi S (2005) Akt phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer 103(2):307–312. https://doi.org/10.1002/cncr.20774
Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M (2007) Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317(5834):121–124. https://doi.org/10.1126/science.1140485
Nishimura T, Kitamura H, Iwakabe K, Yahata T, Ohta A, Sato M, Takeda K, Okumura K, Van Kaer L, Kawano T, Taniguchi M, Nakui M, Sekimoto M, Koda T (2000) The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d-expressing dendritic cells to NKT cells induces the differentiation of antigen-specific cytotoxic T lymphocytes. Int Immunol 12(7):987–994. https://doi.org/10.1093/intimm/12.7.987
Okada Y, Tsuzuki Y, Hokari R, Komoto S, Kurihara C, Kawaguchi A, Nagao S, Miura S (2009) Anti-inflammatory effects of the genus Bifidobacterium on macrophages by modification of phospho-I kappaB and SOCS gene expression. Int J Exp Pathol 90(2):131–140. https://doi.org/10.1111/j.1365-2613.2008.00632.x
Okamoto Y, Shinjo K, Shimizu Y, Sano T, Yamao K, Gao W, Fujii M, Osada H, Sekido Y, Murakami S, Tanaka Y, Joh T, Sato S, Takahashi S, Wakita T, Zhu J, Issa JP, Kondo Y (2014) Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology 146(2):562–572. https://doi.org/10.1053/j.gastro.2013.10.056
Okusaka T, Ikeda M (2018) Immunotherapy for hepatocellular carcinoma: current status and future perspectives. ESMO Open 3(Suppl 1):e000455. https://doi.org/10.1136/esmoopen-2018-000455
Oliveira-Nascimento L, Massari P, Wetzler LM (2012) The role of TLR2 in infection and immunity. Front Immunol 3:79. https://doi.org/10.3389/fimmu.2012.00079
Paquissi FC (2016) Immune imbalances in non-alcoholic fatty liver disease: from general biomarkers and neutrophils to Interleukin-17 Axis activation and new therapeutic targets. Front Immunol 7:490. https://doi.org/10.3389/fimmu.2016.00490
Pardi DS, Loftus EV Jr, Kremers WK, Keach J, Lindor KD (2003) Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 124(4):889–893. https://doi.org/10.1053/gast.2003.50156
Park BS, Lee JO (2013) Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med 45:e66. https://doi.org/10.1038/emm.2013.97
Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M (2010) Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140(2):197–208. https://doi.org/10.1016/j.cell.2009.12.052
Paterlini P, Poussin K, Kew M, Franco D, Brechot C (1995) Selective accumulation of the X transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology 21(2):313–321
Pendyala S, Walker JM, Holt PR (2012) A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 142(5):1100–1101. e1102. https://doi.org/10.1053/j.gastro.2012.01.034
Pennington HL, Hall PM, Wilce PA, Worrall S (1997) Ethanol feeding enhances inflammatory cytokine expression in lipopolysaccharide-induced hepatitis. J Gastroenterol Hepatol 12(4):305–313. https://doi.org/10.1111/j.1440-1746.1997.tb00426.x
Perez MJ, Briz O (2009) Bile-acid-induced cell injury and protection. World J Gastroenterol 15(14):1677–1689. https://doi.org/10.3748/wjg.15.1677
Peterson LW, Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14(3):141–153. https://doi.org/10.1038/nri3608
Petrasek J, Mandrekar P, Szabo G (2010) Toll-like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract 2010:1. https://doi.org/10.1155/2010/710381
Phelan JP, Reen FJ, Caparros-Martin JA, O’Connor R, O’Gara F (2017) Rethinking the bile acid/gut microbiome axis in cancer. Oncotarget 8(70):115736–115747. https://doi.org/10.18632/oncotarget.22803
Pijls KE, Jonkers DM, Elamin EE, Masclee AA, Koek GH (2013) Intestinal epithelial barrier function in liver cirrhosis: an extensive review of the literature. Liver Int 33(10):1457–1469. https://doi.org/10.1111/liv.12271
Pinato DJ, Howlett S, Ottaviani D, Urus H, Patel A, Mineo T, Brock C, Power D, Hatcher O, Falconer A, Ingle M, Brown A, Gujral D, Partridge S, Sarwar N, Gonzalez M, Bendle M, Lewanski C, Newsom-Davis T, Allara E, Bower M (2019) Association of prior antibiotic treatment with survival and response to immune checkpoint inhibitor therapy in patients with Cancer. JAMA Oncol 5:1774. https://doi.org/10.1001/jamaoncol.2019.2785
Pinlaor S, Hiraku Y, Ma N, Yongvanit P, Semba R, Oikawa S, Murata M, Sripa B, Sithithaworn P, Kawanishi S (2004) Mechanism of NO-mediated oxidative and nitrative DNA damage in hamsters infected with Opisthorchis viverrini: a model of inflammation-mediated carcinogenesis. Nitric Oxide 11(2):175–183. https://doi.org/10.1016/j.niox.2004.08.004
Pinzone MR, Celesia BM, Di Rosa M, Cacopardo B, Nunnari G (2012) Microbial translocation in chronic liver diseases. Int J Microbiol 2012:694629. https://doi.org/10.1155/2012/694629
Ponziani FR, Bhoori S, Castelli C, Putignani L, Rivoltini L, Del Chierico F, Sanguinetti M, Morelli D, Paroni Sterbini F, Petito V, Reddel S, Calvani R, Camisaschi C, Picca A, Tuccitto A, Gasbarrini A, Pompili M, Mazzaferro V (2019) Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 69(1):107–120. https://doi.org/10.1002/hep.30036
Purcell RV, Pearson J, Aitchison A, Dixon L, Frizelle FA, Keenan JI (2017) Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS One 12(2):e0171602. https://doi.org/10.1371/journal.pone.0171602
Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ (2008) Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134(2):568–576. https://doi.org/10.1053/j.gastro.2007.10.039
Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, Kang YJ, Keshavarzian A, Rao R, Sartor RB, Swanson C, Turner JR (2008) Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 42(5):349–361. https://doi.org/10.1016/j.alcohol.2008.03.131
Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, Mohan N, Aykut B, Usyk M, Torres LE, Werba G, Zhang K, Guo Y, Li Q, Akkad N, Lall S, Wadowski B, Gutierrez J, Kochen Rossi JA, Herzog JW, Diskin B, Torres-Hernandez A, Leinwand J, Wang W, Taunk PS, Savadkar S, Janal M, Saxena A, Li X, Cohen D, Sartor RB, Saxena D, Miller G (2018) The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 8(4):403–416. https://doi.org/10.1158/2159-8290.CD-17-1134
Quiroz SC, Bucio L, Souza V, Hernandez E, Gonzalez E, Gomez-Quiroz L, Kershenobich D, Vargas-Vorackova F, Gutierrez-Ruiz MC (2001) Effect of endotoxin pretreatment on hepatic stellate cell response to ethanol and acetaldehyde. J Gastroenterol Hepatol 16(11):1267–1273. https://doi.org/10.1046/j.1440-1746.2001.02619.x
Rabot S, Membrez M, Bruneau A, Gerard P, Harach T, Moser M, Raymond F, Mansourian R, Chou CJ (2010) Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J 24(12):4948–4959. https://doi.org/10.1096/fj.10-164921
Racanelli V, Rehermann B (2006) The liver as an immunological organ. Hepatology 43(2 Suppl 1):S54–S62. https://doi.org/10.1002/hep.21060
Raimondi S, Bruno S, Mondelli MU, Maisonneuve P (2009) Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J Hepatol 50(6):1142–1154. https://doi.org/10.1016/j.jhep.2009.01.019
Rajilic-Stojanovic M, de Vos WM (2014) The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev 38(5):996–1047. https://doi.org/10.1111/1574-6976.12075
Ramirez-Ortiz ZG, Specht CA, Wang JP, Lee CK, Bartholomeu DC, Gazzinelli RT, Levitz SM (2008) Toll-like receptor 9-dependent immune activation by unmethylated CpG motifs in Aspergillus fumigatus DNA. Infect Immun 76(5):2123–2129. https://doi.org/10.1128/IAI.00047-08
Rao R (2009) Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 50(2):638–644. https://doi.org/10.1002/hep.23009
Razumilava N, Gores GJ (2014) Cholangiocarcinoma. Lancet 383(9935):2168–2179. https://doi.org/10.1016/S0140-6736(13)61903-0
Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, Xie H, Chen X, Shao L, Zhang R, Xu S, Zhang H, Cui G, Chen X, Sun R, Wen H, Lerut JP, Kan Q, Li L, Zheng S (2019) Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 68(6):1014–1023. https://doi.org/10.1136/gutjnl-2017-315084
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341(6150):1241214. https://doi.org/10.1126/science.1241214
Ridlon JM, Kang DJ, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47(2):241–259. https://doi.org/10.1194/jlr.R500013-JLR200
Ridlon JM, Alves JM, Hylemon PB, Bajaj JS (2013) Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes 4(5):382–387. https://doi.org/10.4161/gmic.25723
Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB (2016) Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7(1):22–39. https://doi.org/10.1080/19490976.2015.1127483
Riviere A, Selak M, Lantin D, Leroy F, De Vuyst L (2016) Bifidobacteria and butyrate-producing colon bacteria: importance and strategies for their stimulation in the Human gut. Front Microbiol 7:979. https://doi.org/10.3389/fmicb.2016.00979
Robinson WS (1994) Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu Rev Med 45:297–323. https://doi.org/10.1146/annurev.med.45.1.297
Robinson MW, Harmon C, O’Farrelly C (2016) Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol 13(3):267–276. https://doi.org/10.1038/cmi.2016.3
Romano KA, Vivas EI, Amador-Noguez D, Rey FE (2015) Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. MBio 6(2):e02481. https://doi.org/10.1128/mBio.02481-14
Ronis MJJ, Pedersen KB, Watt J (2018) Adverse effects of nutraceuticals and dietary supplements. Annu Rev Pharmacol Toxicol 58:583–601. https://doi.org/10.1146/annurev-pharmtox-010617-052844
Roskams T (2006) Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 25(27):3818–3822. https://doi.org/10.1038/sj.onc.1209558
Routy B, Gopalakrishnan V, Daillere R, Zitvogel L, Wargo JA, Kroemer G (2018a) The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol 15(6):382–396. https://doi.org/10.1038/s41571-018-0006-2
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragon L, Jacquelot N, Qu B, Ferrere G, Clemenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, Zitvogel L (2018b) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359(6371):91–97. https://doi.org/10.1126/science.aan3706
Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, Tuohy K (2018) Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr 57(1):1–24. https://doi.org/10.1007/s00394-017-1445-8
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14(2):195–206. https://doi.org/10.1016/j.chom.2013.07.012
Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, Miltiadous O, Walesky C, Deshpande V, Zhu AX, Hezel AF, Yen KE, Straley KS, Travins J, Popovici-Muller J, Gliser C, Ferrone CR, Apte U, Llovet JM, Wong KK, Ramaswamy S, Bardeesy N (2014) Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 513(7516):110–114. https://doi.org/10.1038/nature13441
Santamaria E, Munoz J, Fernandez-Irigoyen J, Sesma L, Mora MI, Berasain C, Lu SC, Mato JM, Prieto J, Avila MA, Corrales FJ (2006) Molecular profiling of hepatocellular carcinoma in mice with a chronic deficiency of hepatic s-adenosylmethionine: relevance in human liver diseases. J Proteome Res 5(4):944–953. https://doi.org/10.1021/pr050429v
Schaap FG, Trauner M, Jansen PL (2014) Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 11(1):55–67. https://doi.org/10.1038/nrgastro.2013.151
Scheppach W, Bartram HP, Richter F (1995) Role of short-chain fatty acids in the prevention of colorectal cancer. Eur J Cancer 31A(7–8):1077–1080. https://doi.org/10.1016/0959-8049(95)00165-f
Schnabl B, Brenner DA (2014) Interactions between the intestinal microbiome and liver diseases. Gastroenterology 146(6):1513–1524. https://doi.org/10.1053/j.gastro.2014.01.020
Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, Zimmermann HW, Vucur M, Huss S, Gassler N, Wasmuth HE, Lira SA, Zender L, Luedde T, Trautwein C, Tacke F (2012) Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut 61(12):1733–1743. https://doi.org/10.1136/gutjnl-2011-301116
Schwabe RF, Seki E, Brenner DA (2006) Toll-like receptor signaling in the liver. Gastroenterology 130(6):1886–1900. https://doi.org/10.1053/j.gastro.2006.01.038
Seitz HK, Homann N (2007) The role of acetaldehyde in alcohol-associated cancer of the gastrointestinal tract. Novartis Found Symp 285:110–119; discussion 119–114, 198–119. https://doi.org/10.1002/9780470511848.ch8
Seki E, Brenner DA (2008) Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48(1):322–335. https://doi.org/10.1002/hep.22306
Sender R, Fuchs S, Milo R (2016) Revised estimates for the number of Human and Bacteria cells in the body. PLoS Biol 14(8):e1002533. https://doi.org/10.1371/journal.pbio.1002533
Sepulveda AR (2013) Helicobacter, inflammation, and gastric Cancer. Curr Pathobiol Rep 1(1):9–18. https://doi.org/10.1007/s40139-013-0009-8
Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, Zhang L, Sharma U, Giri B, Garg B, Ferrantella A, Vickers SM, Banerjee S, Dawra R, Roy S, Ramakrishnan S, Saluja A, Dudeja V (2018) Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology 155(1):33–37. e36. https://doi.org/10.1053/j.gastro.2018.04.001
Setshedi M, Wands JR, Monte SM (2010) Acetaldehyde adducts in alcoholic liver disease. Oxidative Med Cell Longev 3(3):178–185. https://doi.org/10.4161/oxim.3.3.12288
Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, Vrbanac AF, Li W, Perkins A, Matsutani T, Zhong Z, Dhar D, Navas-Molina JA, Xu J, Loomba R, Downes M, Yu RT, Evans RM, Dorrestein PC, Knight R, Benner C, Anstee QM, Karin M (2017) Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551(7680):340–345. https://doi.org/10.1038/nature24302
Shang FM, Liu HL (2018) Fusobacterium nucleatum and colorectal cancer: a review. World J Gastrointest Oncol 10(3):71–81. https://doi.org/10.4251/wjgo.v10.i3.71
Sharpton SR, Ajmera V, Loomba R (2019) Emerging role of the gut Microbiome in nonalcoholic fatty liver disease: from composition to function. Clin Gastroenterol Hepatol 17(2):296–306. https://doi.org/10.1016/j.cgh.2018.08.065
Siegel AB, Zhu AX (2009) Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer 115(24):5651–5661. https://doi.org/10.1002/cncr.24687
Simbolo M, Fassan M, Ruzzenente A, Mafficini A, Wood LD, Corbo V, Melisi D, Malleo G, Vicentini C, Malpeli G, Antonello D, Sperandio N, Capelli P, Tomezzoli A, Iacono C, Lawlor RT, Bassi C, Hruban RH, Guglielmi A, Tortora G, de Braud F, Scarpa A (2014) Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 5(9):2839–2852. https://doi.org/10.18632/oncotarget.1943
Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, Abrouk M, Farahnik B, Nakamura M, Zhu TH, Bhutani T, Liao W (2017) Influence of diet on the gut microbiome and implications for human health. J Transl Med 15(1):73. https://doi.org/10.1186/s12967-017-1175-y
Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R, Lapek JD Jr, Zhang L, Wang WB, Hao S, Flythe MD, Gonzalez DJ, Cani PD, Conejo-Garcia JR, Xiong N, Kennett MJ, Joe B, Patterson AD, Gewirtz AT, Vijay-Kumar M (2018) Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell 175(3):679–694. e622. https://doi.org/10.1016/j.cell.2018.09.004
Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM (2011) Intestinal macrophages and response to microbial encroachment. Mucosal Immunol 4(1):31–42. https://doi.org/10.1038/mi.2010.66
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341(6145):569–573. https://doi.org/10.1126/science.1241165
Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A (2010) The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int 4(4):659–672. https://doi.org/10.1007/s12072-010-9219-x
Sorg JA, Sonenshein AL (2008) Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190(7):2505–2512. https://doi.org/10.1128/JB.01765-07
Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA (2011) Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 140(3):976–986. https://doi.org/10.1053/j.gastro.2010.11.049
Sriamporn S, Pisani P, Pipitgool V, Suwanrungruang K, Kamsa-ard S, Parkin DM (2004) Prevalence of Opisthorchis viverrini infection and incidence of cholangiocarcinoma in Khon Kaen, Northeast Thailand. Tropical Med Int Health 9(5):588–594. https://doi.org/10.1111/j.1365-3156.2004.01234.x
Sripa B, Bethony JM, Sithithaworn P, Kaewkes S, Mairiang E, Loukas A, Mulvenna J, Laha T, Hotez PJ, Brindley PJ (2011) Opisthorchiasis and Opisthorchis-associated cholangiocarcinoma in Thailand and Laos. Acta Trop 120(Suppl 1):S158–S168. https://doi.org/10.1016/j.actatropica.2010.07.006
Streba LA, Vere CC, Rogoveanu I, Streba CT (2015) Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: an open question. World J Gastroenterol 21(14):4103–4110. https://doi.org/10.3748/wjg.v21.i14.4103
Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, Schurch CM, McCoy KD, Kuehne SA, Minton NP, Stecher B, Bernier-Latmani R, Hapfelmeier S (2016) Functional intestinal bile acid 7alpha-Dehydroxylation by Clostridium scindens associated with protection from Clostridium difficile infection in a Gnotobiotic mouse model. Front Cell Infect Microbiol 6:191. https://doi.org/10.3389/fcimb.2016.00191
Sugimoto Y, Narumiya S (2007) Prostaglandin E receptors. J Biol Chem 282(16):11613–11617. https://doi.org/10.1074/jbc.R600038200
Sunami Y, Ringelhan M, Kokai E, Lu M, O’Connor T, Lorentzen A, Weber A, Rodewald AK, Mullhaupt B, Terracciano L, Gul S, Wissel S, Leithauser F, Krappmann D, Riedl P, Hartmann D, Schirmbeck R, Strnad P, Huser N, Kleeff J, Friess H, Schmid RM, Geisler F, Wirth T, Heikenwalder M (2016) Canonical NF-kappaB signaling in hepatocytes acts as a tumor-suppressor in hepatitis B virus surface antigen-driven hepatocellular carcinoma by controlling the unfolded protein response. Hepatology 63(5):1592–1607. https://doi.org/10.1002/hep.28435
Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, Albano E (2014) Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 59(3):886–897. https://doi.org/10.1002/hep.26749
Tada M, Kanai F, Tanaka Y, Tateishi K, Ohta M, Asaoka Y, Seto M, Muroyama R, Fukai K, Imazeki F, Kawabe T, Yokosuka O, Omata M (2008) Down-regulation of hedgehog-interacting protein through genetic and epigenetic alterations in human hepatocellular carcinoma. Clin Cancer Res 14(12):3768–3776. https://doi.org/10.1158/1078-0432.CCR-07-1181
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820. https://doi.org/10.1016/j.cell.2010.01.022
Tanaka M, Tanaka H, Tsukuma H, Ioka A, Oshima A, Nakahara T (2010) Risk factors for intrahepatic cholangiocarcinoma: a possible role of hepatitis B virus. J Viral Hepat 17(10):742–748. https://doi.org/10.1111/j.1365-2893.2009.01243.x
Tarao K, Fujiyama S, Ohkawa S, Miyakawa K, Tamai S, Hirokawa S, Masaki T, Tanaka K (2005) Ursodiol use is possibly associated with lower incidence of hepatocellular carcinoma in hepatitis C virus-associated liver cirrhosis. Cancer Epidemiol Biomark Prev 14(1):164–169
Terabe M, Berzofsky JA (2008) The role of NKT cells in tumor immunity. Adv Cancer Res 101:277–348. https://doi.org/10.1016/S0065-230X(08)00408-9
Testino G, Leone S, Borro P (2014) Alcohol and hepatocellular carcinoma: a review and a point of view. World J Gastroenterol 20(43):15943–15954. https://doi.org/10.3748/wjg.v20.i43.15943
Teufel A, Weinmann A, Centner C, Piendl A, Lohse AW, Galle PR, Kanzler S (2009) Hepatocellular carcinoma in patients with autoimmune hepatitis. World J Gastroenterol 15(5):578–582. https://doi.org/10.3748/wjg.15.578
Thaiss CA, Zmora N, Levy M, Elinav E (2016) The microbiome and innate immunity. Nature 535(7610):65–74. https://doi.org/10.1038/nature18847
Thompson MD, Monga SP (2007) WNT/beta-catenin signaling in liver health and disease. Hepatology 45(5):1298–1305. https://doi.org/10.1002/hep.21651
Thuwajit C, Thuwajit P, Uchida K, Daorueang D, Kaewkes S, Wongkham S, Miwa M (2006) Gene expression profiling defined pathways correlated with fibroblast cell proliferation induced by Opisthorchis viverrini excretory/secretory product. World J Gastroenterol 12(22):3585–3592. https://doi.org/10.3748/wjg.v12.i22.3585
Tiegs G, Lohse AW (2010) Immune tolerance: what is unique about the liver. J Autoimmun 34(1):1–6. https://doi.org/10.1016/j.jaut.2009.08.008
Topping DL, Clifton PM (2001) Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 81(3):1031–1064. https://doi.org/10.1152/physrev.2001.81.3.1031
Toth CA, Thomas P (1992) Liver endocytosis and Kupffer cells. Hepatology 16(1):255–266. https://doi.org/10.1002/hep.1840160137
Toubal A, Nel I, Lotersztajn S, Lehuen A (2019) Mucosal-associated invariant T cells and disease. Nat Rev Immunol 19(10):643–657. https://doi.org/10.1038/s41577-019-0191-y
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344(6184):641–645. https://doi.org/10.1126/science.1251102
Treiber G (2009) mTOR inhibitors for hepatocellular cancer: a forward-moving target. Expert Rev Anticancer Ther 9(2):247–261. https://doi.org/10.1586/14737140.9.2.247
Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, Knight R (2018) The gut-liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 15(7):397–411. https://doi.org/10.1038/s41575-018-0011-z
Tsukamoto H, Lu SC (2001) Current concepts in the pathogenesis of alcoholic liver injury. FASEB J 15(8):1335–1349. https://doi.org/10.1096/fj.00-0650rev
Turnbaugh PJ, Gordon JI (2009) The core gut microbiome, energy balance and obesity. J Physiol 587(Pt 17):4153–4158. https://doi.org/10.1113/jphysiol.2009.174136
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444(7122):1027–1031. https://doi.org/10.1038/nature05414
Turnbaugh PJ, Backhed F, Fulton L, Gordon JI (2008) Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3(4):213–223. https://doi.org/10.1016/j.chom.2008.02.015
Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9(11):799–809. https://doi.org/10.1038/nri2653
Twu JS, Schloemer RH (1987) Transcriptional trans-activating function of hepatitis B virus. J Virol 61(11):3448–3453
Tyson GL, El-Serag HB (2011) Risk factors for cholangiocarcinoma. Hepatology 54(1):173–184. https://doi.org/10.1002/hep.24351
Urdaneta V, Casadesus J (2017) Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med (Lausanne) 4:163. https://doi.org/10.3389/fmed.2017.00163
Van den Abbeele P, Belzer C, Goossens M, Kleerebezem M, De Vos WM, Thas O, De Weirdt R, Kerckhof FM, Van de Wiele T (2013) Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J 7(5):949–961. https://doi.org/10.1038/ismej.2012.158
Villanueva A (2019) Hepatocellular Carcinoma. N Engl J Med 380(15):1450–1462. https://doi.org/10.1056/NEJMra1713263
Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM (2007) Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis 27(1):55–76. https://doi.org/10.1055/s-2006-960171
Vollmar B, Menger MD (2009) The hepatic microcirculation: mechanistic contributions and therapeutic targets in liver injury and repair. Physiol Rev 89(4):1269–1339. https://doi.org/10.1152/physrev.00027.2008
Vos MB, McClain CJ (2009) Fructose takes a toll. Hepatology 50(4):1004–1006. https://doi.org/10.1002/hep.23212
Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D (2008) Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem 19(9):587–593. https://doi.org/10.1016/j.jnutbio.2007.08.002
Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W (2008) Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48(5):1632–1643. https://doi.org/10.1002/hep.22519
Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, Kudo E, Shimada M, Sano T (2010) High expression of toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer 102(5):908–915. https://doi.org/10.1038/sj.bjc.6605558
Wang HX, Liu M, Weng SY, Li JJ, Xie C, He HL, Guan W, Yuan YS, Gao J (2012) Immune mechanisms of Concanavalin a model of autoimmune hepatitis. World J Gastroenterol 18(2):119–125. https://doi.org/10.3748/wjg.v18.i2.119
Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, Auman JT, Hoskins JM, Misher AD, Moser CD, Yourstone SM, Kim JW, Cibulskis K, Getz G, Hunt HV, Thorgeirsson SS, Roberts LR, Ye D, Guan KL, Xiong Y, Qin LX, Chiang DY (2013) Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 32(25):3091–3100. https://doi.org/10.1038/onc.2012.315
Wang L, Yu K, Zhang X, Yu S (2018) Dual functional roles of the MyD88 signaling in colorectal cancer development. Biomed Pharmacother 107:177–184. https://doi.org/10.1016/j.biopha.2018.07.139
Wang Y, Pan CQ, Xing H (2019) Advances in gut microbiota of viral hepatitis cirrhosis. Biomed Res Int 2019:9726786. https://doi.org/10.1155/2019/9726786
Welzel TM, Graubard BI, El-Serag HB, Shaib YH, Hsing AW, Davila JA, McGlynn KA (2007) Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based case-control study. Clin Gastroenterol Hepatol 5(10):1221–1228. https://doi.org/10.1016/j.cgh.2007.05.020
White DL, Thrift AP, Kanwal F, Davila J, El-Serag HB (2017) Incidence of hepatocellular carcinoma in All 50 United States, from 2000 through 2012. Gastroenterology 152(4):812–820 e815. https://doi.org/10.1053/j.gastro.2016.11.020
Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R (2017) Targeting the gut-liver axis in liver disease. J Hepatol 67(5):1084–1103. https://doi.org/10.1016/j.jhep.2017.05.007
Wilson GK, Brimacombe CL, Rowe IA, Reynolds GM, Fletcher NF, Stamataki Z, Bhogal RH, Simoes ML, Ashcroft M, Afford SC, Mitry RR, Dhawan A, Mee CJ, Hubscher SG, Balfe P, McKeating JA (2012) A dual role for hypoxia inducible factor-1alpha in the hepatitis C virus lifecycle and hepatoma migration. J Hepatol 56(4):803–809. https://doi.org/10.1016/j.jhep.2011.11.018
Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, Lorentzen A, Einer C, Schulz S, Clavel T, Protzer U, Thiele C, Zischka H, Moch H, Tschop M, Tumanov AV, Haller D, Unger K, Karin M, Kopf M, Knolle P, Weber A, Heikenwalder M (2014) Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26(4):549–564. https://doi.org/10.1016/j.ccell.2014.09.003
Wolf BJ, Tatituri RV, Almeida CF, Le Nours J, Bhowruth V, Johnson D, Uldrich AP, Hsu FF, Brigl M, Besra GS, Rossjohn J, Godfrey DI, Brenner MB (2015) Identification of a potent microbial lipid antigen for diverse NKT cells. J Immunol 195(6):2540–2551. https://doi.org/10.4049/jimmunol.1501019
Wong VW, Tse CH, Lam TT, Wong GL, Chim AM, Chu WC, Yeung DK, Law PT, Kwan HS, Yu J, Sung JJ, Chan HL (2013) Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis—a longitudinal study. PLoS One 8(4):e62885. https://doi.org/10.1371/journal.pone.0062885
Wongratanacheewin S, Bunnag D, Vaeusorn N, Sirisinha S (1988) Characterization of humoral immune response in the serum and bile of patients with opisthorchiasis and its application in immunodiagnosis. Am J Trop Med Hyg 38(2):356–362. https://doi.org/10.4269/ajtmh.1988.38.356
Woo HG, Lee JH, Yoon JH, Kim CY, Lee HS, Jang JJ, Yi NJ, Suh KS, Lee KU, Park ES, Thorgeirsson SS, Kim YJ (2010) Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res 70(8):3034–3041. https://doi.org/10.1158/0008-5472.CAN-09-2823
Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE (2013) From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol 10(11):627–636. https://doi.org/10.1038/nrgastro.2013.149
Wroblewski LE, Peek RM Jr, Wilson KT (2010) Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23(4):713–739. https://doi.org/10.1128/CMR.00011-10
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD (2011) Linking long-term dietary patterns with gut microbial enterotypes. Science 334(6052):105–108. https://doi.org/10.1126/science.1208344
Wu WR, Shi XD, Zhang R, Zhu MS, Xu LB, Yu XH, Zeng H, Wang J, Liu C (2014) Clinicopathological significance of aberrant notch receptors in intrahepatic cholangiocarcinoma. Int J Clin Exp Pathol 7(6):3272–3279
Xie Y, Xiang Y, Sheng J, Zhang D, Yao X, Yang Y, Zhang X (2018) Immunotherapy for hepatocellular carcinoma: current advances and future expectations. J Immunol Res 2018:8740976. https://doi.org/10.1155/2018/8740976
Xiong Y, Medvedev AE (2011) Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol 90(6):1141–1148. https://doi.org/10.1189/jlb.0611273
Xu Z, Jensen G, Yen TS (1997) Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. J Virol 71(10):7387–7392
Xu M, Jiang Z, Huang W, Yin J, Ou S, Jiang Y, Meng L, Cao S, Yu A, Cao J, Shen Y (2018) Altered gut microbiota composition in subjects infected with Clonorchis sinensis. Front Microbiol 9:2292. https://doi.org/10.3389/fmicb.2018.02292
Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, Han D, Watanabe T, Asano M, Takasawa S, Okamoto H, Shimura S, Karasawa T, Yonekura H, Yamamoto H (2011) Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol 186(5):3248–3257. https://doi.org/10.4049/jimmunol.1002253
Yang L, Seki E (2012) Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms. Front Physiol 3:138. https://doi.org/10.3389/fphys.2012.00138
Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W (2007) Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 67(3):863–867. https://doi.org/10.1158/0008-5472.CAN-06-1078
Yang W, Ma Y, Liu Y, Smith-Warner SA, Simon TG, Chong DQ, Qi Q, Meyerhardt JA, Giovannucci EL, Chan AT, Zhang X (2019) Association of Intake of whole grains and dietary fiber with risk of hepatocellular carcinoma in US adults. JAMA Oncol 5(6):879–886. https://doi.org/10.1001/jamaoncol.2018.7159
Yao ZM, Vance DE (1988) The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem 263(6):2998–3004
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499(7456):97–101. https://doi.org/10.1038/nature12347
Yu LX, Schwabe RF (2017) The gut microbiome and liver cancer: mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol 14(9):527–539. https://doi.org/10.1038/nrgastro.2017.72
Zajonc DM, Girardi E (2015) Recognition of microbial glycolipids by natural killer T cells. Front Immunol 6:400. https://doi.org/10.3389/fimmu.2015.00400
Zender S, Nickeleit I, Wuestefeld T, Sorensen I, Dauch D, Bozko P, El-Khatib M, Geffers R, Bektas H, Manns MP, Gossler A, Wilkens L, Plentz R, Zender L, Malek NP (2013) A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 23(6):784–795. https://doi.org/10.1016/j.ccr.2013.04.019
Zeng H, Chi H (2015) Metabolic control of regulatory T cell development and function. Trends Immunol 36(1):3–12. https://doi.org/10.1016/j.it.2014.08.003
Zhang E, Lu M (2015) Toll-like receptor (TLR)-mediated innate immune responses in the control of hepatitis B virus (HBV) infection. Med Microbiol Immunol 204(1):11–20. https://doi.org/10.1007/s00430-014-0370-1
Zhou Z, Zhong W (2017) Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res 1(4):197–207. https://doi.org/10.1016/j.livres.2017.12.004
Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR (2013) Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57(2):601–609. https://doi.org/10.1002/hep.26093
Zhu J, Wu Y, Tang Q, Leng Y, Cai W (2014) The effects of choline on hepatic lipid metabolism, mitochondrial function and antioxidative status in human hepatic C3A cells exposed to excessive energy substrates. Nutrients 6(7):2552–2571. https://doi.org/10.3390/nu6072552
Zhu Q, Xia P, Zhou X, Li X, Guo W, Zhu B, Zheng X, Wang B, Yang D, Wang J (2019) Hepatitis B virus infection alters gut microbiota composition in mice. Front Cell Infect Microbiol 9:377. https://doi.org/10.3389/fcimb.2019.00377
Zitvogel L, Ayyoub M, Routy B, Kroemer G (2016) Microbiome and anticancer Immunosurveillance. Cell 165(2):276–287. https://doi.org/10.1016/j.cell.2016.03.001
Zou J, Chassaing B, Singh V, Pellizzon M, Ricci M, Fythe MD, Kumar MV, Gewirtz AT (2018) Fiber-mediated nourishment of gut microbiota protects against diet-induced obesity by restoring IL-22-mediated colonic health. Cell Host Microbe 23(1):41–53. e44. https://doi.org/10.1016/j.chom.2017.11.003
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The American Physiological Society
About this chapter

Cite this chapter
McVey, J.C., Zhang, Q., Greten, T.F., Ma, C. (2021). Gut Microbiome and Liver Cancer. In: Sun, J. (eds) Inflammation, Infection, and Microbiome in Cancers. Physiology in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-67951-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-67951-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67950-7
Online ISBN: 978-3-030-67951-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)