
Abstract
The human intestine is believed to contain approximately 100 trillion intestinal (gut) microbiota, comprising about 500–1000 different species. These intestinal microbiota exist in a symbiotic relationship with their host, by metabolizing compounds that the host is unable to utilize and controlling the immune balance of the host’s body. However, the composition of the intestinal microbiota is known to vary, depending on diet, nutrition status, and other factors. The recently developed meta-omics microbial data and the technical progress for the metabolome analysis provide a substantial understanding of the role of intestinal microbes and their metabolism. Interestingly, accumulating evidence suggests that the intestinal microbiota contributes to the onset of colorectal cancer, not only via the pro-carcinogenic activities of specific pathogens but also via the influence of the bacterial metabolites. Moreover, since the gut microbial metabolites circulate in the host’s body, it has been increasingly recognized that the intestinal microbiota are involved in the pathogenesis of diseases not only in the intestine but also in the organs located distant from the intestine. We recently found that metabolites from obesity-induced intestinal microbiota promoted liver cancer, and elucidated the underlying molecular mechanism. In this review, I first summarize the general understanding on the carcinogenic process by bacterial metabolites, and then discuss on the association between intestinal microbiota and colorectal cancer. In the last part, I will introduce our recent findings on liver cancer promotion by a metabolite of the obesity-induced intestinal microbiota.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human intestine is believed to contain approximately 100 trillion intestinal (gut) microbiota, comprising about 500–1000 different species [1]. These intestinal microbiota exist in a symbiotic relationship with their host, by metabolizing compounds that the host is unable to utilize and controlling the immune balance of the host’s body. However, the composition of the intestinal microbiota is known to vary, depending on diet, nutrition status, and other factors. For example, the number of gram-positive bacteria belonging to the phylum Firmicutes is known to increase remarkably during obesity [2, 3]. Recently, it has been increasingly recognized that these intestinal microbiota are involved in the pathogenesis of many diseases [4] not only through their pathogenic activities but also their metabolites. Furthermore, the recently developed meta-omics microbial data provide a substantial understanding of the role of intestinal microbes [5]. In the colon, the synergistic effects of certain types of intestinal microbiota during enteritis [6–8], are known to promote colorectal cancer. Moreover, the production of secondary bile acids by intestinal microbiota with seven-alpha-dehydroxylation activity [9], and/or the decrease of the level of the short-chain fatty acids (SCFA) that facilitate anti-inflammatory effects are known to be involved in the carcinogenesis of colorectal cancer [9, 10].
Furthermore, recent researches have revealed that the intestinal microbiota are also involved in the pathogenesis of diseases in organs located distant from the intestine [4]. For example, changes in the intestinal microbiota and their metabolites have been reported to evoke fatty liver [11], arteriosclerosis [12], and/or neural disease [13, 14]. These and other emerging data have revealed that intestinal microbiota exert various effects not only on the intestine but also on many other organs, thereby greatly affecting the homeostasis of the host’s body. Our recent study focused on obesity-associated pathological conditions, and particularly on the link between the obesity-associated microbiome and cancer. We recently found that metabolites from obesity-induced intestinal microbiota promoted liver cancer, and elucidated the underlying molecular mechanism [15]. In this review, I first summarize the general understandings on bacterial metabolites involved in the carcinogenic process, and then discuss on the carcinogenic microbiota and colorectal cancer. In the last part, our recent findings on liver cancer promoted by a metabolite of the obesity-induced intestinal microbiota will be introduced [15].
Bacterial metabolites and the carcinogenic processes
Many of the undigested dietary components that reach the colon are fermented by the intestinal microbiota to produce a wide range of metabolites. Recently, accumulating evidence suggest that these microbial metabolites have a role in the onset of certain types of cancer by affecting the intestinal inflammation and/or by inducing the DNA damage in the intestinal epithelial cells. Short-chain fatty acids, such as acetate, propionate, and butyrate, have a function in the suppression of inflammation and cancer, whereas other microbial metabolites, such as deoxycholic acid and lithocholic acid produced by seven-alpha dehydoxylation of cholic acid and chenodeoxycholic acid, directly promote cancer through facilitating DNA damage [9]. In this part, these general carcinogenic processes by bacterial metabolites will be discussed.
Anti-inflammatory effect of short-chain fatty acids
The major fermentation products in the intestine are gases and organic acids, and the three short-chain fatty acids (SCFAs) acetate, propionate, and butyrate play an supressive role in the inflammation and cancer in the intestine [9]. These SCFAs are absorbed in the intestine and the intracellular butyrate and propionate inhibit the activity of histone deacetylases (HDACs) in the intestinal epithelial cells and the immune cells, which promotes the hyperacetylation of histones and trans-activates the gene expression [16–18] and cellular differentiation [19], resulting in the downregulation of pro-inflammatory cytokines, such as interleukin-6 (IL-6) and IL-12, in intestinal macrophages [20]. In addition, recent evidence shows that butyrate and propionate induce the differentiation of regulatory T cells that express the transcription factor FOXP3, which have a crucial role in controlling intestinal inflammation [21–23]. It is proposed that butyrate causes increased acetylation of histone H3 in the promoter and enhancer regions of the FOXP3 locus, which results in increased expression of FOXP3 [22]. Moreover, independently of HDAC inhibition, butyrate-driven signaling through GPR109A, receptor for niacin and the butyrate, promotes the anti-inflammatory and anti-carcinogenic effect through inducing the differentiation of regulatory T cells and IL-10-producing T cells [24]. All these anti-inflammatory effects of SCFAs in the intestine play an important role in anti-tumor activity, and the decrease of SCFA-producing bacteria could facilitate colorectal cancer.
DNA damage-inducible bacterial metabolites
The primary bile acids, such as cholic acid and chenodeoxycholic acid which are produced in the liver from cholesterol, are conjugated to glycine or taurine and are excreted into the duodenum to facilitate fat digestion [25]. The primary bile acids are re-absorbed in the distal ileum for enterohepatic circulation [25]. However, approximately 5 % of the total bile acid pool are processed by the bai-operon genes of the microbiota [26, 27] in the large intestine [28] and are converted into several different secondary bile acids. Since these secondary bile acids have the potential to create DNA damage by enhancing the production of reactive oxygen species (ROS) [29], it is possible that unrepaired oncogenic mutations in the colorectal epithelium could remain. The populations of anaerobic bacteria, such as those in the genera of Bacteroides and Clostridium, are reportedly increased in colorectal cancer patients [30]. Some reports have shown that the levels of deoxycholic acid (DCA) and lithocholic acid (LCA), produced during metabolism by these intestinal microbiota with 7α-dehydroxylation activity, are increased in the feces of colorectal cancer patients [30]. In addition, DCA reportedly activates beta-catenin, which enhances cell proliferation [31] thereby promoting the pathogenesis of colorectal cancer through these functions.
A possible role of cellular senescence and SASP for carcinogenesis by intestinal microbiota
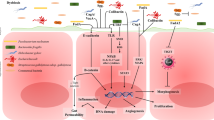
Recent evidence revealed that the DNA damage caused by the secondary bile acid such as DCA has an ability to induce cellular senescence and senescence-associated secretory phenotype (SASP). Cellular senescence is a phenomenon in which normal cells irreversibly cease to proliferate when exposed to an oncogenic impact, such as oncogene activation and/or strong DNA damage. Therefore, cellular senescence has been recognized as a fail-safe mechanism against oncogenesis [32–34]. There is no doubt that cellular senescence functions as an important tumor suppression mechanism. However, recent studies have shown that unlike apoptotic cells, senescent cells do not immediately die, and instead they remain viable for a long period of time. Therefore, the remaining senescent cells may have some biological effects. Of particular biological importance, it has recently become apparent that senescent cells exhibit the increased expression of genes encoding a series of secretory proteins, such as inflammatory cytokines, chemokines, and matrix remodeling factors, which may alter the local tissue environment and/or contribute to chronic inflammation and tumorigenesis [35–38]. This newly identified senescent phenotype, termed the senescence-associated secretory phenotype (SASP) or the senescence-messaging secretome (SMS) (hereafter, referred to as the senescence-associated secretome), is induced by DNA damage and can be beneficial or deleterious, depending on the biological context [35, 39]. For example, IL-6, IL-8, and plasminogen activator inhibitor-1 (PAI-1), which are secreted from senescent fibroblasts, can reportedly reinforce the senescence-associated cell cycle arrest induced by an activated oncogene or oxidative stress [40–42]. On the other hand, IL-6 and IL-8 are also known to promote malignant transformation, in cooperation with certain oncogenes [43–45]. Moreover, the factors secreted from senescent fibroblasts have been shown to induce an epithelial-mesenchymal transition (EMT), an important step in cancer progression [35]. These findings, together with the observations that the proteins secreted by senescent cells can promote hyper-proliferative or metastatic changes in neighboring cells, indicate that the release of the senescence-associated secretome factors results in both beneficial and harmful consequences [35, 46]. The senescence-associated secretome in the microenvironment of cancer may be involved in its latter effects. Indeed, the cancer-associated fibroblasts (CAFs) of human ovarian cancer tissue were found to have the senescent phenotype, implying that the senescence-associated secretome might promote human ovarian cancer [47]. Therefore, in a tissue with an accumulation of senescent cells, it is possible that the deleterious side effects of the senescence-associated secretome may contribute to cancer promotion (Fig. 1), and in this regards, some of the DNA damage-inducible bacterial metabolites such as DCA and LCA could contribute to the formation of cancer-associated microenvironments.

The mechanism of the induction for cellular senescence and senescence-associated secretome. Cellular senescence is a state of irreversible cell proliferation arrest provoked by a variety of DNA damage-inducing signals, and is known to functions as a fail-safe tumor suppression mechanism. However, unlike apoptotic cells, senescent cells remain viable for a long time, and they eventually secrete a series of inflammatory cytokines, chemokines, and matrix remodeling factors that may contribute to form a cancer-promoting microenvironment
Carcinogenic intestinal microbiota
Enterotoxigenic B. fragilis
Some patients with refractory ulcerative colitis develop intercurrent colorectal cancer, but the underlying mechanism is not well understood. Interesting research results have been reported recently, demonstrating that inflammation in the intestine by intestinal microbiota promotes the pathogenesis of colorectal cancer. One example is the Bacteroides fragilis-mediated pathogenesis model of colorectal cancer [6]. There are two types of B. fragilis: enterotoxigenic B. fragilis (ETBF) and nontoxigenic B. fragilis (NTBF), which respectively does and does not secrete B. fragilis toxin (BFT), a zinc-dependent metalloprotease. Using ApcMin/+ mice, which carries a truncation mutation at codon 850 of the Apc gene and readily develop tumors in the small and large intestines, Sears et al. found that the mice inoculated with ETBF in the intestines develop enterocolitis and colorectal cancer, but the mice inoculated with NTBF develop neither. The authors strongly suggested that enterocolitis and tumor formation are caused by the stimulation of intestinal Th17 cells and the Stat3 activation by ETBF-derived BFT [7].
Escherichia coli
E. coli is a normal inhabitant of the human intestine. However, E. coli strains of the phylogenetic group B2 harbor a genomic island called polyketide synthase (pks) that encodes the production of a polyketide-peptide genotoxin, colibactin. There is a report that E. coli harboring the pks island, but not those bearing a pks isogenic mutant, induced the formation of DNA double strand breaks in mouse enterocytes, suggesting that E. coli strains with pks could create oncogenic mutations that promote colorectal cancer [48]. Another report suggests that the colibactin is able to induce senescence-associated secretome to contribute to colon cancer development [49]. This concept of E. coli-mediated colorectal cancer development was strengthened by another report, using IL-10 (anti-inflammatory cytokine)-deficient mice. This report analyzed the types of intestinal microbiota that significantly increased in IL-10-knockout mice treated with azoxymethane (a chemical that promotes colorectal cancer in mice). Among the significantly increased bacteria, E. coli (NC101 strain) was shown to promote highly malignant colorectal cancer [8].
These reports of ETBF-mediated and E. coli-mediated colorectal carcinogenesis have suggested the possibility that enterocolitis and certain types of intestinal microbiota synergistically function to promote colorectal cancer.
Fusobacterium species
The enrichment of Fusobacterium species in human colorectal cancer samples has recently been revealed as a possible cause of colorectal cancer, by metagenomic and meta-16S rRNA gene sequencing [50, 51]. Fusobacterium species were more frequently detected in colon tumor samples than in normal colon tissues. Moreover, recent experimental data further support the colorectal tumor-forming role of Fusobacterium nucleatum. The exposure of ApcMin/+ mice to a F. nucleatum strain, isolated from a patient with inflammatory bowel disease, induced a significant increase in intestinal tumors. This bacterial strain was also detected more frequently in the tumor region than in the normal intestinal mucosa, in an experiment using ApcMin/+ mice [52]. Recently, using periodontal disease-derived F. nucleatum, the carcinogenic properties of these bacteria were found to be derived from the FadA adhesion protein complex (FadAc) [53]. FadAc exists on the surface of the bacteria and binds to the extracellular domain of E-cadherin, thereby promoting the invasion of the tumor cells. In addition, it was reported that FadAc binding to E-cadherin can activate the beta-catenin/Wnt signaling pathway, which promotes cell proliferation. Therefore, Fusobacterium species, which are known as periodontal disease-associated bacteria, are now recognized as possible colorectal cancer-inducing microbes.
Intestinal microbiota and liver cancer
Colorectal cancer has been the primary cancer studied, in terms of the involvement of the intestinal microbiota in cancer. However, common substances may act on both the intestine and the liver, due to enterohepatic circulation (circulation cycle allowing substances absorbed from the intestine to enter the liver via the portal vein and to be excreted from the liver to the intestine with the aid of bile). For example, the bacterial metabolites such as bile acids and fatty acids can circulate throughout the host’s body. Ethanol is also produced by many anaerobic bacteria in the colon, and it can be converted to acetaldehyde, which is toxic and carcinogenic [54]. In addition, ethanol is also known to be involved in the impairment of the intestinal barrier function to circulate bacterial endotoxin that could be involved in the etiology of liver diseases [55].
In this section, we describe our finding that an obesity-induced metabolite of the intestinal microbiota affects the liver through enterohepatic circulation, and promotes liver cancer. Enhanced inflammatory responses due to obesity have previously been suggested to promote cancer [45, 56, 57], but the detailed molecular mechanisms underlying the promotion of cancer by obesity have not been elucidated. We recently found that the enterohepatic circulation of the secondary bile acid DCA, produced by gut bacteria, provokes a cellular senescence phenotype in hepatic stellate cells (HSCs). In turn, HSCs secrete various inflammatory and tumor-promoting factors in the liver, thus facilitating hepatocellular carcinoma (HCC) development in mice [15].
Obesity-induced alteration of intestinal microbiota promotes liver cancer
The population of obese individuals continues to increase worldwide [58], and obesity is known to increase the risk of not only diabetes and cardiovascular diseases, but also various cancers, including colorectal cancer, liver cancer, and prostate cancer [59–62]. Therefore, the growing number of obese people could be a factor responsible for the recent increase in cancer populations in developed countries. Obesity prevention is clearly important for cancer prevention. However, on the other hand, approaches to suppress the onset of these diseases in obese individuals are also needed. The important first step in such approaches is to elucidate the molecular mechanisms that explain how the cancer incidence has increased in the obese population. As the senescence-associated secretome factors, such as IL-6 and PAI-1, are known to be induced in obesity [45], we thought that the senescence-associated secretome may be closely associated with the inflammatory responses and tumorigenesis accompanying obesity. To determine what types of cancer are promoted by obesity, we utilized the chemical carcinogen, 7,12-dimethylbenz (α)anthracene (DMBA), which is known to cause an oncogenic mutation in the H-ras oncogene. We applied this chemical onto the back skin of infant mice (4–5 days old), divided the mice into two groups: high-fat diet and normal diet, and analyzed them after 30 weeks. Very interestingly, all of the mice in the high-fat diet group developed HCC. In contrast, no HCC development was found in the lean mice fed with a normal diet after DMBA application, indicating that obesity promoted liver cancer formation. We also performed a similar experiment using mice that allow the imaging detection of the ongoing cellular senescence in vivo with a luminescent signal [63]. Strong signals were detected in the obesity-associated cancer areas in the liver, demonstrating that cellular senescence was occurring in the cancerous parts.
Enhancement of liver cancer formation with cellular senescence of hepatic stellate cells
Next, immunohistochemical staining was performed to identify the cells in the liver that were undergoing cellular senescence. As a result, the accumulation of DNA damage, a cause of cellular senescence, and the expression of p21 and p16, the important factors that induce cellular senescence, were observed in the stromal hepatic stellate cells in the cancerous part of the liver in the obese mice. In addition, because proliferation markers were observed in cancerous hepatocytes but not in hepatic stellate cells, the hepatic stellate cells seemed to undergo cellular senescence, an irreversible arrest of cell proliferation. Furthermore, the production of various inflammatory cytokines and chemokines, known as senescence-associated secretome factors, was detected in hepatic stellate cells. Based on these results, we hypothesized that obesity-induced DNA damage caused the hepatic stellate cells to become senescent and to secrete the senescence-associate inflammatory cytokines, which in turn promoted the carcinogenesis of the surrounding hepatocytes.
To test this hypothesis, we attempted to suppress the production of senescence-associated secretome factors from hepatic stellate cells. First, we performed the same experiment using knockout mice lacking the gene encoding IL-1β, which is a major senescence-associated secretome factor and a cytokine required for the expression of other downstream inflammatory cytokines. In the IL-1β-deficient mice, we found that the expression of senescence-associated secretome factors was downregulated and the incidence of liver cancer was significantly reduced, while the obesity-induced senescence in hepatic stellate cells was detected at a comparable level to that in wild-type mice. Furthermore, liver tumor development was significantly reduced in obese mice, in which the hepatic stellate cells were selectively removed by knocking down the expression of hepatic stellate cell-specific HSP47 [64]. Based on these results, the induction of the cellular senescence of hepatic stellate cells could be one of the key events to promote hepatocarcinogenesis in their vicinity, by exposure to senescence-associated secretory factors (Fig. 2).

Model for obesity-induced HCC development through senescence-associated secretome. Dietary or genetic obesity induces alteration of gut microbiota, thereby causing promotion of DCA production in the intestinal tract. Elevated levels of DCA provoke senescence-associated secretory phenotype (SASP) factors in HSCs through enterohepatic circulation, which in turn, secretes various inflammatory and tumor-promoting factors in the liver. This event, together with the activation of various oncogenic signaling pathways, results in the promotion of HCC development
Obesity-induced increase of secondary bile acid from intestinal bacteria promotes the pathogenesis of liver cancer
Next, we focused on various obesity-associated changes in vivo, to elucidate the mechanism for obesity-induced cellular senescence in hepatic stellate cells. We particularly focused on the changes in the intestinal microbiota since it has been reportedly involved in obesity and nonalcoholic fatty liver disease (NAFLD). For example, there is a report that the conversion of choline into methylamines by microbiota in strain 129S6 on a high-fat diet reduces the bioavailability of choline and mimics the effect of choline-deficient diets (it can induce hepatic steatosis), causing NAFLD [65]. Also, there are reports that obese induces an increase of phylum of Firmicutes in the intestine in humans and mice [2, 3], and microbiota from obese people can induce obesity in mice [66].
It has also been reported that lipopolysaccharide, a component of gram-negative bacteria in the intestinal microbiota, acts as a ligand of Toll-like receptor 4 (TLR4) to elicit inflammation and promote liver cancer formation, in another liver cancer-inducing mouse model (administration of DEN plus carbon tetrachloride) [67]. Therefore, we administered a mixture of four types of antibiotics to obese mice, to eliminate both the gram-negative and gram-positive intestinal microbiota. We found that the incidence of obesity-induced liver cancer was significantly decreased, and the number of hepatic stellate cells with cellular senescence and the senescence-associated secretome also decreased. These results strongly suggest that the intestinal microbiota play key roles in obesity-induced liver cancer formation. Furthermore, the variable regions of the 16S ribosomal RNA gene sequences of the bacteria in mouse feces were analyzed with a next-generation sequencer, to detect the changes in the intestinal microbiota in obese mice. The analysis revealed that gram-positive bacteria accounted for ≥90 % of the bacteria found in obese mice on a high-fat diet, whereas the intestinal microbiota of mice on a normal diet comprised approximately equal proportions of gram-positive and gram-negative bacteria. In particular, the populations of gram-positive bacteria categorized in Clostridium clusters XI or X IVa were found to be greatly increased in the mice fed a high-fat diet, as compared to those in the mice fed a normal diet. Therefore, we next administered vancomycin to the obese mice, to specifically target the gram-positive bacteria. The vancomycin treatment markedly reduced the formation of obesity-induced liver cancer, accompanied with the simultaneous reductions of the cellular senescence and the senescence-associated secretome of hepatic stellate cells, to the same extents observed upon the treatment with the four antibiotics. Based on these results, we hypothesized that a metabolite or toxin from the obesity-induced gram-positive bacteria in the intestinal microbiota affects the liver via the enterohepatic circulation, and enhances liver cancer formation through the induction of cellular senescence and the senescence-associated secretome of hepatic stellate cells.
We next performed a metabolome analysis, using sera from both normal diet-fed mice and high-fat diet-fed mice, to identify the metabolites that promote obesity-induced liver cancer. We found that deoxycholic acid (DCA), a secondary bile acid, was significantly increased in the serum of obese mice. Primary bile acids produced from cholesterol in vivo are essential for fat emulsification, and are then processed by the 7α-dehydroxylation activity of some intestinal microbiota. A previous study has shown that Clostridium sordellii and Clostridium scindens, which are known to produce DCA, belong to Clostridium clusters XI or X IVa, and their populations are increased in high-fat diet-fed mice [25]. More importantly, a previous study with cultured cells revealed that DCA induces DNA damage via reactive oxygen species, ROS [29, 23], and possibly promotes cancer formation. Based on these results, we thought that DCA is likely to play a critical role in obesity-induced liver cancer formation. Therefore, we performed experiments to reduce the serum DCA level. The incidence of liver cancer and the number of senescent hepatic stellate cells were decreased in obese mice in which the serum DCA levels were reduced by a treatment with di-fructose anhydride III (DFAIII), which inhibits DCA production [68], or a treatment with ursodeoxycholic acid (UDCA) [69], which promotes bile acid excretion. In contrast, when DCA was orally administered to the obese mice treated with the four antibiotics to reduce the intestinal microbiota, the incidence of liver cancer was substantially restored to the level of the standard DMBA-treated obese mice, accompanied by the signs of both cellular senescence and the senescence-associated secretome in the hepatic stellate cells in the cancerous region (Fig. 2). These results revealed that the secondary bile acid DCA, produced by the obesity-induced intestinal microbiota, was transferred to the liver via the enterohepatic circulation, and in turn promoted liver cancer formation therein through the induction of cellular senescence and the senescence-associated secretome of hepatic stellate cells (Fig. 2).
As some obesity-induced gram-positive bacteria belonging to Clostridium clusters XI and X IVa are known to produce DCA, we think that these could be the critical bacteria promoting obesity-induced liver cancer. A detailed phylogenetic analysis of the 16S rRNA gene sequences from obese mice indicated that only one type of bacterium was found in Clostridium cluster XI, and this bacterium proliferated to up to more than 10 % of the entire intestinal microbiota during obesity. In addition, this bacterium was identified as a relative of C. sordellii known to produce DCA; therefore, it is most likely that this bacterium in Clostridium cluster XI is responsible for DCA production in obese mice. Similar results were also observed in an experiment with genetically obese mice fed an excessive amount of a normal diet; thus, the mechanism elucidated in this study may be considered as an effect caused by the pathological condition of obesity, and not simply a consequence of a high-fat diet.
A similar mechanism exists in humans
Next, we tried to examine whether the same cancer-promoting mechanism is involved in human obesity-associated liver cancer formation. IL-1β treatment of cultured human hepatic stellate cells induced the strong expression of senescence-associated secretome factors, such as IL-6 and IL-8. Furthermore, we examined the tumor region of human liver cancer with obesity-associated nonalcoholic steatohepatitis (NASH), and found the induction of cellular senescence of hepatic stellate cells and the production of inflammatory cytokines via the senescence-associated secretome in approximately 30 % of the liver tumors with NASH [70]. These human tumors featured less fibrosis and very prominent fat accumulation in the tumor regions, and are similar to the liver cancer tissue observed in the mouse model in this study. A previous study also indicated that the intestinal microbiota of obese individuals contained a higher number of gram-positive bacteria (a phylum of Firmicutes), and that the number of gram-positive bacteria decreased with a concomitant increase in gram-negative bacteria when the same individuals lost weight [2]. It has also been reported that the DCA concentrations in feces were increased in healthy individuals fed a high-fat diet [71, 72]. Based on these observations, the increased production of DCA by the intestinal microbiota and the induction of the senescence-associated secretome in hepatic stellate cells are strongly suggested to be involved in the formation of a certain type of NASH-based liver cancer in humans.
Conclusion
This review showed the basic understandings of the cellular and molecular mechanisms of microbiota-mediated carcinogenesis via their metabolites, pathogens, toxins, and so on that can alter the inflammatory responses and the cell growth in the hosts. The influence of intestinal microbiota is not only in the host’s intestine but also in other distant organs such as the liver because these bacterial substances circulate throughout the body. Particularly, the short-chain fatty acids, acetate, propionate, and butyrate, function in the suppression of inflammation and cancer, whereas other microbial metabolites, such as secondary bile acids, deoxycholic acid, and lithocholic acid can promote carcinogenesis by increasing the intracellular ROS level that facilitate DNA damage. The further studies on the bacterial metabolites and the function of intestinal microbiota will provide valuable new insights into the development of microbiota-associated cancer and open up new possibilities for its control in the future.
References
Kamada N, Seo SU, Chen GY et al (2013) Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13:321–335
Ley RE, Turnbaugh PJ, Klein S et al (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023
Ley RE, Bäckhed F, Turnbaugh P et al (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102:11070–11075
Holmes E, Li JV, Marchesi JR et al (2012) Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab 16:559–564
Fritz JV, Desai MS, Shah P et al (2013) From meta-omics to causality: experimental models for human microbiome research. Microbiomedicine 1:14
Ou J, Carbonero F, Zoetendal EG et al (2013) Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr 98:111–120
Wu S, Rhee KJ, Albesiano E et al (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15:1016–1022
Arthur JC, Perez-Chanona E, Mühlbauer M et al (2012) Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338:120–123
Louis P, Hold GL, Flint HJ (2014) The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12:661–672
Gonçalves P, Martel F (2013) Butyrate and colorectal cancer: the role of butyrate transport. Curr Drug Metab 14:994–1008
Henao-Mejia J, Elinav E, Jin C et al (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482:179–185
Koeth RA, Wang Z, Levison BS et al (2013) Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19:576–585
Yokote H, Miyake S, Croxford JL et al (2008) NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am J Pathol 173:1714–1723
Collins SM, Surette M, Bercik P (2012) The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol 10:735–742
Yoshimoto S, Loo TM, Atarashi K et al (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499:97–101
Natarajan N, Pluznick JL (2014) From microbe to man: the role of microbial short chain fatty acid metabolites in host cell biology. Am J Physiol Cell Physiol. doi:10.1152/ajpcell.00228.2014
Sleeth ML, Thompson EL, Ford HE et al (2010) Free fatty acid receptor 2 and nutrient sensing: a proposed role for fibre, fermentable carbohydrates and short-chain fatty acids in appetite regulation. Nutr Res Rev 23:135–145
Fung KYV, Cosgrove L, Lockett T et al (2012) A review of the potential mechanism for the lowering of colorectal oncogenesis by butyrate. Br J Nutr 108:820–831
Hamer HM, Jonkers D, Venema K et al (2008) Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27:104–119
Chang PV, Hao L, Offermanns S et al (2014) The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A 111:2247–2252
Smith PM, Howitt MR, Panikov N et al (2013) The microbial metabolites, short-chain fatty acids, regulate colonic T reg cell homeostasis. Science 341:569–573
Furusawa Y, Obata Y, Fukuda S et al (2013) Commensal microbe-derived butyrate induced the differentiation of colonic regulatory T cells. Nature 504:446–450
Arpaia N, Campbell C, Fan X et al (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504:451–455
Singh N, Gurav A, Sivaprakasam S et al (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40:128–139
Ridlon JM, Kang DJ, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259
Zhou H, Hylemon PB (2014) Bile acids are nutrient signaling hormones. Steroids 86C:62–68
Kitazawa S, Denda A, Tsutsumi M et al (1990) Enhanced preneoplastic liver lesion development under ‘selection pressure’ conditions after administration of deoxycholic or lithocholic acid in the initiation phase in rats. Carcinogenesis 11:1323–1328
Ridlon JM, Hylemon PB (2012) Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7a-dehydroxylating intestinal bacterium. J Lipid Res 53:66–76
Payne CM, Weber C, Crowley-Skillicorn C et al (2007) Deoxycholate induces mitochondrial oxidative stress and activates NF-kappaB through multiple mechanisms in HCT-116 colon epithelial cells. Carcinogenesis 28:215–2
Reddy BS, Wynder EL (1973) Large-bowel carcinogenesis: fecal constituents of populations with diverse incidence rates of colon cancer. J Natl Cancer Inst 50:1437–1442
Pai R, Tarnawski AS, Tran T (2004) Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell 15:2156–2163
d’Adda di Fagagna F (2008) Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 8:512–522
Kuilman T, Michaloglou C, Mooi WJ et al (2010) The essence of senescence. Genes Dev 24:2463–2479
Ohtani N, Hara E (2013) Roles and mechanisms of cellular senescence in regulation of tissue homeostasis. Cancer Sci 104:525–530
Coppé JP, Patil CK, Rodier F et al (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6:2853–2868
Rodier F, Coppé JP, Patil CK et al (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11:973–979
Takahashi A, Imai Y, Yamakoshi K et al (2012) DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol Cell 45:123–131
Rodier F, Campisi J (2011) Four faces of cellular senescence. J Cell Biol 192:547–556
Kuilman T, Peeper DS (2009) Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 9:81–94
Acosta JC, O’Loghlen A, Banito A et al (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133:1006–1018
Kuilman T, Michaloglou C, Vredeveld LC et al (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133:1019–1031
Kortlever RM, Higgins PJ, Bernards R (2006) Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 8:877–884
Sparmann A, Bar-Sagi D (2004) Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 6:447–458
Ancrile B, Lim KH, Counter CM (2007) Oncogenic ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev 21:1714–1719
Park EJ, Lee JH, Yu GY et al (2010) Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140:197–208
Ohanna M, Giuliano S, Bonet C et al (2011) Senescent cells develop a PARP-1 and nuclear factor-kB-associated secretome (PNAS). Genes Dev 25:1245–1261
Yang G, Rosen DG, Zhang Z et al (2006) The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A 103:16472–16477
Cuevas-Ramos G, Petit CR, Marcq I et al (2010) Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A 107:11537–11542
Cougnoux A, Dalmasso G, Martinez R et al (2014) Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. doi:10.1136/gutjnl-2013-305257
Castellarin M, Warren RL, Freeman JD et al (2012) Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22:299–306
Kostic AD, Gevers D, Pedamallu CS et al (2012) Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22:292–29
Kostic AD, Chun E, Robertson L et al (2013) Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14:207–215
Rubinstein MR, Wang X, Liu W et al (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 14:195–206
Homann N (2001) Alcohol and upper gastrointestinal tract cancer: the role of local acetaldehyde production. Addict Biol 6:309–323
Bode C, Bode JC (2005) Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res 29:166S–171S
Khandekar MJ, Cohen P, Spiegelman BM (2011) Molecular mechanisms of cancer development in obesity. Nat Rev Cancer 11:886–895
Jais A, Einwallner E, Sharif O et al (2014) Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 158:25–40
Calle EE, Rodriguez C, Walker-Thurmond K et al (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348:1625–1638
Samanic C, Gridley G, Chow WH et al (2004) Obesity and cancer risk among white and black United States veterans. Cancer Causes Control 15:35–43
Møller H, Mellemgaard A, Lindvig K et al (1994) Obesity and cancer risk: a Danish record-linkage study. Eur J Cancer 30A:344–350
Wolk A, Gridley G, Svensson M et al (2001) A prospective study of obesity and cancer risk (Sweden). Cancer Causes Control 12:13–21
Haslam DW, James WP (2005) Obes Lancet 366(9492):1197–1209
Ohtani N, Imamura Y, Yamakoshi K et al (2007) Visualizing the dynamics of p21(Waf1/Cip1) cyclin-dependent kinase inhibitor expression in living animals. Proc Natl Acad Sci U S A 104:15034–15039
Sato Y, Murase K, Kato J et al (2008) Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol 26:431–442
Dumas ME, Barton RH, Toye A et al (2006) Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A 103:12511–12516
Ridaura VK, Faith JJ, Rey FE et al (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341:1241214
Dapito DH, Mencin A, Gwak GY et al (2012) Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21:504–516
Minamida K, Ohashi M, Hara H et al (2006) Effects of ingestion of difructose anhydride III (DFA III) and the DFA III-assimilating bacterium Ruminococcus productus on rat intestine. Biosci Biotechnol Biochem 70:332–339
Beuers U (2006) Drug insight: mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol 3:318–328
Takuma Y, Nouso K (2010) Nonalcoholic steatohepatitis-associated hepatocellular carcinoma: our case series and literature review. World J Gastroenterol 16:1436–1441
Rafter JJ, Child P, Anderson AM et al (1987) Cellular toxicity of fecal water depends on diet. Am J Clin Nutr 45:559–563
David LA, Maurice CF, Carmody RN et al (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505:559–563
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the Special Issue on Microbiome, Immunity and Inflammation - Guest Editor: Hiroshi Ohno
Rights and permissions
About this article

Cite this article
Ohtani, N. Microbiome and cancer. Semin Immunopathol 37, 65–72 (2015). https://doi.org/10.1007/s00281-014-0457-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-014-0457-1