
Abstract
Toll-like receptor 4 (TLR4) is a pattern recognition receptor that functions as lipopolysaccharide (LPS) sensor and whose activation results in the production of several pro-inflammatory, antiviral, and anti-bacterial cytokines. TLR4 is expressed in several cells of healthy liver. Despite the constant confrontation of hepatic TLR4 with gut-derived LPS, the normal liver does not show signs of inflammation due to its low expression of TLR4 and ability to modulate TLR4 signaling. Nevertheless, there is accumulating evidence that altered LPS/TLR4 signaling is a key player in the pathogenesis of many chronic liver diseases (CLD). In this review, we first describe TLR4 structure, ligands, and signaling. Later, we review liver expression of TLR4 and discuss the role of LPS/TLR4 signaling in the pathogenesis of CLD such as alcoholic liver disease, nonalcoholic fatty liver disease, chronic hepatitis C, chronic hepatitis B, primary sclerosing cholangitis, primary biliary cirrhosis, hepatic fibrosis, and hepatocarcinoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The innate immune system recognizes several components of microbes and initiates protective immunological responses. This microbiological recognition is a specific and highly coordinated process involving pattern recognition receptors (PRRs) that identify preserved structures of different pathogens, the so-called pathogen-associated molecular patterns (PAMPs) [1, 2]. Toll-like receptors (TLRs) are the most important family of PRRs, with ten different TLRs being ubiquitously expressed in humans [1, 2]. TLR4 acts as a receptor for lipopolysaccharide (LPS), a cell-wall component of Gram-negative bacteria, promptly inducing the production of several pro-inflammatory, anti-viral, and anti-bacterial cytokines [1, 2].
The TLR4 is expressed in several liver cells, and the liver, due to its anatomic location, is constantly confronted with gut-derived LPS [3]. Despite the constant confrontation of TLR4-expressing liver cells with gut-derived LPS, the normal liver does not show signs of inflammation, which on one hand can be explained by the relatively low expression of TLR4 and its adaptor molecules in the liver [3]. On the other hand, under normal circumstances, the liver negatively regulates TLR4 signaling at different levels, contributing to a process known as “liver tolerance” [3]. A breakdown of liver tolerance, by increased exposure of TLR4 to LPS and/or increased expression or sensitivity of TLR4, may induce an inappropriate immune response which can contribute to chronic inflammatory liver diseases [3]. Recent studies provide evidence for a role of LPS/TLR4 signaling in the pathogenesis of alcoholic liver disease, nonalcoholic fatty liver disease, chronic hepatitis C, chronic hepatitis B, primary sclerosing cholangitis, primary biliary cirrhosis, hepatic fibrosis, and hepatocarcinoma [3].
Herein we first review TLR4 structure, ligands, and signaling pathways. Later, we review liver expression of TLR4 and discuss the role of LPS/TLR4 signaling in the pathogenesis of chronic inflammatory liver diseases.
TLR family
The TLR, originally identified as homologs of Drosophila Toll, belong to the superfamily of interleukin-1 receptors [4]. The human TLR family currently consists of ten members, which are structurally characterized by the presence of a distinct leucine-rich repeat extracellular domain that confers specificity to the receptor, and a conserved toll/interleukin 1 (IL1) receptor (TIR) intracellular domain [5].
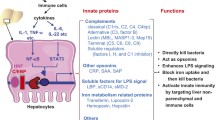
The existence of several TLRs enables the innate immunity system to recognize different groups of pathogens while initiating appropriate and distinct immunological responses, according to the PAMP recognized [3] (Fig. 1). TLR1, TLR2, TLR4, TLR5, and TLR6 are expressed on the cell surface, and TLR3, TLR7, TLR8, and TLR9 are expressed on the endosome–lysosome membrane. TLR1 and TLR6 form heterodimers with TLR2 in order to sense tri-acyl (mycobacterium) and di-acyl lipopeptides (mycoplasma), respectively. TLR4 and TLR5 are the receptors for the Gram-negative bacterial cell wall components, lipopolysaccharide (LPS), and bacterial flagellin, respectively. Intracellular TLRs, TLR3, TLR7/8, and TLR9 detect viral-derived and synthetic double-stranded RNA, viral-related single-stranded RNA, and bacterial unmethylated CpG-DNA, respectively. The ligands for TLR10, TLR12, and TLR13 remain unidentified. TLR8 does not signal in mice. TLR10 is expressed in humans, but not in mice. TLR11, TLR12, and TLR13 are expressed in mice, but not in humans.

Overview of signaling of LPS/TLR4 and other TLRs. LPS recognition is facilitated by LBP and CD14 and is mediated by TLR4/MD-2 receptor complex. TLR4 signaling cascade can be separated into MyD88-dependent and MyD88 independent pathways which mediate the activation of proinflammatory cytokines and IFN-β. These two pathways also mediate the intracellular signaling of other TLRs, enabling interaction between TLR4 and other TLRs at different levels. See text for abbreviations
TLR4 ligands
The TLR4 is expressed on the cell surface and is the receptor for the Gram-negative bacteria cell-wall component, LPS [4]. LPS is composed of hydrophilic polysaccharides of the core and O-antigen and a hydrophobic lipid A component, which corresponds to the conserved molecular pattern of LPS and is the main inducer of biological responses to LPS [4]. Stimulation of TLR4 by LPS is a complex process (Fig. 1), which includes the participation of several molecules [LPS binding protein (LBP), CD14 and MD-2] [6, 7]. LBP (a soluble protein) extracts LPS from the bacterial membrane and shuttles it to CD14 (a glycosylphosphatidylinositol-anchored protein, which also exists in a soluble form). CD14 then transfers the LPS to MD-2 (a soluble protein that non-covalently associates with the extracellular domain of TLR4). Binding of LPS to MD-2 induces a conformational change in MD-2 which then allows the complex MD-2-TLR4 to bind to a second TLR4 receptor, thus achieving TLR4 homo-dimerization and signaling.
Besides LPS, TLR4 also senses endogenous ligands initiating danger signals, such as high-mobility group box-1, hyaluronan, heat shock protein 60, and free fatty acids (C12:0, C14:0, C16:0, and C18:0) [8–10]. Recent reports demonstrated that necrotic cells stimulate TLR4 associated with MyD88 under sterile conditions, thereby pre-emptively inducing an inflammatory response in the absence of microbial challenge [11, 12]. Due to the association of many endogenous ligands with tissue injury, they are termed damage-associated molecular patterns (DAMPs). Interestingly, recent studies show that many of the proposed endogenous TLR4 ligands may also have the capacity to bind and transport LPS and/or enhance the sensitivity of cells to LPS, suggesting that many of these molecules may be more accurately described as PAMP-binding molecules or PAMP-sensitizing molecules, rather than genuine ligands of TLR4 [13].
TLR4 signaling
Binding of ligands to the extracellular domains of TLRs causes a rearrangement of the receptor complex and triggers the recruitment of specific adaptor proteins to the intracellular domain, thus initiating a signaling cascade [6, 7].
TLR4 signals through adaptor molecules such as MyD88, toll/IL-1 receptor domain-containing adaptor protein (TIRAP), toll/IL-1 receptor domain-containing adaptor inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM) to activate transcription factors such as nuclear factor (NF)-κB, activator protein 1 (AP-1), and interferon regulatory factors (IRFs). These transcription factors then initiate the transcription of a specific set of genes involved in proinflammatory, anti-viral, and anti-bacterial responses and genes that control cell survival and apoptosis. TLR4 signaling has been divided into MyD88-dependent (mediated by MyD88) and MyD88-independent (mediated by TRIF) pathways (Fig. 1) [5]. These two pathways also mediate the intracellular signaling of other TLRs, enabling the interaction between TLR4 and other TLRs at different levels from adaptor molecules to transcription factors (Fig. 1). MyD88 is an essential part of the signaling cascade of all TLRs except for TLR3. In contrast, TRIF only interacts with TLR3 and TLR4.
In the MyD88-dependent pathway, TLR4, through TIRAP, recruits MyD88 to activate IL-1R-associated kinase (IRAK)-4 and IRAK-1, which then associate with tumor necrosis receptor-associated factor (TRAF)-6 and transforming growth factor-β-activated kinase 1 (TAK-1). These activate the complex inhibitor of NF-κB kinase (IKK), formed by NEMO, IKKα e IKKβ, which phosphorylates and degrades IκBα (inhibitor of NF-κB), allowing nuclear translocation of NF-κB (normally sequestered in the cytoplasm by ligation to IκBα). NF-κB leads to expression of effectors genes (TNF-α, IL-6, and IL-12). The MyD88-dependent pathway can also activate p38 and c-Jun N-terminal kinase (JNK), leading to AP-1 activation followed by transcription of genes involved in regulation of cell proliferation, morphogenesis, apoptosis, and differentiation.
In the MyD88-independent pathway, TLR4, through TRAM, recruits TRIF. This recruits TRAF3 which associates with TRAF family member associated NF-κB activator (TANK), TBK1 (TANK binding kinase 1) and IKKi with subsequent phosphorylation and nuclear translocation of IRF-3. IRF-3 leads to IFN-β transcription. In MyD88-independent pathway, TRIF also associates with the receptor-interacting serine–threonine kinase (RIP)-1 to activate NF-κB. NF-kB induction in the MyD88-dependent pathway occurs with fast kinetics, whereas NF-kB activation in the MyD88-independent pathway occurs with slower kinetics.
The significance of the two different downstream pathways and the role of distinct adapter molecules of TLR4 activation in liver diseases are largely unknown. Nonetheless, many studies suggest that the activation of the different downstream pathways may be cell- and effect-specific. This may have important implications for developing TLR4 modulators as potential therapeutic agents.
Negative regulation of TLR4 signaling
Because TLR4 stimulation can induce potent inflammatory responses, inhibitory pathways are necessary to protect the host from inflammation-induced damage [14]. The balance is maintained by multiple negative regulators, and the regulation is very precise. TLR4 signaling can be regulated at multiple levels (from receptor level to transcription factors level; Table 1), through many kinds of mechanisms (degradation, deubiquitination, and competition are the most frequently observed). Table 1 describes the targets of each inhibitor. sTLR4 (soluble decoy TLR4), RP105 (radioprotective 105; a homolog of TLR4), SIGIRR (single immunoglobulin IL-1R-related molecule), ST2L (transmembrane form of ST2; homolog of the IL-1 receptor), MyD88s (splice variant of MyD88), SARM (sterile alpha- and armadillo-motif-containing protein), TRAF1, TRAF4, and IRAK-2c (splice variant of IRAK-2) inhibit TLR4 signaling by means of competing with various adaptors and transcription factors for binding sites. TRIAD3A (triad domain-containing protein 3 variant A), SOCS1 (suppressor of cytokine signaling-1), and Pin1 (peptidyl-prolyl isomerase1) inhibit several molecules of TLR4 signaling by means of polyubiquitination and subsequent proteasome-dependent degradation. A20, DUBA (de-ubiquitinating enzyme A), and CYLD (cylindromatosis protein) inhibit several mediators of TLR4 signaling by deubiquitination.
There are many other negative regulators that use different mechanisms to control TLRs signaling pathways. Bcl-3 (B cell leukemia-3) and TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) inhibit activation of NF-κB by stabilization of NF-κBp50 and IκBα, respectively. IRAK-M (a member of IRAK family without kinase activity) inhibits MyD88-mediated signaling by preventing the dissociation of IRAKs from MyD88. ATF3 (activating transcription factor-3) binds to the promoters and recruits histone deacetylase, resulting in altered chromatin structure to limit access to transcription factors (such as NF-κB). Both the Src homology 2 domain-containing protein tyrosine phosphatase (SHP)-1 and SHP-2 are intracellular tyrosine phosphatases, which inhibit IRAK-1 and TBK1, respectively. Tollip constitutively suppresses IRAK by forming a complex that is dissociated after TLR4 activation. MicroRNAs are 21–22-nucleotide, non-coding small RNAs that have been shown to be centrally involved in immune system development and function. Very recently, it was shown that miR-146 expression was increased by LPS stimulation [15], and miR-146 may inhibit IRAK-1 and TRAF6. PI3K (phosphatidylinositol 3-kinase) is a member of the lipid kinase family. Recognition of PAMP by TLRs can activate PI3K, which leads to activation Akt (serine/threonine protein kinase) and subsequent inactivation of glycogen synthase kinase-3β (GSK-3β). Inhibition of GSK-3β decreases NF-kB-dependent production of proinflammatory cytokines.
The expression of most negative regulators (including PI3K, A20, IRAK-M, and miR-146) can be induced by the activation of TLR4 and uses a mode of negative feedback to terminate TLR4 activation. However, there are also some constitutively expressed factors (including Tollip) that could possibly exert their functions only when TLRs are overactivated.
TLR4 expression in the liver
The healthy liver contains low mRNA levels of TLR4 and signaling molecules such as MD-2 and MyD88 in comparison to other organs [16, 17], suggesting that the low expression of TLR4 and signaling molecules may contribute to the high tolerance of the liver to LPS from the intestinal microbiota to which the liver is constantly exposed.
Because of the unique anatomical link between the liver and intestines, Kupffer cells (KC) are the first cell to encounter gut-derived toxins including LPS. Accordingly, Kupffer cells express TLR4 and are responsive to LPS [18]. Upon triggering, TLR4 signaling drives Kupffer cells to produce TNF-α, IL-1β, IL-6, IL-12, IL-18, and anti-inflammatory cytokine IL-10 [19].
Hepatocytes may uptake and eliminate LPS from portal and systemic circulation [20]. Hepatocytes express mRNA for TLR4 and respond to TLR4 ligands although there are contradictory data about the amount of TLR4 mRNA expression and the level of responsiveness to LPS [21, 22].
Activated human hepatic stellate cells (HSCs) express TLR4 and CD14 and respond to LPS [23]. TLR4 directly stimulates HSC to induce proinflammatory features, such as upregulation of chemokines (CCL2, CCL3, and CCL4) and adhesion molecules [vascular cell adhesion molecule 1 (VCAM-1), intercellular cell adhesion molecule 1 (ICAM-1), and E-selectin] and profibrogenic features including the enhancement of TGF-β signaling by the downregulation of TGF-β pseudoreceptor, bone morphogenetic protein and activin membrane-bound inhibitor (BAMBI) [21, 23].
Other liver cells, such as biliary epithelial cells, sinusoidal endothelial cells, and hepatic dendritic cells, express TLR4 and are responsive to LPS, but this expression and response have not been studied in detail [20].
The role of LPS/TLR4 signaling in CLD
There is increasing evidence for a role of LPS/TLR4 signaling in the pathogenesis of alcoholic liver disease, nonalcoholic fatty liver disease, chronic hepatitis C, chronic hepatitis B, primary sclerosing cholangitis, primary biliary cirrhosis, hepatic fibrosis, and hepatocarcinoma (Table 2). The evidence for a role of LPS and TLR4 in these diseases comes from two kinds of studies:
-
1.
Studies showing that LPS/TLR4 signaling is altered as a result of altered portal LPS levels and/or hepatic TLR4 expression in these diseases.
-
2.
Studies showing that modulation of LPS/TLR4 signaling (by suppressing/attenuating LPS production or TLR4 gene expression) influences the pathogenesis of these diseases.
Alcoholic liver disease
Alcoholic liver disease (ALD) is characterized by a spectrum of liver pathology ranging from fatty liver, steatohepatitis, to cirrhosis.
The LPS and TLR4 have been proposed as key players in the pathogenesis of ALD. Chronic ingestion of alcohol leads to a strong elevation of portal and systemic levels of LPS in animal models and humans [24–26]. The elevation of LPS appears to be predominantly caused by two mechanisms. First, alcohol exposure can promote the growth of Gram-negative bacteria in the intestine, which leads to enhanced production of LPS [27]. In addition, alcohol metabolism by Gram-negative bacteria and intestinal epithelial cells can result in accumulation of acetaldehyde, which in turn can increase intestinal permeability by opening intestinal tight junctions. Increased intestinal permeability can lead to increased transfer of LPS from the intestine to portal and systemic circulation [28]. Furthermore, chronic alcohol consumption upregulates hepatic TLR4 and sensitizes it to LPS to enhance TNF-α production [29]. Exposure to LPS during chronic alcohol consumption results in increased production of inflammatory mediators as well as in induction of reactive oxygen species (ROS) [30]. Finally, inhibition of LPS/TLR4 signaling by altering intestinal microbiota and LPS production (antibiotics or probiotics) or suppressing TLR4 gene expression protects against ALD. Indeed, treatment with lactobacillus or antibiotics suppresses alcohol-induced liver injury by reducing LPS circulating levels [31, 32]. TLR4-mutant mice have a strong reduction of alcohol-induced liver injury despite elevated LPS circulating levels [33].
Recent studies have clarified the cellular and molecular pathways by which LPS/TLR4 signaling promotes ALD. Kupffer cells have been established as a crucial cellular target of LPS in alcohol-induced liver injury as demonstrated by a strong reduction of alcoholic liver injury following depletion of Kupffer cells with gadolinium chloride [34]. Moreover, Hritz et al. [35] demonstrated that TLR4-mediated signal in ALD is mediated through a MyD88-independent pathway, most likely through the adapter molecule TRIF. Hepatic alcohol-induced production of inflammatory mediators (TNF-α and IL-6) and TLR4 coreceptors (CD14 and MD2) was prevented by TLR4 deficiency [35]. In addition, ROS production by cytochrome P450 and the nicotinamide adenine dinucleotide phosphate complexes was also prevented by TLR4 deficiency [35]. These data suggest that TLR4-mediated alcoholic liver injury is carried out by increased inflammatory mediators (TNF-α and IL-6) and ROS production. Taken together, these data suggest that activation of TLR4 in Kupffer cells by LPS is a key pathogenetic mediator of ALD through production of inflammatory cytokines and ROS.
Non-alcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) includes a continuum of disease ranging from steatosis to steatohepatitis and cirrhosis and usually develops in the setting of obesity and insulin resistance [36]. Mechanisms involved in the development of NAFLD are not yet fully clarified, and therapeutic options are still limited.
There is accumulating evidence that LPS/TLR4 signaling plays an essential role in the pathogenesis of NAFLD. In different human and animal studies, NAFLD was associated with increased portal LPS levels, through mechanisms involving bacterial overgrowth, and increased intestinal permeability and bacterial translocation [37–41]. Wigg et al. [37] found that bacterial overgrowth was prevalent among 22 patients with NAFLD. Bergheim et al. [38] showed that even the early stages of fructose-induced NAFLD are associated with an increased intestinal translocation of bacterial LPS. NAFLD has also been associated with increased sensitivity to LPS, mainly by increased hepatic TLR4 expression. Leptin or leptin receptor-deficient animals that are genetically obese are highly susceptible to LPS and develop NAFLD after low dose of LPS [42]. Finally, suppression of LPS/TLR4 signaling by alteration of intestinal microbiota (antibiotics or probiotics) or genetic manipulation protects against NAFLD. Selective intestinal decontamination results in decreased LPS levels in mice in a high-fat diet and reduced hepatic triglycerides in mice with diet-induced obesity as well as in leptin-deficient mice [40, 41]. Probiotics diminish non-alcoholic steatohepatitis in leptin-deficient mice [43, 44]. The crucial role for TLR4 signaling in NAFLD was further confirmed in TLR4-mutant mice that display decreased liver injury and lipid accumulation following a methionine- and choline-deficient diet (MCDD) and fructose-induced NAFLD [39, 45].
Besides the role of LPS/TLR4 signaling in the pathogenesis of NAFLD, there is also accumulating evidence showing a bidirectional connection between TLR4 signaling and insulin resistance (to which NALFD is intimately associated). There are several studies showing that LPS/TLR4 activation induces inflammatory signaling pathways which mediate insulin resistance and studies showing that insulin resistance may lead to activation of LPS/TLR4 signaling. Cani et al. [46] demonstrated that subcutaneous infusion of a low dose of LPS resulted in liver insulin resistance in a CD-14-dependent manner. On the other hand, it was shown that free fatty acids, which are often elevated in insulin resistance states, due to increased release from adipose tissue, can induce insulin resistance through activation of TLR4 [10]. Notably, a recent human study demonstrated that TLR4 expression and its ligands (high-mobility group box-1, hyaluronan, heat shock protein 60, and LPS), signaling, and functional activation are increased in recently diagnosed type-2 diabetes and contribute to a proinflammatory state [47].
Recent studies have clarified the mechanisms by which increased LPS/TLR4 signaling promotes NAFLD. Destruction of Kupffer cells with clodronate liposomes blunted histological evidence of non-alcoholic steatohepatitis in a model of MCDD and prevented increasing of TLR4 expression, underscoring a direct link between TLR4 and Kupffer cells within pathogenesis of NAFLD [39]. Hepatic lipid peroxidation, Myd88, and TNF-α levels were significantly decreased in fructose-fed TLR4 mutant mice in comparison to fructose-fed wild-type mice, suggesting that MyD88 may be critical in mediating the effects of TLR4 activation in the promotion of NAFLD, through enhanced ROS and induction of TNF-α [45]. Taken together, these data suggest a major role of TLR4 signaling in the pathogenesis of NAFLD through activation of Kupffer cells and enhanced ROS and TNF-α production.
HCV infection
About 30% of patients chronically infected with hepatitis C virus (HCV) show signs of active hepatic inflammation and are at risk of developing fibrosis, cirrhosis, and HCC [48].
There is an accumulating evidence that LPS and TLR4 play a key role in the pathogenesis of HCV infection. Patients with chronic HCV infection display increased serum levels of LPS even in the absence of significant hepatic fibrosis [49].
Interaction of HCV and TLR4 expression and signaling is robust although complex and may be cell-specific. Machida et al. [50] found that HCV, through the action of its NS5A protein, induces expression of TLR4 on the surface of B cells, leading to enhanced IFN-β and IL-6 production and secretion, particularly in response to LPS. Machida et al. [22] also provided evidence that hepatocyte-specific transgenic expression of the HCV nonstructural protein NS5A upregulates TLR4 expression and signaling. They demonstrated enhanced TAK-1–TRAF-6 and TAK-1–IRAK-1 interactions and phosphorylation of JNK and Iκ-Bα (downstream mediators of TLR4 signaling) in NS5A mice given LPS. Miyazaki et al. [51] found that myeloid dendritic cells from patients with chronic HCV display an increased expression of TLR4, but a decrease in the cytokine production secondary to activation of TLR4 by LPS, thus suggesting the impairment of TLR4 signaling by HCV in myeloid dendritic cells. Abe et al. [52] demonstrated that murine macrophages overexpressing NS3, NS3/4A, NS4B, or NS5A showed a strong suppression of TLR4 signaling. NS5A interacts with MyD88 to prevent IRAK-1 recruitment and cytokine production, such as IL-1, IL-6, and IFN-β response to the ligands for TLR4 [53].
TLR4 signaling itself may regulate HCV replication. Broering et al. [53] found that supernatants from TLR4-stimulated non-parenchymal liver cells (Kupffer cells and sinusoidal epithelial cells) led to potent suppression of HCV replication in murine HCV replicon bearing MH1 cells through IFN-β and induction of IFN-stimulated genes. These novel findings are of particular relevance for the control of HCV replication by the innate immune system of the liver.
Finally, TLR4 has also been associated with many clinical consequences of HCV infection. Machida et al. [22] demonstrated that, in a murine model, synergism between alcohol and HCV in liver damage and tumor formation is mediated by sustained activation of LPS/TLR4 signaling, which results from HCV NS5A-induced hepatic TLR4 expression and alcohol-induced endotoxemia. Recently, in a gene centric functional genome scan in patients with chronic hepatitis C virus, a major CC allele of TLR4 encoding a threonine at amino acid 399 (p.T399I) emerged as the second single nucleotide polymorphism (SNP) with highest ability to predict the risk of developing cirrhosis, indicating a protective role in fibrosis progression of its c.1196C_T (rs4986791) variant at this location (p.T399I), along with another highly cosegregated c.896A_G (rs4986790) SNP located at coding position 299 (p.D299G) [54]. Interestingly, later on, it was shown that these two SNP are associated with reduced TLR4-mediated inflammatory and fibrogenic signaling and lower apoptotic threshold of activated HSCs [55].
Taken together, these data support the hypothesis that HCV selectively influences TLR4 signaling, impairing it in cells that limit HCV replication (dendritic cells and macrophages), while at the same time enhancing it in cells (hepatocytes and B cells) that generate a chronic inflammatory state. Thus, it is likely that the interaction between HCV and TLR4 promotes virus expansion, inflammation, and potentially the progression to fibrosis and cirrhosis.
HBV infection
Hepatitis B virus (HBV) causes a chronic infection in about 10% of adults that may result in cirrhosis and HCC [45].
Recent studies have shown that LPS/TLR4 signaling may have an important role in the pathogenesis of HBV infection. One study reported a 72-fold induction of LPS levels in chronic HBV infection [56]. Moreover, a significant correlation was revealed between systemic LPS levels with virus replication and the degree of basic clinical and laboratory signs in patients with chronic viral hepatitis B [56].
Interaction of HBV and TLR4 expression and signaling is complex. One study demonstrated that TLR4 was downregulated in HBV-infected peripheral blood monocytes, and these cells also had a decreased cytokine response to TLR4 ligands [57]. On the other hand, TLR4 was shown to block HBV replication through its ability to upregulate IFNs. The injection of LPS into HBV transgenic mice reduced HBV replication in an IFN-α/β-dependent manner [58]. These antiviral effects of TLR4 activation are directed at nonparenchymal cells, but not hepatocytes that express low level of TLR4. Further experiments demonstrated that nonparenchymal cell-derived mediators inhibit HBV replication in HBV-Met cells. The supernatants from TLR4-stimulated Kupffer cells inhibit HBV replication independently of MyD88 in vitro, suggesting that TRIF-dependent IFN-β plays a role [59].
Taken together, these data suggest that TLR4 signaling is impaired in HBV infection and that TLR4 agonists can block HBV replication through activation of TRIF-dependent pathway in Kupffer cells.
Hepatic autoimmune disorders
The pathogenesis of hepatic autoimmune disorders remains still largely unknown. It is believed that autoimmunity may develop from genetic predispositions, but the onset of autoimmune tissue injury or disease flare is often triggered by microbial infection. Aberrant innate immune response to infections, providing the necessary inflammatory milieu to activate pre-existing autoreactive cellular repertoire, has the potential to initiate the development of autoimmunity. There is increasing evidence for LPS/TLR4 signaling in the pathogenesis of primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC).
Primary biliary cirrhosis (PBC) is a chronic inflammatory cholestatic disease of unknown origin that affects small and medium intrahepatic bile ducts. Recent studies have demonstrated that significant amounts of LPS accumulate in biliary epithelia of PBC patients [60]. Ballot et al. [61] reported that 64% of PBC sera were positive for IgM antibodies against lipid A, an immunogenic and toxic component of LPS. TLR4 expression is significantly elevated in biliary epithelial cells and periportal hepatocytes of PBC patients [62]. Monocytes from PBC patients appear more sensitive to the ligand for TLR4 (LPS), producing higher levels of proinflammatory cytokines, particularly IL-1β, IL-6, IL-8, and TNF-α [63].
The PSC is characterized by the destruction of hepatic bile duct and a high frequency of antibiliary epithelial cell antibodies (anti-BEC-Ab). One study revealed that, in primary sclerosing cholangitis, LPS gets accumulated abnormally in biliary epithelial cells [60]. Anti-BEC-Ab-stimulated BECs or PSC patient-derived BECs express higher levels of TLR4 and respond to ligands for TLR4 to produce higher levels of inflammatory cytokines (IL-1β, IL-8, IFN-γ, TNF-α, granulocyte–macrophage colony-stimulating factor, and TGF-β) [64].
These data suggest that in CBP and PSC increased accumulation of LPS and TLR4 expression in biliary epithelial cells enhances secretion of selective pro-inflammatory cytokines integral to the inflammatory response that may be critical in the breakdown of self-tolerance and initiation and perpetuation of bile duct injury.
Hepatic fibrosis
The development of hepatic fibrosis and cirrhosis occurs in virtually any type of chronic hepatic injury [65]. In terms of chronic liver injury, several studies have highlighted the role of transforming growth factor-β (TGF-β) in activating hepatic stellate cells (HSC), the main producers of extracellular matrix in the fibrotic liver and the promotion of a fibrogenic phenotype [65, 66]. On the other hand, chronic liver inflammation is a key prerequisite for triggering liver fibrosis [65, 66]. However, until now, the cell-type specific molecular mechanisms linking pathways driving inflammation on one hand and liver fibrogenesis on the other hand have not been defined yet. Recently, there is accumulating evidence that TLR4-induced activation and sensibilization of HSC may constitute the molecular link between hepatic inflammation and fibrogenesis.
The LPS is elevated in experimental models of hepatic fibrosis and in patients with cirrhosis [21, 67]. It is believed that changes in intestinal motility, subsequent alterations of the intestinal microbiota, decreased mucosal integrity, and suppressed immunity in hepatic fibrosis contribute to a failure of the intestinal mucosal barrier, and causes increases in bacterial translocation and LPS levels in later stages of hepatic fibrosis and cirrhosis [68].
Data regarding expression of TLR4 in cirrhotic patients are conflicting, with studies showing increased expression on BEC, maintained or increased expression on hepatocytes, and maintained or decreased expression on PBMC [62–64, 69–71].
Several studies have demonstrated that modulation of the intestinal microbiota in advanced cirrhosis by probiotics or antibiotics is beneficial for the prevention of bacterial translocation and spontaneous bacterial peritonitis [72, 73]. It has been shown that antibiotics prevent hepatic injury and fibrosis induced by CCl4 treatment or a choline-deficient diet, and that LPS enhances hepatic fibrosis induced by a MCCD [74, 75]. Treatment of mice with nonabsorbable broad-spectrum antibiotics also resulted in a clear reduction in the fibrotic response of mice, upon bile duct ligation [21]. Recently, Velayudham et al. showed that VSL#3 (a probiotic) protects against MCDD-induced liver fibrosis, through modulation of collagen expression and inhibition of TGF-β expression and signaling [76].
Recent studies, using TLR4 mutant as well as gut-sterilized, CD14- and LBP-deficient mice, have demonstrated the crucial role for the LPS–TLR4 pathway in hepatic fibrogenesis [21, 77]. TLR4-mutant mice display a profound reduction in hepatic fibrogenesis in three different experimental models of biliary and toxic fibrosis [77].
In a recent study, Seki et al. [21] analyzed the cell-specific molecular mechanism underlying the role of LPS/TLR4 on liver fibrosis. They showed that chimeric mice that contain TLR4-mutant Kupffer cells and TLR4-intact HSCs developed significant fibrosis and the mice that contain TLR4-intact Kupffer cells and TLR4-mutant HSCs developed minimal fibrosis after bile duct ligation, indicating that TLR4 on HSCs, but not on Kupffer cells, is crucial for hepatic fibrosis. Notably, Kupffer cells are essential for fibrosis by producing TGF-β independent of TLR4. TLR4-activated HSCs produce chemokines (CCL2, CCL3, and CCL4) and express adhesion molecules (ICAM-1 and VCAM-1) that recruit Kupffer cells to the site of injury. Simultaneously, TLR4 signaling downregulates the TGF-β decoy receptor (BAMBI) to boost TGF-β signaling and allow for unrestricted activation of HSCs by Kupffer cells, leading to hepatic fibrosis. Finally, by using adenoviral vectors expressing an inhibitor of NF-κB kinase (IκB)-superrepressor and knockout mice for MyD88 and the adapter molecule TRIF, the authors demonstrated that TLR4-dependent downregulation of BAMBI is mediated via a pathway involving MyD88 and NF-κB, but not TRIF. In summary, they demonstrated that LPS/TLR4 signaling acts in a profibrogenic manner via two independent mechanisms: it induces the secretion of chemokines from HSCs and chemotaxis of Kupffer cells which secrete the profibrogenic cytokine TGF-β; additionally, TLR4-dependent signals augment TGF-β signaling on HSCs via downregulation of the TGF-β pseudoreceptor BAMBI.
Recently, Huang et al. [54] conducted a gene centric functional genome scan in patients with chronic hepatitis C virus, which yielded a Cirrhosis Risk Score signature consisting of seven single nucleotide polymorphisms (SNPs) that may predict the risk of developing cirrhosis. Among these, a major CC allele of TLR4 encoding a threonine at amino acid 399 (p.T399I) was the second most predictive SNP among the seven, indicating a protective role in fibrosis progression of its c.1196C>T (rs4986791) variant at this location (p.T399I), along with another highly cosegregated c.896A>G (rs4986790) SNP located at coding position 299 (p.D299G). In a subsequent study, the same group examined the functional linkage of these SNPs to hepatic stellate cell (HSC) responses [55]. They showed both HSCs from TLR4-deficient mice, and a human HSC line (LX-2) reconstituted with either TLR4 D299G and/or T399I complementary DNAs were hyporesponsive to LPS stimulation compared to those expressing wild-type TLR4 as assessed by the expression and secretion of LPS-induced inflammatory and chemotactic cytokines (i.e., monocyte chemoattractant protein-1, IL-6), downregulation of BAMBI expression, and activation of NF-κB-responsive luciferase reporter. In addition, spontaneous apoptosis, as well as apoptosis induced by pathway inhibitors of NF-κB, extracellular signal-regulated kinase (ERK), and phosphatidylinositol 3-kinase were greatly increased in HSCs from either TLR4-deficient or Myd88-deficient mice, as well as in murine HSCs expressing D299G and/or T399I SNPs [55]. Recently, Li et al. expanded the list of TLR4 SNPs that are independently associated with the risk of liver fibrosis progression and the development of cirrhosis [78].
Taken together, these data suggest that LPS/TLR4 signaling in HSC is essential for liver fibrosis development, by stimulating production chemokines that recruit Kupffer cells and at the same time allowing for unrestricted activation of HSCs by Kupffer cells-derived TGF-β.
Hepatocarcinoma
During recent years, evidence has been accumulating to show that inflammation has an important role in initiation, promotion, and progression of tumors [79, 80]. The generation of pro-inflammatory cytokines in the tumor microenvironment provokes activation of NF-κB in cancer cells, leading to protection against pro-apoptotic host immune defense mechanisms [79, 80]. It has been shown that cytokines and growth factors produced by tumor-infiltrating macrophages, lymphocytes, and other cell types in the inflammatory tumor microenvironment influence cell differentiation and exert antiapoptotic and proangiogenic effects which stimulate the growth of cancer cells, tumor invasiveness, and metastasis [79, 80].
Hepatocarcinoma (HCC), a prominent example for inflammation-associated cancer, is a major complication in the end-stage of cirrhosis [81]. In most cases, HCC in humans is the outcome of continuous injury and chronic inflammation; thus, it provides a good and realistic inflammatory-related cancer model to gain insight about the role of TLR4 in the carcinogenesis [81]. Two studies have revealed TLRs, in particular TLR4, as major factors linking hepatic chronic inflammation and hepatocarcinoma.
Diethylnitrosamine (DEN) is a chemical carcinogen used to create a mouse model of HCC [82]. The pathogenesis of HCC in this mouse model differs from that in humans and thus may not be directly comparable to human HCC. Nevertheless, the mouse model of DEN-induced HCC has a histology and genetic signature similar to that of human HCCs with poor prognosis and recapitulates a dependence on inflammation and gender disparity seen in human HCC [83]. Naugler et al. [12] showed in a model of DEN-induced HCC in mice that the tumor appears in 100% of males but only in 13% females. This is correlated with increased liver injury and a higher production of IL-6 in males after toxicant administration. They also showed that IL-6 production after DEN-induced liver injury occurs through TLR4 stimulation and demonstrated the implication of the innate immune response in the hepatocarcinogenic process. They observed that the accumulation of IL-6 mRNA in Kupffer cells incubated with LPS or necrotic hepatocytes was markedly reduced in MyD88 null mice. They also found that liver damage and hepatic IL-6 levels were significantly diminished after DEN administration in MyD88-deficient mice. Importantly, these mice also showed a significant reduction in the number and size of DEN-induced liver tumors.
Clinical and epidemiological evidence implicates long-term alcohol consumption in accelerating HCV-mediated tumorigenesis [84]. A recent study provided evidence that TLR4 mediates the synergism between alcohol and HCV in hepatic oncogenesis. Machida et al. [22] studied the molecular mechanism of synergism between alcohol and HCV, using mice with hepatocyte-specific transgenic expression of the HCV nonstructural protein NS5A, which is known to have a cryptic trans-acting activity for cellular gene promoters. They demonstrated that NS5A and alcohol synergistically induce hepatocellular damage and transformation via accentuated and/or sustained activation of TLR4 signaling, which results from HCV NS5A-induced hepatic TLR4 expression and alcohol-induced endotoxemia. Additionally, Nanog, a stem cell marker, was identified as a novel downstream gene transcriptionally induced by activated TLR4 signaling that is largely responsible for TLR4-mediated liver tumor development.
Taken together, these data suggest that TLR4 signaling in Kupffer cells and hepatocyte may constitute the link between hepatic chronic inflammation and hepatocarcinoma.
Conclusion
TLR4, as the other members of toll-like receptors family, is an essential player of innate immune system. It is activated by LPS, a Gram-negative bacterial cell wall component, as well as endogenous components derived from dying host cell. Activation of TLR4 results in the production of several pro-inflammatory, anti-viral, and anti-bacterial cytokines, which mount a rapid protective response against invading pathogens. Nevertheless, these cytokines may also trigger harmful responses such as cell death, fibrosis, and cancer.
Despite the constant confrontation of hepatic TLR4 with gut-derived LPS, the normal liver does not show signs of inflammation due to its low expression of TLR4 and ability to inhibiting TLR4 signals. Enhanced signaling of TLR4 may lead to persistently elevated inflammatory cytokines, resulting in chronic liver injury (Fig. 2). Indeed, in CLD, such as ALD, NAFLD, PSC, CBP, and fibrosis, it has been shown that LPS/TLR4 signaling is enhanced and is essential for liver injury. Enhanced LPS/TLR4 signaling may result from increased expression and/or sensitivity of TLR4 and, mainly, from increased exposure to LPS. Increased portal levels of LPS have been documented in many CLD and result mainly from increased intestinal permeability. In initial stages of CLD this increase of intestinal permeability may be dependent on etiology of CLD (i.e., alcohol, diet), but later on liver fibrosis and subsequent portal hypertension can become the main inducers of this alteration.

Overview of the role of LPS/TLR4 signaling in chronic liver diseases. Increased expression and/or sensitivity of hepatic TLR4 and increased portal LPS levels (resulting from bacterial overgrowth and intestinal permeability) can lead to enhanced LPS/TLR4 signaling. This can induce anti-viral responses, inflammation, steatosis, fibrosis, and hepatocarcinoma. Hepatic fibrosis contributes to portal hypertension development which further increases bacterial overgrowth and intestinal permeability, creating a positive feedback process
In many of CLD, inhibition of TLR4 has been shown to decrease liver injury, reinforcing the importance of LPS/TLR4 signaling in the pathogenesis of those diseases. Of the many possibilities to suppress TLR4 signaling (modulation of LPS production, TLR and co-receptors expression and downstream signaling molecules), the first appear to be the best as the others may result in systemic suppression of TLR4 disabling it to respond to invading pathogens. Modulation of the intestinal microbiota can be achieved by antibiotics, probiotics, and symbiotics. Probiotics and symbiotics, which already proved to have positive effects in patients with CLD, should be preferred due to their high tolerability and limited side effects.
TLR4 plays also a role in chronic viral hepatitis. Chronic hepatitis B and C viruses lead to a downregulation of antiviral TLR4 signaling pathways. On the other hand, TLR4 was shown to block HBV and HCV replication through its ability to upregulate IFNs. This suggests that TLR4 agonists may boost anti-viral immunity and therefore represent a novel treatment approach for chronic viral hepatitis.
Although we need more studies, mainly in human patients, to translate TLR4 pathogenesis into clinical practice in CLD, we can anticipate that with further research on LPS/TLR4 signaling, this pathway will become an important pharmacological target in CLD.
Abbreviations
- Akt:
-
Serine/threonine protein kinase
- ALD:
-
Alcoholic liver disease
- Anti-BEC-Ab:
-
Antibiliary epithelial cell antibodies
- AP-1:
-
Activator protein 1
- ATF3:
-
Activating transcription factor-3
- BAMBI:
-
Bone morphogenetic protein and activin membrane-bound inhibitor
- Bcl-3:
-
B cell leukemia-3
- BEC:
-
Biliary epithelial cell
- CCL:
-
Chemokine
- CCl4 :
-
Carbon tetrachloride
- CLD:
-
Chronic liver diseases
- CYLD:
-
Cylindromatosis protein
- DAMP:
-
Damage-associated molecular patterns
- DEN:
-
Diethylnitrosamine
- DUBA:
-
De-ubiquitinating enzyme A
- ERK:
-
Extracellular signal-regulated kinase
- GSK-3β:
-
Glycogen synthase kinase-3β
- HBV:
-
Hepatitis B virus
- HCC:
-
Hepatocarcinoma
- HCV:
-
Hepatitis C virus
- HSC:
-
Hepatic stellate cell
- ICAM:
-
Intercellular cell adhesion molecule
- IFN:
-
Interferon
- IKK:
-
Inhibitor of NF-κB kinase
- IL:
-
Interleukin
- IRAK:
-
Interleukin-1 receptor-associated kinase
- IRF:
-
Interferon regulatory factor
- IκBα:
-
Inhibitor of NF-κB
- JNK:
-
C-Jun N-terminal kinase
- KC:
-
Kupffer cells
- LBP:
-
LPS binding protein
- LPS:
-
Lipopolysaccharide
- MCDD:
-
Methionine- and choline-deficient diet
- miR:
-
MicroRNA
- MyD88:
-
Myeloid differentiation factor 88
- MyD88s:
-
Splice variant of MyD88
- NAFLD:
-
Non-alcoholic fatty liver disease
- NEMO:
-
NF-κB essential modifier
- NF-κB:
-
Nuclear factor κB
- PAMP:
-
Pathogen-associated molecular pattern
- PBC:
-
Primary biliary cirrhosis
- PI3K:
-
Phosphatidylinositol 3-kinase
- Pin:
-
Peptidyl-prolyl isomerase
- PRR:
-
Pattern recognition receptor
- PSC:
-
Primary sclerosing cholangitis
- RIP:
-
Receptor-interacting serine–threonine kinase
- ROS:
-
Reactive oxygen species
- RP105:
-
Radioprotective 105
- SARM:
-
Sterile alpha- and armadillo-motif-containing protein
- SHP:
-
Src homology 2 domain-containing protein tyrosine phosphatase
- SIGIRR:
-
Single immunoglobulin IL-1R-related molecule
- SNP:
-
Single nucleotide polymorphism
- SOCS1:
-
Suppressor of cytokine signaling-1
- ST2L:
-
Transmembrane form of ST2
- sTLR4:
-
Soluble decoy TLR4
- TAK:
-
Transforming growth factor-β-activated kinase
- TANK:
-
TRAF family member associated NF-κB activator
- TBK:
-
TANK binding kinase
- TGF:
-
Transforming growth factor
- TIRAP:
-
Toll/IL-1 receptor domain-containing adaptor protein
- TNF:
-
Tumor necrosis factor
- TIR:
-
Toll/interleukin 1 receptor
- Tollip:
-
Toll interacting protein
- TLR:
-
Toll-like receptor
- TRAF:
-
Tumor necrosis receptor-associated factor
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- TRAM:
-
TRIF-related adaptor molecule
- TRIAD3A:
-
Triad domain-containing protein 3 variant A
- TRIF:
-
Toll/IL-1 receptor domain-containing adaptor inducing interferon-β
- VCAM:
-
Vascular cell adhesion molecule
References
Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature 2000;406:782–787
Beutler BA. TLRs and innate immunity. Blood 2009;113:1399–1407
Pimentel-Nunes P, Soares JB, Roncon-Albuquerque R, Dinis-Ribeiro M, Leite-Moreira AF. Toll-like receptors as therapeutic targets in gastrointestinal diseases. Expert Opin Ther Targets 2010;14:347–368
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol 2004;4:499–511
Kim HM, Park BS, Kim J, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee J. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007;130:906–917
Bryant CE, Spring DR, Gangloff M, Gay NJ. The molecular basis of the host response to lipopolysaccharide. Nat Rev Microbiol 2010;8:8–14
Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 2005;201:1135–1143
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 2005;11:1173–1179
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006;116:3015–3025
Chen C, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007;13:851–856
Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007;317:121–124
Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol 2010;87:989–999
Liew FY, Xu D, Brint EK, O’Neill LAJ. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol 2005;5:446–458
Davidson-Moncada J, Papavasiliou FN, Tam W. MicroRNAs of the immune system: roles in inflammation and cancer. Ann N Y Acad Sci 2010;1183:183–194
De Creus A, Abe M, Lau AH, Hackstein H, Raimondi G, Thomson AW. Low TLR4 expression by liver dendritic cells correlates with reduced capacity to activate allogeneic T cells in response to endotoxin. J Immunol 2005;174:2037–2045
Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 2002;168:554–561
Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, Remick DG, Wang SC. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 2000;31:932–936
Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, Adachi K, Futatsugi S, Kuida K, Takeuchi O, Okamura H, Fujimoto J, Akira S, Nakanishi K. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J Immunol 2001;166:2651–2657
Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology 2006;130:1886–1900
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 2007;13:1324–1332
Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, Asahina K, Govindarajan S, Ray R, Ou JJ, Seki E, Deshaies R, Miyake K, Lai MM. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA 2009;106:1548–1553
Paik Y, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003;37:1043–1055
Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol 1991;12:162–169
Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 2000;32:742–747
Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 2000;32:1008–1017
Hauge T, Persson J, Danielsson D. Mucosal bacterial growth in the upper gastrointestinal tract in alcoholics (heavy drinkers). Digestion 1997;58:591–595
Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F, Kang YJ, Keshavarzian A, Rao R, Sartor RB, Swanson C, Turner JR. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 2008;42:349–361
Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Devière J, Le Moine O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology 2006;43:989–1000
Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology 2003;124:778–790
Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 1995;108:218–224
Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease). Proc Soc Exp Biol Med 1994;205:243–247
Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001;34(1):101–108
Adachi Y, Bradford BU, Gao W, Bojes HK, Thurman RG. Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 1994;20:453–460
Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, Kurt-Jones E, Szabo G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008;48:1224–1231
Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 2005;42:44–52
Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001;48:206–211
Bergheim I, Weber S, Vos M, Krämer S, Volynets V, Kaserouni S, McClain CJ, Bischoff SC. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol 2008;48:983–992
Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 2007;47:571–579
Brun P, Castagliuolo I, Di Leo V, Buda A, Pinzani M, Palù G, Martines D. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2007;292:G518–G525
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008;57:1470–1481
Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA 1997;94:2557–2562
Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, Desimone C, Song X, Diehl AM. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003;37:343–350
Solga SF, Diehl AM. Non-alcoholic fatty liver disease: lumen-liver interactions and possible role for probiotics. J Hepatol 2003;38:681–687
Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009;50:1094–1104
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–1772
Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010;33:861–868
Boonstra A, Woltman AM, Janssen HLA. Immunology of hepatitis B and hepatitis C virus infections. Best Pract Res Clin Gastroenterol 2008;22:1049–1061
Dolganiuc A, Norkina O, Kodys K, Catalano D, Bakis G, Marshall C, Mandrekar P, Szabo G. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology 2007;133:1627–1636
Machida K, Cheng KTH, Sung VM, Levine AM, Foung S, Lai MMC. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol 2006;80:866–874
Miyazaki M, Kanto T, Inoue M, Itose I, Miyatake H, Sakakibara M, Yakushijin T, Kakita N, Hiramatsu N, Takehara T, Kasahara A, Hayashi N. Impaired cytokine response in myeloid dendritic cells in chronic hepatitis C virus infection regardless of enhanced expression of Toll-like receptors and retinoic acid inducible gene-I. J Med Virol 2008;80:980–988
Abe T, Kaname Y, Hamamoto I, Tsuda Y, Wen X, Taguwa S, Moriishi K, Takeuchi O, Kawai T, Kanto T, Hayashi N, Akira S, Matsuura Y. Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J Virol 2007;81:8953–8966
Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, Szczeponek A, Gerken G, Schlaak JF. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol 2008;48:914–922
Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, Layden TJ, Wright TL, White T, Cheung RC. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology 2007;46:297–306
Guo J, Loke J, Zheng F, Hong F, Yea S, Fukata M, Tarocchi M, Abar OT, Huang H, Sninsky JJ, Friedman SL. Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of Toll-like receptor 4 to hepatic stellate cell responses. Hepatology 2009;49:960–968
Sozinov AS. Systemic endotoxemia during chronic viral hepatitis. Bull Exp Biol Med 2002;133:153–155
Chen Z, Cheng Y, Xu Y, Liao J, Zhang X, Hu Y, Zhang Q, Wang J, Zhang Z, Shen F, Yuan Z. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin Immunol 2008;128:400–408
Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol 2005;79:7269–7272
Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, Schlaak JF. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology 2007;46:1769–1778
Sasatomi K, Noguchi K, Sakisaka S, Sata M, Tanikawa K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol 1998;29:409–416
Ballot E, Bandin O, Chazouilleres O, Johanet C, Poupon R. Immune response to lipopolysaccharide in primary biliary cirrhosis and autoimmune diseases. J Autoimmun 2004;22:153–158
Wang A, Migita K, Ito M, Takii Y, Daikoku M, Yokoyama T, Komori A, Nakamura M, Yatsuhashi H, Ishibashi H. Hepatic expression of toll-like receptor 4 in primary biliary cirrhosis. J Autoimmun 2005;25:85–91
Mao TK, Lian Z, Selmi C, Ichiki Y, Ashwood P, Ansari AA, Coppel RL, Shimoda S, Ishibashi H, Gershwin ME. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology 2005;42:802–808
Karrar A, Broomé U, Södergren T, Jaksch M, Bergquist A, Björnstedt M, Sumitran-Holgersson S. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology 2007;132:1504–1514
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209–218
Henderson NC, Iredale JP. Liver fibrosis: cellular mechanisms of progression and resolution. Clin Sci 2007;112:265–280
Chan CC, Hwang SJ, Lee FY, Wang SS, Chang FY, Li CP, Chu CJ, Lu RH, Lee SD. Prognostic value of plasma endotoxin levels in patients with cirrhosis. Scand J Gastroenterol 1997;32:942–946
Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005;41:422–433
Manigold T, Böcker U, Hanck C, Gundt J, Traber P, Antoni C, Rossol S. Differential expression of toll-like receptors 2 and 4 in patients with liver cirrhosis. Eur J Gastroenterol Hepatol 2003;15:275–282
Riordan SM, Skinner N, Nagree A, McCallum H, McIver CJ, Kurtovic J, Hamilton JA, Bengmark S, Williams R, Visvanathan K. Peripheral blood mononuclear cell expression of toll-like receptors and relation to cytokine levels in cirrhosis. Hepatology 2003;37:1154–1164
Tazi KA, Quioc J, Saada V, Bezeaud A, Lebrec D, Moreau R. Upregulation of TNF-alpha production signaling pathways in monocytes from patients with advanced cirrhosis: possible role of Akt and IRAK-M. J Hepatol 2006;45:280–289
Lata J, Novotný I, Príbramská V, Juránková J, Fric P, Kroupa R, Stibůrek O. The effect of probiotics on gut flora, level of endotoxin and Child-Pugh score in cirrhotic patients: results of a double-blind randomized study. Eur J Gastroenterol Hepatol 2007;19:1111–1113
Fernández J, Navasa M, Planas R, Montoliu S, Monfort D, Soriano G, Vila C, Pardo A, Quintero E, Vargas V, Such J, Ginès P, Arroyo V. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology 2007;133:818–824
Luckey TD, Reyniers JA, Gyorgy P, Forbes M. Germfree animals and liver necrosis. Ann N Y Acad Sci 1954;57:932–935
Rutenburg AM, Sonnenblick E, Koven I, Aprahamian HA, Reiner L, Fine J. The role of intestinal bacteria in the development of dietary cirrhosis in rats. J Exp Med 1957;106:1–14
Velayudham A, Dolganiuc A, Ellis M, Petrasek J, Kodys K, Mandrekar P, Szabo G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009;49:989–997
Isayama F, Hines IN, Kremer M, Milton RJ, Byrd CL, Perry AW, McKim SE, Parsons C, Rippe RA, Wheeler MD. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am J Physiol Gastrointest Liver Physiol 2006;290:G1318–G1328
Li Y, Chang M, Abar O, Garcia V, Rowland C, Catanese J, Ross D, Broder S, Shiffman M, Cheung R, Wright T, Friedman SL, Sninsky J. Multiple variants in toll-like receptor 4 gene modulate risk of liver fibrosis in Caucasians with chronic hepatitis C infection. J Hepatol 2009;51:750–757
Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005;5:749–759
Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 2006;441:431–436
Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology 2004;127:S5–S16
Maeda S, Kamata H, Luo J, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005;121:977–990
Lee J, Chu I, Mikaelyan A, Calvisi DF, Heo J, Reddy JK, Thorgeirsson SS. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet 2004;36:1306–1311
Brechot C, Nalpas B, Feitelson MA. Interactions between alcohol and hepatitis viruses in the liver. Clin Lab Med 1996;16:273–287
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Soares, JB., Pimentel-Nunes, P., Roncon-Albuquerque, R. et al. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int 4, 659–672 (2010). https://doi.org/10.1007/s12072-010-9219-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-010-9219-x