
Abstract
Innate immunity against Mycobacterium tuberculosis is a critical early response to prevent the establishment of the infection. Despite recent advances in understanding the host-pathogen dialogue in the early stages of tuberculosis (TB), much has yet to be learnt. The nature and consequences of this dialogue ultimately determine the path of infection: namely, either early clearance of M. tuberculosis, or establishment of M. tuberculosis infection leading to active TB disease and/or latent TB infection. On the frontline in innate immunity are pattern recognition receptors (PRRs), with soluble factors (e.g. collectins and complement) and cell surface factors (e.g. Toll-like receptors and other C-type lectin receptors (Dectin 1/2, Nod-like receptors, DC-SIGN, Mincle, mannose receptor, and MCL) that play a central role in recognising M. tuberculosis and facilitating its clearance. However, in a ‘double-edged sword’ scenario, these factors can also be involved in enhancement of pathogenesis as well. Furthermore, innate immunity is also a critical bridge in establishing the subsequent adaptive immune response, which is also responsible for granuloma formation that cordons off M. tuberculosis infection, establishing latency and acting as a reservoir for bacterial persistence and dissemination of future disease. This chapter discusses the current understanding of pattern recognition of M. tuberculosis by innate immunity and the role this plays in the pathogenesis and protection against TB.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
Tuberculosis (TB), predominately caused by the bacterium Mycobacterium tuberculosis, remains one of the world’s most significant infectious diseases, with a worldwide yearly burden of approximately 8.7 million new cases of active TB, 1.4 million deaths and a third of the world’s population with latent TB infection (LTBI) (WHO 2019). The major burden for TB disease in still bared by developing countries, with Asia (e.g. China and India) having the highest number of cases of the disease. The epidemiological trend in developed countries continues to be that the majority of TB cases have originated from recent immigrants that have come from TB endemic areas of the world (Zumla et al. 2013; Zaheen and Bloom 2020). There are also significant numbers of TB patients co-infected with Human immunodeficiency virus (HIV), particularly in sub-Saharan Africa, resulting in the highest rates of TB cases per capita (WHO 2019). The outbreak of the COVID-19 pandemic may also result in a similar dangerous synergy with TB, although the impact of co-infection of M. tuberculosis with the SARS-CoV-2 virus in patients remains to be determined (Ong et al. 2020). The frequency of multi-drug resistant tuberculosis (MDR-TB) to the main drugs used for treatment (e.g. rifampicin and isoniazid) is still worryingly high, particularly in India, Russia, China, Pakistan and South Africa (WHO 2019; Zaheen and Bloom 2020). Of further concern is the rise of extensively drug resistant (XDR-TB) in several countries to all current second and third-line therapies (Zaheen and Bloom 2020).
There continues to be a strong concerted effort to develop new interventions and therapies against TB with a particularly important focus on understanding innate immunity against TB particularly in the early stages of infection and the granuloma. It is well know that M. tuberculosis is able to persist as an intracellular parasite for years in the host as LTBI, mainly because of its ability to persist in the host macrophage by manipulating phagolysosome maturation, providing a favourable niche for it to be able to reside (Russell 2001; Gupta et al. 2012). M. tuberculosis-infected macrophages in LTBI are predominantly present within the granuloma, which is a complex structure of T cells, B cells, and giant epithelioid cells among others and a re-sectioning of tissue to contain these infected macrophages (Gupta et al. 2012). The formation and maintenance of this immune cordon against TB infection is crucial in preventing disseminated disease and transmission of the infection to other hosts. It is still not fully understood how the granuloma is formed and maintained in TB infection and the full extent of the contribution played by innate immunity. Here, we will discuss the latest advancements in the understanding of innate immune recognition of M. tuberculosis and how these contribute to both downstream protection and pathogenesis of TB.
The primary interactions of M. tuberculosis with the host upon inhalation remain to be determined fully, particularly the targeting and recognition by the innate immune response. The lungs are the main route of entry to the host for M. tuberculosis and is the main anatomical site for infection and pathogenesis, but not entirely as extra-pulmonary TB (EPTB) also relatively commonly occurs in 10–42% of cases (Caws et al. 2008). The establishment and dissemination of M. tuberculosis infection is dependent on several host and pathogen factors with the pathogen able to alter and circumvent facets of both the innate and adaptive immune responses. Initially, M. tuberculosis bacilli, within aerosol droplet nuclei (on average 4–7μm in size), are inhaled into the pulmonary alveoli where they come into primary contact and are phagocytosed by alveolar macrophages (Fennelly et al. 2004; Fennelly 2020). During this interaction, most of the bacilli are killed, but some can endure within the macrophage (Russell 2001; Gupta et al. 2012). The recognition and uptake of M. tuberculosis by the host is govern by several soluble and cell-bound factors such as pattern recognition receptors (PRRs) that recognise pathogen-associated molecular patterns (PAMPs) that are present on the surface of microbes and normally absent on host cells. Examples of PAMPs include lipopolysaccharide (LPS), porins, peptidoglycan, lipoteichoic acid (LTA), mannose-rich glycans, flagellin, bacterial and viral genomes, mycolic acid, and lipoarabinomannan (LAM). PRRs include phagocytic PRRs and signalling PRRs. Examples of phagocytic PRRs include C-type lectins receptors (CTLRs) (e.g. collectins such as surfactant protein A (SP-A), surfactant protein D (SP-D), mannose receptor (MR), Dectin-1), scavenger receptors (e.g. CD-36, CD68, and SRB-1), opsonic receptors (e.g. plasma acute phase proteins like mannose binding lectin (MBL), C-reactive protein (CRP)) and complement proteins (e.g. C3b, iC3b, factor H and properdin). Signalling PRRs are either present on cell surface (e.g. Toll-like receptors (TLRs), CD14, on intracellular membranes (e.g. endosomes, lysosomes) or in the cytoplasm (e.g. nucleotide-binding oligomerization domain (NOD)-like receptors. In microbial infection and in particular M. tuberculosis infection the type of interaction of PRRs with PAMPs and innate immune cells (e.g. macrophages) also determine the subsequent cell signalling pathways (leading to production of cytokines/chemokines), which initiates inflammation and tissue modification (e.g. granuloma formation) (Feng et al. 2006; Lockhart et al. 2006; Eum et al. 2010). Furthermore, the formation of the granuloma occurs without the requirement for specific immunity (North and Izzo 1993; Hansch et al. 1996; Smith et al. 1997), with both tumour necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ) being the foremost signalling cytokines for cell infiltration, although they are not needed to begin the process of granuloma formation (Flynn et al. 1995; Smith et al. 1997).
After M. tuberculosis enters the alveoli space, the bacteria are internalized into alveolar macrophages by phagocytosis, a process triggered by receptor-ligand engagement. M. tuberculosis tends to target binding to macrophages in cholesterol-rich regions of the host cell membrane (Gatfield and Pieters 2000). Mycobacteria can be targeted via a wide variety of receptors that recognise opsonised and non-opsonised bacilli. These include collectins (SP-A, SP-D, conglutinin), MR (CD207), dendritic-cell-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN), dectin-1, complement receptors (CR), surfactant protein (SP) receptors, scavenger receptors, and glycosylphosphatidylinositol (GPI)-anchored receptors such as CD14 (Schlesinger et al. 1990; Schlesinger 1993; Stokes et al. 1993; Hirsch et al. 1994; Zimmerli et al. 1996; Ehlers and Daffe 1998; Ernst 1998; Peyron et al. 2000; Rooyakkers and Stokes 2005). Various TLRs have also been shown to play important roles in M. tuberculosis interactions on the surface and within phagocytic cells (Means et al. 1999). In addition, M. tuberculosis can also recruit several host cell molecules on its surface that enhance its uptake by phagocytes.
2 Collectins and Mycobacteria
Collectins are a group of soluble C-type (calcium-dependent) lectins, characterised by an N-terminal collagen region, an alpha helical coil neck motif, and a C-terminal carbohydrate recognition domain (CRD). Collectins can recognise and bind to PAMPs on variety of microbes via their CRD region and have a crucial role in their neutralisation and clearance but are also a critical bridge between the innate immunity and adaptive immunity (Kishore et al. 2006). The mammalian collectin family comprises of SP-A and SP-D, MBL, liver collectin (CL-L1), kidney collectin (CL-K1), CL-LK (composed of CL-L1 and Cl-K1) and placenta collectin (CL-P1), conglutinin, CL-43 and CL-46, with the latter three found in Bovidae (Murugaiah et al. 2020; Tsolaki and Kishore 2020). Several of these collectins have a role in mycobacterial infection and pathogenesis.
Both SP-A and SP-D are the most relevant collectins for pulmonary TB as they are important components of pulmonary surfactant which is essential for the physiology of alveoli (Murugaiah et al. 2020). Furthermore, early observations also showed that pulmonary surfactant had anti-microbial properties being able to enhance clearance of Staphylococcus aureus by alveolar macrophages (AM) (Laforce et al. 1973). In fact, both SP-A and SP-D can target Gram-negative and Gram-positive bacteria enhancing their clearance through phagocytosis by AM (Pikaar et al. 1995).
Both SP-A and SP-D can bind and agglutinate mycobacteria but seem to have opposing effects on the phagocytosis of M. tuberculosis. SP-A targets the putative surface adhesin Apa glycoprotein on M. tuberculosis (Ragas et al. 2007), whilst SP-D can also bind to LTA and peptidoglycan and to lipoarabinomannan (LAM) from M. tuberculosis and Mycobacterium avium (Ferguson et al. 1999; Van De Wetering et al. 2001; Kudo et al. 2004). SP-A can facilitate uptake of M. tuberculosis and M. avium by enhancing the expression of macrophage MR (Gaynor et al. 1995; Beharka et al. 2002; Kudo et al. 2004) (Fig. 9.1). Similarly, SP-A enhances expression of scavenger receptor A (SR-A), increasing the phagocytosis of Streptococcus pneumoniae by AM (Kuronuma et al. 2004). Additionally, bound SP-A can also facilitate uptake of Mycobacterium bovis bacillus Calmette-Guérin (BCG) by binding to specific 210-kDa SP-A receptor (SPR210) in U937 macrophages and rat AM (Chroneos et al. 1996; Weikert et al. 1997). Furthermore, this interaction led to increased mycobacterial killing and production of TNF-α and nitric oxide (Weikert et al. 2000). In contrast, SP-D inhibits phagocytosis of M. tuberculosis by blocking the interaction of LAM with macrophage MR, and is independent of agglutination by SP-D (Ferguson et al. 1999; Ferguson et al. 2002) (Fig. 9.1). Gene knockout mice (SP-A−/−, SP-D−/−, and SP-A/D−/−) infected with M. tuberculosis, still processed the ability for phagocytosis and bacterial clearance, suggesting that both SP-A and SP-D are not crucial for protection in this animal model for TB (Lemos et al. 2011). SP-A and SP-D can also influence the intracellular environment post phagocytosis, by stimulating and enhancing reactive oxygen and nitrogen species enabling the killing of intracellular pathogens such as mycobacteria (Fig. 9.1). SP-A enhances the intracellular killing of M. bovis BCG by enhancing nitric oxide (NO) levels and releasing TNF-α (Weikert et al. 2000). However, in M. tuberculosis and M. avium-infected AM primed by IFN-γ, SP-A was able to supress intracellular NO levels by inhibiting TNF-α production and nuclear factor-kappa B (NF-κB) activation (Pasula et al. 1999; Hussain, 2003). Thus, SP-A facilitates the intracellular survival of M. tuberculosis (Gaynor et al. 1995; Pasula et al. 1999). Moreover, HIV-1 infected patients, who had raised levels of pulmonary SP-A, had a significantly greater susceptibility to M. tuberculosis infection (Downing et al. 1995). Thus, SP-A appears to have pleiotropic effects being able to both enhance inflammation in the presence of infected macrophages and inhibit inflammation in uninfected macrophages, thus possibly acting as a protective molecule against lung tissue damage from excessive and non-specific inflammation (Gold et al. 2004).


Role of collectins in recognising M. tuberculosis and subsequent consequences. SP-A and SP-D can bind and agglutinate mycobacteria but have opposing effects on phagocytosis. SP-A binds to Apa glycoprotein, whilst SP-D binds to lipoteichoic acid, peptidoglycan and to lipoarabinomannan (LAM) on M. tuberculosis. SP-A enhances the expression of macrophage mannose receptor (MR) facilitating uptake of M. tuberculosis, increasing mycobacterial killing and production of inflammatory components TNF-α and IL-6. SP-D inhibits phagocytosis of M. tuberculosis by blocking the interaction of LAM with macrophage MR
A number of genetic polymorphisms in the SP-A and SP-D genes are associated with TB susceptibility and protection in humans. SP-A is secreted as two distinct variants (SP-A1 and SP-A2) which are coded for by distinct genes. In individuals from Mexico, Ethiopia, India and China, mutations within and flanking the SP-A1 and SP-A2 genes are linked with protection or susceptibility toward pulmonary TB (Floros et al. 2000; Madan et al. 2002; Malik et al. 2006; Vaid et al. 2006; Yang et al. 2014). A study of Indian individuals identified a single mutation in the SP-D gene that was significantly associated with TB susceptibility (Vaid et al. 2006).
MBL is a serum protein and has a similar overall structure to SP-A and can target PAMPs on the surface of several Gram-positive and Gram-negative bacteria (Ip et al. 2009; Lugo-Villarino et al. 2011). MBL that is bound to microbial surfaces can activate complement via MBL-associated serine proteases (MASPs) of the lectin complement pathway, resulting in deposition of complement components (e.g. C3 and C4) that facilitates microbial phagocytosis and clearance. MBL also possesses complement-independent activity, acting directly as an opsonin, and an inhibitor of bacterial adhesion (Kuhlman et al. 1989; Polotsky et al. 1997; Jack et al. 2005). MBL can also bind to peptidoglycan and LTA from Staphylococcus aureus (Polotsky et al. 1996; Nadesalingam et al. 2005). In mycobacteria, MBL can bind to LAM from M. avium (Polotsky et al. 1997), antigen 85 (Ag85) of M. tuberculosis (Swierzko et al. 2016), and mannosylated lipoarabinomannan (ManLAM) from several mycobacterial species (M. tuberculosis, M. bovis, M. kansasii, M. gordonae and M. smegmatis) (Bartlomiejczyk et al. 2014). MBL can also enhance the uptake of mycobacteria by macrophages (Polotsky et al. 1997). Both normal and elevated levels in serum MBL have been associated with recurrent infection with M. tuberculosis and M. leprae, probably driven by enhanced complement-mediated phagocytosis (Garred et al. 1994, 1997). Genetic polymorphisms associated with MBL serum-deficiency are common and some of these are linked to susceptibility to TB and other inflammatory diseases in several ethnicities (Takahashi and Ezekowitz 2005; Thiel et al. 2006; Goyal et al. 2016). Among the minor collectins, CL-L1, CL-K1 and CL-P1 bind to bacteria, with CL-K1 being able to bind M. tuberculosis (Troegeler et al. 2015). The heteromeric form CL-LK binds to ManLAM of M. tuberculosis, but not M. smegmatis because of the absence of capped mannose on its LAM (Troegeler et al. 2015). Furthermore, serum levels of CL-LK in TB patients are almost depleted, compared to normal healthy controls (Troegeler et al. 2015).
Of the bovine collectins, conglutinin has protective activity against several microbes, including mycobacteria. Conglutinin has a similar structure to SP-D (which targets mycobacterial LAM) (Murugaiah et al. 2020) but is predominantly a serum protein synthesised by the liver (Holmskov et al. 1998). Conglutinin has anti-microbial properties; low serum levels of conglutinin are linked with acute infections (e.g. pneumonia, metritis and other respiratory infection (Ingram and Mitchell 1971; Holmskov et al. 1998). Conglutinin is able to bind to Gram-positive bacteria such as mycobacteria (Dec et al. 2012; Mehmood et al. 2019), and uniquely to complement C3 fragment iC3b, via the mannose residues (Laursen et al. 1994). A recombinant truncated form of conglutinin (rfBC), containing the α-helical neck region and the CRD of conglutinin (Wang et al. 1995), is able to bind to M. bovis BCG and inhibit phagocytosis of the bacterium both in the presence and absence of complement deposition (Mehmood et al. 2019). Furthermore, there is a modulation of the inflammatory response with the elevation of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, IL-12) and suppression of anti-inflammatory cytokines (TGF-β and IL-10) (Mehmood et al. 2019). Thus, it is probable that conglutinin interferes with the phagocytosis of M. bovis BCG by macrophages through two separate mechanisms: firstly, inhibiting binding of mycobacterial LAM (like SP-D) with mannose receptor, and secondly, inhibiting binding of iC3b with complement receptors CR3 and CR4 (Mehmood et al. 2019).
3 Complement and Mycobacteria
The complement system is a major arm of the innate immune response and is crucial for clearing microbial infection. The interactions between complement system and mycobacteria are not fully understood and this is probably more important in EPTB and disseminated disease than pulmonary TB. The complement system is composed of nearly 50 different proteins that are involved in three distinct pathways for activation: Alternative, Classical and Lectin (Carroll and Sim 2011) (Figs. 9.2a, b). Complement can be activated through several target surfaces including pathogens and altered-self cells or indirectly by pathogen-bound antibodies and results in the covalent binding of C3b component to the targeted cell, and subsequent cell lysis via the assembly of the membrane attack complex (MAC) (Carroll and Sim 2011). The classical pathway is activated by C1q binding to a target ligand either directly or to bound IgG/IgM antibodies (Fig. 9.2a). The lectin pathway is activated by the binding of MBL or ficolins to a target ligand (Matsushita and Fujita 1992; Matsushita et al. 2000; Matsushita and Fujita 2001) (Fig. 9.2a). However, the alternative pathway is activated differently and does not require an initiator like C1q or MBL but instead relies on the constant spontaneous hydrolysis of C3 to C3(H2O). The consequences of complement activation by any pathway is the formation of C3 convertase and the deposition of C3b on target surfaces to prompt opsonisation, the formation of the MAC and several other immunological functions (Carroll and Sim 2011) (Fig. 9.2b). There are also other complement regulatory proteins such and properdin (CFP) and factor H (CFH), where the latter is also a cofactor for factor I that is involved in the cleavage of C3b to iC3b (Whaley and Ruddy 1976a; Sim et al. 1993) (Fig. 9.2b).


Role of complement activation and complement proteins in recognising M. tuberculosis and subsequent consequences. (a) The classical pathway is activated by C1q binding to a target ligand either directly or to bound or anti-mycobacterial antibodies. The lectin pathway is activated by the binding of MBL or ficolins to target mycobacterial ligands. (b) The alternative pathway is activated via the constant spontaneous hydrolysis of C3 to C3(H2O). Properdin and factor H both act as patten recognition receptors (PRR) and have complement independent functions on mycobacteria, being able to inhibit phagocytosis and alter the subsequent inflammatory response
In the classical pathway, C1q seems to bind in the presence of IgG and IgM from serum, presumably because of M. bovis BCG vaccination (Carroll et al. 2009). Experiments using C1q-deficient serum result in a reduction of C3 binding to mycobacteria (Ferguson et al. 2004). The levels of C1q are significantly elevated in the lungs (determined by bronchoalveolar lavage (BAL) and sera of active TB patients), compared to control patients and those with LTBI, indicating that C1q is an important biomarker for TB (Lubbers et al. 2018). The classical pathway may be more relevant in EPTB where C1q is predominantly a serum protein, however, local pulmonary synthesis occurs in the lungs during active TB accounting for the raised levels of C1q observed. Furthermore, systemic and local C1q levels are raised significantly upon vaccination with BCG in non-human primates (Dijkman et al. 2020). Complement receptor CR3 deficient mice (CR3−/−), infected with M. tuberculosis, had a lower percentage of infected macrophages at 2 h but not at 4 h post infection, suggesting the opsonisation and uptake via complement and receptors may be key during the early stages of infection (Hu et al. 2000). Genetic polymorphisms in complement receptor CR1 have been reported to increase susceptibility to Mycobacterium leprae infection and TB disease (Fitness et al. 2004a, b; Kretzschmar et al. 2018), whilst a congenital deficiency of the classical pathway did not seem to affect susceptibility to TB (Kumararatne 1997). In contrast, a recent study has shown that polymorphisms in the C1q gene cluster are significantly associated with TB susceptibility and differing plasma levels of C1qA in South African TB patients (Bruiners et al. 2020).
The alternative pathway differs from the classical and lectin pathways because it does not need a specific stimulus for activation, since the alternative pathway is constantly active at low levels and able to target pathogens promptly (Kouser et al. 2013). Properdin (CFP) is a key regulatory protein of the alternative pathway and contains thrombospondin (TSR) type 1 repeats (TSR1-TSR6), which are crucial for its function; TSR4 stabilises C3bBb; whilst TSR5 binds to C3b (Higgins et al. 1995; Kouser et al. 2013). Both CFP and recombinant TSR4 + 5 are able to bind to M. bovis BCG, inhibiting its uptake by macrophages (Al-Mozaini et al. 2018). In contrast, CFH downregulates complement activation but has also been shown to bind to M. bovis BCG and inhibit its uptake by macrophages in a similar manner (Carroll et al. 2009; Abdul-Aziz et al. 2016). Both CFP and CFH are also able to modulate the immune response from the macrophage during M. bovis BCG phagocytosis by enhancing the pro-inflammatory response (TNF-α, IL-1β, IL-6 and IL-12) and dampening the anti-inflammatory response (TGF-β and IL-10). This suggests consequences for the adaptive immune response to follow against M. tuberculosis infection, particularly in the formation and maintenance of the protective granuloma.
The lectin pathway is primarily triggered by MBL and ficolins that recognise terminal sugar residues on the surface of bacteria (e.g. mannose, fucose and N-acetyl-glucosamine) and subsequently activates MASPs resulting in the cleavage of complement components C4 and C2 to C4bC2a. There are three human ficolins: L-ficolin and H-ficolin, which are synthesised by the liver and predominantly circulate in the serum and M-ficolin which exist in granules of monocytes, neutrophils, and type II alveolar epithelial cells. All three human ficolin can associate with MASPs and activate the complement cascade (Liu et al. 2005). MBL and ficolins can bind to several mycobacteria (M. tuberculosis, M. gordonae, M. kansasii and M. smegmatis) (Bartlomiejczyk et al. 2014). Direct binding of L-ficolin from human serum to M. bovis BCG and subsequent MASP-2 activation has been reported, but no binding was detected for H-ficolin (Carroll et al. 2009). L-ficolin is also able to bind with higher affinity to M. tuberculosis than to non-virulent mycobacteria and inhibit infection of human lung A549 epithelial cells (Luo et al. 2013). In mice, exogenously administered L-ficolin had a significant protective effect against virulent M. tuberculosis infection, whilst Ficolin-A (homologous to human L-ficolin in mouse) demonstrated increased susceptibility to M. tuberculosis infection (Luo et al. 2013). Furthermore, L-ficolin also modulates the immune response against M. tuberculosis infection by partially activating c-Jun N-terminal kinase (JNK) phosphorylation, stimulating the secretion of IFN-γ, IL-17, IL-6, TNF-α, and NO production by macrophages (Luo et al. 2013). Clinically, L-ficolin serum levels in pulmonary TB patients are much lower than compared to healthy controls (Luo et al. 2013), suggesting an important role for L-ficolin in M. tuberculosis infection.
The cell wall of mycobacteria is complex and is composed of a thick peptidoglycan layer, which covers the bacteria plasma membrane and is the scaffold to which various components are covalently attached (e.g. LAM, arabinogalactans, arabinomannans, glycolipids and mycolic acids) (Daffe and Draper 1998). Furthermore, there is also a capsule layer surrounding the mycolates made up of additional proteins, polysaccharides and lipids (e.g. phospholipids and glycolipids) (Daffe and Etienne 1999). One of these components is trehalose dimycolate (TDM) (also known as cord factor), which activates complement (Ramanathan et al. 1980). The complex mycobacterial cell wall has evolved to protect the bacterium from immunological attack (particularly intracellularly), but also plays a major role in determining ant-mycobacterial drug efficacy (Besra 1998). Several bacteria have evolved strategies to circumvent the immune response by interfering and inhibiting complement activation by either producing bacterial complement inhibitors, inactivating host complement inhibitors (e.g. CFH), or secreting bacterial proteases that break-down complement proteins (e.g. Salmonella enterica and Porphyromonas gingivalis) (Wingrove et al. 1992; Jagels et al. 1996; Ramu et al. 2007). M. tuberculosis is a highly evolved intracellular pathogen, being able to persistently reside in the phagosome of macrophages. Nevertheless, the interaction of complement and mycobacteria and the implications for pathogenesis and protection against TB are not well understood.
M. bovis BCG can activate the classical, lectin and alternative pathways (Ramanathan et al. 1980; Ferguson et al. 2004; Carroll et al. 2009). Activation via the alternative pathway also occurred in the absence of antibody, but intriguingly CFH was also found to bind to the mycobacterial surface, possibly indicating a means for complement moderation (Carroll et al. 2009). Although C3b is deposited on the mycobacterial surface (Carroll et al. 2009), it is not clear if MAC formation occurs. The fixation of complement proteins on the mycobacterial surface may enhance the phagocytosis of mycobacteria via complement receptors. Several studies have reported C3b deposition on mycobacteria and its role in phagocytosis via complement receptors CR1, CR3 or CR4 on macrophages (Hetland and Wiker 1994; Schlesinger and Horwitz 1994; Cywes et al. 1996; Hetland et al. 1998; Mueller-Ortiz et al. 2001; Ferguson et al. 2004). However, the specific importance of C3b and iC3b deposition on mycobacteria is not well understood. Complement activation by classical and alternative pathways has been shown on M. tuberculosis and M. bovis BCG resulting in C3b and iC3b deposition, but the target ligands are not known (Ferguson et al. 2004; Carroll et al. 2009). During the alternative pathway, CFH plays a major role in the cleavage of C3b to iC3b by acting as a cofactor of factor I, whilst also controlling the formation of the C3 and C5 convertases (Whaley and Ruddy 1976b; Whaley et al. 1976). C3b component is essential for the complement cascade to proceed to the terminal MAC, whilst iC3b is unable to facilitate this. Also, both C3b and iC3b have different complement receptors (C3b is a ligand for CR1; iC3b is a ligand for CR3 and CR4) (Ross 1986). iC3b exits as a cleavage product from C3b produced by factor I with cofactors CFH and CR1 (Figueroa and Densen 1991). Thus, both opsonic C3b or iC3b complement components may facilitate phagocytosis of host cells by mycobacteria, either promoting clearance or intracellular persistence. Indeed, a recent study showed enhanced uptake of complement-deposited M. bovis BCG by THP-1 macrophages compared to non-deposited M. bovis BCG (Mehmood et al. 2019).This same study also observed that phagocytosis of complement-deposited M. bovis BCG bacteria are inhibited from phagocytosis by THP-1 macrophages by rfBC (a recombinant truncated form of bovine conglutinin) which uniquely binds to iC3b (Mehmood et al. 2019). These observations suggest that the blocking of iC3b by conglutinin may be indicative of a protective mechanism against mycobacterial infection in the bovine host, by inhibiting phagocytosis via macrophage receptors CR3 and CR4 (Mehmood et al. 2019).
Both properdin (CFP) and factor H (CFH) are complement components that have also been observed to bind to mycobacteria in a in a dose-dependent manner and independently of C3b deposition (Carroll et al. 2009; Abdul-Aziz et al. 2016; Al-Mozaini et al. 2018). Both CFP and CFH have been shown to be PRRs for mycobacteria that have complement-independent functions. M. bovis BCG bound with CFP or CFH are inhibited for phagocytosis by THP-1 macrophages compared to M. bovis BCG alone (Al-Mozaini et al. 2018; Abdul-Aziz et al. 2016). Moreover, the subsequent macrophage inflammatory response was altered in terms of enhanced secretion of TNF-α, IL-1β, IL-6 and IL-12, whilst simultaneously dampening anti-inflammatory cytokines (TGF-β and IL-10) (Al-Mozaini et al. 2018 Abdul-Aziz et al. 2016). CFH binding has been reported in other bacteria where it serves to circumvent complement activation and thus opsonisation and killing through MAC (e.g. S. pyogenes, Streptococcus pneumoniae, Yersinia enterocolitica, Haemophilus influenza, Neisseria gonorrhoea and N. meningitidis (China et al. 1993; Diaz et al. 1997; Ram et al. 1998a, b; Dave et al. 2001; Meri et al. 2002; Schneider et al. 2006). In the case of mycobacterial infection, the ability to bind CFH may serve its immune evasion by activating C3 and using C3b opsonisation to enhance phagocytosis by macrophages via complement receptors (Schlesinger et al. 1990; Ferguson et al. 2004). These intriguing results describe potentially novel mechanisms in shaping the adaptive immune response against mycobacterial infection. For M. tuberculosis, there is a fine balance in activating complement to an optimum limited level to allow for enhanced opsonisation and uptake into macrophages, whilst avoiding being killed. Thus, the complex interactions between M. tuberculosis and complement is a major mechanism through which mycobacteria can evade the immune response by persistently intracellularly in the macrophage.
CR3 is an integrin (also known as αMβ2; CD11b/CD18), commonly expressed on neutrophils, macrophages, NK cells, and monocytes and is involved in both opsonic and non-opsonic phagocytosis (Le Cabec et al. 2002; Velasco-Velazquez et al. 2003). CR3 can bind iC3b (particularly on complement-deposited mycobacteria), mycobacterial LAM, Ag85C, PIMs, ICAM-1, several bacterial ligands and other carbohydrate residues (e.g. β-glucan, glucose, N-acetylglucosamine (GlcNAc) (Arnaout 1990; Ehlers and Daffe 1998; Velasco-Velazquez et al. 2003; Villeneuve et al. 2005). Elevated levels of CR3 in tuberculosis patients have been reported in several studies, particularly in phagocytic cells in the peripheral blood and AMs, suggesting a probable role in pathogenesis (Yassin and Hamblin 1994; Kuo et al. 1996; Juffermans et al. 2001). Indeed, complement activation via classical pathway in the lungs may also be a major mechanism for opsonin-mediated uptake of M. tuberculosis by AMs (Watford et al. 2000; Ferguson et al. 2004). However, CR3 deficiency in mice does not appear to affect the intracellular killing mechanisms (induction of reactive oxygen and nitrogen intermediates), or on the survival of the mycobacteria inside the cell, but it did result in reduced opsonisation and phagocytosis (Hu et al. 2000; Melo et al. 2000; Rooyakkers and Stokes 2005). CR3 has been found associated with several GPI-anchored proteins localized in cholesterol-rich rafts of the plasma membrane in neutrophils and is involved in the uptake of Mycobacterium kansasii (Peyron et al. 2000). Moreover, the existence of host plasma membrane cholesterol appears to be critical for CR3-mediated uptake of M. tuberculosis (Gatfield and Pieters 2000; Peyron et al. 2000). M. tuberculosis may also use cholesterol as an energy source during intracellular survival in macrophages (Van Der Geize et al. 2007). Furthermore, survival of mycobacteria within the macrophage may depend on the receptor involved in phagocytosis, since pro-inflammatory responses and respiratory burst occurs when mycobacteria are phagocytosed via Fc receptors (Russell 2001), whilst macrophage activation is inhibited when mycobacteria are phagocytosed via CR3 receptors (Caron and Hall 1998).
CR3 mediates the phagocytosis of ~80% of complement-opsonized M. tuberculosis (Schlesinger et al. 1990). CR3 is also able to facilitate phagocytosis of non-opsonized mycobacteria (Velasco-Velazquez et al. 2003). CR3 is mainly expressed on the cell surface of macrophages, neutrophils, monocytes, and natural killer cells. In lung alveolar macrophages, expression of CR3 is relatively low, whilst in vitro, differentiated macrophages have increased expression of CR3, enhancing their capacity to bind mycobacteria (Stokes et al. 1998). Several mycobacterial ligands are recognised by CR3, including Ag85C and LAM from M. tuberculosis, with the latter being the main ligand for CR3 (Velasco-Velazquez et al. 2003). Whilst CR3 plays a major role in facilitating the phagocytosis of M. tuberculosis, it does not necessarily result in the intracellular killing of the pathogen (Velasco-Velazquez et al. 2003; Rooyakkers and Stokes 2005). Furthermore, it may also not be essential in protection against M. tuberculosis infection, since CR3-deficient and wild-type mice are equally resistant to M. tuberculosis infection, suggesting that M. tuberculosis phagocytosis may occur efficiently through alternative receptors (Hu et al. 2000). Therefore, the role of CR3 in TB pathogenesis may be redundant. To date, no genetic polymorphisms in the CR3 genes have been associated with susceptibility to TB.
4 Toll-like Receptors (TLRs) and Mycobacteria
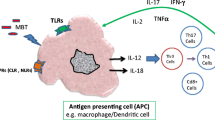
TLRs are key signalling PRRs present on several immune and non-immune cells (e.g. monocytes/macrophages, B and T cells, dendritic cells, neutrophils, epithelial and endothelial cells). TLRs recognise a wide variety of microbial ligands (PAMPs) and host danger signals (DAMPs). TLRs have key roles in innate immunity and are an important bridge to adaptive immunity (Fig. 9.3). 13 TLRs have been described in human and mouse so far. TLRs are transmembrane proteins that have ligand sensing N-terminal leucine-rich extracellular domains and a cytoplasmic Toll/IL-1R (TIR) C- terminal domain. The TIR domain mediates interactions between TLRs and adaptor proteins (e.g. myeloid differentiation primary response protein (MyD88), TIR domain-containing adaptor inducing IFN-β (TRIF), TIRAP/MAL, and TRAM) (Lim and Staudt 2013). Several kinases are also activated and involved in signalling, e.g. Interleukin-1 receptor-associated kinases (IRAK4, IRAK1, IRAK2), IκB kinase-ε (IKKε) and TANK-binding kinase-1 (TBK1), and ubiquitin ligases TNF receptor associated factor 6 (TRAF6) and Pellino-1. Upon ligand recognition, TLR signalling progresses via two distinct signalling pathways: either MyD88-dependent or TRIF-dependent pathway. Of the two, MyD88 is the most involved in TLR signalling. The triggering of the MyD88-dependent pathways ultimately results in the translocation of transcription factors NF-κB (RelA/p50) and activator protein 1 (AP1), inducing pro-inflammatory cytokine production (IL-6, TNF-α, and IL-1β). For the TRIF-dependent pathway (most relevant for TLR3 and TLR4), its signalling involves either 1) interaction with TRAF6 which goes on to activate transforming growth factor-β-activated kinase (TAK1) complex that in turn activates NF-κB and mitogen-activated protein kinases (MAPKs), or 2) interaction of TRAF3 which induces activation of interferon-regulatory factor 3 (IRF3) transcription factor that leads to the production of type I interferon (IFN-α and IFN-β) (Kawai and Akira 2010).


C-type lectin receptors (CTLRs) involved in the recognition of M. tuberculosis and subsequent consequences. Several mycobacterial ligands are recognised by a variety of host CTLR PRRs that can stimulate a multitude of signalling pathways involved in mycobacterial phagocytosis, clearance, and inflammatory responses
In humans, TLR1, 2, 4, 5, 6, and 10 are found on the host cell surface and mainly target microbial surface components (e.g. membrane of cell wall ligands), whilst TLR3, 7, 8, and 9 are found intracellularly in the endolysosomal membrane compartments and target nucleic acids (Akira et al. 2006; Triantafilou et al. 2006; Seo et al. 2018). TLRs are key downstream signalling molecules which can stimulate the production of pro-inflammatory cytokines, chemokines, and interferons (type I IFN) (Kawai and Akira 2010). These pathways are sometimes over-activated, in an uncontrolled manner, in response to stimuli, generating severe immunopathology (Vijay 2018).
TLRs play several important roles in TB. In blood samples from patients with active pulmonary TB, the expression of several TLRs are upregulated (Chang et al. 2006). TLRs recognise M. tuberculosis or a variety of its components and can initiate a set of innate and adaptive immune responses (Jo et al. 2007). The main TLRs involved in host-pathogen interaction in TB are TLR2, TLR4, TLR9 and TLR1/TLR6 (Jo et al. 2007; Kim et al. 2019). The precise nature and consequence of the signalling pathways induced by mycobacteria remain to be fully understood (Berrington and Hawn 2007; Holscher et al. 2008). Although TLRs target M. tuberculosis, this does not occur directly, but is triggered intracellularly by TLR signalling via MyD88-dependant pathway (Quesniaux et al. 2004). This also results in the activation of pro- and anti-inflammatory responses via enhanced NF-κB expression and MAPKs activation generating secretion of TNF-α, IL-1β and IL-12, and production of nitric oxide (Yamamoto et al. 2003; Jo et al. 2007; Xu et al. 2007; Jo 2008; Garlanda et al. 2007).
TLR2 plays a key role in recognising mycobacteria PAMPs and is central to activating the intracellular signalling that triggers NF-κB and MAPKs pathways, inducing secretion of pro-inflammatory cytokines and chemokines and initiating phagocytosis, intracellular killing of M. tuberculosis, and antigen presentation. TLR2 also works together with TLR4 and TLR9 during M. tuberculosis infection (Jung et al. 2006). TLR2 can bind to several mycobacterial ligands, such as LpqH, LprA, LprG, LAM, lipomannan (LM), 38-kDa lipoprotein, 19-kDa lipoprotein, phosphatidylinositol mannoside (PIMs) (Quesniaux et al. 2004; Kawai and Akira 2011; Kleinnijenhuis et al. 2011; Basu et al. 2012; Kim et al. 2019) (Fig. 9.3). However, TLR2 does not seem to be necessary for protection in mice during acute M. tuberculosis infection (Reiling et al. 2002; Sugawara et al. 2003; Mcbride et al. 2011). However, TLRs are important for the long-term control of the M. tuberculosis infection in mice (Abel et al. 2002; Drennan et al. 2004). TLR2 knockout mice (but not TLR6 knockout mice) have an impaired ability to clear M. tuberculosis infection and form granulomas compared to wild-type animals; TLR2-deficient mice have significantly lower pro-inflammatory cytokine production in response to M. tuberculosis infection (Reiling et al. 2002; Sugawara et al. 2003; Drennan et al. 2004). TLR2 knockout mice also exhibit increased M. tuberculosis bacterial load and impaired neutrophil inflammation via the downregulation of CXCL5 during infection (Gopalakrishnan et al. 2019). During M. tuberculosis infection, TLR2 is critical for the expression of TNF-α (Underhill et al. 1999), whilst both TLR2 and TLR6 are key in the expression IL-1β via MyD88 (Kleinnijenhuis et al. 2009). Another key cytokine in TB, IL-12, which is also dependent on TLR2 in macrophages and dendritic cells (Pompei et al. 2007). Indeed, the production of TNF-α and IL-12 is mainly dependent on TLR2 rather than TLR4 signalling during M. tuberculosis infection (Means et al. 2001), with TLR2 and TLR9 also being involved in controlling dendritic cell-derived IL-12 secretion in mice infected with M. tuberculosis (Bafica et al. 2005). Furthermore, in monocytes, reactive oxygen species (ROS) production and the expression of CXCL8 and CCL2 is also dependant on TLR2 during M. tuberculosis infection (Lee et al. 2009a). In dendritic cells, TLR2 induces ROS production, facilitating dendritic cell maturation and subsequent lymphocyte proliferation during M. tuberculosis infection (Romero et al. 2016). M. tuberculosis lipoproteins induce significant signalling of TLR2 which inhibits macrophage major histocompatibility complex (MHC) class II (MHC-II) expression and antigen presentation (Fulton et al. 2004; Pai et al. 2004), resulting in poor activation of CD4+ T cell responses (Noss et al. 2001; Jo 2008). TLRs gene polymorphisms also seem to have an influence on the immune response in TB (Mukherjee et al. 2019; Zhang et al. 2013b; Sun et al. 2015). A single nucleotide polymorphism (SNP) in the TLR2 gene resulting in an amino acid change (T597C) has been reported to be associated with the development of TB meningitis and miliary TB, suggesting that TLR2 may have relevance for the dissemination of M. tuberculosis infection (Thuong et al. 2007). Another gene polymorphism (rs5743708) in the TLR2 gene is also associated with higher risk for TB (Guo and Xia 2015).
TLR4 is an important sensor for bacterial endotoxins, particularly those derived from Gram-negative bacteria (e.g. LPS) (Pandey et al. 2014). In mycobacterial infection, the TLR4 signalling pathway plays a central role in immune response (Sepehri et al. 2019). Blocking interaction of M. tuberculosis with TLR4, using anti-TLR4 antibody and an endotoxin antagonist, inhibits macrophage-dependent killing of intracellular bacteria as well as the pro-inflammatory response (Means et al. 2001; Lv et al. 2017). TLR4 can target several M. tuberculosis ligands, such as heat shock proteins GrpE, Hsp65 and Resuscitation promoting factor (RpfB) (Kim et al. 2019). Additionally, mycobacterial LM can modulate macrophages inflammatory response via the TLR4 signalling (Doz et al. 2007). Both TLR4 and TLR2 expression is significantly upregulated in lymphocytes from patients with active pulmonary TB compared to healthy controls (Chang et al. 2006). Furthermore, increased expression of TLR4, CD14 and MR on monocytes (but not TLR2) was observed in M. bovis BCG vaccinated individuals compared to those who were not vaccinated; BCG-vaccinated individuals showed elevated Th1 and Th17 immune responses (Kleinnijenhuis et al. 2014). However, M. tuberculosis H37Rv strain was able to significantly enhance the expression of TLR4, TNF-α, and scavenger receptors in neutrophils when compared to mycobacterial vaccine strains (Hilda et al. 2012). The importance of TLR4 in protecting mice from TB infection is controversial. TLR4-mutant mice were observed to be more susceptible to pulmonary TB than wild-type mice, and had a reduced capacity to produce IFN-γ (Branger et al. 2004). After infection with M. tuberculosis, TLR4-mutant mice were observed to have lower pulmonary expression of TNF-α, IL-12p40, and monocyte chemoattractant protein 1, compared with the wild-type controls (Abel et al. 2002). In mice, cooperation between TLR4- and TLR2-dependent signalling is critical in macrophage apoptosis induced by M. tuberculosis infection, with the absence of TLR4 favouring necrosis instead (Sanchez et al. 2010). However, there are studies that report no significant difference in protection to M. tuberculosis infection between wild-type and TLR4-mutant mice (Shim et al. 2003; Gopalakrishnan et al. 2019). Thus, the precise role of TLR4 in TB remains to be fully determined (Reiling et al. 2002; Shim et al. 2003).
It is well known that TLR9 recognizes bacterial DNA, including M. tuberculosis DNA, with TLR9 signalling subsequently activating the macrophage pro-inflammatory response and induction of T-cell differentiation (Hemmi et al. 2000; Latz et al. 2004; Jo et al. 2007; Rahman et al. 2009). Cooperation between TLR9 and TLR2 have a protective role against M. tuberculosis infection, with TLR2/TLR9 knockout mice showing significantly enhanced susceptibility to infection, coupled together with supressed levels of IL-12p40 and IFN- γ production (Bafica et al. 2005). Interestingly, TLR9 knockout mice have modest susceptibility to M. tuberculosis infection compared to the TLR2/TLR9 double knockout mice (Bafica et al. 2005). Macrophages, pre-treated with vitamin D, were able to significantly up-regulate TLR9 expression, which boosted the pro-inflammatory response to DNA from different evolutionary lineages of M. tuberculosis (Cervantes et al. 2019). TLR9 genetic polymorphisms in the human population may be linked to susceptibility to TB. In a meta-analysis of 1745 scientific articles, a single TLR9 polymorphism (rs352139) was identified that may be associated with decreased TB risk in Indonesians individuals, whilst increased risk in Mexican individuals (Chen et al. 2015). In a study of Vietnamese individuals, two further polymorphisms were identified, with the first (rs352142) strongly associated with meningeal TB, and the second (rs352143) associated with pulmonary TB (Graustein et al. 2015). Another single-nucleotide polymorphism (rs187084) has been associated with susceptibility to pulmonary TB amongst an Indian tribe (Bharti et al. 2014).
Other TLRs that may have a significant role in TB include TLR7 and TLR8. The upregulation of TLR7 was observed to eliminate intracellular M. tuberculosis through autophagy (Bao et al. 2017). TLR7 in M. tuberculosis infected macrophages was upregulated and this also increase viability of infected host cells, whilst down-regulation of TLR7 decrease cell viability (Bao et al. 2017). Furthermore, the autophagosome was significantly increased in the M. tuberculosis-infected macrophages after upregulation of TLR7, but in contrast, the autophagosome was not observed in macrophages following down-regulation of TLR7 (Bao et al. 2017). Interestingly, TLR8 expression is also upregulated in M. bovis BCG infected THP-1 macrophages (Davila et al. 2008), whilst TLR8 expression is significantly upregulated in pulmonary TB patients during the acute phase of disease (Davila et al. 2008). Genetic polymorphisms in TLR7 and TLR8 genes are associated with increased susceptibility to M. tuberculosis infection as a result of impaired phagocytosis and TLR signalling (Davila et al. 2008; Lai et al. 2016).
The overall role of TLRs in TB pathogenesis and protection is complex. TLR-mediated signalling in TB results in an inflammatory and protective immune response, instead of a M. tuberculosis LAM-(host receptor)-mediated signalling involving C-type lectins such as MR and DC-SIGN, which tends to result in a more anti-inflammatory and suppressive immune response (Kaufmann and Schaible 2003). Furthermore, M. tuberculosis ManLAM, which is predominantly recognised by MR and DC-SIGN, results in an anti-inflammatory response and is not recognized by any TLR, suggesting that the type of cap modification on the LAM antigen has an important effect on the downstream immune response against mycobacterial infection (Quesniaux et al. 2004). An optimum IFN-γ secretion in M. tuberculosis infection requires crosstalk between TLR2, TLR4 and MR (Mukhopadhyay et al. 2004).
5 Other PRRs and Mycobacteria
5.1 Dendritic Cell-Associated C-Type Lectin (Dectin)
5.1.1 Dectin-1
Dectin-1, coded by the CLEC7 gene, is a non-TLR PRR and a type II transmembrane receptor involved in cellular activation; it is expressed on macrophages, dendritic cells, neutrophils, eosinophils, B cells, and mast cells in the lung (Brown 2006). Dectin-1 tends to target β-glucans on fungal pathogens but can also interact with M. tuberculosis, although its specific mycobacterial ligands are not known. During the recognition of fungal ligands, Dectin-1 can induce production of cytokines/chemokines, intracellular killing, phagocytosis, and DC maturation (Brown 2006) (Fig. 9.3). Downstream signalling by Dectin-1 occurs via Spleen tyrosine kinase (Syk)-dependent or -independent mechanisms involving several transcription factors (e.g. NF-κB, MAPK, NFAT, IRF1, IRF5) and the intracellular sensor NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3), central to the NLRP3 inflammasome (Kerrigan and Brown 2011; Dambuza and Brown 2015). Dectin-1 can also associate with TLR2 when recognising several mycobacteria facilitating the production of pro-inflammatory cytokines (Yadav and Schorey 2006; Shin et al. 2008; Romero et al. 2016). Dectin-1 is necessary for the TLR2-dependent production of TNF-α, IL-6, RANTES, and GM-CSF by murine macrophages infected with non-pathogenic mycobacteria (M. tuberculosis H37Ra, M. smegmatis and M. bovis BCG), but not for M. tuberculosis H37Rv (Yadav and Schorey 2006). In DCs derived from TLR2−/− mice, M. tuberculosis-induced IL-12p40 was dampened by inhibition of Dectin-1 by laminarin and by the inhibition of Syk (Rothfuchs et al. 2007). Similarly, enhanced phagocytosis and expression of Dectin-1, Src kinase, and induction of ROS occurs via TLR2 in M. tuberculosis-infected human lung epithelial cells (Lee et al. 2009b). M. tuberculosis-induced ROS production in human DCs occurs via Dectin-1 associating with TLR2 (Romero et al. 2016). M. tuberculosis:Dectin-1 interaction also appears to be the key in inducing Th1/Th17 responses in human monocyte derived DCs, but is inhibited by MR and Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN) co-expression in the cell (Zenaro et al. 2009). In human PBMCs, M. tuberculosis induction of Th17 responses is mediated by Dectin-1 and TLR4, but not TLR2, with IL-17A production requiring the IL-1 pathway (Van De Veerdonk et al. 2010). Thus, Dectin-1 plays a role in the innate immune response against M. tuberculosis. However, in knockout (Dectin-1−/−) mice, there does not seem to be a difference in survival to M. tuberculosis infection compared to wild type animals (Marakalala et al. 2011). Although a genetic deficiency resulting in a truncated Dectin-1 has been associated with susceptibility to several fungal infections (Rosentul et al. 2011; Sainz et al. 2012), no polymorphisms in the Dectin-1 gene have been reported to be involved in TB susceptibility.
5.1.2 Dectin-2
Dectin-2, coded by the Clec4n gene, is also a CTLR similar in structure to Dectin-1, composed of an N-terminal cytoplasmic domain, a transmembrane domain, and a C-terminal extracellular Ca2+- dependant CRD region (Ariizumi et al. 2000; Kanazawa et al. 2004; Sato et al. 2006). Dectin-2 is predominantly expressed in the lungs, but its expression has also been reported in spleen and lymph tissues and on DCs, monocytes, macrophages and B cells (Kanazawa et al. 2004; Sancho et al. 2012). Dectin-2 acts as an adaptor molecule recognising the γ-chain of Fc receptor triggering the activation of cells (Sato et al. 2006). Dectin-2 expression can be influenced by different ligands, with its CRD region targeting mannose residues (Gavino et al. 2005; Taylor et al. 2005; Mcgreal et al. 2006). Moreover, soluble recombinant Dectin-2 has been reported to bind to M. tuberculosis (Mcgreal et al. 2006) via ManLAM, although dectin-2 does not bind mycobacteria lacking mannose-capped LAM (Yonekawa et al. 2014; Decout et al. 2018) (Fig. 9.3). Expression of Dectin-2 on macrophages is upregulated by TNF (Decout et al. 2018). Dectin-2 elicits pro- and anti-inflammatory cytokine production (e.g. IL-6, TNF-α, MIP-2, IL-2, and IL-10) in bone marrow-derived DCs and seems to be important for DC maturation and IL-17 secretion (Yonekawa et al. 2014). This effect of ManLAM was completely negated in Clec4n−/− bone marrow-derived DCs, whilst Clec4n−/− mice infected with M. tuberculosis showed significantly greater lung pathology than wild-type mice (Yonekawa et al. 2014). To date, no polymorphisms in the human population have been described in the Clec4n gene that are linked to TB susceptibility. Thus, the role of Dectin-2 receptor in TB pathogenesis remains intriguing.
5.2 Macrophage-Inducible C-Type Lectin (Mincle)
The Macrophage-inducible C-type lectin (Mincle), coded by the CLEC4E gene, is a PRR that is found on the surface of macrophages, myeloid DCs, monocytes, neutrophils, and certain B cells and binds to several target PAMPs (e.g. mannose and fucose, among others) (Lee et al. 2011; Kerscher et al. 2013). Mincle is an LPS inducible transcriptional target in macrophages and is able to stimulate pro-inflammatory cytokines via the Syk-CARD9 pathway (Matsumoto et al. 1999; Yamasaki et al. 2008; Schoenen et al. 2010) (Fig. 9.3). Mincle can bind to trehalose dimycolate (cord factor), a key component of the mycobacterial cell wall that has also been implicated in lung granuloma formation in mice (Ishikawa et al. 2009). Trehalose dimycolate can inhibit phagosome maturation, promoting intracellular persistence and interfering with antigen presentation (Spargo et al. 1991; Actor et al. 2002; Indrigo et al. 2003; Hunter et al. 2006; Axelrod et al. 2008). Mincle, being a key receptor for the trehalose dimycolate, regulates Th1/Th17 responses in mice (Schoenen et al. 2010). In neutrophils, trehalose dimycolate-induced Mincle signalling increased cell adherence (important in early stages of granuloma formation), CR3 (CD11b/CD18) expression, together with TLR2 activation leading to reactive oxygen species and TNF-α production (Lee et al. 2012). Mincle−/− mice had impaired immune responses when challenged by aerosol M. tuberculosis, and exhibited increased inflammation and mycobacterial load than wild-type mice (Lee et al. 2012). Neutrophil depletion (using anti-Ly6G antibody) showed inhibition of IL-6 and MCP-1 (monocyte chemotactic protein-1) following trehalose dimycolate treatment, thus reducing immune cell recruitment (Lee et al. 2012). Therefore, Mincle may modulate neutrophils during the early stage of mycobacterial infection. However, another study concluded that Mincle was not essential for controlling M. tuberculosis; Mincle−/− mice could still form granulomas, had Th1 and Th17 responses, and a similar mycobacterial burden after aerosol infection to wild-type mice (Heitmann et al. 2013). Another study using Mincle−/− mice found that inoculation of mycobacteria (M. bovis BCG) intravenously, rather than intratracheally, resulted in higher mycobacterial burden in the lungs and other tissues, suggesting Mincle may play a greater role in systemic mycobacterial infection (Behler et al. 2012). Interestingly, in Mincle−/− mice, DCs induced Th1 responses in the spleen, but not in the liver, suggesting a role in systemic mycobacterial infection (Behler et al. 2015). The interaction of Mincle with trehalose dimycolate and M. bovis BCG can also promote anti-inflammatory IL-10 but conversely alter pro-inflammatory IL-12p40 secretion from murine bone-derived macrophages in vitro (Patin et al. 2016).
Mincle recognises trehalose-6,6-dibehenate (TDB) (a synthetic analogue of trehalose dimycolate), which is involved in NLRP3 inflammasome activation and Myd88-dependent Th1 and Th17 responses through IL-1R-signalling in mice bone-derived DCs (Desel et al. 2013; Schweneker et al. 2013; Shenderov et al. 2013). Mincle appears to be a crucial switch for macrophages to shift from cytokine expression to high nitric oxide (NO) production. Mincle can have dual functions in mycobacterial infection: 1) having a stimulatory role on TLR-mediated transcription, and 2) enhancing the translation of key genes required for NO synthesis, thus in the promotion NO production and subsequent resolution of inflammation and the granuloma (Lee et al. 2016b). In fact, in resting murine macrophages, Mincle is expressed at low levels but is upregulated by LPS (a TLR ligand), leading to Myd88-dependent NO production (Matsumoto et al. 1999; Schoenen et al. 2014; Kerscher et al. 2016a). Together with TLR4, Mincle has been reported to induce autophagy through Myd88, which facilitates M. tuberculosis intracellular growth (Pahari et al. 2020).
Much of the above data on Mincle has come from the mouse model of M. tuberculosis infection, but there are several studies that show similar immune responses in humans. Human antigen presenting cells have a similar response to trehalose dimycolate/TDB, inducing various cytokines via Syk-signalling (Ostrop et al. 2015), whilst the CRDs of human and mouse Mincle are similar in structure, having comparable affinity to trehalose dimycolate, but not other mycobacterial ligands (Rambaruth et al. 2015; Richardson et al. 2015; Van Der Peet et al. 2015). The downstream signalling resulting from trehalose dimycolate-Mincle interaction seems to be more complex. A recent study used quantitative phosphoproteome analysis and showed substantial reprogramming of macrophages by trehalose dimycolate and revealed both Mincle-dependent and Mincle-independent signalling mechanisms (Hansen et al. 2019).
There have been a few reports of genetic polymorphisms in the CLEC4E gene and susceptibility to TB in the human population. In one study in South African, 4 SNPs (rs10841845, rs10841847, rs10841856 and rs4620776) were described in the CLEC4E gene, but no association was found with TB susceptibility (Bowker et al. 2016). However, two of the SNPs in CLEC4E (rs10841845 and rs10841847) described earlier, were found to be associated with increased individual protection against pulmonary TB in the northern Chinese population (Kabuye et al. 2019). Furthermore, SNP rs10841847 in the CLEC4E gene was also associated with pulmonary TB risk in a study population from Guinea-Bissau (West Africa) (Olvany et al. 2020).
Mincle remains a fascinating PPR and its involvement in tuberculosis pathogenesis remains to be fully elucidated. Further studies are needed on Mincle’s involvement with genetically diverse M. tuberculosis strains, other mycobacterial ligands and in resolving the complex Mincle-dependent and Mincle-independent intracellular pathways that can be elicited in immune cells.
5.3 Macrophage C-Type Lectin (MCL)
Macrophage C-type lectin (MCL; also known as Clecsf8, Dectin-3 and CD368) is a membrane-bound PRR coded by the CLEC4D gene. First described in mice (Balch et al. 1998), MCL was subsequently characterised in humans as a type II membrane glycoprotein composed of an N-terminal cytoplasmic region lacking the consensus signalling motifs and an extracellular C-terminal region with a single CRD (Arce et al. 2004). MCL is commonly expressed on myeloid cells but it is also found on neutrophils, monocytes and DCs (Graham et al. 2012). MCL expression is downregulated upon DC maturation or monocyte/macrophage differentiation (Graham et al. 2012). The CLEC4D gene is proximal to the CLEC4E gene, and thus, the MCL gene may have originated from Mincle gene duplication. Like Mincle, MCL can also bind to trehalose dimycolate (but with lower affinity) as well as some fungal species (Arce et al. 2004; Miyake et al. 2013; Zhu et al. 2013) (Fig. 9.3).
The expression of MCL and Mincle are co-regulated, induced via Myd88 (Lobato-Pascual et al. 2013; Miyake et al. 2015; Kerscher et al. 2016a). Thus, MCL is closely linked with Mincle function, with the FcRγ region of MCL being essential for inducing Mincle expression upon binding to trehalose dimycolate (Graham et al. 2012). Furthermore, MCL cross-linking can lead to initiation of phagocytosis, intracellular respiratory burst, and cytokine secretion via Syk-signalling (Graham et al. 2012). In contrast, MCL knockout mice (Clec4d−/−) have compromised trehalose dimycolate-induced responses, cytokine production and a reduced ability to form granulomas (Miyake et al. 2013; Zhao et al. 2014). An alternative idea is that MCL and Mincle do not co-associate, but instead, MCL’s function is to induce initial Mincle expression (Zhao et al. 2014).
MCL appears to be a key, non-redundant PRR in anti-mycobacterial immunity; MCL knockout mice (Clec4d−/−) show significantly higher mycobacterial loads and increased mortality after M. tuberculosis infection (Wilson et al. 2015), concomitant with enhanced pulmonary inflammation and neutrophil recruitment (Wilson et al. 2015). Phagocytes derived from MCL knockout mice show impaired phagocytosis of mycobacteria, but this defect is restored when MCL-opsonized mycobacteria are challenged (Wilson et al. 2015).
A single genetic polymorphism (rs4304840) in MCL in humans (Indonesian cohort) has been associated with an increased susceptibility to pulmonary TB (Wilson et al. 2015). MCL seems to play a central role, together with Mincle, in the protective anti-mycobacterial immune response.
5.4 Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Non-integrin (DC-SIGN)
DC-SIGN (encoded by CD209 gene; Geijtenbeek et al. 2000) is a type II transmembrane receptor expressed predominantly on some macrophages (alveolar), DCs (myeloid) cells and activated B lymphocytes (Rappocciolo et al. 2006; Lugo-Villarino et al. 2011). DC-SIGN recognizes PAMPs such as N-linked high-mannose and branched fucosylated residues. DC-SIGN has a key role in the clearance of microbial infections, but conversely, pathogens can also manipulate DC-SIGN to alter DCs in their favour for their survival. DC-SIGN is made up of four domains: the N-terminal cytoplasmic domain, transmembrane domain, extracellular domain comprising the neck region, and a single C-terminal CRD (Garcia-Vallejo and Van Kooyk 2013).
DC-SIGN is a PRR for several microbes, most notably HIV-1 (Curtis et al. 1992; Geijtenbeek et al. 2002), but can also bind to bacterial and fungal species (Van Kooyk and Geijtenbeek 2003). DC-SIGN recognises and binds the ManLAM from M. tuberculosis (Appelmelk et al. 2003; Maeda et al. 2003), and enhances the internalization of both M. bovis BCG and M. tuberculosis (Geijtenbeek et al. 2003; Tailleux et al. 2003). Interestingly, mycobacteria are able to subvert DC-SIGN function by altering TLR-mediated activation of DCs. Mycobacteria are strong inducers of the Th1 response and can also facilitate the expression of downstream co-stimulatory molecules and cytokines (e.g. IL-12) by DCs via TLR2 and TLR4 PRRs (Nigou et al. 2001). Despite alveolar macrophages being the predominate targets of mycobacteria in the lungs, the role of DCs is becoming increasingly key in understanding the pathogenesis of TB since DCs expressing DC-SIGN are present in the airway mucosa and interstitial sites of the respiratory system (Soilleux et al. 2002; Tailleux et al. 2003).
The importance of DC-SIGN in TB pathogenesis is also shown in several studies involving transgenic mice. In fact, mice have eight different DC-SIGN homologues (SIGNR1-8). Gene knockout studies have shown that SIGNR3 (the most similar to human DC-SIGN) has a key role in resistance to early M. tuberculosis infection (Tanne et al. 2009; Tanne and Neyrolles 2010; Lugo-Villarino et al. 2011). Furthermore, transgenic mice expressing human DC-SIGN showed decreased pathology and prolonged survival following mycobacterial infection (Schaefer et al. 2008).
Capped ManLAM is the main PAMP for DC-SIGN (Geijtenbeek et al. 2003; Kaufmann and Schaible 2003; Maeda et al. 2003). DC-SIGN does not bind to non-capped LAM (AraLAM), which is present on fast-growing mycobacterial species (M. smegmatis, M. fortuitum and M. chelonae) (Geijtenbeek et al. 2003; Tailleux et al. 2003). DC-SIGN appears to be the main DC receptor for mycobacteria (Geijtenbeek et al. 2003); competitive inhibition using anti-DC-SIGN antibodies inhibited M. bovis BCG and ManLAM binding by 80% (Geijtenbeek et al. 2003). DC-SIGN also binds to other mycobacterial PAMPs (mannosylated and α-glucan cell wall components, and PIMs). However, mycobacteria can be phagocytosed by DCs in a non-DC-SIGN dependent manner, showing a degree of redundancy in the host–pathogen interaction (Gagliardi et al. 2005; Pitarque et al. 2005; Appelmelk et al. 2008; Driessen et al. 2012; Geurtsen 2009 #972).
DC-SIGN-mediated DC responses requires prior activation of NF-κB via TLR signalling (Geijtenbeek and Gringhuis 2009; Gringhuis et al. 2009; Sancho et al. 2012; Garcia-Vallejo and Van Kooyk 2013), whilst several different PAMPs can trigger a variety of intracellular signalling from DC-SIGN (Gringhuis et al. 2009; Sancho et al. 2012) (Fig. 9.3). DC-SIGN-ManLAM interaction results in Raf-1 phosphorylation and then phosphorylation of transcription factor NF-κB, inducing cytokine production (e.g. IL-12, IL-10, IL-6, and CXCL8) and other co-stimulatory molecules (e.g. CD80, CD83 and CD86) (Gringhuis et al. 2007, 2009). Infection of immature monocyte-derived DCs by M. tuberculosis facilitated the maturation of DCs, producing TNF-α, IL-1β, IL-6, and IL-23, and stimulated CD4+ T cells to produce IFN-γ and IL-17 (Zenaro et al. 2009). Furthermore, DC-SIGN interferes negatively with the pro-inflammatory responses and control of M. tuberculosis intracellular growth in human macrophages mediated by Dectin-1 (Lugo-Villarino et al. 2018).
In immature DCs, internalisation of M. tuberculosis via ManLAM-DC-SIGN interaction results in the pathogen being directed to the late endosomes/lysosomes and suppression of LPS-induced IL-12 secretion (Nigou et al. 2001). ManLAM-DC-SIGN interaction on immature DCs also interferes with TLR4 signalling, since LPS binding and signalling is via TLR4 (Akira et al. 2001). M. tuberculosis interferes between the balance of TLR signalling (DC maturation and inflammation) and DC-SIGN signalling (inhibition of DC maturation and immunosuppression) (Nigou et al. 2001; Engering et al. 2002a, b; Geijtenbeek et al. 2003). Both M. tuberculosis-infected DCs and macrophages can secrete the ManLAM that can bind to DC-SIGN on other proximal DCs (Sada et al. 1990; Chatterjee and Khoo 1998); this interferes with the TLR-signalling, inhibiting DC maturation and inducing anti-inflammatory IL-10 cytokine production (Tsuji et al. 2000; Geijtenbeek et al. 2003). Thus, M. tuberculosis is able to modulate the DC response to immune suppression to facilitate its intracellular survival (Fortsch et al. 2000; Jiao et al. 2002).
Two genetic polymorphisms have been reported in the DC-SIGN promoter region (-336A/G and -871A/G) but it is unclear as to their effect on TB susceptibility. The polymorphism -336G results in reduced expression of DC-SIGN, which also correlates with the severity of dengue disease (Despres et al. 2005). In a meta-analysis study, polymorphisms (-336A/G, -871A/G) were found not to substantially contribute to TB susceptibility, except that the genotype -336G/G might be associated with increased TB susceptibility for the Asians population (Chang et al. 2012). In another meta-analysis study, the -871A/G polymorphism was associated with decreased susceptibility to pulmonary TB, whilst the -336A/G polymorphism was associated with increased susceptibility of pulmonary TB in the Asian population (Yi et al. 2015). However, an additional polymorphism (-139G/A) was not found to be associated with susceptibility to pulmonary TB (Yi et al. 2015). Moreover, two other genotypes (-871G and -336A) seem to be associated with protection against TB and may have an increased frequency in non-African populations, possibly due to host genetic adaptation as a result of longer history of exposure to M. tuberculosis (Barreiro et al. 2006). In the Russian population, −336A genotypes were more sensitive to infection with an M. tuberculosis lineage 2 (Beijing/W) strain, whilst those with the -336G genotype and M. tuberculosis lineage 2 genotype had increased frequency of death due to pulmonary TB (Ogarkov et al. 2012).
DC-SIGN plays a key role in host-pathogen interactions in TB. Whether DC-SIGN plays a protective role for the host, or is manipulated by the M. tuberculosis to circumvent immune responses needs further study. Further data is needed from GWAS as to the genetic susceptibility to TB from CD209 polymorphisms in the human population. Further studies are also required that investigate the interaction of DC-SIGN with different phylogeographic lineages of M. tuberculosis strains.
5.5 NOD-like Receptors (NLRs)
NOD-Like Receptors (NLRs) are a large family of intracellular PRRs that contain a nucleotide binding oligomerization domain (NOD). Structurally, NLRs have a variable N-terminal interaction domain, a central NACHT domain (NTPase domain that is evolutionarily conserved), and a C-terminal leucine-rich repeat domain (Fritz et al. 2006; Werts et al. 2006; Franchi et al. 2008) (Fig. 9.3). NLRs are cytosolic sensors that tend to target bacterial cell wall components such as peptidoglycan (containing N-acetylglucosamine and N-acetylmuramic acid) and muramyl dipeptide (MDP) (Girardin et al. 2003a, b; Chen et al. 2009; Franchi et al. 2009). Some NLRs have an amino-terminal caspase recruitment domain (CARD), which is critical to initiate NF-κB signalling, resulting in the release of pro-inflammatory cytokines (e.g. IL-1β, IL-6, TNF-α, and IL-8), antimicrobial peptides (β-defensin 2), other chemokines, NO and upregulation of adhesins (Darcissac et al. 1996; Heinzelmann et al. 2000; Chin et al. 2002; Guo et al. 2006; Kramer et al. 2006; Uehara et al. 2007). Some of the most prominent members involved in innate immune detection of M. tuberculosis in the cytosol are NOD1, NOD2, NLRP3 and NLR family CARD domain containing 4 (NLRC4). This stems from the ability of M. tuberculosis to escape from phagosomes into macrophage cytosol via the early secretory antigenic target-6 (ESAT-6) secretion system-1 (ESX-1) mechanism (Simeone et al. 2012). NOD2−/− knockout mice have impaired resistance to M. tuberculosis infection because of decreased production of type 1 cytokines and reduced recruitment of CD8+ and CD4+ T cells; there is a higher bacterial burden in the lungs, 6 months after infection than wild-type controls (Divangahi et al. 2008). MDP treatment of AMs also activates NOD2, which enhances the control of intracellular growth of M. tuberculosis and the release of TNF-α, IL-6 and bactericidal LL37 (Juarez et al. 2012). Furthermore, an increase in autophagy proteins (e.g. IRGM, LC3 and ATG16L1) was observed in the mycobacteria-containing autophagosome, suggesting a PRR-dependent mechanism for autophagy activation (Juarez et al. 2012). The CARD9 domain plays a central role in NOD2-mediated activation of p38 and JNK signalling during innate immune responses to intracellular pathogens (Hsu et al. 2007). NOD2 can act in synergy with TLR2 to induce inflammatory cytokines during M. tuberculosis infection, and this synergism is lost in mononuclear cells defective in either TLR2 or NOD2, suggesting a non-redundant recognition mechanisms (Ferwerda et al. 2005). Similarly, NOD2 and TLR4 also work synergistically in stimulating the activity of DCs, enhancing T cell recruitment by inducing autophagy and bolstering IL-12p40/70, IL-6, IFN-γ and CD40, CD80 and CD86 co-stimulatory molecules (Khan et al. 2016b). Activating DCs through NOD2 and TLR4 restricts M. tuberculosis intracellular survival through strong release of cytokines, nitric oxide, autophagy and enhanced DC migration to lymph nodes (Khan et al. 2016a). NOD1 seems to co-operate with NOD2 or TLRs to produce cytokines (IL-6 and IL-1β) in bone-marrow derived macrophages in response to M. tuberculosis infection (Lee et al. 2016a). Similarly, NOD1 is involved in AM and MDM innate responses, which include pro-inflammatory cytokines (e.g. IL-1β, IL-6, IL-8, and TNF-α) and autophagy (Juarez et al. 2014). Intriguingly, an approach using adjunct therapy (with ligands of NOD2 and TLR4) to treat M. tuberculosis-infected mice in conjunction with isoniazid, improved drug efficacy against M. tuberculosis (Khan et al. 2016a). A therapeutic role for NOD-2 has also be suggested in augmenting T cells responses to M. tuberculosis infection (Pahari et al. 2017).
ESAT-6 is a potent activator of the NLRP3/ASC inflammasome and NLRs and CARD proteins play a central role in IL-1β secretion during M. tuberculosis infection, via an NLRP3, ASC and caspase-1 infection-inducible inflammasome complex (Mishra et al. 2010). Mycobacterial PPE13 triggers the inflammasome-response in macrophages, by binding to the LRR and NATCH domains of NLRP3 via its MPTR domain (Yang et al. 2020). In DCs, PPE60 was observed to activate the NLRP3 inflammasome, followed by caspase-1-dependent IL-1β and IL-18 synthesis (Su et al. 2018). However, NLRP3 may not be essential for survival in the early stages of M. tuberculosis infection or in granuloma formation (Allen et al. 2010; Mcelvania Tekippe et al. 2010; Walter et al. 2010).
Mutations in the NLR genes suggest their importance in protection against several microbial infections, granulomatous inflammatory disorders and inflammatory bowel disease (e.g. Crohn’s disease) (Hugot et al. 2001; Miceli-Richard et al. 2001; Ogura et al. 2001). Several polymorphisms in NLR genes linked to TB susceptibility have been reported. Two polymorphism in the NOD1 gene (rs751770147 and chr7:30477156(T)) are associated with TB progression in the Ethiopian population (Mekonnen et al. 2018). Three polymorphisms (Pro268Ser, Arg702Trp, and Ala725Gly) in the NOD2 gene are significantly associated with TB disease in African-American subjects in the USA (Austin et al. 2008). Another polymorphism (Arg587Arg) in the NOD2 gene has been associated with TB susceptibility in the Chinese population but not in the Uyghur and Kazak populations (Zhao et al. 2012). In a recent meta-analysis of NOD2 polymorphisms, no significant association was found between the Arg587Arg polymorphisms and TB risk; however, Arg702Trp polymorphism was found to be likely associated with protection against TB (Wang et al. 2013). For NLRP3, a single polymorphism (rs34298354) was associated with protection against TB (Liu et al. 2020). A single polymorphism (Q705K) in the NLRP3 gene was associated with poor TB treatment outcome in the Ethiopia population (Abate et al. 2019). Interestingly, in TB/HIV patients from Botswana, a NLRP3 polymorphism (rs10754558-G) was associated with an increased risk for early mortality after starting initiating anti-retroviral therapy (ART), suggesting that these patients may benefit from interventions that decrease inflammasome-mediated inflammation (Ravimohan et al. 2018).
NLRs have given significant insight into the innate immune recognition of M. tuberculosis in the cytosol. The role of the inflammasome in protection/pathogenesis is unclear and its activation may be triggered by M. tuberculosis as a means of latent infection.
5.6 Mannose Receptor (MR)
Mannose receptor (MR; CD206), coded for by the MRC1 gene, is a type I transmembrane glycoprotein of 165 kDa made up of a C-terminal cytoplasmic domain containing a tyrosine-based motif and three types of extracellular domains (an N-terminal cysteine-rich R-type domain, a fibronectin type II repeat (FNII), and eight consecutive CRDs) (Taylor et al. 1990; Stahl and Ezekowitz 1998). MR is mainly expressed on the surface of macrophages (particularly AMs), monocyte-derived DCs and other cells (e.g. non-vascular endothelial cells) (Martinez-Pomares 2012). MR is also commonly found in intracellular membranes; only 10–30% is constitutively expressed at the cell surface, which reflects its role in recycling and internalization (Schweizer et al. 2000). MR is unique in that its multiple CRDs recognise different PAMPs. The R-type domain can bind to glycans (without the need for Ca2+) (Leteux et al. 2000), whilst the FNII domain binds to collagens (Martinez-Pomares et al. 2006). MR is able to bind to mannose via CRDs 4 to 8, with CRD4 having the primary preference for terminal mannose-containing glycoconjugates, fucose, and N-acetylglucosamine, but less well to glucose (Lennartz et al. 1987; Taylor et al. 1990). In contrast, CRD5 and CRD7 are involved in binding to mannose-containing glycans, whilst CRDs 1 to 3 seem to pay less of a role in binding sugars (Kery et al. 1992; Taylor and Drickamer 1993).
MR recognises complex glycoproteins or glycolipids with multiple sugar moieties endogenously and exogenously. MR may interact with an additional receptor, or soluble MR (as a result of proteolytic cleavage) to facilitate phagocytosis (Le Cabec et al. 2005; Martinez-Pomares 2012) (Fig. 9.3). Intriguingly, pulmonary TB patients with poor prognosis show significantly higher levels of serum soluble MR; pathological analysis revealed enhanced levels of soluble MR in the lung and pleural tissues with caseating granulomas (Suzuki et al. 2018).
ManLAM is a major ligand for MR and this interaction on DCs initiates uptake of the mycobacterium, with probable antigen presentation via CD1b and the major histocompatibility complex class II (MHC-II) (Prigozy et al. 1997). In addition to ManLAM, MR can also bind to PIM, lipomannan (LM), and other mannosylated proteins on M. tuberculosis (Schlesinger et al. 1994; Diaz-Silvestre et al. 2005; Torrelles et al. 2006). MR is a major macrophage phagocytic receptor for virulent M. tuberculosis strains (H37Rv and Erdman) but not the attenuated strain H37Ra (Schlesinger 1993). Additionally, structural differences in LAM from different M. tuberculosis strains seem to alter adherence during the initial interactions with macrophage MR (Schlesinger et al. 1996).
Binding and phagocytosis of ManLAM or mannosylated beads via MR can inhibit phagosome-lysosome fusion, facilitating intracellular persistence of M. tuberculosis (Astarie-Dequeker et al. 1999, 2002; Kang et al. 2005). In DCs, ManLAM facilitates intracellular persistence of M. tuberculosis and M. bovis BCG by inhibiting IL-12 responses via interfering with the LPS-induced signalling from TLR2 (Nigou et al. 2001). This indicates a cross-linking between MR and TLR2 when binding to ManLAM (Nigou et al. 2001). Cross-linking of MR using a specific anti-MR monoclonal antibody during binding of ManLAM inhibited IL-12 production, but also induced the production of anti-inflammatory IL-10, IL-1R antagonist, and IL-1R type II in DCs (Chieppa et al. 2003). A recurring theme during TB host-pathogen interaction is the degree of cross-linking between various PRR in the recognition of M. tuberculosis, via its several PAMPs. In addition to TLRs, MR and DC-SIGN co-stimulation inhibits Dectin-1-induced Th17 responses, whilst enhancing the Th1 responses in M. tuberculosis-infected DCs (Zenaro et al. 2009). In macrophages, binding of M. tuberculosis mannosylated ligands to MR results in receptor-mediated signalling mechanisms (modulation of cytoskeleton, activation of protein kinases, and transcriptional activation by AP-1), leading to production of matrix metalloproteinase-9 (MMP-9) that may contribution to lung tissue pathology during TB in vivo (Rivera-Marrero et al. 2002). SP-D is able to bind to M. tuberculosis and inhibit its MR-mediated uptake by macrophages (Ferguson et al. 2002), suggesting that SP-D may be masking mycobacterial ligands and inhibiting phagocytosis of mycobacteria by macrophages. MR may benefit M. tuberculosis intracellular persistence; however, in mouse models of TB infection, MR does not seem to be implicated in determining survival or disease severity (Court et al. 2010).
The frequency of a polymorphism of the MRC1 gene (rs34039386), allele G1186A, was higher in individuals with pulmonary TB than healthy controls (Zhang et al. 2012), including in the Uygur population (Zhang et al. 2013a). The G1186A polymorphisms (in exon 7 for CRD2 of MR) may affect the affinity of MR binding to mycobacterial ligands (Zhang et al. 2013a). MR is undoubtedly a major phagocytotic receptor for M. tuberculosis, but its importance is overshadowed by many other PRRs. However, entry of M. tuberculosis via MR may be a key route for the pathogen to manipulate and circumvent the immune response and prolong its intracellular survival.
5.7 CD14
CD14 receptor is a lipid-anchored glycan-linked protein lacking transmembrane and cytoplasmic domains and is mainly expressed on myeloid monocytic cells. CD14 can bind to M. tuberculosis LAM, resulting in the macrophage production of IL-8 (Pugin et al. 1994). CD14 binding of bacterial ligands (LPS, lipoteichoic acid and peptidoglycan) requires co-interaction with other host receptor and cell surface components (TLRs) to facilitate phagocytosis, cell activation and cytokine secretion (Dziarski et al. 2000; Kaisho and Akira 2000). CD14 has also been shown to mediate uptake of non-opsonised M. tuberculosis by microglia cells, suggesting that this may be important in the pathogenesis of cerebral TB (Peterson et al. 1995). In AMs expressing high levels of CD14, the phagocytosis of M. bovis was enhanced (Khanna et al. 1996); however, M. tuberculosis merely up-regulates CD14 expression in macrophages without mediating phagocytosis (Shams et al. 2003). M. tuberculosis molecular chaperone chaperonin 60.1 protein partially activates human peripheral blood mononuclear cells via a CD14-mediated mechanism (Lewthwaite et al. 2001).
In mice, CD14 deficiency seems to be protective against chronic M. tuberculosis infection by supressing inflammatory responses. Mouse bone marrow derived macrophages deficient in CD14 exhibited a significant reduction in TNF-α secretion when infected with M. avium compared to controls, but infection of CD14-deficient mice with M. avium or M. tuberculosis showed no difference in controlling mycobacterial infection compared to controls (Reiling et al. 2001, 2002). However, another study found CD14−/− mice survive chronic M. tuberculosis infection, although their wild type counterparts succumbed to infection due to reduced pulmonary inflammation (Wieland et al. 2008).
Soluble CD14, produced from proteolytic cleavage of membrane CD14, seems to be significantly elevated in patients with pulmonary TB (Hoheisel et al. 1995). A SNP in the promoter region (C(−159)T) of the CD14 gene has been found to be associated with high levels of soluble CD14 and increased probably of developing pulmonary TB in the Mexican population (Rosas-Taraco et al. 2007). In another study, the -159TT allele in the CD14 promoter was also significantly associated with TB risk in the Korean population, probably from higher promoter activity resulting in higher level of soluble CD14, but also decreased IFN-γ secretion in individuals with this genotype (Kang et al. 2009).
6 Concluding Remarks
The nature of host-pathogen interaction is complex in tuberculosis. At the very heart of this is the host receptor-mycobacterial ligand interaction, which is the critical molecular dialogue in the early stages of M. tuberculosis infection. Understanding this molecular dialogue is profoundly important in determining the infection outcome. In vitro studies have proven an essential first step, but they often only involve one receptor-ligand interaction. In vivo, the host-pathogen communication is undoubtedly more complex involving an array of mycobacteria ligands that interact with several host PRRs, both soluble and membrane bound. There is indeed redundancy in both the mycobacterial ligands and host PRRs. In vivo, internalisation of M. tuberculosis involves multiple routes of cellular entry and crosstalk and co-operation between different PRRs. There seems to be a balance between TLR and CTLR entry with TLR often favouring a pro-inflammatory response, whereas CTLR favouring an anti-inflammatory response. Furthermore, the favoured target cell of M. tuberculosis (macrophage or dendritic cell) adds an additional layer of complexity. Fully understanding the host-pathogen dialogue in the early stages of infection is the ‘holy grail’ in preventing tuberculosis, because only then can we devise strategies to fully block mycobacterial interaction and entry into human cells.
References
Abate E, Blomgran R, Verma D, Lerm M, Fredrikson M, Belayneh M, Soderkvist P, Stendahl O, Schon T (2019) Polymorphisms in CARD8 and NLRP3 are associated with extrapulmonary TB and poor clinical outcome in active TB in Ethiopia. Sci Rep 9:3126
Abdul-Aziz M, Tsolaki AG, Kouser L, Carroll MV, Al-Ahdal MN, Sim RB, Kishore U (2016) Complement factor H interferes with Mycobacterium bovis BCG entry into macrophages and modulates the pro-inflammatory cytokine response. Immunobiology 221:944–952
Abel B, Thieblemont N, Quesniaux VJ, Brown N, Mpagi J, Miyake K, Bihl F, Ryffel B (2002) Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J Immunol 169:3155–3162
Actor JK, Indrigo J, Beachdel CM, Olsen M, Wells A, Hunter RL Jr, Dasgupta A (2002) Mycobacterial glycolipid cord factor trehalose 6,6′-dimycolate causes a decrease in serum cortisol during the granulomatous response. Neuroimmunomodulation 10:270–282
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2:675–680
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Allen IC, Tekippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207:1045–1056
Al-Mozaini MA, Tsolaki AG, Abdul-Aziz M, Abozaid SM, Al-Ahdal MN, Pathan AA, Murugaiah V, Makarov EM, Kaur A, Sim RB, Kishore U, Kouser L (2018) Human Properdin modulates macrophage: Mycobacterium bovis BCG interaction via thrombospondin repeats 4 and 5. Front Immunol 9:533
Appelmelk BJ, Van Die I, Van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, Van Kooyk Y (2003) Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol 170:1635–1639
Appelmelk BJ, Den Dunnen J, Driessen NN, Ummels R, Pak M, Nigou J, Larrouy-Maumus G, Gurcha SS, Movahedzadeh F, Geurtsen J, Brown EJ, Eysink Smeets MM, Besra GS, Willemsen PT, Lowary TL, Van Kooyk Y, Maaskant JJ, Stoker NG, Van Der Ley P, Puzo G, Vandenbroucke-Grauls CM, Wieland CW, Van Der Poll T, Geijtenbeek TB, Van Der Sar AM, Bitter W (2008) The mannose cap of mycobacterial lipoarabinomannan does not dominate the mycobacterium-host interaction. Cell Microbiol 10:930–944
Arce I, Martinez-Munoz L, Roda-Navarro P, Fernandez-Ruiz E (2004) The human C-type lectin CLECSF8 is a novel monocyte/macrophage endocytic receptor. Eur J Immunol 34:210–220
Ariizumi K, Shen GL, Shikano S, Ritter R 3rd, Zukas P, Edelbaum D, Morita A, Takashima A (2000) Cloning of a second dendritic cell-associated C-type lectin (dectin-2) and its alternatively spliced isoforms. J Biol Chem 275:11957–11963
Arnaout MA (1990) Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood 75:1037–1050
Astarie-Dequeker C, N’diaye EN, Le Cabec V, Rittig MG, Prandi J, Maridonneau-Parini I (1999) The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect Immun 67:469–477
Astarie-Dequeker C, Carreno S, Cougoule C, Maridonneau-Parini I (2002) The protein tyrosine kinase Hck is located on lysosomal vesicles that are physically and functionally distinct from CD63-positive lysosomes in human macrophages. J Cell Sci 115:81–89
Austin CM, Ma X, Graviss EA (2008) Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J Infect Dis 197:1713–1716
Axelrod S, Oschkinat H, Enders J, Schlegel B, Brinkmann V, Kaufmann SH, Haas A, Schaible UE (2008) Delay of phagosome maturation by a mycobacterial lipid is reversed by nitric oxide. Cell Microbiol 10:1530–1545
Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A (2005) TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med 202:1715–1724
Balch SG, Mcknight AJ, Seldin MF, Gordon S (1998) Cloning of a novel C-type lectin expressed by murine macrophages. J Biol Chem 273:18656–18664
Bao M, Yi Z, Fu Y (2017) Activation of TLR7 inhibition of Mycobacterium Tuberculosis survival by autophagy in RAW 264.7 macrophages. J Cell Biochem 118:4222–4229
Barreiro LB, Neyrolles O, Babb CL, Tailleux L, Quach H, Mcelreavey K, Helden PD, Hoal EG, Gicquel B, Quintana-Murci L (2006) Promoter variation in the DC-SIGN-encoding gene CD209 is associated with tuberculosis. PLoS Med 3:e20
Bartlomiejczyk MA, Swierzko AS, Brzostek A, Dziadek J, Cedzynski M (2014) Interaction of lectin pathway of complement-activating pattern recognition molecules with mycobacteria. Clin Exp Immunol 178:310–319
Basu J, Shin DM, Jo EK (2012) Mycobacterial signaling through toll-like receptors. Front Cell Infect Microbiol 2:145
Beharka AA, Gaynor CD, Kang BK, Voelker DR, Mccormack FX, Schlesinger LS (2002) Pulmonary surfactant protein A up-regulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J Immunol 169:3565–3573
Behler F, Steinwede K, Balboa L, Ueberberg B, Maus R, Kirchhof G, Yamasaki S, Welte T, Maus UA (2012) Role of Mincle in alveolar macrophage-dependent innate immunity against mycobacterial infections in mice. J Immunol 189:3121–3129
Behler F, Maus R, Bohling J, Knippenberg S, Kirchhof G, Nagata M, Jonigk D, Izykowski N, Magel L, Welte T, Yamasaki S, Maus UA (2015) Macrophage-inducible C-type lectin Mincle-expressing dendritic cells contribute to control of splenic Mycobacterium bovis BCG infection in mice. Infect Immun 83:184–196
Berrington WR, Hawn TR (2007) Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? Immunol Rev 219:167–186
Besra GS (1998) Preparation of cell-wall fractions from mycobacteria. Methods Mol Biol 101:91–107
Bharti D, Kumar A, Mahla RS, Kumar S, Ingle H, Shankar H, Joshi B, Raut AA, Kumar H (2014) The role of TLR9 polymorphism in susceptibility to pulmonary tuberculosis. Immunogenetics 66:675–681
Bowker N, Salie M, Schurz H, Van Helden PD, Kinnear CJ, Hoal EG, Moller M (2016) Polymorphisms in the pattern recognition receptor Mincle gene (CLEC4E) and association with tuberculosis. Lung 194:763–767
Branger J, Leemans JC, Florquin S, Weijer S, Speelman P, Van Der Poll T (2004) Toll-like receptor 4 plays a protective role in pulmonary tuberculosis in mice. Int Immunol 16:509–516
Brown GD (2006) Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol 6:33–43
Bruiners N, Schurz H, Daya M, Salie M, Van Helden PD, Kinnear CJ, Hoal EG, Moller M, Gey Van Pittius NC (2020) A regulatory variant in the C1Q gene cluster is associated with tuberculosis susceptibility and C1qA plasma levels in a south African population. Immunogenetics 72:305–314
Caron E, Hall A (1998) Identification of two distinct mechanisms of phagocytosis controlled by different rho GTPases. Science 282:1717–1721
Carroll MV, Sim RB (2011) Complement in health and disease. Adv Drug Deliv Rev 63:965–975
Carroll MV, Lack N, Sim E, Krarup A, Sim RB (2009) Multiple routes of complement activation by Mycobacterium bovis BCG. Mol Immunol 46:3367–3378
Caws M, Thwaites G, Dunstan S, Hawn TR, Lan NT, Thuong NT, Stepniewska K, Huyen MN, Bang ND, Loc TH, Gagneux S, Van Soolingen D, Kremer K, Van Der Sande M, Small P, Anh PT, Chinh NT, Quy HT, Duyen NT, Tho DQ, Hieu NT, Torok E, Hien TT, Dung NH, Nhu NT, Duy PM, Van Vinh Chau N, Farrar J (2008) The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog 4:e1000034
Cervantes JL, Oak E, Garcia J, Liu H, Lorenzini PA, Batra D, Chhabra A, Salazar JC, Roca X (2019) Vitamin D modulates human macrophage response to Mycobacterium tuberculosis DNA. Tuberculosis (Edinb) 116S:S131–S137
Chang JS, Huggett JF, Dheda K, Kim LU, Zumla A, Rook GA (2006) Myobacterium tuberculosis induces selective up-regulation of TLRs in the mononuclear leukocytes of patients with active pulmonary tuberculosis. J Immunol 176:3010–3018
Chang K, Deng S, Lu W, Wang F, Jia S, Li F, Yu L, Chen M (2012) Association between CD209 -336A/G and -871A/G polymorphisms and susceptibility of tuberculosis: a meta-analysis. PLoS One 7:e41519
Chatterjee D, Khoo KH (1998) Mycobacterial lipoarabinomannan: an extraordinary lipoheteroglycan with profound physiological effects. Glycobiology 8:113–120
Chen G, Shaw MH, Kim YG, Nunez G (2009) NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol 4:365–398
Chen Z, Wang W, Liang J, Wang J, Feng S, Zhang G (2015) Association between toll-like receptors 9 (TLR9) gene polymorphism and risk of pulmonary tuberculosis: meta-analysis. BMC Pulm Med 15:57
Chieppa M, Bianchi G, Doni A, Del Prete A, Sironi M, Laskarin G, Monti P, Piemonti L, Biondi A, Mantovani A, Introna M, Allavena P (2003) Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J Immunol 171:4552–4560
Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G (2002) Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature 416:190–194
China B, Sory MP, N'guyen BT, De Bruyere M, Cornelis GR (1993) Role of the YadA protein in prevention of opsonization of Yersinia enterocolitica by C3b molecules. Infect Immun 61:3129–3136
Chroneos ZC, Abdolrasulnia R, Whitsett JA, Rice WR, Shepherd VL (1996) Purification of a cell-surface receptor for surfactant protein A. J Biol Chem 271:16375–16383
Court N, Vasseur V, Vacher R, Fremond C, Shebzukhov Y, Yeremeev VV, Maillet I, Nedospasov SA, Gordon S, Fallon PG, Suzuki H, Ryffel B, Quesniaux VF (2010) Partial redundancy of the pattern recognition receptors, scavenger receptors, and C-type lectins for the long-term control of Mycobacterium tuberculosis infection. J Immunol 184:7057–7070
Curtis BM, Scharnowske S, Watson AJ (1992) Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc Natl Acad Sci U S A 89:8356–8360
Cywes C, Godenir NL, Hoppe HC, Scholle RR, Steyn LM, Kirsch RE, Ehlers MR (1996) Nonopsonic binding of Mycobacterium tuberculosis to human complement receptor type 3 expressed in Chinese hamster ovary cells. Infect Immun 64:5373–5383
Daffe M, Draper P (1998) The envelope layers of mycobacteria with reference to their pathogenicity. Adv Microb Physiol 39:131–203
Daffe M, Etienne G (1999) The capsule of mycobacterium tuberculosis and its implications for pathogenicity. Tuber Lung Dis 79:153–169
Dambuza IM, Brown GD (2015) C-type lectins in immunity: recent developments. Curr Opin Immunol 32:21–27
Darcissac EC, Bahr GM, Parant MA, Chedid LA, Riveau GJ (1996) Selective induction of CD11a,b,c/CD18 and CD54 expression at the cell surface of human leukocytes by muramyl peptides. Cell Immunol 169:294–301
Dave S, Brooks-Walter A, Pangburn MK, Mcdaniel LS (2001) PspC, a pneumococcal surface protein, binds human factor H. Infect Immun 69:3435–3437
Davila S, Hibberd ML, Hari Dass R, Wong HE, Sahiratmadja E, Bonnard C, Alisjahbana B, Szeszko JS, Balabanova Y, Drobniewski F, Van Crevel R, Van De Vosse E, Nejentsev S, Ottenhoff TH, Seielstad M (2008) Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet 4:e1000218
Dec M, Wernicki A, Puchalski A, Urban-Chmiel R, Radej S (2012) Effect of conglutinin on phagocytic activity of bovine granulocytes. Pol J Vet Sci 15:455–462
Decout A, Silva-Gomes S, Drocourt D, Blattes E, Riviere M, Prandi J, Larrouy-Maumus G, Caminade AM, Hamasur B, Kallenius G, Kaur D, Dobos KM, Lucas M, Sutcliffe IC, Besra GS, Appelmelk BJ, Gilleron M, Jackson M, Vercellone A, Tiraby G, Nigou J (2018) Deciphering the molecular basis of mycobacteria and lipoglycan recognition by the C-type lectin Dectin-2. Sci Rep 8:16840
Desel C, Werninghaus K, Ritter M, Jozefowski K, Wenzel J, Russkamp N, Schleicher U, Christensen D, Wirtz S, Kirschning C, Agger EM, Prazeres Da Costa C, Lang R (2013) The Mincle-activating adjuvant TDB induces MyD88-dependent Th1 and Th17 responses through IL-1R signaling. PLoS One 8:e53531
Despres P, Sakuntabhai A, Julier C (2005) [A variant in the CD209 (DC-SIGN) promoter is associated with severity of dengue disease]. Med Sci (Paris) 21:905–906
Diaz A, Ferreira A, Sim RB (1997) Complement evasion by Echinococcus granulosus: sequestration of host factor H in the hydatid cyst wall. J Immunol 158:3779–3786
Diaz-Silvestre H, Espinosa-Cueto P, Sanchez-Gonzalez A, Esparza-Ceron MA, Pereira-Suarez AL, Bernal-Fernandez G, Espitia C, Mancilla R (2005) The 19-kDa antigen of Mycobacterium tuberculosis is a major adhesin that binds the mannose receptor of THP-1 monocytic cells and promotes phagocytosis of mycobacteria. Microb Pathog 39:97–107
Dijkman K, Lubbers R, Borggreven NV, Ottenhoff THM, Joosten SA, Trouw LA, Verreck, F.a.W. (2020) Systemic and pulmonary C1q as biomarker of progressive disease in experimental non-human primate tuberculosis. Sci Rep 10:6290
Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, Kobayashi KS, Flavell RA, Gros P, Behr MA (2008) NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol 181:7157–7165
Downing JF, Pasula R, Wright JR, Twigg HL 3rd, Martin WJ 2nd (1995) Surfactant protein a promotes attachment of Mycobacterium tuberculosis to alveolar macrophages during infection with human immunodeficiency virus. Proc Natl Acad Sci U S A 92:4848–4852
Doz E, Rose S, Nigou J, Gilleron M, Puzo G, Erard F, Ryffel B, Quesniaux VF (2007) Acylation determines the toll-like receptor (TLR)-dependent positive versus TLR2-, mannose receptor-, and SIGNR1-independent negative regulation of pro-inflammatory cytokines by mycobacterial lipomannan. J Biol Chem 282:26014–26025
Drennan MB, Nicolle D, Quesniaux VJ, Jacobs M, Allie N, Mpagi J, Fremond C, Wagner H, Kirschning C, Ryffel B (2004) Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol 164:49–57
Driessen NN, Boshoff HI, Maaskant JJ, Gilissen SA, Vink S, van der Sar AM, Vandenbroucke-Grauls CM, Bewley CA, Appelmelk BJ, Geurtsen J (2012) Cyanovirin-N
Dziarski R, Ulmer AJ, Gupta D (2000) Interactions of CD14 with components of gram-positive bacteria. Chem Immunol 74:83–107
Ehlers MR, Daffe M (1998) Interactions between mycobacterium tuberculosis and host cells: are mycobacterial sugars the key? Trends Microbiol 6:328–335
Engering A, Geijtenbeek TB, Van Kooyk Y (2002a) Immune escape through C-type lectins on dendritic cells. Trends Immunol 23:480–485
Engering A, Geijtenbeek TB, Van Vliet SJ, Wijers M, Van Liempt E, Demaurex N, Lanzavecchia A, Fransen J, Figdor CG, Piguet V, Van Kooyk Y (2002b) The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol 168:2118–2126
Ernst JD (1998) Macrophage receptors for Mycobacterium tuberculosis. Infect Immun 66:1277–1281
Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, Cho SN, Via LE, Barry CE 3rd (2010) Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137:122–128
Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, Wynn TA, Sher A (2006) NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J Immunol 177:7086–7093
Fennelly KP (2020) Particle sizes of infectious aerosols: implications for infection control. Lancet Respir Med 8:914–924
Fennelly KP, Martyny JW, Fulton KE, Orme IM, Cave DM, Heifets LB (2004) Cough-generated aerosols of Mycobacterium tuberculosis: a new method to study infectiousness. Am J Respir Crit Care Med 169:604–609
Ferguson JS, Voelker DR, Mccormack FX, Schlesinger LS (1999) Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J Immunol 163:312–321
Ferguson JS, Voelker DR, Ufnar JA, Dawson AJ, Schlesinger LS (2002) Surfactant protein D inhibition of human macrophage uptake of Mycobacterium tuberculosis is independent of bacterial agglutination. J Immunol 168:1309–1314
Ferguson JS, Weis JJ, Martin JL, Schlesinger LS (2004) Complement protein C3 binding to Mycobacterium tuberculosis is initiated by the classical pathway in human bronchoalveolar lavage fluid. Infect Immun 72:2564–2573
Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, De Jong DJ, Langenberg DM, Van Crevel R, Adema GJ, Ottenhoff TH, Van Der Meer JW, Netea MG (2005) NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog 1:279–285
Figueroa JE, Densen P (1991) Infectious diseases associated with complement deficiencies. Clin Microbiol Rev 4:359–395
Fitness J, Floyd S, Warndorff DK, Sichali L, Malema S, Crampin AC, Fine PE, Hill AV (2004a) Large-scale candidate gene study of tuberculosis susceptibility in the Karonga district of northern Malawi. Am J Trop Med Hyg 71:341–349
Fitness J, Floyd S, Warndorff DK, Sichali L, Mwaungulu L, Crampin AC, Fine PE, Hill AV (2004b) Large-scale candidate gene study of leprosy susceptibility in the Karonga district of northern Malawi. Am J Trop Med Hyg 71:330–340
Floros J, Lin HM, Garcia A, Salazar MA, Guo X, DiAngelo S, Montano M, Luo J, Pardo A, Selman M (2000) Surfactant
Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR (1995) Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2:561–572
Fortsch D, Rollinghoff M, Stenger S (2000) IL-10 converts human dendritic cells into macrophage-like cells with increased antibacterial activity against virulent Mycobacterium tuberculosis. J Immunol 165:978–987
Franchi L, Park JH, Shaw MH, Marina-Garcia N, Chen G, Kim YG, Nunez G (2008) Intracellular NOD-like receptors in innate immunity, infection and disease. Cell Microbiol 10:1–8
Franchi L, Warner N, Viani K, Nunez G (2009) Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev 227:106–128
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE (2006) Nod-like proteins in immunity, inflammation and disease. Nat Immunol 7:1250–1257
Fulton SA, Reba SM, Pai RK, Pennini M, Torres M, Harding CV, Boom WH (2004) Inhibition of major histocompatibility complex II expression and antigen processing in murine alveolar macrophages by Mycobacterium bovis BCG and the 19-kilodalton mycobacterial lipoprotein. Infect Immun 72:2101–2110
Gagliardi MC, Teloni R, Giannoni F, Pardini M, Sargentini V, Brunori L, Fattorini L, Nisini R (2005) Mycobacterium bovis Bacillus Calmette-Guerin infects DC-SIGN- dendritic cell and causes the inhibition of IL-12 and the enhancement of IL-10 production. J Leukoc Biol 78:106–113
Garcia-Vallejo JJ, Van Kooyk Y (2013) The physiological role of DC-SIGN: a tale of mice and men. Trends Immunol 34:482–486
Garlanda C, Di Liberto D, Vecchi A, La Manna MP, Buracchi C, Caccamo N, Salerno A, Dieli F, Mantovani A (2007) Damping excessive inflammation and tissue damage in Mycobacterium tuberculosis infection by Toll IL-1 receptor 8/single Ig IL-1-related receptor, a negative regulator of IL-1/TLR signaling. J Immunol 179:3119–3125
Garred P, Harboe M, Oettinger T, Koch C, Svejgaard A (1994) Dual role of mannan-binding protein in infections: another case of heterosis? Eur J Immunogenet 21:125–131
Garred P, Richter C, Andersen AB, Madsen HO, Mtoni I, Svejgaard A, Shao J (1997) Mannan-binding lectin in the sub-Saharan HIV and tuberculosis epidemics. Scand J Immunol 46:204–208
Gatfield J, Pieters J (2000) Essential role for cholesterol in entry of mycobacteria into macrophages. Science 288:1647–1650
Gavino AC, Chung JS, Sato K, Ariizumi K, Cruz PD Jr (2005) Identification and expression profiling of a human C-type lectin, structurally homologous to mouse dectin-2. Exp Dermatol 14:281–288
Gaynor CD, Mccormack FX, Voelker DR, Mcgowan SE, Schlesinger LS (1995) Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J Immunol 155:5343–5351
Geijtenbeek TB, Gringhuis SI (2009) Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9:465–479
Geijtenbeek TB, Torensma R, Van Vliet SJ, Van Duijnhoven GC, Adema GJ, Van Kooyk Y, Figdor CG (2000) Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 100:575–585
Geijtenbeek TB, Van Duijnhoven GC, Van Vliet SJ, Krieger E, Vriend G, Figdor CG, Van Kooyk Y (2002) Identification of different binding sites in the dendritic cell-specific receptor DC-SIGN for intercellular adhesion molecule 3 and HIV-1. J Biol Chem 277:11314–11320
Geijtenbeek TB, Van Vliet SJ, Koppel EA, Sanchez-Hernandez M, Vandenbroucke-Grauls CM, Appelmelk B, Van Kooyk Y (2003) Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med 197:7–17
Geurtsen J, Chedammi S, Mesters J, Cot M, Driessen NN, Sambou T, Kakutani R, Ummels R, Maaskant J, Takata H, Baba O, Terashima T, Bovin N, Vandenbroucke-Grauls CM, Nigou J, Puzo G, Lemassu A, Daffe M, Appelmelk BJ (2009) Identification of mycobacterial alpha-glucan as a novel ligand for DCSIGN: involvement of mycobacterial capsular polysaccharides in host immune modulation. J Immunol 183(8):5221–5231
Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, Tedin K, Taha MK, Labigne A, Zahringer U, Coyle AJ, Distefano PS, Bertin J, Sansonetti PJ, Philpott DJ (2003a) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587
Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, Sansonetti PJ, Mengin-Lecreulx D (2003b) Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem 278:41702–41708
Gold JA, Hoshino Y, Tanaka N, Rom WN, Raju B, Condos R, Weiden MD (2004) Surfactant protein A modulates the inflammatory response in macrophages during tuberculosis. Infect Immun 72:645–650
Gopalakrishnan A, Dietzold J, Verma S, Bhagavathula M, Salgame P (2019) Toll-like receptor 2 prevents neutrophil-driven immunopathology during infection with Mycobacterium tuberculosis by curtailing CXCL5 production. Infect Immun 87:e00760–e00718
Goyal S, Klassert TE, Slevogt H (2016) C-type lectin receptors in tuberculosis: what we know. Med Microbiol Immunol 205:513–535
Graham LM, Gupta V, Schafer G, Reid DM, Kimberg M, Dennehy KM, Hornsell WG, Guler R, Campanero-Rhodes MA, Palma AS, Feizi T, Kim SK, Sobieszczuk P, Willment JA, Brown GD (2012) The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J Biol Chem 287:25964–25974
Graustein AD, Horne DJ, Arentz M, Bang ND, Chau TT, Thwaites GE, Caws M, Thuong NT, Dunstan SJ, Hawn TR (2015) TLR9 gene region polymorphisms and susceptibility to tuberculosis in Vietnam. Tuberculosis (Edinb) 95:190–196
Gringhuis SI, Den Dunnen J, Litjens M, Van Het Hof B, Van Kooyk Y, Geijtenbeek TB (2007) C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 26:605–616
Gringhuis SI, Den Dunnen J, Litjens M, Van Der Vlist M, Geijtenbeek TB (2009) Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat Immunol 10:1081–1088
Guo XG, Xia Y (2015) The rs5743708 gene polymorphism in the TLR2 gene contributes to the risk of tuberculosis disease. Int J Clin Exp Pathol 8:11921–11928
Guo LH, Guo KT, Wendel HP, Schluesener HJ (2006) Combinations of TLR and NOD2 ligands stimulate rat microglial P2X4R expression. Biochem Biophys Res Commun 349:1156–1162
Gupta A, Kaul A, Tsolaki AG, Kishore U, Bhakta S (2012) Mycobacterium tuberculosis: immune evasion, latency and reactivation. Immunobiology 217:363–374
Hansch HC, Smith DA, Mielke ME, Hahn H, Bancroft GJ, Ehlers S (1996) Mechanisms of granuloma formation in murine Mycobacterium avium infection: the contribution of CD4+ T cells. Int Immunol 8:1299–1310
Hansen M, Peltier J, Killy B, Amin B, Bodendorfer B, Hartlova A, Uebel S, Bosmann M, Hofmann J, Buttner C, Ekici AB, Kuttke M, Franzyk H, Foged C, Beer-Hammer S, Schabbauer G, Trost M, Lang R (2019) Macrophage phosphoproteome analysis reveals MINCLE-dependent and -independent mycobacterial cord factor Signaling. Mol Cell Proteomics 18:669–685
Heinzelmann M, Polk HC Jr, Chernobelsky A, Stites TP, Gordon LE (2000) Endotoxin and muramyl dipeptide modulate surface receptor expression on human mononuclear cells. Immunopharmacology 48:117–128
Heitmann L, Schoenen H, Ehlers S, Lang R, Holscher C (2013) Mincle is not essential for controlling Mycobacterium tuberculosis infection. Immunobiology 218:506–516
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745
Hetland G, Wiker HG (1994) Antigen 85C on Mycobacterium bovis, BCG and M. tuberculosis promotes monocyte-CR3-mediated uptake of microbeads coated with mycobacterial products. Immunology 82:445–449
Hetland G, Wiker HG, Hogasen K, Hamasur B, Svenson SB, Harboe M (1998) Involvement of antilipoarabinomannan antibodies in classical complement activation in tuberculosis. Clin Diagn Lab Immunol 5:211–218
Higgins JM, Wiedemann H, Timpl R, Reid KB (1995) Characterization of mutant forms of recombinant human properdin lacking single thrombospondin type I repeats. Identification of modules important for function. J Immunol 155:5777–5785
Hilda JN, Selvaraj A, Das SD (2012) Mycobacterium tuberculosis H37Rv is more effective compared to vaccine strains in modulating neutrophil functions: an in vitro study. FEMS Immunol Med Microbiol 66:372–381
Hirsch CS, Ellner JJ, Russell DG, Rich EA (1994) Complement receptor-mediated uptake and tumor necrosis factor-alpha-mediated growth inhibition of Mycobacterium tuberculosis by human alveolar macrophages. J Immunol 152:743–753
Hoheisel G, Zheng L, Teschler H, Striz I, Costabel U (1995) Increased soluble CD14 levels in BAL fluid in pulmonary tuberculosis. Chest 108:1614–1616
Holmskov U, Jensenius JC, Tornoe I, Lovendahl P (1998) The plasma levels of coglutinin are heritable in cattle and low levels predispose to infection. Immunology 93:431–436
Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, Lenz I, Sonntag T, Kroger S, Akira S, Mossmann H, Kirschning CJ, Wagner H, Freudenberg M, Ehlers S (2008) Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur J Immunol 38:680–694
Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X (2007) The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol 8:198–205
Hu C, Mayadas-Norton T, Tanaka K, Chan J, Salgame P (2000) Mycobacterium tuberculosis infection in complement receptor 3-deficient mice. J Immunol 165:2596–2602
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411:599–603
Hunter RL, Olsen M, Jagannath C, Actor JK (2006) Trehalose 6,6′-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol 168:1249–1261
Hussain S, Wright JR, Martin WJ 2nd (2003) Surfactant protein A decreases nitric oxide production by macrophages in a tumor necrosis factor-alpha-dependent mechanism. Am J Respir Cell Mol Biol 28(4):520–527
Indrigo J, Hunter RL, Actor JK (2003) Cord factor trehalose 6,6′-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology (Reading) 149:2049–2059
Ingram DG, Mitchell WR (1971) Conglutinin level in dairy cattle: changes associated with disease. Am J Vet Res 32:875–878
Ip WK, Takahashi K, Ezekowitz RA, Stuart LM (2009) Mannose-binding lectin and innate immunity. Immunol Rev 230:9–21
Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, Kinoshita T, Akira S, Yoshikai Y, Yamasaki S (2009) Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med 206:2879–2888
Jack DL, Lee ME, Turner MW, Klein NJ, Read RC (2005) Mannose-binding lectin enhances phagocytosis and killing of Neisseria meningitidis by human macrophages. J Leukoc Biol 77:328–336
Jagels MA, Ember JA, Travis J, Potempa J, Pike R, Hugli TE (1996) Cleavage of the human C5A receptor by proteinases derived from Porphyromonas gingivalis: cleavage of leukocyte C5a receptor. Adv Exp Med Biol 389:155–164
Jiao X, Lo-Man R, Guermonprez P, Fiette L, Deriaud E, Burgaud S, Gicquel B, Winter N, Leclerc C (2002) Dendritic cells are host cells for mycobacteria in vivo that trigger innate and acquired immunity. J Immunol 168:1294–1301
Jo EK (2008) Mycobacterial interaction with innate receptors: TLRs, C-type lectins, and NLRs. Curr Opin Infect Dis 21:279–286
Jo EK, Yang CS, Choi CH, Harding CV (2007) Intracellular signalling cascades regulating innate immune responses to Mycobacteria: branching out from Toll-like receptors. Cell Microbiol 9:1087–1098
Juarez E, Carranza C, Hernandez-Sanchez F, Leon-Contreras JC, Hernandez-Pando R, Escobedo D, Torres M, Sada E (2012) NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol 42:880–889
Juarez E, Carranza C, Hernandez-Sanchez F, Loyola E, Escobedo D, Leon-Contreras JC, Hernandez-Pando R, Torres M, Sada E (2014) Nucleotide-oligomerizing domain-1 (NOD1) receptor activation induces pro-inflammatory responses and autophagy in human alveolar macrophages. BMC Pulm Med 14:152
Juffermans NP, Dekkers PE, Verbon A, Speelman P, Van Deventer SJ, Van Der Poll T (2001) Concurrent upregulation of urokinase plasminogen activator receptor and CD11b during tuberculosis and experimental endotoxemia. Infect Immun 69:5182–5185
Jung SB, Yang CS, Lee JS, Shin AR, Jung SS, Son JW, Harding CV, Kim HJ, Park JK, Paik TH, Song CH, Jo EK (2006) The mycobacterial 38-kilodalton glycolipoprotein antigen activates the mitogen-activated protein kinase pathway and release of proinflammatory cytokines through Toll-like receptors 2 and 4 in human monocytes. Infect Immun 74:2686–2696
Kabuye D, Chu Y, Lao W, Jin G, Kang H (2019) Association between CLEC4E gene polymorphism of mincle and pulmonary tuberculosis infection in a northern Chinese population. Gene 710:24–29
Kaisho T, Akira S (2000) Critical roles of Toll-like receptors in host defense. Crit Rev Immunol 20:393–405
Kanazawa N, Tashiro K, Inaba K, Lutz MB, Miyachi Y (2004) Molecular cloning of human dectin-2. J Invest Dermatol 122:1522–1524
Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, Desjardin LE, Schlesinger LS (2005) The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med 202:987–999
Kang YA, Lee HW, Kim YW, Han SK, Shim YS, Yim JJ (2009) Association between the -159C/T CD14 gene polymorphism and tuberculosis in a Korean population. FEMS Immunol Med Microbiol 57:229–235
Kaufmann SH, Schaible UE (2003) A dangerous liaison between two major killers: Mycobacterium tuberculosis and HIV target dendritic cells through DC-SIGN. J Exp Med 197:1–5
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650
Kerrigan AM, Brown GD (2011) Syk-coupled C-type lectins in immunity. Trends Immunol 32:151–156
Kerscher B, Willment JA, Brown GD (2013) The Dectin-2 family of C-type lectin-like receptors: an update. Int Immunol 25:271–277
Kerscher B, Dambuza IM, Christofi M, Reid DM, Yamasaki S, Willment JA, Brown GD (2016a) Signalling through MyD88 drives surface expression of the mycobacterial receptors MCL (Clecsf8, Clec4d) and Mincle (Clec4e) following microbial stimulation. Microbes Infect 18:505–509
Kerscher B, Wilson GJ, Reid DM, Mori D, Taylor JA, Besra GS, Yamasaki S, Willment JA, Brown GD (2016b) Mycobacterial receptor, Clec4d (CLECSF8, MCL), is coregulated with Mincle and upregulated on mouse myeloid cells following microbial challenge. Eur J Immunol 46:381–389
Kery V, Krepinsky JJ, Warren CD, Capek P, Stahl PD (1992) Ligand recognition by purified human mannose receptor. Arch Biochem Biophys 298:49–55
Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN (2016a) NOD-2 and TLR-4 Signaling reinforces the efficacy of dendritic cells and reduces the dose of TB drugs against Mycobacterium tuberculosis. J Innate Immun 8:228–242
Khan N, Vidyarthi A, Pahari S, Negi S, Aqdas M, Nadeem S, Agnihotri T, Agrewala JN (2016b) Signaling through NOD-2 and TLR-4 bolsters the T cell priming capability of dendritic cells by inducing autophagy. Sci Rep 6:19084
Khanna KV, Choi CS, Gekker G, Peterson PK, Molitor TW (1996) Differential infection of porcine alveolar macrophage subpopulations by nonopsonized Mycobacterium bovis involves CD14 receptors. J Leukoc Biol 60:214–220
Kim JS, Kim YR, Yang CS (2019) Latest comprehensive knowledge of the crosstalk between TLR signaling and mycobacteria and the antigens driving the process. J Microbiol Biotechnol 29:1506–1521
Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, Bernal AL, Reid KB, Madan T (2006) Chakraborty, Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol Immunol 43(9):1293–1315
Kleinnijenhuis J, Joosten LA, Van De Veerdonk FL, Savage N, Van Crevel R, Kullberg BJ, Van Der Ven A, Ottenhoff TH, Dinarello CA, Van Der Meer JW, Netea MG (2009) Transcriptional and inflammasome-mediated pathways for the induction of IL-1beta production by Mycobacterium tuberculosis. Eur J Immunol 39:1914–1922
Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R (2011) Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol 2011:405310
Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C, Van Loenhout J, Xavier RJ, Aaby P, Van Der Meer JW, Van Crevel R, Netea MG (2014) Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun 6:152–158
Kouser L, Abdul-Aziz M, Nayak A, Stover CM, Sim RB, Kishore U (2013) Properdin and factor h: opposing players on the alternative complement pathway “see-saw”. Front Immunol 4:93
Kramer M, Netea MG, De Jong DJ, Kullberg BJ, Adema GJ (2006) Impaired dendritic cell function in Crohn’s disease patients with NOD2 3020insC mutation. J Leukoc Biol 79:860–866
Kretzschmar GC, Oliveira LC, Nisihara RM, Velavan TP, Stinghen ST, Stahlke ERS, Petzl-Erler ML, Messias-Reason IJT, Boldt ABW (2018) Complement receptor 1 (CR1, CD35) association with susceptibility to leprosy. PLoS Negl Trop Dis 12:e0006705
Kudo K, Sano H, Takahashi H, Kuronuma K, Yokota S, Fujii N, Shimada K, Yano I, Kumazawa Y, Voelker DR, Abe S, Kuroki Y (2004) Pulmonary collectins enhance phagocytosis of Mycobacterium avium through increased activity of mannose receptor. J Immunol 172:7592–7602
Kuhlman M, Joiner K, Ezekowitz RA (1989) The human mannose-binding protein functions as an opsonin. J Exp Med 169:1733–1745
Kumararatne DS (1997) Tuberculosis and immunodeficiency—of mice and men. Clin Exp Immunol 107:11–14
Kuo HP, Ho TC, Wang CH, Yu CT, Lin HC (1996) Increased production of hydrogen peroxide and expression of CD11b/CD18 on alveolar macrophages in patients with active pulmonary tuberculosis. Tuber Lung Dis 77:468–475
Kuronuma K, Sano H, Kato K, Kudo K, Hyakushima N, Yokota S, Takahashi H, Fujii N, Suzuki H, Kodama T, Abe S, Kuroki Y (2004) Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J Biol Chem 279:21421–21430
Laforce FM, Kelly WJ, Huber GL (1973) Inactivation of staphylococci by alveolar macrophages with preliminary observations on the importance of alveolar lining material. Am Rev Respir Dis 108:784–790
Lai YF, Lin TM, Wang CH, Su PY, Wu JT, Lin MC, Eng HL (2016) Functional polymorphisms of the TLR7 and TLR8 genes contribute to Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 98:125–131
Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT (2004) TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol 5:190–198
Laursen SB, Thiel S, Teisner B, Holmskov U, Wang Y, Sim RB, Jensenius JC (1994) Bovine conglutinin binds to an oligosaccharide determinant presented by iC3b, but not by C3, C3b or C3c. Immunology 81:648–654
Le Cabec V, Carreno S, Moisand A, Bordier C, Maridonneau-Parini I (2002) Complement receptor 3 (CD11b/CD18) mediates type I and type II phagocytosis during nonopsonic and opsonic phagocytosis, respectively. J Immunol 169:2003–2009
Le Cabec V, Emorine LJ, Toesca I, Cougoule C, Maridonneau-Parini I (2005) The human macrophage mannose receptor is not a professional phagocytic receptor. J Leukoc Biol 77:934–943
Lee HM, Shin DM, Kim KK, Lee JS, Paik TH, Jo EK (2009a) Roles of reactive oxygen species in CXCL8 and CCL2 expression in response to the 30-kDa antigen of Mycobacterium tuberculosis. J Clin Immunol 29:46–56
Lee HM, Yuk JM, Shin DM, Jo EK (2009b) Dectin-1 is inducible and plays an essential role for mycobacteria-induced innate immune responses in airway epithelial cells. J Clin Immunol 29:795–805
Lee RT, Hsu TL, Huang SK, Hsieh SL, Wong CH, Lee YC (2011) Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities. Glycobiology 21:512–520
Lee WB, Kang JS, Yan JJ, Lee MS, Jeon BY, Cho SN, Kim YJ (2012) Neutrophils promote mycobacterial Trehalose Dimycolate-induced lung inflammation via the Mincle pathway. PLoS Pathog 8:e1002614
Lee JY, Hwang EH, Kim DJ, Oh SM, Lee KB, Shin SJ, Park JH (2016a) The role of nucleotide-binding oligomerization domain 1 during cytokine production by macrophages in response to Mycobacterium tuberculosis infection. Immunobiology 221:70–75
Lee WB, Kang JS, Choi WY, Zhang Q, Kim CH, Choi UY, Kim-Ha J, Kim YJ (2016b) Mincle-mediated translational regulation is required for strong nitric oxide production and inflammation resolution. Nat Commun 7:11322
Lemos MP, Mckinney J, Rhee KY (2011) Dispensability of surfactant proteins A and D in immune control of Mycobacterium tuberculosis infection following aerosol challenge of mice. Infect Immun 79:1077–1085
Lennartz MR, Cole FS, Shepherd VL, Wileman TE, Stahl PD (1987) Isolation and characterization of a mannose-specific endocytosis receptor from human placenta. J Biol Chem 262:9942–9944
Leteux C, Chai W, Loveless RW, Yuen CT, Uhlin-Hansen L, Combarnous Y, Jankovic M, Maric SC, Misulovin Z, Nussenzweig MC, Feizi T (2000) The cysteine-rich domain of the macrophage mannose receptor is a multispecific lectin that recognizes chondroitin sulfates A and B and sulfated oligosaccharides of blood group Lewis(a) and Lewis(x) types in addition to the sulfated N-glycans of lutropin. J Exp Med 191:1117–1126
Lewthwaite JC, Coates AR, Tormay P, Singh M, Mascagni P, Poole S, Roberts M, Sharp L, Henderson B (2001) Mycobacterium tuberculosis chaperonin 60.1 is a more potent cytokine stimulator than chaperonin 60.2 (Hsp 65) and contains a CD14-binding domain. Infect Immun 69:7349–7355
Lim KH, Staudt LM (2013) Toll-like receptor signaling. Cold Spring Harb Perspect Biol 5:a011247
Liu Y, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I, Inoue K, Munakata M, Fujita T (2005) Human M-ficolin is a secretory protein that activates the lectin complement pathway. J Immunol 175:3150–3156
Liu CW, Lin CJ, Hu HC, Liu HJ, Chiu YC, Lee SW, Wu LS (2020) The association of inflammasome and TLR2 gene polymorphisms with susceptibility to tuberculosis in the Han Taiwanese population. Sci Rep 10:10184
Lobato-Pascual A, Saether PC, Fossum S, Dissen E, Daws MR (2013) Mincle, the receptor for mycobacterial cord factor, forms a functional receptor complex with MCL and FcepsilonRI-gamma. Eur J Immunol 43:3167–3174
Lockhart E, Green AM, Flynn JL (2006) IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol 177:4662–4669
Lubbers R, Sutherland JS, Goletti D, De Paus RA, Van Moorsel CHM, Veltkamp M, Vestjens SMT, Bos WJW, Petrone L, Del Nonno F, Bajema IM, Dijkman K, Verreck FAW, Walzl G, Gelderman KA, Groeneveld GH, Geluk A, Ottenhoff THM, Joosten SA, Trouw LA (2018) Complement component C1q as serum biomarker to detect active tuberculosis. Front Immunol 9:2427
Lugo-Villarino G, Hudrisier D, Tanne A, Neyrolles O (2011) C-type lectins with a sweet spot for Mycobacterium tuberculosis. Eur J Microbiol Immunol (Bp) 1:25–40
Lugo-Villarino G, Troegeler A, Balboa L, Lastrucci C, Duval C, Mercier I, Benard A, Capilla F, Al Saati T, Poincloux R, Kondova I, Verreck FAW, Cougoule C, Maridonneau-Parini I, Sasiain MDC, Neyrolles O (2018) The C-type lectin receptor DC-SIGN has an anti-inflammatory role in human M(IL-4) macrophages in response to Mycobacterium tuberculosis. Front Immunol 9:1123
Luo F, Sun X, Wang Y, Wang Q, Wu Y, Pan Q, Fang C, Zhang XL (2013) Ficolin-2 defends against virulent Mycobacteria tuberculosis infection in vivo, and its insufficiency is associated with infection in humans. PLoS One 8:e73859
Lv J, He X, Wang H, Wang Z, Kelly GT, Wang X, Chen Y, Wang T, Qian Z (2017) TLR4-NOX2 axis regulates the phagocytosis and killing of Mycobacterium tuberculosis by macrophages. BMC Pulm Med 17:194
Madan T, Saxena S, Murthy KJ, Muralidhar K,Sarma PU (2002) Association of polymorphisms in the collagen region of human SP-A1 and SP-A2 genes with pulmonary tuberculosis in Indian population. Clin Chem Lab Med 40(10):1002–1008
Malik S, Greenwood CM, Eguale T, Kifle A, Beyene J, Habte A, Tadesse A, Gebrexabher H, Britton S, Schurr E (2006) Variants of the SFTPA1 and SFTPA2 genes and susceptibility to tuberculosis in Ethiopia. Hum Genet 118(6):752–759
Maeda N, Nigou J, Herrmann JL, Jackson M, Amara A, Lagrange PH, Puzo G, Gicquel B, Neyrolles O (2003) The cell surface receptor DC-SIGN discriminates between Mycobacterium species through selective recognition of the mannose caps on lipoarabinomannan. J Biol Chem 278:5513–5516
Marakalala MJ, Guler R, Matika L, Murray G, Jacobs M, Brombacher F, Rothfuchs AG, Sher A, Brown GD (2011) The Syk/CARD9-coupled receptor Dectin-1 is not required for host resistance to Mycobacterium tuberculosis in mice. Microbes Infect 13:198–201
Martinez-Pomares L (2012) The mannose receptor. J Leukoc Biol 92:1177–1186
Martinez-Pomares L, Wienke D, Stillion R, Mckenzie EJ, Arnold JN, Harris J, Mcgreal E, Sim RB, Isacke CM, Gordon S (2006) Carbohydrate-independent recognition of collagens by the macrophage mannose receptor. Eur J Immunol 36:1074–1082
Matsumoto M, Tanaka T, Kaisho T, Sanjo H, Copeland NG, Gilbert DJ, Jenkins NA, Akira S (1999) A novel LPS-inducible C-type lectin is a transcriptional target of NF-IL6 in macrophages. J Immunol 163:5039–5048
Matsushita M, Fujita T (1992) Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J Exp Med 176:1497–1502
Matsushita M, Fujita T (2001) Ficolins and the lectin complement pathway. Immunol Rev 180:78–85
Matsushita M, Endo Y, Fujita T (2000) Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J Immunol 164:2281–2284
Mcbride A, Bhatt K, Salgame P (2011) Development of a secondary immune response to Mycobacterium tuberculosis is independent of Toll-like receptor 2. Infect Immun 79:1118–1123
Mcelvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, Mccann JR, Sandor M, Braunstein M, Ting JP (2010) Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS One 5:e12320
Mcgreal EP, Rosas M, Brown GD, Zamze S, Wong SY, Gordon S, Martinez-Pomares L, Taylor PR (2006) The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology 16:422–430
Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ (1999) Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J Immunol 163:3920–3927
Means TK, Jones BW, Schromm AB, Shurtleff BA, Smith JA, Keane J, Golenbock DT, Vogel SN, Fenton MJ (2001) Differential effects of a Toll-like receptor antagonist on Mycobacterium tuberculosis-induced macrophage responses. J Immunol 166:4074–4082
Mehmood A, Kouser L, Kaur A, Holmskov U, Al-Ahdal MN, Sim RB, Kishore U, Tsolaki AG (2019) Complement dependent and independent interaction between bovine conglutinin and Mycobacterium bovis BCG: implications in bovine tuberculosis. Front Immunol 9:3159
Mekonnen E, Bekele E, Stein CM (2018) Novel polymorphisms in TICAM2 and NOD1 associated with tuberculosis progression phenotypes in Ethiopian populations. Glob Health Epidemiol Genom 3:e1
Melo MD, Catchpole IR, Haggar G, Stokes RW (2000) Utilization of CD11b knockout mice to characterize the role of complement receptor 3 (CR3, CD11b/CD18) in the growth of Mycobacterium tuberculosis in macrophages. Cell Immunol 205:13–23
Meri T, Hartmann A, Lenk D, Eck R, Wurzner R, Hellwage J, Meri S, Zipfel PF (2002) The yeast Candida albicans binds complement regulators factor H and FHL-1. Infect Immun 70:5185–5192
Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, Chamaillard M, Zouali H, Thomas G, Hugot JP (2001) CARD15 mutations in Blau syndrome. Nat Genet 29:19–20
Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, Anes E (2010) Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol 12:1046–1063
Miyake Y, Toyonaga K, Mori D, Kakuta S, Hoshino Y, Oyamada A, Yamada H, Ono K, Suyama M, Iwakura Y, Yoshikai Y, Yamasaki S (2013) C-type lectin MCL is an FcRgamma-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity 38:1050–1062
Miyake Y, Masatsugu OH, Yamasaki S (2015) C-type lectin receptor MCL facilitates mincle expression and signaling through complex formation. J Immunol 194:5366–5374
Mueller-Ortiz SL, Wanger AR, Norris SJ (2001) Mycobacterial protein HbhA binds human complement component C3. Infect Immun 69:7501–7511
Mukherjee S, Huda S, Sinha Babu SP (2019) Toll-like receptor polymorphism in host immune response to infectious diseases: a review. Scand J Immunol 90:e12771
Mukhopadhyay S, Herre J, Brown GD, Gordon S (2004) The potential for Toll-like receptors to collaborate with other innate immune receptors. Immunology 112:521–530
Murugaiah V, Tsolaki AG, Kishore U (2020) Collectins: innate immune pattern recognition molecules. Adv Exp Med Biol 1204:75–127
Nadesalingam J, Dodds AW, Reid KB, Palaniyar N (2005) Mannose-binding lectin recognizes peptidoglycan via the N-acetyl glucosamine moiety, and inhibits ligand-induced proinflammatory effect and promotes chemokine production by macrophages. J Immunol 175:1785–1794
Nigou J, Zelle-Rieser C, Gilleron M, Thurnher M, Puzo G (2001) Mannosylated lipoarabinomannans inhibit IL-12 production by human dendritic cells: evidence for a negative signal delivered through the mannose receptor. J Immunol 166:7477–7485
North RJ, Izzo AA (1993) Granuloma formation in severe combined immunodeficient (SCID) mice in response to progressive BCG infection. Tendency not to form granulomas in the lung is associated with faster bacterial growth in this organ. Am J Pathol 142:1959–1966
Noss EH, Pai RK, Sellati TJ, Radolf JD, Belisle J, Golenbock DT, Boom WH, Harding CV (2001) Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J Immunol 167:910–918
Ogarkov O, Mokrousov I, Sinkov V, Zhdanova S, Antipina S, Savilov E (2012) ‘Lethal’ combination of Mycobacterium tuberculosis Beijing genotype and human CD209 -336G allele in Russian male population. Infect Genet Evol 12:732–736
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411:603–606
Olvany JM, Sausville LN, White MJ, Tacconelli A, Tavera G, Sobota RS, Ciccacci C, Bohlbro AS, Wejse C, Williams SM, Sirugo G (2020) CLEC4E (Mincle) genetic variation associates with pulmonary tuberculosis in Guinea-Bissau (West Africa). Infect Genet Evol 85:104560
Ong CWM, Migliori GB, Raviglione M, Macgregor-Skinner G, Sotgiu G, Alffenaar JW, Tiberi S, Adlhoch C, Alonzi T, Archuleta S, Brusin S, Cambau E, Capobianchi MR, Castilletti C, Centis R, Cirillo DM, D'ambrosio L, Delogu G, Esposito SMR, Figueroa J, Friedland JS, Ho BCH, Ippolito G, Jankovic M, Kim HY, Rosales Klintz S, Kodmon C, Lalle E, Leo YS, Leung CC, Martson AG, Melazzini MG, Najafi Fard S, Penttinen P, Petrone L, Petruccioli E, Pontali E, Saderi L, Santin M, Spanevello A, Van Crevel R, Van Der Werf MJ, Visca D, Viveiros M, Zellweger JP, Zumla A, Goletti D (2020) Epidemic and pandemic viral infections: impact on tuberculosis and the lung: a consensus by the World Association for Infectious Diseases and Immunological Disorders (WAidid), Global Tuberculosis Network (GTN), and members of the European Society of Clinical Microbiology and Infectious Diseases Study Group for Mycobacterial Infections (ESGMYC). Eur Respir J 56:2001727
Ostrop J, Jozefowski K, Zimmermann S, Hofmann K, Strasser E, Lepenies B, Lang R (2015) Contribution of MINCLE-SYK Signaling to activation of primary human APCs by mycobacterial cord factor and the novel adjuvant TDB. J Immunol 195:2417–2428
Pahari S, Kaur G, Aqdas M, Negi S, Chatterjee D, Bashir H, Singh S, Agrewala JN (2017) Bolstering immunity through pattern recognition receptors: a unique approach to control tuberculosis. Front Immunol 8:906
Pahari S, Negi S, Aqdas M, Arnett E, Schlesinger LS, Agrewala JN (2020) Induction of autophagy through CLEC4E in combination with TLR4: an innovative strategy to restrict the survival of Mycobacterium tuberculosis. Autophagy 16:1021–1043
Pai RK, Pennini ME, Tobian AA, Canaday DH, Boom WH, Harding CV (2004) Prolonged toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infect Immun 72:6603–6614
Pandey S, Kawai T, Akira S (2014) Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb Perspect Biol 7:a016246
Pasula R, Wright JR, Kachel DL, Martin WJ 2nd (1999) Surfactant protein A suppresses reactive nitrogen intermediates by alveolar macrophages in response to Mycobacterium tuberculosis. J Clin Invest 103(4):483–490
Patin EC, Willcocks S, Orr S, Ward TH, Lang R, Schaible UE (2016) Mincle-mediated anti-inflammatory IL-10 response counter-regulates IL-12 in vitro. Innate Immun 22:181–185
Peterson PK, Gekker G, Hu S, Sheng WS, Anderson WR, Ulevitch RJ, Tobias PS, Gustafson KV, Molitor TW, Chao CC (1995) CD14 receptor-mediated uptake of nonopsonized Mycobacterium tuberculosis by human microglia. Infect Immun 63:1598–1602
Peyron P, Bordier C, N'diaye EN, Maridonneau-Parini I (2000) Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J Immunol 165:5186–5191
Pikaar JC, Voorhout WF, Van Golde LM, Verhoef J, Van Strijp JA, Van Iwaarden JF (1995) Opsonic activities of surfactant proteins A and D in phagocytosis of gram-negative bacteria by alveolar macrophages. J Infect Dis 172:481–489
Pitarque S, Herrmann JL, Duteyrat JL, Jackson M, Stewart GR, Lecointe F, Payre B, Schwartz O, Young DB, Marchal G, Lagrange PH, Puzo G, Gicquel B, Nigou J, Neyrolles O (2005) Deciphering the molecular bases of Mycobacterium tuberculosis binding to the lectin DC-SIGN reveals an underestimated complexity. Biochem J 392:615–624
Polotsky VY, Fischer W, Ezekowitz RA, Joiner KA (1996) Interactions of human mannose-binding protein with lipoteichoic acids. Infect Immun 64:380–383
Polotsky VY, Belisle JT, Mikusova K, Ezekowitz RA, Joiner KA (1997) Interaction of human mannose-binding protein with Mycobacterium avium. J Infect Dis 175:1159–1168
Pompei L, Jang S, Zamlynny B, Ravikumar S, Mcbride A, Hickman SP, Salgame P (2007) Disparity in IL-12 release in dendritic cells and macrophages in response to Mycobacterium tuberculosis is due to use of distinct TLRs. J Immunol 178:5192–5199
Prigozy TI, Sieling PA, Clemens D, Stewart PL, Behar SM, Porcelli SA, Brenner MB, Modlin RL, Kronenberg M (1997) The mannose receptor delivers lipoglycan antigens to endosomes for presentation to T cells by CD1b molecules. Immunity 6:187–197
Pugin J, Heumann ID, Tomasz A, Kravchenko VV, Akamatsu Y, Nishijima M, Glauser MP, Tobias PS, Ulevitch RJ (1994) CD14 is a pattern recognition receptor. Immunity 1:509–516
Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, Bihl F, Erard F, Botha T, Drennan M, Soler MN, Le Bert M, Schnyder B, Ryffel B (2004) Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect 6:946–959
Ragas A, Roussel L, Puzo G, Riviere M (2007) The Mycobacterium tuberculosis cell-surface glycoprotein apa as a potential adhesin to colonize target cells via the innate immune system pulmonary C-type lectin surfactant protein A. J Biol Chem 282:5133–5142
Rahman AH, Taylor DK, Turka LA (2009) The contribution of direct TLR signaling to T cell responses. Immunol Res 45:25–36
Ram S, Mcquillen DP, Gulati S, Elkins C, Pangburn MK, Rice PA (1998a) Binding of complement factor H to loop 5 of porin protein 1A: a molecular mechanism of serum resistance of nonsialylated Neisseria gonorrhoeae. J Exp Med 188:671–680
Ram S, Sharma AK, Simpson SD, Gulati S, Mcquillen DP, Pangburn MK, Rice PA (1998b) A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med 187:743–752
Ramanathan VD, Curtis J, Turk JL (1980) Activation of the alternative pathway of complement by mycobacteria and cord factor. Infect Immun 29:30–35
Rambaruth ND, Jegouzo SA, Marlor H, Taylor ME, Drickamer K (2015) Mouse mincle: characterization as a model for human mincle and evolutionary implications. Molecules 20:6670–6682
Ramu P, Tanskanen R, Holmberg M, Lahteenmaki K, Korhonen TK, Meri S (2007) The surface protease PgtE of Salmonella enterica affects complement activity by proteolytically cleaving C3b, C4b and C5. FEBS Lett 581:1716–1720
Rappocciolo G, Piazza P, Fuller CL, Reinhart TA, Watkins SC, Rowe DT, Jais M, Gupta P, Rinaldo CR (2006) DC-SIGN on B lymphocytes is required for transmission of HIV-1 to T lymphocytes. PLoS Pathog 2:e70
Ravimohan S, Nfanyana K, Tamuhla N, Tiemessen CT, Weissman D, Bisson GP (2018) Common variation in NLRP3 is associated with early death and elevated inflammasome biomarkers among advanced HIV/TB co-infected patients in Botswana. Open Forum Infect Dis 5:ofy075
Reiling N, Klug K, Krallmann-Wenzel U, Laves R, Goyert S, Taylor ME, Lindhorst TK, Ehlers S (2001) Complex encounters at the macrophage-mycobacterium interface: studies on the role of the mannose receptor and CD14 in experimental infection models with Mycobacterium avium. Immunobiology 204:558–571
Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, Ehlers S (2002) Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol 169:3480–3484
Richardson MB, Torigoe S, Yamasaki S, Williams SJ (2015) Mycobacterium tuberculosis beta-gentiobiosyl diacylglycerides signal through the pattern recognition receptor Mincle: total synthesis and structure activity relationships. Chem Commun (Camb) 51:15027–15030
Rivera-Marrero CA, Schuyler W, Roser S, Ritzenthaler JD, Newburn SA, Roman J (2002) M. tuberculosis induction of matrix metalloproteinase-9: the role of mannose and receptor-mediated mechanisms. Am J Physiol Lung Cell Mol Physiol 282:L546–L555
Romero MM, Basile JI, Corra Feo L, Lopez B, Ritacco V, Aleman M (2016) Reactive oxygen species production by human dendritic cells involves TLR2 and dectin-1 and is essential for efficient immune response against Mycobacteria. Cell Microbiol 18:875–886
Rooyakkers AW, Stokes RW (2005) Absence of complement receptor 3 results in reduced binding and ingestion of Mycobacterium tuberculosis but has no significant effect on the induction of reactive oxygen and nitrogen intermediates or on the survival of the bacteria in resident and interferon-gamma activated macrophages. Microb Pathog 39:57–67
Rosas-Taraco AG, Revol A, Salinas-Carmona MC, Rendon A, Caballero-Olin G, Arce-Mendoza AY (2007) CD14 C(-159)T polymorphism is a risk factor for development of pulmonary tuberculosis. J Infect Dis 196:1698–1706
Rosentul DC, Plantinga TS, Oosting M, Scott WK, Velez Edwards DR, Smith PB, Alexander BD, Yang JC, Laird GM, Joosten LA, Van Der Meer JW, Perfect JR, Kullberg BJ, Netea MG, Johnson MD (2011) Genetic variation in the dectin-1/CARD9 recognition pathway and susceptibility to candidemia. J Infect Dis 204:1138–1145
Ross GD (1986) Opsonization and membrane complement receptors. In: Ross GD (ed) Immunobiology of the complement system: an introduction for research and clinical medicine. Academic, Orlando, pp 87–114
Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, Brown GD, Sher A (2007) Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol 179:3463–3471
Russell DG (2001) Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol 2:569–577
Sada E, Brennan PJ, Herrera T, Torres M (1990) Evaluation of lipoarabinomannan for the serological diagnosis of tuberculosis. J Clin Microbiol 28:2587–2590
Sainz J, Lupianez CB, Segura-Catena J, Vazquez L, Rios R, Oyonarte S, Hemminki K, Forsti A, Jurado M (2012) Dectin-1 and DC-SIGN polymorphisms associated with invasive pulmonary Aspergillosis infection. PLoS One 7:e32273
Sanchez D, Rojas M, Hernandez I, Radzioch D, Garcia LF, Barrera LF (2010) Role of TLR2- and TLR4-mediated signaling in Mycobacterium tuberculosis-induced macrophage death. Cell Immunol 260:128–136
Sancho D, Reis E, Sousa C (2012) Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol 30:491–529
Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, Kimberly RP, Underhill D, Cruz PD Jr, Ariizumi K (2006) Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem 281:38854–38866
Schaefer M, Reiling N, Fessler C, Stephani J, Taniuchi I, Hatam F, Yildirim AO, Fehrenbach H, Walter K, Ruland J, Wagner H, Ehlers S, Sparwasser T (2008) Decreased pathology and prolonged survival of human DC-SIGN transgenic mice during mycobacterial infection. J Immunol 180:6836–6845
Schlesinger LS (1993) Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J Immunol 150:2920–2930
Schlesinger LS, Horwitz MA (1994) A role for natural antibody in the pathogenesis of leprosy: antibody in nonimmune serum mediates C3 fixation to the Mycobacterium leprae surface and hence phagocytosis by human mononuclear phagocytes. Infect Immun 62:280–289
Schlesinger LS, Bellinger-Kawahara CG, Payne NR, Horwitz MA (1990) Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J Immunol 144:2771–2780
Schlesinger LS, Hull SR, Kaufman TM (1994) Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J Immunol 152:4070–4079
Schlesinger LS, Kaufman TM, Iyer S, Hull SR, Marchiando LK (1996) Differences in mannose receptor-mediated uptake of lipoarabinomannan from virulent and attenuated strains of Mycobacterium tuberculosis by human macrophages. J Immunol 157:4568–4575
Schneider MC, Exley RM, Chan H, Feavers I, Kang YH, Sim RB, Tang CM (2006) Functional significance of factor H binding to Neisseria meningitidis. J Immunol 176:7566–7575
Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, Agger EM, Stenger S, Andersen P, Ruland J, Brown GD, Wells C, Lang R (2010) Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol 184:2756–2760
Schoenen H, Huber A, Sonda N, Zimmermann S, Jantsch J, Lepenies B, Bronte V, Lang R (2014) Differential control of Mincle-dependent cord factor recognition and macrophage responses by the transcription factors C/EBPbeta and HIF1alpha. J Immunol 193:3664–3675
Schweizer A, Stahl PD, Rohrer J (2000) A di-aromatic motif in the cytosolic tail of the mannose receptor mediates endosomal sorting. J Biol Chem 275:29694–29700
Schweneker K, Gorka O, Schweneker M, Poeck H, Tschopp J, Peschel C, Ruland J, Gross O (2013) The mycobacterial cord factor adjuvant analogue trehalose-6,6'-dibehenate (TDB) activates the Nlrp3 inflammasome. Immunobiology 218:664–673
Seo JY, Choi JW, Lee JY, Park YS, Park YI (2018) Enzyme hydrolysates of ginseng Marc polysaccharides promote the phagocytic activity of macrophages Via activation of TLR2 and Mer tyrosine kinase. J Microbiol Biotechnol 28:860–873
Sepehri Z, Kiani Z, Kohan F, Ghavami S (2019) Toll-like receptor 4 as an immune receptor against Mycobacterium tuberculosis: A systematic review. Lab Med 50:117–129
Shams H, Wizel B, Lakey DL, Samten B, Vankayalapati R, Valdivia RH, Kitchens RL, Griffith DE, Barnes PF (2003) The CD14 receptor does not mediate entry of Mycobacterium tuberculosis into human mononuclear phagocytes. FEMS Immunol Med Microbiol 36:63–69
Shenderov K, Barber DL, Mayer-Barber KD, Gurcha SS, Jankovic D, Feng CG, Oland S, Hieny S, Caspar P, Yamasaki S, Lin X, Ting JP, Trinchieri G, Besra GS, Cerundolo V, Sher A (2013) Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J Immunol 190:5722–5730
Shim TS, Turner OC, Orme IM (2003) Toll-like receptor 4 plays no role in susceptibility of mice to Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 83:367–371
Shin DM, Yang CS, Yuk JM, Lee JY, Kim KH, Shin SJ, Takahara K, Lee SJ, Jo EK (2008) Mycobacterium abscessus activates the macrophage innate immune response via a physical and functional interaction between TLR2 and dectin-1. Cell Microbiol 10:1608–1621
Sim RB, Day AJ, Moffatt BE, Fontaine M (1993) Complement factor I and cofactors in control of complement system convertase enzymes. Methods Enzymol 223:13–35
Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J (2012) Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8:e1002507
Smith D, Hansch H, Bancroft G, Ehlers S (1997) T-cell-independent granuloma formation in response to Mycobacterium avium: role of tumour necrosis factor-alpha and interferon-gamma. Immunology 92:413–421
Soilleux EJ, Morris LS, Leslie G, Chehimi J, Luo Q, Levroney E, Trowsdale J, Montaner LJ, Doms RW, Weissman D, Coleman N, Lee B (2002) Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc Biol 71:445–457
Spargo BJ, Crowe LM, Ioneda T, Beaman BL, Crowe JH (1991) Cord factor (alpha,alpha-trehalose 6,6'-dimycolate) inhibits fusion between phospholipid vesicles. Proc Natl Acad Sci U S A 88:737–740
Stahl PD, Ezekowitz RA (1998) The mannose receptor is a pattern recognition receptor involved in host defense. Curr Opin Immunol 10:50–55
Stokes RW, Haidl ID, Jefferies WA, Speert DP (1993) Mycobacteria-macrophage interactions. Macrophage phenotype determines the nonopsonic binding of Mycobacterium tuberculosis to murine macrophages. J Immunol 151:7067–7076
Stokes RW, Thorson LM, Speert DP (1998) Nonopsonic and opsonic association of Mycobacterium tuberculosis with resident alveolar macrophages is inefficient. J Immunol 160:5514–5521
Su H, Zhang Z, Liu Z, Peng B, Kong C, Wang H, Xu Y (2018) Mycobacterium tuberculosis PPE60 antigen drives Th1/Th17 responses via Toll-like receptor 2-dependent maturation of dendritic cells. J Biol Chem 293:10287–10302
Sugawara I, Yamada H, Li C, Mizuno S, Takeuchi O, Akira S (2003) Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol Immunol 47:327–336
Sun Q, Zhang Q, Xiao HP, Bai C (2015) Toll-like receptor polymorphisms and tuberculosis susceptibility: a comprehensive meta-analysis. J Huazhong Univ Sci Technolog Med Sci 35:157–168
Suzuki Y, Shirai M, Asada K, Yasui H, Karayama M, Hozumi H, Furuhashi K, Enomoto N, Fujisawa T, Nakamura Y, Inui N, Shirai T, Hayakawa H, Suda T (2018) Macrophage mannose receptor, CD206, predict prognosis in patients with pulmonary tuberculosis. Sci Rep 8:13129
Swierzko AS, Bartlomiejczyk MA, Brzostek A, Lukasiewicz J, Michalski M, Dziadek J, Cedzynski M (2016) Mycobacterial antigen 85 complex (Ag85) as a target for ficolins and mannose-binding lectin. Int J Med Microbiol 306:212–221
Tailleux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A, Legres L, Dreher D, Nicod LP, Gluckman JC, Lagrange PH, Gicquel B, Neyrolles O (2003) DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J Exp Med 197:121–127
Takahashi K, Ezekowitz RA (2005) The role of the mannose-binding lectin in innate immunity. Clin Infect Dis 41(Suppl 7):S440–S444
Tanne A, Neyrolles O (2010) C-type lectins in immune defense against pathogens: the murine DC-SIGN homologue SIGNR3 confers early protection against Mycobacterium tuberculosis infection. Virulence 1:285–290
Tanne A, Ma B, Boudou F, Tailleux L, Botella H, Badell E, Levillain F, Taylor ME, Drickamer K, Nigou J, Dobos KM, Puzo G, Vestweber D, Wild MK, Marcinko M, Sobieszczuk P, Stewart L, Lebus D, Gicquel B, Neyrolles O (2009) A murine DC-SIGN homologue contributes to early host defense against Mycobacterium tuberculosis. J Exp Med 206:2205–2220
Taylor ME, Drickamer K (1993) Structural requirements for high affinity binding of complex ligands by the macrophage mannose receptor. J Biol Chem 268:399–404
Taylor ME, Conary JT, Lennartz MR, Stahl PD, Drickamer K (1990) Primary structure of the mannose receptor contains multiple motifs resembling carbohydrate-recognition domains. J Biol Chem 265:12156–12162
Taylor PR, Reid DM, Heinsbroek SE, Brown GD, Gordon S, Wong SY (2005) Dectin-2 is predominantly myeloid restricted and exhibits unique activation-dependent expression on maturing inflammatory monocytes elicited in vivo. Eur J Immunol 35:2163–2174
Thiel S, Frederiksen PD, Jensenius JC (2006) Clinical manifestations of mannan-binding lectin deficiency. Mol Immunol 43:86–96
Thuong NT, Hawn TR, Thwaites GE, Chau TT, Lan NT, Quy HT, Hieu NT, Aderem A, Hien TT, Farrar JJ, Dunstan SJ (2007) A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun 8:422–428
Torrelles JB, Azad AK, Schlesinger LS (2006) Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J Immunol 177:1805–1816
Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, Triantafilou K (2006) Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem 281:31002–31011
Troegeler A, Lugo-Villarino G, Hansen S, Rasolofo V, Henriksen ML, Mori K, Ohtani K, Duval C, Mercier I, Benard A, Nigou J, Hudrisier D, Wakamiya N, Neyrolles O (2015) Collectin CL-LK is a novel soluble pattern recognition receptor for Mycobacterium tuberculosis. PLoS One 10:e0132692
Tsolaki AG, Kishore U (2020) Bovine collectins: role in health and disease. In: Kishore U, editor. Collectin protein family and its multiple biological activities. Springer Nature
Tsuji S, Matsumoto M, Takeuchi O, Akira S, Azuma I, Hayashi A, Toyoshima K, Seya T (2000) Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette-Guerin: involvement of toll-like receptors. Infect Immun 68:6883–6890
Uehara A, Fujimoto Y, Fukase K, Takada H (2007) Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol 44:3100–3111
Underhill DM, Ozinsky A, Smith KD, Aderem A (1999) Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A 96:14459–14463
Vaid M, Kaur S, Madan T, Singh H, Gupta VK, Murthy KJR, Sarma PU (2006) Association of SP-D, MNL and I-NOS genetic variants with pulmonary tuberculosis. Indian J Hum Genet 12:105–110
Van De Veerdonk FL, Teirlinck AC, Kleinnijenhuis J, Kullberg BJ, Van Crevel R, Van Der Meer JW, Joosten LA, Netea MG (2010) Mycobacterium tuberculosis induces IL-17A responses through TLR4 and dectin-1 and is critically dependent on endogenous IL-1. J Leukoc Biol 88:227–232
Van De Wetering JK, Van Eijk M, Van Golde LM, Hartung T, Van Strijp JA, Batenburg JJ (2001) Characteristics of surfactant protein A and D binding to lipoteichoic acid and peptidoglycan, 2 major cell wall components of gram-positive bacteria. J Infect Dis 184:1143–1151
Van Der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, Sim E, Dijkhuizen L, Davies JE, Mohn WW, Eltis LD (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 104:1947–1952
Van Der Peet PL, Gunawan C, Torigoe S, Yamasaki S, Williams SJ (2015) Corynomycolic acid-containing glycolipids signal through the pattern recognition receptor Mincle. Chem Commun (Camb) 51:5100–5103
Van Kooyk Y, Geijtenbeek TB (2003) DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol 3:697–709
Velasco-Velazquez MA, Barrera D, Gonzalez-Arenas A, Rosales C, Agramonte-Hevia J (2003) Macrophage--Mycobacterium tuberculosis interactions: role of complement receptor 3. Microb Pathog 35:125–131
Vijay K (2018) Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol 59:391–412
Villeneuve C, Gilleron M, Maridonneau-Parini I, Daffe M, Astarie-Dequeker C, Etienne G (2005) Mycobacteria use their surface-exposed glycolipids to infect human macrophages through a receptor-dependent process. J Lipid Res 46:475–483
Walter K, Holscher C, Tschopp J, Ehlers S (2010) NALP3 is not necessary for early protection against experimental tuberculosis. Immunobiology 215:804–811
Wang JY, Kishore U, Reid KB (1995) A recombinant polypeptide, composed of the alpha-helical neck region and the carbohydrate recognition domain of conglutinin, self-associates to give a functionally intact homotrimer. FEBS Lett 376:6–10
Wang C, Chen ZL, Pan ZF, Wei LL, Xu DD, Jiang TT, Zhang X, Ping ZP, Li ZJ, Li JC (2013) NOD2 polymorphisms and pulmonary tuberculosis susceptibility: a systematic review and meta-analysis. Int J Biol Sci 10:103–108
Watford WT, Ghio AJ, Wright JR (2000) Complement-mediated host defense in the lung. Am J Physiol Lung Cell Mol Physiol 279:L790–L798
Weikert LF, Edwards K, Chroneos ZC, Hager C, Hoffman L, Shepherd VL (1997) SP-A enhances uptake of bacillus Calmette-Guerin by macrophages through a specific SP-A receptor. Am J Phys 272:L989–L995
Weikert LF, Lopez JP, Abdolrasulnia R, Chroneos ZC, Shepherd VL (2000) Surfactant protein A enhances mycobacterial killing by rat macrophages through a nitric oxide-dependent pathway. Am J Physiol Lung Cell Mol Physiol 279:L216–L223
Werts C, Girardin SE, Philpott DJ (2006) TIR, CARD and PYRIN: three domains for an antimicrobial triad. Cell Death Differ 13:798–815
Whaley K, Ruddy S (1976a) Modulation of C3b hemolytic activity by a plasma protein distinct from C3b inactivator. Science 193:1011–1013
Whaley K, Ruddy S (1976b) Modulation of the alternative complement pathways by beta 1 H globulin. J Exp Med 144:1147–1163
Whaley K, Schur PH, Ruddy S (1976) C3b inactivator in the rheumatic diseases. Measurement by radial immunodiffusion and by inhibition of formation of properdin pathway C3 convertase. J Clin Invest 57:1554–1563
WHO (2019) Global tuberculosis report 2019. World Health Organisation, Geneva
Wieland CW, Van Der Windt GJ, Wiersinga WJ, Florquin S, Van Der Poll T (2008) CD14 contributes to pulmonary inflammation and mortality during murine tuberculosis. Immunology 125:272–279
Wilson GJ, Marakalala MJ, Hoving JC, Van Laarhoven A, Drummond RA, Kerscher B, Keeton R, Van De Vosse E, Ottenhoff TH, Plantinga TS, Alisjahbana B, Govender D, Besra GS, Netea MG, Reid DM, Willment JA, Jacobs M, Yamasaki S, Van Crevel R, Brown GD (2015) The C-type lectin receptor CLECSF8/CLEC4D is a key component of anti-mycobacterial immunity. Cell Host Microbe 17:252–259
Wingrove JA, Discipio RG, Chen Z, Potempa J, Travis J, Hugli TE (1992) Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem 267:18902–18907
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27:135–144
Yadav M, Schorey JS (2006) The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108:3168–3175
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301:640–643
Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T (2008) Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol 9:1179–1188
Yang Y, Xu P, He P, Shi F, Tang Y, Guan C, Zeng H, Zhou Y, Song Q, Zhou B, Jiang S, Shao C, Sun J, Wang X, Song H (2020) Mycobacterial PPE13 activates inflammasome by interacting with the NATCH and LRR domains of NLRP3. FASEB J 34(9):12820–12833
Yang HY, Li H, Wang YG, Xu CY, Zhao YL, Ma XG, Li XW, Chen H (2014) Correlation analysis between single nucleotide polymorphisms of pulmonary surfactant protein A gene and pulmonary tuberculosis in the Han population in China. Int J Infect Dis 26:31–36
Yassin RJ, Hamblin AS (1994) Altered expression of CD11/CD18 on the peripheral blood phagocytes of patients with tuberculosis. Clin Exp Immunol 97:120–125
Yi L, Zhang K, Mo Y, Zhen G, Zhao J (2015) The association between CD209 gene polymorphisms and pulmonary tuberculosis susceptibility: a meta-analysis. Int J Clin Exp Pathol 8:12437–12445
Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, Inoue H, Tanaka M, Yoneyama M, Oh-Hora M, Akashi K, Yamasaki S (2014) Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity 41:402–413
Zaheen A, Bloom BR (2020) Tuberculosis in 2020—new approaches to a continuing global health crisis. N Engl J Med 382:e26
Zenaro E, Donini M, Dusi S (2009) Induction of Th1/Th17 immune response by Mycobacterium tuberculosis: role of dectin-1, mannose receptor, and DC-SIGN. J Leukoc Biol 86:1393–1401
Zhang X, Jiang F, Wei L, Li F, Liu J, Wang C, Zhao M, Jiang T, Xu D, Fan D, Sun X, Li JC (2012) Polymorphic allele of human MRC1 confer protection against tuberculosis in a Chinese population. Int J Biol Sci 8:375–382
Zhang X, Li X, Zhang W, Wei L, Jiang T, Chen Z, Meng C, Liu J, Wu F, Wang C, Li F, Sun X, Li Z, Li JC (2013a) The novel human MRC1 gene polymorphisms are associated with susceptibility to pulmonary tuberculosis in Chinese Uygur and Kazak populations. Mol Biol Rep 40:5073–5083
Zhang Y, Jiang T, Yang X, Xue Y, Wang C, Liu J, Zhang X, Chen Z, Zhao M, Li JC (2013b) Toll-like receptor -1, -2, and -6 polymorphisms and pulmonary tuberculosis susceptibility: a systematic review and meta-analysis. PLoS One 8:e63357
Zhao M, Jiang F, Zhang W, Li F, Wei L, Liu J, Xue Y, Deng X, Wu F, Zhang L, Zhang X, Zhang Y, Fan D, Sun X, Jiang T, Li JC (2012) A novel single nucleotide polymorphism within the NOD2 gene is associated with pulmonary tuberculosis in the Chinese Han, Uygur and Kazak populations. BMC Infect Dis 12:91
Zhao XQ, Zhu LL, Chang Q, Jiang C, You Y, Luo T, Jia XM, Lin X (2014) C-type lectin receptor dectin-3 mediates trehalose 6,6'-dimycolate (TDM)-induced Mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-kappaB activation. J Biol Chem 289:30052–30062
Zhu LL, Zhao XQ, Jiang C, You Y, Chen XP, Jiang YY, Jia XM, Lin X (2013) C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39:324–334
Zimmerli S, Edwards S, Ernst JD (1996) Selective receptor blockade during phagocytosis does not alter the survival and growth of Mycobacterium tuberculosis in human macrophages. Am J Respir Cell Mol Biol 15:760–770
Zumla A, Raviglione M, Hafner R, Von Reyn CF (2013) Tuberculosis. N Engl J Med 368:745–755
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tsolaki, A.G., Varghese, P.M., Kishore, U. (2021). Innate Immune Pattern Recognition Receptors of Mycobacterium tuberculosis: Nature and Consequences for Pathogenesis of Tuberculosis. In: Kishore, U. (eds) Microbial Pathogenesis. Advances in Experimental Medicine and Biology, vol 1313. Springer, Cham. https://doi.org/10.1007/978-3-030-67452-6_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-67452-6_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67451-9
Online ISBN: 978-3-030-67452-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)