
Abstract
Sphingosine-1-phosphate (S1P), together with other phosphosphingolipids, has been found to regulate complex cellular function in the tumor microenvironment (TME) where it acts as a signaling molecule that participates in cell–cell communication. S1P, through intracellular and extracellular signaling, was found to promote tumor growth, angiogenesis, chemoresistance, and metastasis; it also regulates anticancer immune response, modulates inflammation, and promotes angiogenesis. Interestingly, cancer cells are capable of releasing S1P and thus modifying the behavior of the TME components in a way that contributes to tumor growth and progression. Therefore, S1P is considered an important therapeutic target, and several anticancer therapies targeting S1P signaling are being developed and tested in clinics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sphingosine-1 phosphate (S1P)
- Tumor microenvironment
- Sphingosine kinase (SphK)
- Sphingosine-1 phosphate receptors (S1PR)
- Cell motility
- Chemotaxis
- Immunomodulation
- Macrophage polarization
- TAM/M2 macrophages
- Tumor angiogenesis
- Cancer metastasis
- Tumor growth
- Hypoxia-inducible factor 1α (HIF1α)
- Nuclear factor-κB (NF-κB)
- Inflammation
7.1 Introduction
The tumor microenvironment (TME) plays an important role in cancer biology contributing to tumor initiation, progression, metastasis, and responses to treatment. Cancer cells within a solid tumor influence the surrounding microenvironment through the release of extracellular signals in the form of cytokines, chemokines, and lipid mediators. These signals work to control immune responses, inflammation, as well as angiogenesis. Sphingosine-1-phosphate (S1P), a bioactive sphingolipid, has emerged over the last few decades as a new player in the TME and cancer progression. It can be produced and released into the TME from cancerous and noncancerous tissues and acts to regulate the interactions between tumor, immune, and mesenchymal cells that are present within the TME. In this chapter, we summarize the mechanisms through which S1P, present in the TME, participates in tumor progression, inhibits antitumor immune response, modulates inflammation, regulates response to hypoxic conditions, and facilitates the recruitment of mesenchymal cells to increase tumor angiogenesis. Additionally, we will discuss therapeutic strategies that target S1P signaling in cancer patients.
7.2 Metabolism of S1P
S1P is generated by the conversion of ceramide to sphingosine, which is catalyzed by ceramidase, and subsequent phosphorylation of sphingosine by sphingosine kinases (SphK1 and SphK2) (Fig. 7.1). SphK1 is localized mainly in the cytosol [1], whereas SphK2 can be found in the nucleus and internal membranes of the endoplasmic reticulum, Golgi, and mitochondria [2, 3] which suggest the distinct function of generated S1P. Both enzymes can be translocated to different cell compartments in response to specific signals. For example, SphK1 can be recruited to the plasma membrane in response to growth stimulating factors such as epidermal growth factor (EGF) or phorbol 12-myristate 13-acetate [4, 5] and targeted to Golgi apparatus by phosphatidic acid [6], whereas SphK2 can be translocated from the nucleus to the cytoplasm dependent on PKD-mediated phosphorylation [7].

Metabolism of sphingosine-1 phosphate (S1P). Enzymes and substrates involved in the synthesis and degradation of S1P. Solid lines represent confirmed pathways, dotted line represents the pathway identified in vitro but not yet confirmed in vivo
Interestingly, S1P levels and SphK1/2 expression and/or activity were found to be increased in distinct cancer types including acute lymphoblastic leukemia [8], astrocytoma [9], breast cancer [10, 11], colon cancer [12, 13], gastric cancer [14], glioblastoma [15, 16], lung cancer [17], non-Hodgkin’s lymphoma [18], prostate cancer [19], thyroid cancer [20, 21], and many others [22,23,24]. Several reports also indicate that increased expression of SphK1 correlated with disease progression, cancer recurrence, and reduced patient survival [9, 10, 14, 15, 23, 24] as well as invasion and lymph node metastasis [25]. In contrast, reduced expression of SphK1 and subsequently lower level of S1P in plasma were found in prostate cancer patients [26]. Moreover, S1P level correlates with patients’ survival, and downregulation of SphK in erythrocytes could have implication in cancer-induced anemia [26].
It has been shown that S1P can also be generated by autotaxin (ATX) through hydrolysis of sphingosylphosphorylcholine (SPC) [27]; however, it is uncertain whether this pathway is active in vivo. First of all, the reported Km value of ATX for SPC (~23 mM) [27] is much higher than normal SPC levels in plasma/serum (0.03–0.13 μM) [28, 29]. Moreover, in mice with downregulated Autotaxin, the level of S1P was not changed when compared with wild-type animals, in contrast to the main autotaxin metabolite, lysophosphatidic acid, which was decreased by ~50% [30]. This suggests that the in vivo contribution of Autotaxin to the total pool of S1P is limited.
S1P levels are the result of the balance between its synthesis and reversible conversion to sphingosine or irreversible degradation. Dephosphorylation of S1P is catalyzed by specific S1P phosphatases (SPP1 and SPP2), or lipid phosphate phosphatases (LPP1–3) and subsequent sphingosine conversion to ceramide by ceramide synthase [31] or through irreversible degradation by S1P lyase (SPL) that cleaves S1P to hexadecenal and phosphoethanolamine [32]. Similarly to SphK, enzymes responsible for S1P degradation were also found to be dysregulated in malignant tissues; lower expression of SPL was found in colon [33, 34], prostate [35], and pancreatic cancers [35], and was shown to have implications in chemo and radiotherapy resistance and cancer cells metastasis [35]. Downregulation of SPP was found in colon cancer [31], gastric cancer [31], and glioblastoma [16], and its expression was correlated with lymph node metastasis and gastric cancer patient’s survival [31].
7.3 Sources of S1P in TME
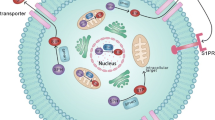
S1P is present in the components of the TME such as blood , lymph, and interstitial fluid. In circulation, S1P is bound to plasma proteins, mainly high-density lipoprotein (HDL) [36] apolipoprotein M [37], and to a lesser extent to albumin [38]. The main source of plasma S1P was thought to be platelets, which are characterized by high SphK activity and lack of SPL which allows them to accumulate large amounts of S1P, up to nine- fold more than erythrocytes. Although erythrocytes produce less S1P than platelets, at the same time they constitute about 95% of total blood cell number, thus their contribution to the S1P pool in the blood is considerably much higher and is estimated to be 75%. Other important contributors of plasma S1P are the vascular endothelium and endothelial cells, and in lymph, lymphatic endothelial cells, thus suggesting that stromal cells, could synthesize and release endogenous S1P also to TME. Recently, it has been shown that cancer cells themselves can also secrete high levels of S1P [39,40,41] hence contributing to the total S1P pool present in TME, which could explain high level of S1P in ascites fluids from ovarian cancer [42, 43] and additional observation that plasma S1P level decrease in patients after ovarian cancer surgery [44]. Moreover, S1P can also be released from dying cells (necrotic or apoptotic) and damaged tissues [45,46,47,48]. This can have important implication in anticancer therapies since it was shown that S1P levels were increased in several organs after γ-irradiation or chemotherapy, creating an unwanted prometastatic environment as a side effect of the treatment [46] (Fig. 7.2).

Sphingosine-1 phosphate (S1P) plays a role in the formation of the prometastatic environment as a side effect of radio/chemotherapy. S1P released from damaged tissues (malignant and nonmalignant) induces migration of tumor cells that survive initial anticancer treatment. Such cells metastasize to distant locations where they can form secondary tumors
The structure of S1P and its relatively high solubility in water unable S1P to diffuse over the membranes to the extracellular compartments. Therefore to act as a signaling molecule, S1P has to be either generated in extracellular compartments directly or synthesized intracellularly and transported outside the cells by specific transporters. SphK1 was found to be constitutively released from endothelial cells in quantities that allow for the synthesis of extracellular S1P and obtain its physiologically relevant concentrations [49, 50]. Moreover, SphK1 was also found to be released from histiocytic lymphoma U937 cells in response to stimulation with oxidized LDL immune complexes [51] thus indicating that extracellular synthesis of S1P might be regulated by additional signaling factors. Several transporters of S1P that allows for the autocrine/paracrine signaling of S1P have been identified including Spinster 2 (SPNS2) [52, 53] and several members of the ABC-type lipid transporters family, namely ABCC1, ABCC2, and ABCA1 [54]. This diversity in the type of transporters might suggest their importance in the regulation of S1P levels in different tissues. Indeed, recently, it has been shown that SNPS2 transporter was necessary for secretion of S1P to the lymph, but it did not play an important role in the regulation of S1P levels in plasma [53, 55]. Moreover, Spns2 is not expressed in murine erythrocytes, and the level of S1P in the blood is not affected in Spns2 knockout mice [56]. On the other hand, several in vitro studies have revealed that ABC transporters mediate S1P release in different types of cells, including mast cells [57], erythrocytes [58], breast cancer cells [59], astrocytes [60], and also platelets [61]. Additionally, since S1P is present in the circulation, mainly in complex with HDL particles [38], it might suggest that the S1P export may be coupled with ABCA1-dependent lipoprotein formation [60].
What is worth to note, changes in SPNS2 expression were found in non–small cell lung cancer (NSCLC) patients’ samples, and in vitro studies indicate that the overexpression of SPNS2 induced apoptosis, whereas its knockdown enhanced NSCLC cells migration [62]. Moreover, alterations of SPNS2 affected the expression of several enzymes involved in S1P metabolism, including SphK, SPP, and SPL1 [62], thus indicating a cross talk between the pathways involved in S1P synthesis/degradation and extracellular transport of S1P.
7.4 S1P Signaling
S1P has been shown to regulate cellular functions both via intracellular (Fig. 7.3) and extracellular (Fig. 7.4) mechanisms. Intracellular S1P was first identified as an activator of intracellular calcium channels via an inositol triphosphate-independent pathway [63], but a target ion channel has not been identified. However, several studies support this observation by demonstrating that increased level of S1P also upregulates intracellular calcium concentrations [64]. In the nucleus, S1P was found to play a role in the regulation of gene expression by binding to histone deacetylases (HDAC1 and HDAC2), and inhibiting their activity thus regulating transcription of several genes including the cyclin-dependent kinase inhibitor p21 [65] (Fig. 7.3). In mitochondria, the interaction of S1P with the prohibitin 2 (PHB2) protein was found to be important for cytochrome c oxidase assembly and mitochondrial respiration [66]. Interestingly, in both cases, SphK2 was found to be the main enzyme involved in the synthesis of S1P in a particular cellular compartment [65, 66]. On the other hand, S1P generated by SphK1 was found to act as a cofactor for the TNF receptor-associated factor 2 (TRAF2) E3 ubiquitin ligase complex by which it regulates the activity of the nuclear factor-κB (NF-κB) signaling involved in inflammatory, antiapoptotic, and immune processes [67]. NF-κB activation was observed also in response to endoplasmic reticulum (ER) stress which resulted in S1P increase and subsequent interaction with HSP90 and/or GRP94 protein to form a signaling complex with an ER stress responsive protein, IRE1α, TRAF2, and RIP1 [68]. Of note, NF-κB activation may also be induced by extracellular S1P [69, 70], suggesting that S1P acts upon several stages within the same signaling cascade. S1P has also been shown to enhance the cellular inhibitor of apoptosis 2 (cIAP2)-mediated K63-linked polyubiquitination of interferon regulatory factor-1 (IRF-1), which is essential for IL-1-induced production of chemokines CXCL10 and CCL5 [71]. Moreover, S1P interacts with the transcription factor peroxisome proliferator-activated receptor γ (PPARγ) through which it can regulate angiogenesis [72].

Intracellular sphingosine-1 phosphate (S1P) signaling. Intracellular S1P regulates the nuclear factor-κB (NF-κB) signaling pathway by targeting TNF receptor-associated factor 2 (TRAF2) or heat shock protein 90 (Hsp90)/glucose-regulated protein 94 (GRP94). In mitochondria, it interacts with prohibitin 2 (PHB2) thus regulating mitochondrial respiration, whereas in the nucleus it regulates the activity of histone deacetylases (HDACs) and nuclear transcription factor PPARγ

Extracellular sphingosine-1 phosphate (S1P) signaling. Intracellularly generated S1P is exported to the extracellular compartments by Spinster 2 (SPNS2) and members of the ABC transporter family thus allowing for autocrine/paracrine signaling of S1P. Extracellular S1P binds to the specific G-coupled S1P receptors designated as S1PR1–5, regulating mitogen-activated protein kinase (MAPK), Rac, Rho, phosphatidylinositol 3-kinase (PI3K), phospholipase C (PLC), and adenyl cyclase (AC) pathways
Extracellular S1P acts through binding to G-protein-coupled receptors (S1PR1–S1PR5) by which it regulates several cell processes, including cell survival and migration [73] (Fig. 7.4). Expression patterns of S1PRs vary between tissues and can change during development and aging. S1PR1–S1PR3 are essentially ubiquitously expressed, whereas expression of S1PR4 and S1PR5 is restricted to distinct cell types [74]. S1PRs can activate several different signaling pathways. S1PR1 is coupled with Gi protein and activates Ras, mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), protein kinase B (AKT), and phospholipase C (PLC) pathways. Both S1PR2 and S1PR3 are coupled to Gi, Gq, and G12/13 and can activate Ras, MAPK, PI3K, AKT, PLC, and Rho-dependent pathways [75]. Through coupling with Gi and G12/13, S1PR4 mediates cell shape change and motility via a Rho-dependent pathway [75], whereas S1PR5 appears to activate the G12/13 protein and the subsequent Rho/ROCK signaling pathway [76] and through coupling with Gi inhibits adenyl cyclase (AC) [77]. These studies strongly suggest that S1P can mediate diverse functions through activation of different signal transduction pathways in different cell types, as well as within the same cell, depending on the patterns of S1PR expression. What is worth noting, it has been suggested that only S1P generated by SphK1 and not SphK2 can activate S1PRs [78].
7.5 S1P as a Modulator of Cancer Biology
Extra- and intracellular S1P can activate various signaling cascades that are implicated in cancer cell proliferation, survival, migration, inhibition of apoptosis, and chemoresistance. SphK1 was found to be involved in the regulation of S1P-dependent proliferation of several cancers, including gastric [79] and colorectal cancers [80]. In contrast to SphK1, the role of SphK2 in the regulation of cell growth seems to be more context-dependent. In some cells, upregulation of SphK2 levels was found to cause cell cycle arrest, caspase-3 activation, cytochrome c release, and thus inhibited cell growth [78, 81]. Whereas in others, such as glioblastoma, colorectal cancer, or prostate cancer, downregulation of SphK2 was associated with decreased proliferation of malignant cells in vitro and in vivo [82, 83]. Surprisingly, a study testing new SphK inhibitors on a wide variety of cancer cell lines including breast cancer, glioblastoma, melanoma, cervix, and colon cancers revealed that SphK activity was not required for tumor cell viability both in vivo and in vitro [84]. Thus, these discrepancies with opposing findings require further investigation. Several studies were able to identify receptors through which S1P affects cancer cell proliferation. In human prostate cancer PC-3 cells and glioma cells S1P attenuates cell proliferation through activation of S1PR5 [85, 86], S1PR2 is involved in growth of hepatocellular carcinoma and Willim’s tumor [87, 88], downregulation of S1PR1 was associated with decreased growth of rhabdomyosarcoma xenografts [46], whereas in breast cancer higher S1PR1 expression correlates with decreased cell proliferation [89]. Interestingly, in glioblastoma S1PR1–3 were found to be involved in the stimulation of proliferation [90], while activation of S1PR5 has an opposite effect [91]. Taken together, these observations suggest that both exogenous and intracellular S1P are important for tumor growth and indicate that attenuation of S1P signaling pathway could be a promising strategy in cancer treatment.
Extracellular S1P has been found to act a potent chemoattractant or chemokinetic factor for different types of cancers, including gastric cancer [92], leukemia [93], lung cancers [94], glioblastoma [95], ovarian cancer [96], or rhabdomyosarcoma [46], thus indicating the role of S1P in tumor metastasis. In the majority of cells, S1P stimulates the motility of cancer cells through S1PR1 or S1PR3, mainly through activation of the ERK1/2 signaling pathway. However, some evidence also indicates that a S1PR1–RAC1–CDC42-dependent pathway involving the tyrosine phosphorylation of membrane-type matrix metalloproteinase 1 might play a role [97, 98]. Moreover, in some cells, such as ovarian cancer, calcium mobilization might accompany S1P-mediated invasion [96]. By contrast, S1P can inhibit cancer cell motility through a S1PR2-dependent regulation of Rho [95]. The specific effect of S1P is partially determined by the type of the receptors expressed in particular tumors, e.g., exposure to S1P of gastric cancer cell lines that dominantly expressed S1PR3 induced their migration, whereas the opposite effect was observed in gastric tumor cells that mainly expressed S1PR2 [92]. The effects of S1P on the motility of cancer cells can also be context-dependent. For example, activation of the S1P–S1PR2 axis increased glioma invasiveness by enhancing the expression of secreted, angiogenic matricellular protein CCN1 [99]. The metastatic behavior of tumor cells can also depend on S1P metabolic enzymes’ pattern of expression. It has been found that SphK levels positively correlated with migration and invasion of ovarian cancer [100], breast cancer, and kidney carcinoma cells [101]. In addition to S1P-induced motility of cancer cells, this sphingolipid was also found to be involved in the regulation of invasion processes. S1P was found to induce overexpression of MMP-2 through the MAPK/ERK1/2 and NF-κB pathways and is responsible for cancer cell invasion in endothelial cells [102]. Upregulation of S1P can also lead to overexpression of MMP2, which has a major role in initiating cell invasion via H-Ras signaling in human breast cancer [103]. In the same cells, S1P was also found to induce MMP9, and this effect was S1PR3-dependent.
Angiogenesis, a process of formation of new blood vessels, which is necessary for tumor growth and invasion, can also be regulated by S1P signaling. Increased angiogenesis in tumor tissue can be at least partially explained by the S1P-induced migration of endothelial cells [104] and vascular smooth muscle cells [105, 106] which participate in the formation of new blood vessels in cancerous tissue. However, it was also shown that cross talk between S1P and VEGF signaling can additionally contribute to increase angiogenesis. S1P was found to upregulate VEGF in human prostate cancer [75] and thyroid cancer [107], whereas in bladder cancer, VEGF was found to stimulate SphK1 leading to the increase of intracellular levels of S1P [107]. S1PR1 was found to be involved in S1P-induced regulation of vascularization in thyroid cancer [107], breast cancer cells [108], and Lewis lung carcinoma [106]. Angiogenesis of tumors was also found to correlate with SphK1 level in hepatoma cells [109], Lewis lung carcinoma [106], as well as in glioma cells [110].
Accumulating evidence indicates that factors present in the TME, including S1P, might play a role in acquiring of chemoresistance by cancer cells. Overexpression of SpK1 was found to be associated with the chemoresistance of leukemic cells to imatinib [111] and daunorubicin [112], prostate cancer to docetaxel [113], renal cancer to sunitinib [114], and gastroesophageal cancer to oxaliplatin, cisplatin, and docetaxel [115]. It has been suggested that at least part of this effect is the result of an imbalance between ceramide and S1P [116]. Also, other S1P metabolizing enzymes were shown to be involved in chemoresistance, e.g., overexpression SphK2 has been correlated to gefitinib chemoresistance in non-small cell lung cancer cells [117] and hormone-independent breast cancer [118], whereas depletion of SPL caused cisplatin resistance [119]. Interestingly, the treatment of prostate cancer cells with camptothecin upregulates both SK1 and S1P receptors, suggesting that resistance to camptothecin could involve autocrine/paracrine mechanisms [120]. Some studies also indicate the involvement of specific S1P receptors in the acquired chemoresistance of cancer cells; for example, the use of S1PR1 antagonist was found to sensitize neuroblastoma cells to etoposide [121].
Taken together, these data strongly indicate that modulation of S1P-related signaling may constitute a promising anticancer therapy; however, due to high heterogeneity between the tumors and observed opposing effects in response to S1P stimulation, more work has to be done to better understand the mechanisms of its action in cancer cells.
7.6 S1P as a Modulator of the Immune Response
The composition and characteristics of the TME vary between different types of tumors, but it is crucial in determining the antitumor response. Several different populations of immune cells are capable of playing a role in antitumor immune response, including macrophages, natural killer cells (NKs), dendritic cells (DCs), and effector T cells. However, at the same time, tumor cells protect themselves from destruction by induction of an immunosuppressive microenvironment, thus promoting the development of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells. Accumulating evidence indicates that S1P might be one of the components of the TME that can regulate this interplay between cancer and immune cells (Fig. 7.5). The best-studied effect of S1P on immune cells includes its role in the regulation of migration and polarization of macrophages. Macrophages originate from the myeloid lineage and belong to the innate immune system. They are derived from blood monocytes that migrate into cancer tissue in response to specific chemokines present in the TME. One of their main functions is the removal of microbes and cell debris through phagocytosis, but they also play a crucial role in the initiation of inflammation [122, 123]. Based on signals from the microenvironment, macrophages can exhibit different phenotypes, and the process of acquiring different functional programs is known as polarization. Two major subsets have been proposed: M1 and M2 macrophages which correspond to the extreme phenotypes of the opposite spectrum. M1 macrophages (classically activated macrophages) are aggressive and highly phagocytic, produce large amounts of reactive oxygen and nitrogen species, secrete high levels of IL-12 and IL-23, and induce the activation, and clonal expansion of T-helpers cells thus contributes to inflammation. In contrast, M2 macrophages (alternatively activated macrophages) are anti-inflammatory and aid in the process of angiogenesis and tissue repair. In the TME, M1 macrophages are involved in the elimination of tumor cells, whereas M2 macrophages stimulate tumor growth by releasing angiogenic factors and anti-inflammatory cytokines [124]. Tumor-associated macrophages (TAMs) display an M2-like phenotype, and several studies indicate a correlation between TAM density and poor prognosis of cancer patients. Moreover, new evidence connect TAMs with chemotherapy resistance.

Sphingosine-1 phosphate (S1P) is a key regulator of cell–cell interaction and modulator of the anticancer immune response in the tumor microenvironment (TME). S1P released from cancer cells chemoattract monocytes and induce their polarization into TAM/M2 macrophages, which in turn secrete growth factors and cytokines that stimulate tumor growth. S1P also inhibits cytotoxic activity of natural killer (NK) cells and promotes regulatory T-cell (Treg) expansion, migration, and accumulation in malignant tissue which results in immunosuppression of anticancer response. In the presence of S1P, NK-mediated cell lysis of immature monocyte-derived dendritic cells (DCs) is inhibited. Moreover, S1P enhances endocytosis and induces migration of mature DCs which could potentially increase immune response toward cancer cells. Mast cells respond to S1P stimulation with increased motility and degranulation which result in the release of growth factors and cytokines that depending on the context can stimulate or inhibit tumor growth and progression. Similarly, fibroblasts, in the presence of S1P, secrete growth factors, proteases, and also S1P that accelerate tumor growth, angiogenesis, and invasion. Additionally, S1P induces angiogenesis and tumor growth by enhancing migration of endothelial cells
S1P has been identified to be a potent chemoattractant for many different types of normal and malignant cells. Interestingly, S1P released from apoptotic leukemic cells not only was found to attract monocytes, but its effect was comparable with monocyte chemoattractant protein-1 (MCP-1/CCL2) [45]. These results were confirmed in several other models including breast cancer [47, 48] and acute T-cell leukemia [125, 126], and more detailed studies indicate the involvement of S1P-S1PR1 axis in monocyte migration [125]. There are also some suggestions that SphKs are involved in the release of S1P from apoptotic cells; however, the results are contradictory; some studies showed that activation of SphK1 is necessary to facilitate this process [45, 126], whereas the other pointed out to the involvement of SphK2 [126].
Accumulating evidence point to a crucial role of S1P in macrophage polarization. In several models, including breast cancer, S1P was found to induce a M2 phenotype in macrophages which was characterized by decreased tumor necrosis factor (TNF)-alpha, IL-12, and nitric oxide synthase production, but increased formation of IL-4, IL-8, IL-10, TGF-β1, and, interestingly, SphK1 [47, 48, 127, 128]. More detailed studies indicated that this process involved suppression of NF-κB, p38 MAPK, and JNK signaling pathways [47, 128, 129], as well as activation of tyrosine kinase receptor A and ERK1/2 [48, 128]. Moreover, S1P activity was dependent on the expression of SphK2, S1PR1/3 but not S1PR2 [47, 126, 129, 130]. Additionally, IL-10, a potent, anti-inflammatory cytokine was found to stimulate macrophages to secrete prostaglandin E2 (PGE2) that induced migration of endothelial cells and increased angiogenesis, a seal of tumor progression [130]. S1P released from cancer cells also induced Bcl-X(L) and Bcl-2 upregulation, which protected macrophages from cell death [126].
The identification of S1P function in monocyte recruitment and polarization of macrophages toward the less aggressive M2 phenotype suggest that S1P present in the TME can have a positive effect on tumor growth not only by direct stimulation of cell proliferation, invasiveness, and chemoresistance but also by allowing them to evade the tumor-killing response elicited by cytotoxic macrophages. Therefore, a better understanding of S1P role in the regulation of macrophage production and polarization could lead to the development of new therapies allowing for the reprogramming of TAMs toward an M1 phenotype and activation of their antitumor response.
Even though most of the literature focus on the crucial role of S1P in monocyte/macrophage recruitment, survival, and polarization, it was also found that this phosphosphingolipid can modulate the function of other immune cells present in the TME such as regulatory T cells (Tregs) or natural killer cells (NK). Tregs are a subpopulation of T cells that act to suppress the immune response, thus maintaining homeostasis and self-tolerance. In cancers, however, their suppressive activity toward other immune cells promotes tumor progression. S1PR1 was found to be involved in the regulation of Tregs functions since the permanent deletion of this receptor from Treg cells resulted in autoimmunity [131]. Moreover, activation of S1PR1 signaling leads to Tregs accumulation in cancerous tissues and promotes tumor growth, through activation of STAT3 [132]. However, in other studies, S1PRs agonist FTY720 (fingolimod) was found to inhibit IL-2-induced STAT-5 phosphorylation, paralleled by a loss of forkhead box protein 3 (FoxP3) expression, which resulted in decreased Treg cells proliferation, both, in vitro and in vivo [133, 134]. FTY720 is an agonist of four S1PRs (S1PR1, S1PR3, S1PR4, and S1PR5) [135], therefore the discrepancy between studies might indicate that although S1PR1 can stimulate the suppressive nature of Tregs, this effect can be counteracted by stimulation of other S1P receptors. Therefore more studies have to be done to better characterize the role of S1P and its receptors in Tregs regulation. Interestingly, S1PR1 present on cancer cells was found to regulate a cross talk between bladder cancer cells and Tregs. Overexpression of this receptor in bladder cancer cells promoted the generation of bladder cancer-induced Tregs by activation TGF-β signaling pathway, leading to the secretion of TGF-β and IL-10 from tumor cells [134].
Recently S1P was identified as a potent chemoattractant for natural killers cells (NK) that play a crucial role in antitumor immunity [136, 137]. However, at the same time it was shown that S1P stimulation activates the phosphatidylinositol 3-kinase (PI3K)-dependent signaling pathway, protein kinase A (PKA), protein kinase B (PKB/AKT), glycogen synthase kinase-3beta (GSK-3beta) and increases the level of cAMP, thus inhibiting the cytotoxic activity of NK cells [136]. Further studies confirmed that S1P can act as an anti-inflammatory molecule that significantly reduced the release of IL-17A and IFN-gamma from NK cells in an S1PR1-independent manner [138]. At the same time, it was found that S1P can modulate the interaction between NK and tumor cells since activation of S1PR1 on human myeloid leukemia K562 cells protected them from NK cells-induced lysis [138]. Interestingly, S1P was also found to modulate the interaction of NK cells with immature dendritic cells (DCs) [138]. DCs are antigen-presenting cells that play a central role in the initiation of adaptive immune responses. In the presence of S1P, NK-mediated cell lysis of immature monocyte-derived DCs was inhibited, which may favor antigen presentation to T cells [138]. Moreover, S1P enhances endocytosis and induce migration of mature DCs in an S1PR3--dependent but not S1PR1-dependent manner [139], which could potentially increase immune response toward cancer cells. This duality in S1P action cells requires more studies to better understand the effect of its signaling on migration and phenotypic modulation of NK and DCs cells.
One of the best-studied effects of S1P is its role in migration and egress of lymphocytes from lymphoid organs. However, less attention was put in resolving the role of S1P in the regulation of B and T lymphocytes in cancer progression. In diffuse large B-cell lymphoma (DLBCL) cell lines, expression of S1PR2 inversely correlated with the oncogenic transcription of FOXP1, resulting in reduced tumor growth in S1PR2 overexpressing cells [140]. Moreover, low S1PR2 expression was found to be a strong negative prognostic factor of patient survival, especially in combination with high FOXP1 expression [140]. Interestingly, different B-cell populations express different combinations of S1PRs; S1PR1 was found to promote migration, whereas S1PR4 modulates and S1PR2 inhibits S1PR1 signals [141]. Moreover, the expression of CD69 in activated B lymphocytes and B cells from patients with chronic lymphocytic leukemia (CLL) inhibited S1P-induced migration [141]. Studying B-cell lines, normal B lymphocytes, and B cells from patients with primary immunodeficiencies, Bruton’s tyrosine kinase, β-arrestin 2, LPS-responsive beige-like anchor protein, dedicator of cytokinesis 8, and Wiskott-Aldrich syndrome protein were found to be critical signaling components downstream of S1PR1 [141]. S1PR1 is expressed at low levels in CLL lymph nodes as compared with normal B cells [142], increased expression of S1PR1 correlates with STAT3 activation and survival of B-cell lymphoma cells [143]. Furthermore, downregulated expression of S1PR1 in CLL B cells impairs their egress from the peripheral lymphoid organs and enhances their survival [144]. S1P was also identified as a molecule that inhibits T-cell proliferation [145].
Mast cells (MCs) are immune cells of the myeloid lineage and are present in connective tissues throughout the body. The activation and degranulation of mast cells modulate many aspects of physiological and pathological conditions in various settings [146]. Immuno-modulating action of mast cells is related to the production and release of several multi-potent molecules including S1P [147, 148], Interestingly, MCs have been found to act as both tumor promotors and tumor suppressors [147, 149, 150], and this effect can differ within the same tumor depending on the tissue compartment, e.g., in prostate tumors intratumoral MCs negatively regulate angiogenesis and tumor growth, whereas peritumoral MCs stimulate the expansion of cancer cells [150]. High numbers of mast cells have been found in several tumors including colorectal [151], pancreatic [152], melanoma, [153], NSCLC [154], squamous cell carcinomas (SCC) of the esophagus [155], mouth [156], and lip [157]. Interestingly, an elevated number of MCs was correlated with good [150, 158, 159] and poor prognosis [158, 160]. Fr example, in prostate cancer, patients with higher MCs counts had a better prognosis than the patients with lower MSc counts [159, 160]. Moreover, the MCs numbers correlated well with the clinical stages of tumors [160]. Similarly to other immune cells, S1P was found to stimulate motility of MCs [161], and this effect was S1PR1-dependent [161]. On the other hand, S1PR2 showed an inhibitory effect on MCs migration; however, it was necessary for S1P-induced degranulation [161]. Moreover, SphK1 but not SphK2 was found to be critical in MCs for antigen-induced degranulation, chemokine secretion, and migration, while both isozymes are essential for cytokine secretion [162]. Studies with MCs also led to the discovery that ABCC1 promotes the export of S1P across the plasma membrane independent of MCs degranulation [57]. Interestingly, exposure of MCs to S1P can lead to increased release of proteinases involved in tumor growth and metastasis, as well as pro-angiogenic VEGF [92, 148].
The complexity of S1P signaling that regulates immune cells in the TME (Fig. 7.5) requires further studies especially that not only S1P can directly act on different subpopulations of cells but also modulate cross talk between immune cells and tumor. Nevertheless, some of the studies indicate that the modulation of S1P-dependent signaling to increase the antitumor response of the immune system can be a promising target for anticancer therapies.
7.7 S1P as a Modulator in Inflammatory Pathways
Several studies indicate the involvement of S1P or enzymes controlling its metabolism in the regulation of inflammation. As mentioned earlier, intracellular S1P can activate NF-κB; however, recent studies indicate that this activation can also be mediated by S1PR (mainly S1PR1–3), thus involving an extracellular pool of S1P [69, 163, 164]. Interestingly, some results indicate a link between TNF signaling and NF-κB activation that involves S1P as a signaling molecule and/or as a cofactor of TRAF2 E3 ubiquitin ligase [67]. In melanoma cells, activation of NF-κB by extracellular S1P was found to be irreversibly correlated with expression of actin-binding protein FlnA [164], an interacting partner of SphK1 [165] and TRAF2 [166]. Moreover, it was also found that TRAF-interacting protein (TRIP), a binding partner of TRAF2, abrogated TNF-induced NF-κB activation by inhibiting binding of S1P to TRAF2 and thus suppressing its E3 ubiquitin ligase activity [67]. In inflammation-associated colon cancer, S1P was found to be essential for the production of the NF-κB-regulated cytokine, IL-6, crucial for persistent activation of transcription factor STAT3. This leads to upregulation of S1PR1 and reciprocally, enhanced S1PR1 expression activates STAT3 and upregulates IL-6 gene expression, thus accelerating tumor growth and metastasis in a STAT3-dependent manner [167]. The connection between S1PR1 and STAT3 activation was found to be crucial in distinct tumors, including lymphoma, adenocarcinoma, melanoma, breast, and prostate cancers [167] and decreased expression of STAT3-regulated genes by targeting S1PR1 was found to inhibit tumor progression [143]. What is interesting is S1P-induced STAT3 activation in colitis-induced colon cancer was correlated with upregulation of SphK1 or decreased level of SPL. A similar association was observed in animal models of inflammation, as S1P levels were increased in mice with dextran sulfate-induced colitis, but not in mice lacking the SphK1 gene [168, 169]. The connection between S1P-metabolized enzymes and S1P-induced activation of STAT3 was also observed in cholangiocarcinoma cells where inhibition of SphK2 abrogated STAT3 phosphorylation and decreased cells’ proliferation [170]. Also, in ER-negative breast cancer cells, SphK1 knockdown led to a significant reduction in leptin-induced STAT3 phosphorylation [171]. SphK1 and S1P were also found to be required for TNF-α-induced cyclooxygenase 2 (COX2), and prostaglandin E2 (PEG2) production [172]. Additionally, S1P in cooperation with lipopolysaccharide (LPS) was found to increase the expression of pro-inflammatory molecules such as IL-6, cyclooxygenase-2 (COX2), and prostacyclin in human endothelial cells [163]. Moreover, S1P was able to induce COX2 expression in Wilms tumor and this effect was mediated by S1PR2 [173].
Increased levels of S1P suggesting its involvement in controlling inflammation was observed in several nonmalignant conditions such as inflammatory arthritis [174, 175] multiple sclerosis [176], or asthma [177]. In contrast, increased expression of SPL and SPP2, thus indicating a decreased level of S1P, was found in the skin of patients and animals with psoriasis [178] and atopic dermatitis [179, 180], which could suggest that S1P exert anti-inflammatory actions in the skin. This hypothesis was confirmed in animal models of dermatitis or psoriasis, where the topical application of S1P reduced skin hyperproliferation and swelling [181, 182]. This mechanism of suppression involved inhibition of Langerhans cells migration to the lymph nodes or reduced antigen processing through S1PR2 activation [183]. Additionally, in a model of allergic asthma, inhalation of S1P suppressed airway by altering dendritic cell function [184]. Interestingly, only HDL-bound S1P, but not albumin-bound S1P, restrained lymphopoiesis and neuroinflammation in mice [185]. Although the described mechanisms were found in noncancerous tissues, one cannot exclude that S1P can regulate similar processes in tumors.
7.8 S1P as a Regulator of Cells’ Interaction
The role of S1P signaling in the regulation of the interaction between tumor and the immune system has been described in previous paragraphs; however, S1P can also modulate the interaction between other cell types present in the TME (Fig. 7.5). In S1PR2 knockout mice, the lack of this receptor on endothelial, vascular smooth muscle, and DC11b-positive bone marrow cells resulted in accelerated angiogenesis and growth of Lewis lung carcinoma and B16 melanoma cells [186]. The opposite effect was observed for S1PR1, whose downregulation in endothelial cells resulted in inhibited endothelial cell migration in vitro and the growth of neovessels into subcutaneous implants of Matrigel in vivo, thus leading to dramatic suppression of tumor growth [106]. In a model of melanoma, inhibition of SphK1 activity in dermal fibroblast enhanced tumor growth, whereas factors released from Sphk1 expressing melanoma cells were necessary for fibroblast differentiation into myofibroblasts [67, 187]. Moreover, myofibroblasts were found to release S1P and metalloproteinases that additionally increased melanoma growth and metastasis [67, 187]. Similarly, SphK1 expressed in the tumor stroma of serous ovarian cancer was required for the differentiation and the tumor-promoting function of cancer-associated fibroblasts through activation of TGF-β-signaling pathways via transactivation of S1PR2 and S1PR3 [188]. The importance of stromal SphK1 in tumorigenesis was confirmed in vivo in SphK1 knockout mice, where reduced tumor growth and decreased metastasis were observed [188]. S1P also mediate interactions in the pancreas between tumor and stromal cells where pancreatic cancer cell-derived S1P activates pancreatic stellate cells to release paracrine factors, including matrix metalloproteinase-9 (MMP9), which in turn promotes tumor cell migration and invasion, both in vitro and in vivo [189]. Interestingly, it has been proposed that communication between the host organism and cancer cells in a lung cancer model is transduced by S1P generated systemically rather than via tumor-derived S1P, and that lung colonization and cancer metastasis requires S1PR2 activation [190]. Recently, some additional mechanism has also been described where stromal–cancer interaction has been facilitating through microvesicles [191]. It has been found that S1PR2 can be shed from breast cancer cells in exosomes present in conditioned medium. Moreover, when combining with fibroblast, S1PR2 was proteolyzed to produce a constitutively active form which promoted proliferation of these cells through activation of the ERK1/2 pathway [191]. S1PR2 was also found to be important in the regulation of epithelial defense against cancer (EDAC), a process in which epithelial cells eliminate neighboring cancer cells [192]. Altogether, these results strongly support the hypothesis that S1P in the TME may regulate the communication between cancerous and stromal cells to enhance tumor development (Fig. 7.5).
7.9 S1P and Hypoxia
Hypoxia is a non-physiological low level of oxygen in a tissue, and this phenomenon is observed in a majority of malignant tumors. It is the result of intensive proliferation and expansion of tumor tissue in which oxygen demand is surpassed by oxygen supply [193]. Decreased oxygenation may lead to either cancer cell death or cancer cell survival, and the type of response partially depends on the time of exposure to hypoxia [193]. Hypoxia induces several intracellular signaling pathways, including the hypoxia-inducible factor (HIF) pathway. The SphK1 promoter has two hypoxia-inducible factor-responsive elements (HREs) and both hypoxic-inducible factors HIF1α and HIF2α have been shown to regulate transcription of SphK1 [194, 195] (Fig. 7.6). Interestingly, in glioma cells, HIF1α and HIF2α had the opposite effect on SphK1 expression; while downregulation of HIF2α decreased expression of SphK1 and S1P levels, silencing of HIF1α increased SphK1 synthesis [196]. At the same time, both SphK1 and SphK2 were found to be necessary to stabilize HIF1α in normal and malignant cells [197, 198] and SphK1 was also found to control HIF1α expression through a phospholipase D-driven mechanism [196]. In lung cancer, hypoxia was found to enhance SphK2 activity and lead to sphingosine 1-phosphate-mediated chemoresistance through an autocrine/paracrine mechanism that includes activation of S1PR1 and S1PR3 in cancer cells [199]. Hypoxia-induced SphK1 also promotes endothelial cell migration in [195] and increases S1P production and release from glioma cells [200]. Additionally, conditioned medium from hypoxia-treated tumor cells resulted in neoangiogenesis in human umbilical vein endothelial cells in a S1PR-dependent manner thus providing evidence of a link between S1P production as a potent angiogenic agent and the hypoxic phenotype observed in many tumors [200]. S1P was also found to be involved in the activation of HIF1α in macrophages [201], and migration of endothelial cells [202]. Taken together, presented results indicate that regulation of S1P signaling in response to hypoxic conditions could be a potential therapeutic target leading to decreased angiogenesis in growing tumors.

Role of sphingosine-1-phosphate (S1P) in hypoxia. At the molecular level (lower panel), hypoxia induces expression of hypoxia-inducible factor 1α (HIF1α) that regulates expression of sphingosine kinase 1 (SphK1). At the same time, SphK1 stabilizes HIF1α and controls its expression through a phospholipase D-driven mechanism. At the cellular level (upper panel), S1P released from cancer cells promotes endothelial cell migration and subsequent angiogenesis. S1P was also found to induce TAM/M2 macrophage polarization and is involved in the activation of HIF1α in macrophages. Moreover, stabilization of HIF1α in macrophages induces vascular endothelial growth factor (VEGF) release that additionally stimulates angiogenesis
7.10 S1P-Targeting Anticancer Therapies
Several strategies have been applied to the inhibition of S1P signaling in cancers targeting either metabolizing enzymes (mainly SphK1 and SphK2), specific S1P receptors, or S1P itself. The majority of SphK-targeting inhibitors either, rapidly and reversibly, inhibit catalytic activity [203] of SphK or induce ubiquitin-proteasomal degradation of SphK [204, 205], which results in a significant reduction in SphK levels in cancer cells. Unfortunately, some of them were found to be not isoform-specific, inhibit enzymes other than SphK (e.g., protein kinase C and ceramide kinase) [206, 207] or, despite showing efficacy in cancer models [208,209,210,211,212], were characterized with high toxicity [209]. Interestingly, some natural products like B-5354c [213], S-15183a, and S-15183b [214] have been shown to inhibit SphK in vitro and were found to reduce tumor growth in vivo [120]. Nevertheless, the efficacy and toxicity of three compounds that target SphK have been or are being assessed in clinical trials. Safingol (L-threo-dihydrosphingosine), which decreases the activity of SphK1, but also acts on protein kinase C [215], was shown to effectively downregulate the levels of S1P. However, although reversible, dose-dependent hepatic toxicity was observed. Currently, Safingol is being tested in patients with relapsed malignancies. Good tolerance and effectiveness in decreasing S1P levels were also shown for the selective SphK2 inhibitor ABC294640 (YELIVA) in Phase I studies in patients with advanced solid tumors [216]. Now, YELIVA is being evaluated in Phase II studies for the treatment of cholangiocarcinoma and hepatocellular carcinoma. A third compound, phenoxodiol, which was shown to reduce the activation of SphK1 [217] was assessed in clinical trials for the treatment of ovarian and prostate cancers [218,219,220]. However, the effect of phenoxodiol on SphK is indirect, and it also downregulates antiapoptotic proteins, induces AKT downregulation, and inhibits topoisomerase II.
Multiple S1PR-selective agents are available on the market [135], and although some of them were tested in animals models, none of them were or are being evaluated in clinical trials as anticancer treatments. One of the explanations might be the heterogeneous pattern of S1PR expression in cancer, which can differ not only between the same types of cancer from different patients but also between cells within the same tumor, thus limiting the efficacy of S1PR treatments.
Another approach for inhibition of S1P-related signaling includes the development of monoclonal antibodies (mAbs) that bind and neutralize S1P from blood and other compartments. Sphingomab (LT1009), an S1P-specific antibody, was found to reduce tumor progression, metastasis and, in some cases, eliminated tumors in mouse xenograft and allograft models [221]. This effect was attributed to its anti-angiogenic properties since in vitro studies indicated that anti-S1P mAbs blocked endothelial cell migration and resulting capillary formation and in vivo observation indicated a reduction in tumor blood flow [221]. On the other hand, in in vivo prostate, Sphingomab blocked the activity of HIF-1α in cancer exposed to hypoxia and modified vessel architecture, thus increasing intratumoral blood perfusion [222]. This transient vascular normalization of tumor vessels sensitized it to chemotherapeutic treatment leading to decreased tumor growth and metastasis [222]. Two monoclonal S1P-specific antibodies, LT1002 and Sonepcizumab (Asonep), a humanized form of sphingomab, were shown to have high specificity for S1P but not to other structurally related lipids [223]. Sonepcizumab has recently completed Phase I clinical trials for the treatment of solid tumors, but results are not yet available. However, results from Phase II clinical study of Sonepcizumab in patients with metastatic renal cell carcinoma showed encouraging overall survival and favorable safety profile suggesting further investigation of this agent in combination with VEGF-directed agents or checkpoint inhibitors [224].
Binding and sequestering of S1P from the environment can be also obtained using Spiegelmers (Spiegel = German word for mirror) which are biostable oligonucleotides made from nonnatural mirror-image l-nucleotides that adopt complex three-dimensional structures and bind targets in a fashion comparable to antibodies [225]. NOX-S93, a high-affinity inhibitor of S1P, was shown to reduce angiogenesis in in vitro assay [225]. Moreover, in in vivo experiments, administration of NOX-S93 decreased S1P-induced spread of rhabdomyosarcoma cells [46].
7.11 Conclusions
There is no doubt that S1P plays an essential role in the regulation of the TME and modulates interactions between its components. S1P released from tumor cells allows them to inhibit anticancer immune response, increase angiogenesis, and adapt to hypoxic conditions. Cells stimulated by S1P within the TME can, in turn, secrete growth factors and cytokines that orchestrate cancer progression and chemoresistance. Thus, S1P signaling in the TME should be taken into account when designing novel therapeutic strategies for cancer patients.
References
Pyne NJ, El Buri A, Adams DR, Pyne S (2018) Sphingosine 1-phosphate and cancer. Adv Biol Regul 68:97–106
Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R et al (2005) SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem 280:37118–37129
Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem 278:46832–46839
Johnson KR, Becker KP, Facchinetti MM, Hannun YA, Obeid LM (2002) PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane. Extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate (PMA). J Biol Chem 277:35257–35262
Paugh BS, Paugh SW, Bryan L, Kapitonov D, Wilczynska KM, Gopalan SM et al (2008) EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells. FASEB J 22:455–465
Pyne S, Lee SC, Long J, Pyne NJ (2009) Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell Signal 21:14–21
Ding G, Sonoda H, Yu H, Kajimoto T, Goparaju SK, Jahangeer S et al (2007) Protein kinase D-mediated phosphorylation and nuclear export of sphingosine kinase 2. J Biol Chem 282:27493–27502
Wallington-Beddoe CT, Powell JA, Tong D, Pitson SM, Bradstock KF, Bendall LJ (2014) Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing MYC expression. Cancer Res 74:2803–2815
Li J, Guan HY, Gong LY, Song LB, Zhang N, Wu J et al (2008) Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin Cancer Res 14:6996–7003
Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S et al (2008) Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat 112:41–52
Watson C, Long JS, Orange C, Tannahill CL, Mallon E, McGlynn LM et al (2010) High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am J Pathol 177:2205–2215
Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J et al (2009) Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J 23:405–414
Long J, Xie Y, Yin J, Lu W, Fang S (2016) SphK1 promotes tumor cell migration and invasion in colorectal cancer. Tumour Biol 37:6831–6836
Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ et al (2009) Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res 15:1393–1399
Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW (2005) Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol 64:695–705
Abuhusain HJ, Matin A, Qiao Q, Shen H, Kain N, Day BW et al (2013) A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J Biol Chem 288:37355–37364
Johnson KR, Johnson KY, Crellin HG, Ogretmen B, Boylan AM, Harley RA et al (2005) Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J Histochem Cytochem 53:1159–1166
Bayerl MG, Bruggeman RD, Conroy EJ, Hengst JA, King TS, Jimenez M et al (2008) Sphingosine kinase 1 protein and mRNA are overexpressed in non-Hodgkin lymphomas and are attractive targets for novel pharmacological interventions. Leuk Lymphoma 49:948–954
Malavaud B, Pchejetski D, Mazerolles C, de Paiva GR, Calvet C, Doumerc N et al (2010) Sphingosine kinase-1 activity and expression in human prostate cancer resection specimens. Eur J Cancer 46:3417–3424
Guan H, Liu L, Cai J, Liu J, Ye C, Li M et al (2011) Sphingosine kinase 1 is overexpressed and promotes proliferation in human thyroid cancer. Mol Endocrinol 25:1858–1866
Qiu W, Yang Z, Fan Y, Zheng Q (2016) MicroRNA-613 inhibits cell growth, migration and invasion of papillary thyroid carcinoma by regulating SphK2. Oncotarget 7:39907–39915
French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL et al (2003) Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res 63:5962–5969
Li J, Wu H, Li W, Yin L, Guo S, Xu X et al (2016) Downregulated miR-506 expression facilitates pancreatic cancer progression and chemoresistance via SPHK1/Akt/NF-kappaB signaling. Oncogene 35:5501–5514
Uranbileg B, Ikeda H, Kurano M, Enooku K, Sato M, Saigusa D et al (2016) Increased mRNA levels of Sphingosine kinases and S1P Lyase and reduced levels of S1P were observed in hepatocellular carcinoma in association with poorer differentiation and earlier recurrence. PLoS One 11:e0149462
Kim HS, Yoon G, Ryu JY, Cho YJ, Choi JJ, Lee YY et al (2015) Sphingosine kinase 1 is a reliable prognostic factor and a novel therapeutic target for uterine cervical cancer. Oncotarget 6:26746–26756
Nunes J, Naymark M, Sauer L, Muhammad A, Keun H, Sturge J et al (2012) Circulating sphingosine-1-phosphate and erythrocyte sphingosine kinase-1 activity as novel biomarkers for early prostate cancer detection. Br J Cancer 106:909–915
Clair T, Aoki J, Koh E, Bandle RW, Nam SW, Ptaszynska MM et al (2003) Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res 63:5446–5453
Liliom K, Sun G, Bunemann M, Virag T, Nusser N, Baker DL et al (2001) Sphingosylphosphocholine is a naturally occurring lipid mediator in blood plasma: a possible role in regulating cardiac function via sphingolipid receptors. Biochem J 355:189–197
El-Najjar N, Orso E, Wallner S, Liebisch G, Schmitz G (2015) Increased levels of Sphingosylphosphorylcholine (SPC) in plasma of metabolic syndrome patients. PLoS One 10:e0140683
Tanaka M, Okudaira S, Kishi Y, Ohkawa R, Iseki S, Ota M et al (2006) Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem 281:25822–25830
Gao XY, Li L, Wang XH, Wen XZ, Ji K, Ye L et al (2015) Inhibition of sphingosine-1-phosphate phosphatase 1 promotes cancer cells migration in gastric cancer: clinical implications. Oncol Rep 34:1977–1987
Serra M, Saba JD (2010) Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv Enzym Regul 50:349–362
Oskouian B, Sooriyakumaran P, Borowsky AD, Crans A, Dillard-Telm L, Tam YY et al (2006) Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc Natl Acad Sci U S A 103:17384–17389
Degagne E, Pandurangan A, Bandhuvula P, Kumar A, Eltanawy A, Zhang M et al (2014) Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest 124:5368–5384
Brizuela L, Ader I, Mazerolles C, Bocquet M, Malavaud B, Cuvillier O (2012) First evidence of sphingosine 1-phosphate lyase protein expression and activity downregulation in human neoplasm: implication for resistance to therapeutics in prostate cancer. Mol Cancer Ther 11:1841–1851
Hammad SM, Al Gadban MM, Semler AJ, Klein RL (2012) Sphingosine 1-phosphate distribution in human plasma: associations with lipid profiles. J Lipids 2012:180705
Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M et al (2011) Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A 108:9613–9618
Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A et al (2000) Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J 352(Pt 3):809–815
Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Yamada A et al (2012) Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res 72:726–735
Nagahashi M, Yamada A, Miyazaki H, Allegood JC, Tsuchida J, Aoyagi T et al (2016) Interstitial fluid sphingosine-1-phosphate in murine mammary gland and cancer and human breast tissue and cancer determined by novel methods. J Mammary Gland Biol Neoplasia 21:9–17
Nagahashi M, Tsuchida J, Moro K, Hasegawa M, Tatsuda K, Woelfel IA et al (2016) High levels of sphingolipids in human breast cancer. J Surg Res 204:435–444
Xu Y, Xiao YJ, Baudhuin LM, Schwartz BM (2001) The role and clinical applications of bioactive lysolipids in ovarian cancer. J Soc Gynecol Investig 8:1–13
Wang D, Zhao Z, Caperell-Grant A, Yang G, Mok SC, Liu J et al (2008) S1P differentially regulates migration of human ovarian cancer and human ovarian surface epithelial cells. Mol Cancer Ther 7:1993–2002
Sutphen R, Xu Y, Wilbanks GD, Fiorica J, Grendys EC Jr, LaPolla JP et al (2004) Lysophospholipids are potential biomarkers of ovarian cancer. Cancer Epidemiol Biomark Prev 13:1185–1191
Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R et al (2008) Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J 22:2629–2638
Schneider G, Bryndza E, Abdel-Latif A, Ratajczak J, Maj M, Tarnowski M et al (2013) Bioactive lipids S1P and C1P are prometastatic factors in human rhabdomyosarcoma, and their tissue levels increase in response to radio/chemotherapy. Mol Cancer Res 11:793–807
Weigert A, Tzieply N, von Knethen A, Johann AM, Schmidt H, Geisslinger G et al (2007) Tumor cell apoptosis polarizes macrophages role of sphingosine-1-phosphate. Mol Biol Cell 18:3810–3819
Ley S, Weigert A, Weichand B, Henke N, Mille-Baker B, Janssen RA et al (2013) The role of TRKA signaling in IL-10 production by apoptotic tumor cell-activated macrophages. Oncogene 32:631–640
Venkataraman K, Thangada S, Michaud J, Oo ML, Ai Y, Lee YM et al (2006) Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem J 397:461–471
Ancellin N, Colmont C, Su J, Li Q, Mittereder N, Chae SS et al (2002) Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J Biol Chem 277:6667–6675
Hammad SM, Taha TA, Nareika A, Johnson KR, Lopes-Virella MF, Obeid LM (2006) Oxidized LDL immune complexes induce release of sphingosine kinase in human U937 monocytic cells. Prostaglandins Other Lipid Mediat 79:126–140
Osborne N, Brand-Arzamendi K, Ober EA, Jin SW, Verkade H, Holtzman NG et al (2008) The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr Biol 18:1882–1888
Mendoza A, Breart B, Ramos-Perez WD, Pitt LA, Gobert M, Sunkara M et al (2012) The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep 2:1104–1110
Takabe K, Spiegel S (2014) Export of sphingosine-1-phosphate and cancer progression. J Lipid Res 55:1839–1846
Nagahashi M, Kim EY, Yamada A, Ramachandran S, Allegood JC, Hait NC et al (2013) Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels, and the lymphatic network. FASEB J 27:1001–1011
Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T et al (2012) The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest 122:1416–1426
Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S (2006) Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci U S A 103:16394–16399
Kobayashi N, Kobayashi N, Yamaguchi A, Nishi T (2009) Characterization of the ATP-dependent sphingosine 1-phosphate transporter in rat erythrocytes. J Biol Chem 284:21192–21200
Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, Nagahashi M et al (2010) Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem 285:10477–10486
Sato K, Malchinkhuu E, Horiuchi Y, Mogi C, Tomura H, Tosaka M et al (2007) Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J Neurochem 103:2610–2619
Kobayashi N, Nishi T, Hirata T, Kihara A, Sano T, Igarashi Y et al (2006) Sphingosine 1-phosphate is released from the cytosol of rat platelets in a carrier-mediated manner. J Lipid Res 47:614–621
Bradley E, Dasgupta S, Jiang X, Zhao X, Zhu G, He Q et al (2014) Critical role of Spns2, a sphingosine-1-phosphate transporter, in lung cancer cell survival and migration. PLoS One 9:e110119
Ghosh TK, Bian J, Gill DL (1990) Intracellular calcium release mediated by sphingosine derivatives generated in cells. Science 248:1653–1656
Himmel HM, Meyer zu Heringdorf D, Windorfer B, van Koppen CJ, Ravens U, Jakobs KH (1998) Guanine nucleotide-sensitive inhibition of L-type Ca2+ current by lysosphingolipids in RINm5F insulinoma cells. Mol Pharmacol 53:862–869
Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK et al (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325:1254–1257
Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC et al (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J 25:600–612
Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY et al (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465:1084–1088
Park K, Ikushiro H, Seo HS, Shin KO, Kim YI, Kim JY et al (2016) ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex. Proc Natl Acad Sci U S A 113:E1334–E1342
Blom T, Bergelin N, Meinander A, Lof C, Slotte JP, Eriksson JE et al (2010) An autocrine sphingosine-1-phosphate signaling loop enhances NF-kappaB-activation and survival. BMC Cell Biol 11:45
Siehler S, Wang Y, Fan X, Windh RT, Manning DR (2001) Sphingosine 1-phosphate activates nuclear factor-kappa B through Edg receptors. Activation through Edg-3 and Edg-5, but not Edg-1, in human embryonic kidney 293 cells. J Biol Chem 276:48733–48739
Harikumar KB, Yester JW, Surace MJ, Oyeniran C, Price MM, Huang WC et al (2014) K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat Immunol 15:231–238
Parham KA, Zebol JR, Tooley KL, Sun WY, Moldenhauer LM, Cockshell MP et al (2015) Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-gamma that regulates neoangiogenesis. FASEB J 29:3638–3653
Blaho VA, Hla T (2014) An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res 55:1596–1608
Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH (2010) International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev 62:579–587
Huang YL, Huang WP, Lee H (2011) Roles of sphingosine 1-phosphate on tumorigenesis. World J Biol Chem 2:25–34
Novgorodov AS, El-Alwani M, Bielawski J, Obeid LM, Gudz TI (2007) Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J 21:1503–1514
Im DS, Heise CE, Ancellin N, O’Dowd BF, Shei GJ, Heavens RP et al (2000) Characterization of a novel sphingosine 1-phosphate receptor, Edg-8. J Biol Chem 275:14281–14286
Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H et al (2003) Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem 278:40330–40336
Xia J, Wu Z, Yu C, He W, Zheng H, He Y et al (2012) miR-124 inhibits cell proliferation in gastric cancer through down-regulation of SPHK1. J Pathol 227:470–480
Ju T, Gao D, Fang ZY (2016) Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem Biophys Res Commun 470:728–734
Don AS, Rosen H (2009) A lipid binding domain in sphingosine kinase 2. Biochem Biophys Res Commun 380:87–92
Xun C, Chen MB, Qi L, Tie-Ning Z, Peng X, Ning L et al (2015) Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J Exp Clin Cancer Res 34:94
Schrecengost RS, Keller SN, Schiewer MJ, Knudsen KE, Smith CD (2015) Downregulation of critical oncogenes by the selective SK2 inhibitor ABC294640 hinders prostate cancer progression. Mol Cancer Res 13:1591–1601
Rex K, Jeffries S, Brown ML, Carlson T, Coxon A, Fajardo F et al (2013) Sphingosine kinase activity is not required for tumor cell viability. PLoS One 8:e68328
Chang CL, Ho MC, Lee PH, Hsu CY, Huang WP, Lee H (2009) S1P(5) is required for sphingosine 1-phosphate-induced autophagy in human prostate cancer PC-3 cells. Am J Physiol Cell Physiol 297:C451–C458
Rodgers A, Mormeneo D, Long JS, Delgado A, Pyne NJ, Pyne S (2009) Sphingosine 1-phosphate regulation of extracellular signal-regulated kinase-1/2 in embryonic stem cells. Stem Cells Dev 18:1319–1330
Cheng JC, Wang EY, Yi Y, Thakur A, Tsai SH, Hoodless PA (2018) S1P stimulates proliferation by upregulating CTGF expression through S1PR2-mediated YAP activation. Mol Cancer Res 16:1543–1555
Li MH, Sanchez T, Pappalardo A, Lynch KR, Hla T, Ferrer F (2008) Induction of antiproliferative connective tissue growth factor expression in Wilms’ tumor cells by sphingosine-1-phosphate receptor 2. Mol Cancer Res 6:1649–1656
Lei FJ, Cheng BH, Liao PY, Wang HC, Chang WC, Lai HC et al (2018) Survival benefit of sphingosin-1-phosphate and receptors expressions in breast cancer patients. Cancer Med 7:3743–3754
Van Brocklyn J, Letterle C, Snyder P, Prior T (2002) Sphingosine-1-phosphate stimulates human glioma cell proliferation through Gi-coupled receptors: role of ERK MAP kinase and phosphatidylinositol 3-kinase beta. Cancer Lett 181:195–204
Hu WM, Li L, Jing BQ, Zhao YS, Wang CL, Feng L et al (2010) Effect of S1P5 on proliferation and migration of human esophageal cancer cells. World J Gastroenterol 16:1859–1866
Chumanevich A, Wedman P, Oskeritzian CA (2016) Sphingosine-1-phosphate/sphingosine-1-phosphate receptor 2 axis can promote mouse and human primary mast cell Angiogenic potential through upregulation of vascular endothelial growth factor-a and matrix metalloproteinase-2. Mediat Inflamm 2016:1503206
Abdelbaset-Ismail A, Cymer M, Borkowska-Rzeszotek S, Brzezniakiewicz-Janus K, Rameshwar P, Kakar SS et al (2019) Bioactive phospholipids enhance migration and adhesion of human leukemic cells by inhibiting heme oxygenase 1 (HO-1) and inducible nitric oxygenase synthase (iNOS) in a p38 MAPK-dependent manner. Stem Cell Rev 15:139–154
Schneider G, Sellers ZP, Bujko K, Kakar SS, Kucia M, Ratajczak MZ (2017) Novel pleiotropic effects of bioactive phospholipids in human lung cancer metastasis. Oncotarget 8:58247–58263
Malchinkhuu E, Sato K, Maehama T, Mogi C, Tomura H, Ishiuchi S et al (2008) S1P(2) receptors mediate inhibition of glioma cell migration through rho signaling pathways independent of PTEN. Biochem Biophys Res Commun 366:963–968
Park KS, Kim MK, Lee HY, Kim SD, Lee SY, Kim JM et al (2007) S1P stimulates chemotactic migration and invasion in OVCAR3 ovarian cancer cells. Biochem Biophys Res Commun 356:239–244
Fisher KE, Pop A, Koh W, Anthis NJ, Saunders WB, Davis GE (2006) Tumor cell invasion of collagen matrices requires coordinate lipid agonist-induced G-protein and membrane-type matrix metalloproteinase-1-dependent signaling. Mol Cancer 5:69
Nyalendo C, Michaud M, Beaulieu E, Roghi C, Murphy G, Gingras D et al (2007) Src-dependent phosphorylation of membrane type I matrix metalloproteinase on cytoplasmic tyrosine 573: role in endothelial and tumor cell migration. J Biol Chem 282:15690–15699
Young N, Van Brocklyn JR (2007) Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp Cell Res 313:1615–1627
Zhang H, Wang Q, Zhao Q, Di W (2013) MiR-124 inhibits the migration and invasion of ovarian cancer cells by targeting SphK1. J Ovarian Res 6:84
Gao P, Smith CD (2011) Ablation of sphingosine kinase-2 inhibits tumor cell proliferation and migration. Mol Cancer Res 9:1509–1519
Wu WT, Chen CN, Lin CI, Chen JH, Lee H (2005) Lysophospholipids enhance matrix metalloproteinase-2 expression in human endothelial cells. Endocrinology 146:3387–3400
Moon A, Kim MS, Kim TG, Kim SH, Kim HE, Chen YQ et al (2000) H-ras, but not N-ras, induces an invasive phenotype in human breast epithelial cells: a role for MMP-2 in the H-ras-induced invasive phenotype. Int J Cancer 85:176–181
Wang F, Van Brocklyn JR, Hobson JP, Movafagh S, Zukowska-Grojec Z, Milstien S et al (1999) Sphingosine 1-phosphate stimulates cell migration through a G(i)-coupled cell surface receptor. Potential involvement in angiogenesis. J Biol Chem 274:35343–35350
Boguslawski G, Grogg JR, Welch Z, Ciechanowicz S, Sliva D, Kovala AT et al (2002) Migration of vascular smooth muscle cells induced by sphingosine 1-phosphate and related lipids: potential role in the angiogenic response. Exp Cell Res 274:264–274
Chae SS, Paik JH, Furneaux H, Hla T (2004) Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J Clin Invest 114:1082–1089
Shu X, Wu W, Mosteller RD, Broek D (2002) Sphingosine kinase mediates vascular endothelial growth factor-induced activation of ras and mitogen-activated protein kinases. Mol Cell Biol 22:7758–7768
Boucharaba A, Guillet B, Menaa F, Hneino M, van Wijnen AJ, Clezardin P et al (2009) Bioactive lipids lysophosphatidic acid and sphingosine 1-phosphate mediate breast cancer cell biological functions through distinct mechanisms. Oncol Res 18:173–184
Lu Z, Zhang W, Gao S, Jiang Q, Xiao Z, Ye L et al (2015) MiR-506 suppresses liver cancer angiogenesis through targeting sphingosine kinase 1 (SPHK1) mRNA. Biochem Biophys Res Commun 468:8–13
Guillermet-Guibert J, Davenne L, Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C et al (2009) Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol Cancer Ther 8:809–820
Baran Y, Salas A, Senkal CE, Gunduz U, Bielawski J, Obeid LM et al (2007) Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J Biol Chem 282:10922–10934
Sobue S, Nemoto S, Murakami M, Ito H, Kimura A, Gao S et al (2008) Implications of sphingosine kinase 1 expression level for the cellular sphingolipid rheostat: relevance as a marker for daunorubicin sensitivity of leukemia cells. Int J Hematol 87:266–275
Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V et al (2005) Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res 65:11667–11675
Gao H, Deng L (2014) Sphingosine kinase-1 activation causes acquired resistance against Sunitinib in renal cell carcinoma cells. Cell Biochem Biophys 68:419–425
Matula K, Collie-Duguid E, Murray G, Parikh K, Grabsch H, Tan P et al (2015) Regulation of cellular sphingosine-1-phosphate by sphingosine kinase 1 and sphingosine-1-phosphate lyase determines chemotherapy resistance in gastroesophageal cancer. BMC Cancer 15:762
Okamoto H, Takuwa N, Yokomizo T, Sugimoto N, Sakurada S, Shigematsu H et al (2000) Inhibitory regulation of Rac activation, membrane ruffling, and cell migration by the G protein-coupled sphingosine-1-phosphate receptor EDG5 but not EDG1 or EDG3. Mol Cell Biol 20:9247–9261
Liu W, Ning J, Li C, Hu J, Meng Q, Lu H et al (2016) Overexpression of Sphk2 is associated with gefitinib resistance in non-small cell lung cancer. Tumour Biol 37:6331–6336
Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S et al (2011) Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther 11:678–689
Min J, Stegner AL, Alexander H, Alexander S (2004) Overexpression of sphingosine-1-phosphate lyase or inhibition of sphingosine kinase in Dictyostelium discoideum results in a selective increase in sensitivity to platinum-based chemotherapy drugs. Eukaryot Cell 3:795–805
Pchejetski D, Doumerc N, Golzio M, Naymark M, Teissie J, Kohama T et al (2008) Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Mol Cancer Ther 7:1836–1845
Lifshitz V, Priceman SJ, Li W, Cherryholmes G, Lee H, Makovski-Silverstein A et al (2017) Sphingosine-1-phosphate receptor-1 promotes environment-mediated and acquired chemoresistance. Mol Cancer Ther 16:2516–2527
Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M (2013) Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 229:176–185
Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D et al (2009) Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A 106:14978–14983
Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21:309–322
Weichand B, Weis N, Weigert A, Grossmann N, Levkau B, Brune B (2013) Apoptotic cells enhance sphingosine-1-phosphate receptor 1 dependent macrophage migration. Eur J Immunol 43:3306–3313
Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, Brune B (2006) Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 108:1635–1642
He H, Zhang S, Tighe S, Son J, Tseng SC (2013) Immobilized heavy chain-hyaluronic acid polarizes lipopolysaccharide-activated macrophages toward M2 phenotype. J Biol Chem 288:25792–25803
Park SJ, Lee KP, Kang S, Lee J, Sato K, Chung HY et al (2014) Sphingosine 1-phosphate induced anti-atherogenic and atheroprotective M2 macrophage polarization through IL-4. Cell Signal 26:2249–2258
Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC (2008) Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res 102:950–958
Brecht K, Weigert A, Hu J, Popp R, Fisslthaler B, Korff T et al (2011) Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J 25:2408–2417
Eken A, Duhen R, Singh AK, Fry M, Buckner JH, Kita M et al (2017) S1P1 deletion differentially affects TH17 and regulatory T cells. Sci Rep 7:12905
Priceman SJ, Shen S, Wang L, Deng J, Yue C, Kujawski M et al (2014) S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. Cell Rep 6:992–999
Wolf AM, Eller K, Zeiser R, Durr C, Gerlach UV, Sixt M et al (2009) The sphingosine 1-phosphate receptor agonist FTY720 potently inhibits regulatory T cell proliferation in vitro and in vivo. J Immunol 183:3751–3760
Liu YN, Zhang H, Zhang L, Cai TT, Huang DJ, He J et al (2019) Sphingosine 1 phosphate receptor-1 (S1P1) promotes tumor-associated regulatory T cell expansion: leading to poor survival in bladder cancer. Cell Death Dis 10:50
Park SJ, Im DS (2017) Sphingosine 1-phosphate receptor modulators and drug discovery. Biomol Ther (Seoul) 25:80–90
Lagadari M, Lehmann K, Ziemer M, Truta-Feles K, Berod L, Idzko M et al (2009) Sphingosine-1-phosphate inhibits the cytotoxic activity of NK cells via Gs protein-mediated signalling. Int J Oncol 34:287–294
Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L et al (2007) Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol 8:1337–1344
Rolin J, Sand KL, Knudsen E, Maghazachi AA (2010) FTY720 and SEW2871 reverse the inhibitory effect of S1P on natural killer cell mediated lysis of K562 tumor cells and dendritic cells but not on cytokine release. Cancer Immunol Immunother 59:575–586
Maeda Y, Matsuyuki H, Shimano K, Kataoka H, Sugahara K, Chiba K (2007) Migration of CD4 T cells and dendritic cells toward sphingosine 1-phosphate (S1P) is mediated by different receptor subtypes: S1P regulates the functions of murine mature dendritic cells via S1P receptor type 3. J Immunol 178:3437–3446
Flori M, Schmid CA, Sumrall ET, Tzankov A, Law CW, Robinson MD et al (2016) The hematopoietic oncoprotein FOXP1 promotes tumor cell survival in diffuse large B-cell lymphoma by repressing S1PR2 signaling. Blood 127:1438–1448
Sic H, Kraus H, Madl J, Flittner KA, von Munchow AL, Pieper K et al (2014) Sphingosine-1-phosphate receptors control B-cell migration through signaling components associated with primary immunodeficiencies, chronic lymphocytic leukemia, and multiple sclerosis. J Allergy Clin Immunol 134:420–428
Till KJ, Pettitt AR, Slupsky JR (2015) Expression of functional sphingosine-1 phosphate receptor-1 is reduced by B cell receptor signaling and increased by inhibition of PI3 kinase delta but not SYK or BTK in chronic lymphocytic leukemia cells. J Immunol 194:2439–2446
Liu Y, Deng J, Wang L, Lee H, Armstrong B, Scuto A et al (2012) S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood 120:1458–1465
Capitani N, Patrussi L, Trentin L, Lucherini OM, Cannizzaro E, Migliaccio E et al (2012) S1P1 expression is controlled by the pro-oxidant activity of p66Shc and is impaired in B-CLL patients with unfavorable prognosis. Blood 120:4391–4399
Jin Y, Knudsen E, Wang L, Bryceson Y, Damaj B, Gessani S et al (2003) Sphingosine 1-phosphate is a novel inhibitor of T-cell proliferation. Blood 101:4909–4915
Krystel-Whittemore M, Dileepan KN, Wood JG (2015) Mast cell: a multi-functional master cell. Front Immunol 6:620
Oskeritzian CA (2015) Mast cell plasticity and sphingosine-1-phosphate in immunity, inflammation and cancer. Mol Immunol 63:104–112
Price MM, Oskeritzian CA, Milstien S, Spiegel S (2008) Sphingosine-1-phosphate synthesis and functions in mast cells. Future Lipidol 3:665–674
Marichal T, Tsai M, Galli SJ (2013) Mast cells: potential positive and negative roles in tumor biology. Cancer Immunol Res 1:269–279
Johansson A, Rudolfsson S, Hammarsten P, Halin S, Pietras K, Jones J et al (2010) Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am J Pathol 177:1031–1041
Gulubova M, Vlaykova T (2009) Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. J Gastroenterol Hepatol 24:1265–1275
Strouch MJ, Cheon EC, Salabat MR, Krantz SB, Gounaris E, Melstrom LG et al (2010) Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res 16:2257–2265
Toth-Jakatics R, Jimi S, Takebayashi S, Kawamoto N (2000) Cutaneous malignant melanoma: correlation between neovascularization and peritumor accumulation of mast cells overexpressing vascular endothelial growth factor. Hum Pathol 31:955–960
Carlini MJ, Dalurzo MC, Lastiri JM, Smith DE, Vasallo BC, Puricelli LI et al (2010) Mast cell phenotypes and microvessels in non-small cell lung cancer and its prognostic significance. Hum Pathol 41:697–705
Elpek GO, Gelen T, Aksoy NH, Erdogan A, Dertsiz L, Demircan A et al (2001) The prognostic relevance of angiogenesis and mast cells in squamous cell carcinoma of the oesophagus. J Clin Pathol 54:940–944
Iamaroon A, Pongsiriwet S, Jittidecharaks S, Pattanaporn K, Prapayasatok S, Wanachantararak S (2003) Increase of mast cells and tumor angiogenesis in oral squamous cell carcinoma. J Oral Pathol Med 32:195–199
Rojas IG, Spencer ML, Martinez A, Maurelia MA, Rudolph MI (2005) Characterization of mast cell subpopulations in lip cancer. J Oral Pathol Med 34:268–273
Pittoni P, Tripodo C, Piconese S, Mauri G, Parenza M, Rigoni A et al (2011) Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res 71:5987–5997
Fleischmann A, Schlomm T, Kollermann J, Sekulic N, Huland H, Mirlacher M et al (2009) Immunological microenvironment in prostate cancer: high mast cell densities are associated with favorable tumor characteristics and good prognosis. Prostate 69:976–981
Nonomura N, Takayama H, Nishimura K, Oka D, Nakai Y, Shiba M et al (2007) Decreased number of mast cells infiltrating into needle biopsy specimens leads to a better prognosis of prostate cancer. Br J Cancer 97:952–956
Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J et al (2004) Transactivation of sphingosine-1-phosphate receptors by FcepsilonRI triggering is required for normal mast cell degranulation and chemotaxis. J Exp Med 199:959–970
Oskeritzian CA, Alvarez SE, Hait NC, Price MM, Milstien S, Spiegel S (2008) Distinct roles of sphingosine kinases 1 and 2 in human mast-cell functions. Blood 111:4193–4200
Fernandez-Pisonero I, Duenas AI, Barreiro O, Montero O, Sanchez-Madrid F, Garcia-Rodriguez C (2012) Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce inflammatory molecules and leukocyte adhesion in endothelial cells. J Immunol 189:5402–5410
Campos LS, Rodriguez YI, Leopoldino AM, Hait NC, Lopez Bergami P, Castro MG et al (2016) Filamin a expression negatively regulates sphingosine-1-phosphate-induced NF-kappaB activation in melanoma cells by inhibition of Akt signaling. Mol Cell Biol 36:320–329
Maceyka M, Alvarez SE, Milstien S, Spiegel S (2008) Filamin a links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Mol Cell Biol 28:5687–5697
Leonardi A, Ellinger-Ziegelbauer H, Franzoso G, Brown K, Siebenlist U (2000) Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. J Biol Chem 275:271–278
Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A et al (2010) STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med 16:1421–1428
Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA et al (2009) A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J 23:143–152
Pyne NJ, Ohotski J, Bittman R, Pyne S (2014) The role of sphingosine 1-phosphate in inflammation and cancer. Adv Biol Regul 54:121–129
Ding X, Chaiteerakij R, Moser CD, Shaleh H, Boakye J, Chen G et al (2016) Antitumor effect of the novel sphingosine kinase 2 inhibitor ABC294640 is enhanced by inhibition of autophagy and by sorafenib in human cholangiocarcinoma cells. Oncotarget 7:20080–20092
Alshaker H, Wang Q, Frampton AE, Krell J, Waxman J, Winkler M et al (2015) Sphingosine kinase 1 contributes to leptin-induced STAT3 phosphorylation through IL-6/gp130 transactivation in oestrogen receptor-negative breast cancer. Breast Cancer Res Treat 149:59–67
Pettus BJ, Bielawska A, Spiegel S, Roddy P, Hannun YA, Chalfant CE (2003) Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J Biol Chem 278:38206–38213
Li MH, Sanchez T, Milne GL, Morrow JD, Hla T, Ferrer F (2009) S1P/S1P2 signaling induces cyclooxygenase-2 expression in Wilms tumor. J Urol 181:1347–1352
Lai WQ, Melendez AJ, Leung BP (2010) Role of sphingosine kinase and sphingosine-1-phosphate in inflammatory arthritis. World J Biol Chem 1:321–326
Alvarez SE, Milstien S, Spiegel S (2007) Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab 18:300–307
Kulakowska A, Zendzian-Piotrowska M, Baranowski M, Kononczuk T, Drozdowski W, Gorski J et al (2010) Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci Lett 477:149–152
Ammit AJ, Hastie AT, Edsall LC, Hoffman RK, Amrani Y, Krymskaya VP et al (2001) Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J 15:1212–1214
Mechtcheriakova D, Wlachos A, Sobanov J, Kopp T, Reuschel R, Bornancin F et al (2007) Sphingosine 1-phosphate phosphatase 2 is induced during inflammatory responses. Cell Signal 19:748–760
Seo EY, Park GT, Lee KM, Kim JA, Lee JH, Yang JM (2006) Identification of the target genes of atopic dermatitis by real-time PCR. J Invest Dermatol 126:1187–1189
Baumer W, Rossbach K, Mischke R, Reines I, Langbein-Detsch I, Luth A et al (2011) Decreased concentration and enhanced metabolism of sphingosine-1-phosphate in lesional skin of dogs with atopic dermatitis: disturbed sphingosine-1-phosphate homeostasis in atopic dermatitis. J Invest Dermatol 131:266–268
Reines I, Kietzmann M, Mischke R, Tschernig T, Luth A, Kleuser B et al (2009) Topical application of sphingosine-1-phosphate and FTY720 attenuate allergic contact dermatitis reaction through inhibition of dendritic cell migration. J Invest Dermatol 129:1954–1962
Schaper K, Dickhaut J, Japtok L, Kietzmann M, Mischke R, Kleuser B et al (2013) Sphingosine-1-phosphate exhibits anti-proliferative and anti-inflammatory effects in mouse models of psoriasis. J Dermatol Sci 71:29–36
Japtok L, Schaper K, Baumer W, Radeke HH, Jeong SK, Kleuser B (2012) Sphingosine 1-phosphate modulates antigen capture by murine Langerhans cells via the S1P2 receptor subtype. PLoS One 7:e49427
Idzko M, Hammad H, van Nimwegen M, Kool M, Muller T, Soullie T et al (2006) Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest 116:2935–2944
Blaho VA, Galvani S, Engelbrecht E, Liu C, Swendeman SL, Kono M et al (2015) HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 523:342–346
Du W, Takuwa N, Yoshioka K, Okamoto Y, Gonda K, Sugihara K et al (2010) S1P(2), the G protein-coupled receptor for sphingosine-1-phosphate, negatively regulates tumor angiogenesis and tumor growth in vivo in mice. Cancer Res 70:772–781
Albinet V, Bats ML, Huwiler A, Rochaix P, Chevreau C, Segui B et al (2014) Dual role of sphingosine kinase-1 in promoting the differentiation of dermal fibroblasts and the dissemination of melanoma cells. Oncogene 33:3364–3373
Beach JA, Aspuria PJ, Cheon DJ, Lawrenson K, Agadjanian H, Walsh CS et al (2016) Sphingosine kinase 1 is required for TGF-beta mediated fibroblastto- myofibroblast differentiation in ovarian cancer. Oncotarget 7:4167–4182
Bi Y, Li J, Ji B, Kang N, Yang L, Simonetto DA et al (2014) Sphingosine-1-phosphate mediates a reciprocal signaling pathway between stellate cells and cancer cells that promotes pancreatic cancer growth. Am J Pathol 184:2791–2802
Ponnusamy S, Selvam SP, Mehrotra S, Kawamori T, Snider AJ, Obeid LM et al (2012) Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol Med 4:761–775
El Buri A, Adams DR, Smith D, Tate RJ, Mullin M, Pyne S et al (2018) The sphingosine 1-phosphate receptor 2 is shed in exosomes from breast cancer cells and is N-terminally processed to a short constitutively active form that promotes extracellular signal regulated kinase activation and DNA synthesis in fibroblasts. Oncotarget 9:29453–29467
Yamamoto S, Yako Y, Fujioka Y, Kajita M, Kameyama T, Kon S et al (2016) A role of the sphingosine-1-phosphate (S1P)-S1P receptor 2 pathway in epithelial defense against cancer (EDAC). Mol Biol Cell 27:491–499
Muz B, de la Puente P, Azab F, Azab AK (2015) The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 3:83–92
Ahmad M, Long JS, Pyne NJ, Pyne S (2006) The effect of hypoxia on lipid phosphate receptor and sphingosine kinase expression and mitogen-activated protein kinase signaling in human pulmonary smooth muscle cells. Prostaglandins Other Lipid Mediat 79:278–286
Schwalm S, Doll F, Romer I, Bubnova S, Pfeilschifter J, Huwiler A (2008) Sphingosine kinase-1 is a hypoxia-regulated gene that stimulates migration of human endothelial cells. Biochem Biophys Res Commun 368:1020–1025
Bouquerel P, Gstalder C, Muller D, Laurent J, Brizuela L, Sabbadini RA et al (2016) Essential role for SphK1/S1P signaling to regulate hypoxia-inducible factor 2alpha expression and activity in cancer. Oncogene 5:e209
Ader I, Brizuela L, Bouquerel P, Malavaud B, Cuvillier O (2008) Sphingosine kinase 1: a new modulator of hypoxia inducible factor 1alpha during hypoxia in human cancer cells. Cancer Res 68:8635–8642
Michaud MD, Robitaille GA, Gratton JP, Richard DE (2009) Sphingosine-1-phosphate: a novel nonhypoxic activator of hypoxia-inducible factor-1 in vascular cells. Arterioscler Thromb Vasc Biol 29:902–908
Schnitzer SE, Weigert A, Zhou J, Brune B (2009) Hypoxia enhances sphingosine kinase 2 activity and provokes sphingosine-1-phosphate-mediated chemoresistance in A549 lung cancer cells. Mol Cancer Res 7:393–401
Anelli V, Gault CR, Cheng AB, Obeid LM (2008) Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. J Biol Chem 283:3365–3375
Herr B, Zhou J, Werno C, Menrad H, Namgaladze D, Weigert A et al (2009) The supernatant of apoptotic cells causes transcriptional activation of hypoxia-inducible factor-1alpha in macrophages via sphingosine-1-phosphate and transforming growth factor-beta. Blood 114:2140–2148
Williams PA, Stilhano RS, To VP, Tran L, Wong K, Silva EA (2015) Hypoxia augments outgrowth endothelial cell (OEC) sprouting and directed migration in response to sphingosine-1-phosphate (S1P). PLoS One 10:e0123437
Baek DJ, MacRitchie N, Anthony NG, Mackay SP, Pyne S, Pyne NJ et al (2013) Structure-activity relationships and molecular modeling of sphingosine kinase inhibitors. J Med Chem 56:9310–9327
Loveridge C, Tonelli F, Leclercq T, Lim KG, Long JS, Berdyshev E et al (2010) The sphingosine kinase 1 inhibitor 2-(p-hydroxyanilino)-4-(p-chlorophenyl)thiazole induces proteasomal degradation of sphingosine kinase 1 in mammalian cells. J Biol Chem 285:38841–38852
Byun HS, Pyne S, Macritchie N, Pyne NJ, Bittman R (2013) Novel sphingosine-containing analogues selectively inhibit sphingosine kinase (SK) isozymes, induce SK1 proteasomal degradation and reduce DNA synthesis in human pulmonary arterial smooth muscle cells. Medchemcomm 4. https://doi.org/10.1039/C3MD00201B
Igarashi Y, Hakomori S, Toyokuni T, Dean B, Fujita S, Sugimoto M et al (1989) Effect of chemically well-defined sphingosine and its N-methyl derivatives on protein kinase C and src kinase activities. Biochemistry 28:6796–6800
Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S et al (2002) Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem 277:23294–23300
Endo K, Igarashi Y, Nisar M, Zhou QH, Hakomori S (1991) Cell membrane signaling as target in cancer therapy: inhibitory effect of N,N-dimethyl and N,N,N-trimethyl sphingosine derivatives on in vitro and in vivo growth of human tumor cells in nude mice. Cancer Res 51:1613–1618
Okoshi H, Hakomori S, Nisar M, Zhou QH, Kimura S, Tashiro K et al (1991) Cell membrane signaling as target in cancer therapy. II: inhibitory effect of N,N,N-trimethylsphingosine on metastatic potential of murine B16 melanoma cell line through blocking of tumor cell-dependent platelet aggregation. Cancer Res 51:6019–6024
Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK et al (2009) Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res 69:6915–6923
Shirahama T, Sweeney EA, Sakakura C, Singhal AK, Nishiyama K, Akiyama S et al (1997) In vitro and in vivo induction of apoptosis by sphingosine and N, N-dimethylsphingosine in human epidermoid carcinoma KB-3-1 and its multidrug-resistant cells. Clin Cancer Res 3:257–264
French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD (2006) Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther 318:596–603
Kono K, Tanaka M, Ogita T, Kohama T (2000) Characterization of B-5354c, a new sphingosine kinase inhibitor, produced by a marine bacterium. J Antibiot (Tokyo) 53:759–764
Kono K, Tanaka M, Ono Y, Hosoya T, Ogita T, Kohama T (2001) S-15183a and b, new sphingosine kinase inhibitors, produced by a fungus. J Antibiot (Tokyo) 54:415–420
Schwartz GK, Ward D, Saltz L, Casper ES, Spiess T, Mullen E et al (1997) A pilot clinical/pharmacological study of the protein kinase C-specific inhibitor safingol alone and in combination with doxorubicin. Clin Cancer Res 3:537–543
Britten CD, Garrett-Mayer E, Chin SH, Shirai K, Ogretmen B, Bentz TA et al (2017) A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin Cancer Res 23:4642–4650
Gamble JR, Xia P, Hahn CN, Drew JJ, Drogemuller CJ, Brown D et al (2006) Phenoxodiol, an experimental anticancer drug, shows potent antiangiogenic properties in addition to its antitumour effects. Int J Cancer 118:2412–2420
Miyamoto M, Takano M, Aoyama T, Soyama H, Ishibashi H, Kato K et al (2018) Phenoxodiol increases cisplatin sensitivity in ovarian clear cancer cells through XIAP down-regulation and autophagy inhibition. Anticancer Res 38:301–306
Kelly MG, Mor G, Husband A, O’Malley DM, Baker L, Azodi M et al (2011) Phase II evaluation of phenoxodiol in combination with cisplatin or paclitaxel in women with platinum/taxane-refractory/resistant epithelial ovarian, fallopian tube, or primary peritoneal cancers. Int J Gynecol Cancer 21:633–639
McPherson RA, Galettis PT, de Souza PL (2009) Enhancement of the activity of phenoxodiol by cisplatin in prostate cancer cells. Br J Cancer 100:649–655
Zhang L, Wang X, Bullock AJ, Callea M, Shah H, Song J et al (2015) Anti-S1P antibody as a novel therapeutic strategy for VEGFR TKI-resistant renal cancer. Clin Cancer Res 21:1925–1934
Ader I, Gstalder C, Bouquerel P, Golzio M, Andrieu G, Zalvidea S et al (2015) Neutralizing S1P inhibits intratumoral hypoxia, induces vascular remodelling and sensitizes to chemotherapy in prostate cancer. Oncotarget 6:13803–13821
O’Brien N, Jones ST, Williams DG, Cunningham HB, Moreno K, Visentin B et al (2009) Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies. J Lipid Res 50:2245–2257
Pal SK, Drabkin HA, Reeves JA, Hainsworth JD, Hazel SE, Paggiarino DA et al (2017) A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 123:576–582
Purschke WG, Hoehlig K, Buchner K, Zboralski D, Schwoebel F, Vater A et al (2014) Identification and characterization of a mirror-image oligonucleotide that binds and neutralizes sphingosine 1-phosphate, a central mediator of angiogenesis. Biochem J 462:153–162
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Schneider, G. (2020). S1P Signaling in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1223. Springer, Cham. https://doi.org/10.1007/978-3-030-35582-1_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-35582-1_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-35581-4
Online ISBN: 978-3-030-35582-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)