
Abstract
In this chapter, roles of bioactive sphingolipids, specifically sphingosine kinase 1 (SK1) and 2 (SK2) and their product—sphingosine 1-phosphate (S1P)—will be reviewed with respect to regulation of cancer growth, metastasis, chemotherapeutics, and drug resistance. Sphingolipids are known to be key bioeffector molecules that regulate cancer proliferation, angiogenesis, and cell death. Sphingolipid molecules such as ceramide and S1P have been shown to control cancer cell death and proliferation, respectively. Roles of S1P have been described with respect to their intracellular and extracellular pro-survival and drug resistance functions mostly through S1P receptor (S1PR1-5) engagement. Identification of novel intracellular SK/S1P targets has broadened the existing complex regulatory roles of bioactive sphingolipids in cancer pathogenesis and therapeutics. Thus, deciphering the biochemical and molecular regulation of SK/S1P/S1PR signaling could permit development of novel therapeutic interventions to improve cancer therapy and/or overcome drug resistance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Sphingolipids are structural and functional components of biological membranes (Ogretmen and Hannun 2004; Ponnusamy et al. 2010), which contribute to maintenance of membrane fluidity and subdomain structure. They are also implicated in bioeffector roles in cancer pathogenesis (Hannun and Obeid 2008; Ogretmen and Hannun 2004). Bioactive sphingolipids such as ceramide, sphingosine, and S1P are important in cell death pathways (apoptosis, necrosis, autophagy, anoikis), cancer proliferation, migration, inflammation, and drug resistance (Hannun and Obeid 2008; Ogretmen and Hannun 2004; Ponnusamy et al. 2010; Saddoughi et al. 2008). This chapter will focus on the roles of SK/S1P/S1PR signaling in cancer cell growth, therapeutics, drug resistance, and metastasis.
2 Sphingolipid Metabolism
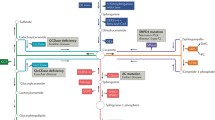
The de novo sphingolipid synthesis pathway (Fig. 1) begins with the condensation of serine and palmitoyl-coA catalyzed by serine palmitoyl transferase (SPT) (Dolgachev et al. 2004; Reynolds et al. 2004) leading to 3-ketosphinganine generation, which is rapidly reduced to dihydrosphingosine. Dihydrosphingosine is then N-acylated by a family of six dihydroceramide synthases (CerS1-6, also known as longevity associated gene, LAG, homologues, LASS1-6), which show preference for varying fatty acyl chain length specificity to synthesize dihydroceramide (Futerman and Hannun 2004). Then, a double bond is introduced between carbons 4 and 5 of the sphingosine backbone to generate ceramide (Kraveka et al. 2007; Michel et al. 1997). Ceramide is at the center of the sphingolipid metabolism, displaying mainly antiproliferative and pro-apoptotic roles (Ogretmen and Hannun 2004; Ponnusamy et al. 2010). Ceramide can be deacylated by ceramidases to generate sphingosine, which is rapidly phosphorylated by SK1 and SK2 to generate S1P, a pleiotropic lipid elucidating pro-survival, anti-apoptotic, metastatic, and/or chemoresistance functions in various cancers (Spiegel and Milstien 2003). S1P can be dephosphorylated by two S1P phosphatases (S1PP1 and S1PP2) in a reversible reaction to generate sphingosine (Mandala 2001; Mao et al. 1999; Spiegel and Milstien 2003), or S1P can be irreversibly cleaved by S1P lyase to form ethanolamine-1-phosphate and hexadecenal (Ikeda et al. 2004). Recently, S1P and hexadecenal have been shown to promote MOMP (mitochondrial outer membrane permeabilization). Interestingly, S1P and hexadecenal have been shown to cooperate with BAK and BAX, respectively, to regulate MOMP and cellular responses to apoptosis (Chipuk et al. 2012).

De novo sphingolipid synthetic pathway. The initial step is the condensation of serine and palmitoyl-coA by serine palmitoyl transferase (SPT) followed by the action of ceramide synthases (CerS) and desaturase (DES) to generate ceramide. Ceramide is also generated by the degradation of sphingomyelin (SM) by sphingomyelinase (SMase) or by the action of cerebrosidase (CRS) on glycosphingolipids also by the action of ceramide 1-phosphate phosphatase (C1PP). Ceramide is further metabolized by ceramidase (CDase) to yield sphingosine, which is used as a substrate by SK1 and SK2 to generate S1P. S1P can be dephosphorylated by S1P phosphatases (S1PP) to generate sphingosine, or it can be irreversibly cleaved by S1P lyase into ethanolamine 1-phosphate and C18 fatty aldehyde (hexadecenal). Ceramide is further metabolized to generate complex glucosyl and galactosyl-ceramide or glycolipids. Ceramide can be acted upon by sphingomyelin synthase to generate sphingomyelin or by ceramide kinase to generate ceramide 1-phosphate (C1P). C1P ceramide 1-phosphate, C1PP ceramide 1-phosphate phosphatase, CDase ceramidase, CerS ceramide synthase, CRS cerebrosidase, DES desaturase, GCS glucosyl ceramide synthase, S1P sphingosine 1-phosphate, S1PP S1P phosphatase, SK1 sphingosine kinase 1, SK2 sphingosine kinase 2, SM sphingomyelin, SMase sphingomyelinase, SPT serine palmitoyl transferase
3 Ceramide/S1P Rheostat in Cancer
The fate of a cell is determined by the balance between ceramide and S1P signaling (not necessarily a quantitative ratio for the amount of lipids, but a biological/metabolic balance between these two signaling arms of sphingolipids with opposing functions), which is often referred to as the ceramide/S1P rheostat (Fig. 2). There exists a dynamic balance between ceramide and S1P signaling, and when a shift towards ceramide is achieved by stress signaling such as radiation, heat, and chemotherapy treatment, this drives cells to undergo cell death and antiproliferation (Hannun and Obeid 2008). On the other hand, when the balance shifts towards S1P accumulation, cells exert pro-survival, anti-apoptosis, and/or chemoresistance (Ponnusamy et al. 2010). Increases in endogenous ceramide by chemotherapeutic agents, TNF-α, CD95, hypoxia, DNA damage, and heat stress can activate cell death pathways. Also, increases in ceramide via inhibiting ceramide metabolism or overexpressing CerS lead to cell death, in general. Moreover, overexpression of bacterial SMase, which generates ceramide by degradation of sphingomyelin, was shown to induce apoptosis (Meyers-Needham et al. 2012a). In contrast, inhibition of de novo ceramide generation by fumonisin B1 blocks ceramide-mediated cell death by chemotherapeutic drugs.

Ceramide-S1P rheostat in cancer and therapy. There exists a balance or rheostat in ceramide to S1P signaling in cancer. A shift towards ceramide accumulation leads to pro-apoptotic, autophagic, ER stress response, and anti-survival effects, whereas a dynamic shift towards S1P accumulation leads to pro-survival, anti-apoptotic, metastatic, and drug-resistant phenotypes. Potential therapeutic approaches will be to increase ceramide and decrease S1P in cancer cells by chemotherapy, radiation, monoclonal antibody, and other molecular approaches. ER endoplasmic reticulum, S1P sphingosine 1-phosphate
Ceramide has various established intracellular targets such as PP1, PP2A, I2PP2A, cathepsin D, and protein kinase Cζ, which mediate its apoptotic/cell death functions (Fox et al. 2007; Heinrich et al. 2000; Ogretmen and Hannun 2004; Wang et al. 2005). Our laboratory identified telomerase to be a nuclear target of ceramide (exogenous C6-ceramide or C18-ceramide generated by CerS1) which decreased c-Myc-mediated activation of hTERT promoter in lung cancer cells (Ogretmen et al. 2001). In fact, ceramide deacetylates Sp3 transcription factor and deacetylated Sp3 recruits HDAC1 to the hTERT promoter, thereby decreasing hTERT expression/activity (Wooten-Blanks et al. 2007; Wooten and Ogretmen 2005). We also showed that ceramide binds I2PP2A and relieves PP2A-mediated tumor suppression functions in A549 lung cancer cells. Ceramide mediates the degradation of c-Myc by PP2A activation via prevention of I2PP2A-dependent inhibition of PP2A (Mukhopadhyay et al. 2009).
Recently, ceramides with different fatty acid chain lengths were suggested to have distinct functions. For example, in head and neck cancer cells, CerS1-generated C18-ceramide suppressed tumor growth, whereas CerS6-generated C16-ceramide increased tumor growth/proliferation (Senkal et al. 2011). These distinct and unexpected functions of endogenous ceramides might be due to their downstream targets regulated via ceramide/protein interactions and/or their subcellular localization at distinct biological membranes or membrane microdomains. Although mechanisms that regulate CerS expression and function are largely unknown for de novo ceramide generation, new insights about the modulation of CerS expression suggested that epigenetic and posttranscriptional control by concerted functions of HDAC1 and microRNA-574-5-mediated targeting of CerS1 mRNA is involved in its repression in HNSCC (Meyers-Needham et al. 2012b). In addition, dimerization of CerS proteins has been shown to regulate their function for the generation of ceramide (Laviad et al. 2012).
In contrast to ceramide, S1P plays a pro-survival function. Sphingosine kinases and S1P-phosphatases/lyase are important players in the regulation of S1P generation/metabolism. SiRNA-mediated knockdown of SK1 inhibits cell proliferation and increases ceramide/S1P rheostat in pancreatic, prostate, and leukemia cancer cells (Akao et al. 2006; Baran et al. 2007; Guillermet-Guibert et al. 2009; Pchejetski et al. 2005). In contrast, overexpression of SK1 leads to increased cell proliferation by inducing G1/S phase transition and DNA synthesis (Olivera et al. 1999). Exogenous S1P addition was found to significantly inhibit apoptosis via increased Bcl-2 (Sauer et al. 2005) and Mcl-1 expression (Li et al. 2008) or decreased BAD and BAX expression (Avery et al. 2008). In particular, exogenous S1P prevents BAD/BAX translocation to mitochondria, thereby inhibiting the intrinsic cell death (mitochondrial) pathway (Betito and Cuvillier 2006). Also, SK1 overexpression has been shown to inhibit cytochrome c release and caspase-3 activation by the regulation of BCL-XL, MCL1, and BIM in chronic myelogenous leukemia (CML) cells (Bonhoure et al. 2008).
Moreover, important in vivo findings indicate the involvement of S1P in cancer progression. For example, breast and prostate cancer cells that overexpressed SK1 formed tumors in mice, and ceramide/S1P was altered in these tumors (Nava et al. 2002; Pchejetski et al. 2005), which were resistant to doxorubicin and had increased neovascularization and decreased ceramide/S1P (Nava et al. 2002; Pchejetski et al. 2005). Interestingly, CML and leukemia cells, which are sensitive to imatinib and daunorubicin, respectively, had higher ceramide/S1P compared to resistant cells. Furthermore, increased SK1/S1P in response to camptothecin treatment in prostate cancer cells suggests a role for S1P signaling in chemoresistance (Fig. 3).

SK1/S1P signaling and intracellular targets of S1P. SK/S1P signaling pathway involves the activation of SK1 by agonist-mediated receptor action. The activated SK1 translocates to the inner leaflet of the plasma membrane to utilize the substrate sphingosine to generate S1P. The S1P generated inside the cells are transported outside the cell by S1P transporters and furthermore engage in an autocrine or paracrine fashion to five G protein-coupled receptors specific for S1P (S1PR1–5) to induce an array of downstream mechanisms involved in cell motility, survival, migration, and/or proliferation. S1P generated by SK1 has been reported to play intracellular function by binding with TRAF2 protein to regulate NF-κB function. S1P generated by SK2 has been shown to interact with HDAC1/2 in the nucleus to regulate transcription of p21 and c-Fos genes. Moreover, SK2-generated S1P also binds to prohibitin 2 (PHB2), mitochondrial protein which in turn regulates cytochrome c oxidase (Cox-2) in respiration complex assembly and function. HDAC histone deacetylase, NF-κB nuclear factor kappa light chain enhance of activated B cells, PHB2 prohibitin 2; TRAF2 TNF receptor associated factor 2
4 S1P Signaling
S1P generated by SK1 has been shown to be secreted and engaged with S1P receptors (S1PR1–5) to elicit various downstream responses involved in inflammation, cell migration, angiogenesis, and/or lymphocyte trafficking (Spiegel and Milstien 2003; Strub et al. 2010). S1PR1 is important in vascular maturation, immune cell trafficking, endothelial barrier function, and angiogenesis as displayed in S1PR1-null mice. Also the S1P–S1PR1 interaction reveals receptor tyrosine kinase (RTK) activation such as EGF- and PDGF-mediated pathways that dictate cellular growth and migration. Importantly, ATP-dependent ABC transport proteins such as ABCC1, ABCA1, and ABCG2 are involved in S1P transport (Mitra et al. 2006; Sato et al. 2007; Takabe et al. 2010). Also, ABCC1 is involved in S1P export from mast cells independent of degranulation (Mitra et al. 2006). Recently, TOH/SPNS2 (two of hearts protein) has been identified as an S1P transporter in zebra fish, and interestingly TOH/SPNS2 acts upstream of MIL, an orthologue of S1PR2 involved in heart development (Kawahara et al. 2009; Osborne et al. 2008). SPNS2 was shown to be important for the transport of phosphorylated form of FTY-720, an immunomodulatory drug used for lymphocyte egress in multiple sclerosis. Importantly SPNS2-null mice had increased accumulation of mature T cells and a decreased T cell population in blood and secondary lymphoid organs (Fukuhara et al. 2012; Hisano et al. 2011). Thus, S1P-mediated signaling (Fig. 3) is important in various cellular processes, and how this mechanism is dysregulated in cancer needs further investigation. Recently, the structure of ligand-bound S1PR1 was solved, and data suggest that S1P might engage with the receptor within the plasma membrane (Hanson et al. 2012), indicating that lateral movements of S1P within the membrane or ligand swapping between S1PR1 and other receptors might be involved in this process.
5 Roles of SK in Cancer Pathogenesis
Sphingosine kinases (SK1 and SK2) are novel lipid kinases, which are evolutionarily conserved as diverse as in humans, mice, yeast, and plants. SK1 and SK2 are encoded by SPHK1 and SPHK2 genes in humans (Pitson 2011; Strub et al. 2010). SphK1 and SphK2 belong to the diacylglycerol kinase family and have five conserved domains C1–C5 (Spiegel and Milstien 2003). The unique catalytic domain is present between domains C1 and C3. SphK1 and SphK2 differ by the presence of a TM (transmembrane domain) present only in SphK2 and not in SphK1. Both SK1 and SK2 enzymes show substrate specificity towards D-e-sphingosine and D-e-dihydrosphingosine. Interestingly, SK1 and SK2 show different tissue distribution with SK1 expression higher in lung and spleen, whereas SK2 levels were found to be higher in the liver and the heart. SK1 is predominantly cytosolic in nature, whereas SK2 has been shown to localize both in the cytoplasm and nucleus (Kohama et al. 1998; Liu et al. 2000, 2002; Pitson 2011). Moreover, SK1 expression was detected at embryonic day 7 (E7) of mice and SK2 at E11 indicating the distinct functions of both of these isoenzymes. The functions of SK1 and SK2 seem redundant during development; SK1 and SK2 null mice survive, whereas the SK double knockout is embryonically lethal (Mizugishi et al. 2005; Pitson 2011; Spiegel and Milstien 2003).
5.1 SK1 and Cancer
SK1 has well-established pro-survival functions in various cancers. Indeed, by virtue of its transformation potential, SK1 is considered to be a bona fide oncogene (Vadas et al. 2008). Upon transfection with SK1, untransformed NIH3T3 cells undergo transformation with the ability to form colonies in soft agar and tumors in nude mice (Xia et al. 2000). Activation of SK1 by agonists of GPCRs, protein kinases, proinflammatory cytokines, and small GTPases leads to its translocation to plasma membrane where it catalyzes the conversion of sphingosine to S1P. Importantly, phosphorylation of SK1 at Ser 225 by ERK1 and ERK2 is important for its activation and translocation to the plasma membrane (Pitson et al. 2003). This finding was further confirmed by S225A mutant of SK1 that lacks the phosphorylation site (Pitson et al. 2005). When SK1 S225A mutant was targeted to the plasma membrane by tagging with a myristoyl or palmitoyl moiety, the cells became transformed, via targeting to plasma membrane (Pitson et al. 2005). The membrane affinity and plasma membrane selectivity are determined by Thr54 and Asn89 residues of human SK1, wherein these residues interact specifically with phosphatidyl serine in the plasma membrane, thus making sphingosine available to generate S1P, which can be secreted outside the cell and engage with S1PRs to induce pro-survival functions (Stahelin et al. 2005). Interestingly, SK1 expression was found also to be important for Ras-mediated transformation. In cancers, such as breast, lung, ovary, stomach, and kidney, SK1 mRNA increased approximately twofold compared to paired normal tissues (French et al. 2003; Johnson et al. 2005; Kawamori et al. 2006). Also immunohistochemical studies with lung, colon, and breast cancer tissues were positive for SK1 expression in tumor tissues and/or carcinoma cells (Johnson et al. 2005; Kawamori et al. 2006; Kohno et al. 2006; Ruckhaberle et al. 2008). Moreover, microarray data show elevated SK1 in squamous cell carcinoma (Nindl et al. 2006), melanoma cancer (Talantov et al. 2005), N-methyl-N-nitrosourea-induced rat breast cancer (Chan et al. 2005), recurrent breast cancer after tamoxifen treatment (Ma et al. 2004), cervical cancer (Wong et al. 2003), head and neck cancer, and leukemia (Andersson et al. 2007; Ginos et al. 2004; Pyeon et al. 2007). Interestingly, siRNA-mediated downregulation of SK1 showed decreased migration of EGF, prolactin, and E2-induced migration of MCF-7 breast cancer cells (Doll et al. 2007; Sarkar et al. 2005), whereas downregulation of both SK1 and SK2 inhibited migration of MDA-MB-453 cells (Hait et al. 2005), indicating a possible redundancy/overlap in functions between these isoenzymes in cancer cell migration.
Novel intracellular targets of SK1 have been recently established: SK1-generated S1P has been shown to bind to TRAF2 protein, an E3 ubiquitin ligase, modulating TNF-α-induced activation of NF-κB signaling (Alvarez et al. 2010). SK1 has been shown to bind TRAF2 to induce K63-mediated polyubiquitination of RIP1 leading to IκB degradation and subsequent stimulation of the NF-κB pathway (Fig. 3). These findings suggest SK1/S1P to be important driver of cancer progression.
Remarkably, a recent finding from our laboratory suggests that serum S1P generated by SK1, and not tumor S1P, was important for tumor metastasis to the lungs (Fig. 4). In this study, genetic ablation of systemic SK1 leads to increased breast cancer metastasis suppressor 1 (Brms1) expression via alterations of S1PR2 signaling in tumor cells, leading to suppression of metastasis. Furthermore, treatment with anti-S1P monoclonal antibody (Sphingomab) decreased lung metastasis in this model by neutralizing circulating/systemic S1P, inducing cancer Brms1 expression, and thus further proving the importance of SK1-generated systemic S1P in regulating tumor metastasis (Ponnusamy et al., 2012). Recently, the role of SK1 in the regulation of tumor metastasis has been also shown in a breast cancer model (Nagahashi et al. 2012). These data suggest that inhibition of systemic SK1/S1P and/or cancer S1PR2 signaling might inhibit tumor metastasis.

Role of systemic SK1/S1P in the modulation of S1PR2/Brms1 expression in lung tumor metastasis. S1P generated in response to cancer cell exposure to bloodstream elevates systemic S1P levels thereby leading to the induction of S1PR2 expression and further leading to the suppression of Brms1, a master metastatic suppressor gene in cancer cells, inducing their metastatic potential. Use of pharmacologic inhibitors or antibody-based therapeutic tools (Sphingomab, Lpath Inc.) effectively modulates serum S1P via inhibition of cancer cell S1PR2 signaling to elevate tumor Brms1 levels and to suppress lung metastasis. Brms1 breast cancer metastasis suppressor 1, SK1 sphingosine kinase 1, S1P sphingosine 1 phosphate, S1PR2 S1P receptor 2
5.2 SK2 and Cancer
SK2 has been shown to predominantly localize to the nucleus, although cytoplasmic and ER localizations were also reported (Igarashi et al. 2003; Maceyka et al. 2005). SK2 localizes to the nucleoplasm and causes cell cycle arrest. Importantly, SK2 retains a nuclear export signal, and activation with phorbol myristate acetate (PMA) leads to protein kinase D-mediated phosphorylation and export from the nucleus. SK2, unlike SK1, possesses a BH-3 domain in its sequence that sequesters Bcl-XL to diminish its anti-apoptotic functions (Liu et al. 2003). Interestingly, when SK2 is overexpressed, it induces apoptosis, cell cycle arrest, and/or caspase-3 activation (Liu et al. 2003). In contrast to the apoptotic roles, SK2 also displays anti-apoptotic functions. Knockdown of SK2 in HCT116 colon cancer cells and MCF7 breast cancer cells prevents doxorubicin-induced p21 expression and G2/M cell cycle arrest in a p53 independent manner (Sankala et al. 2007). SK2 knockdown decreased glioblastoma cell proliferation, whereas SK1 knockdown did not have any effects (Van Brocklyn et al. 2005). EGF activates SK2 at Ser351 and Thr578 (Hait et al. 2007) residues leading to EGF-mediated migration of MDA-MB-453 breast cancer cells. These studies elucidate SK2’s anti-apoptotic and pro-proliferative effects.
Most importantly, the first identified intracellular function of S1P generated by SK2 was in the nucleus to inhibit HDAC1/2 enzymatic activity thereby preventing deacetylation of histone H3 (Hait et al. 2009). The SK2/HDAC repressor complex was found to be associated with promoter regions of P21 (CDKN1) and c-Fos genes thereby modulating their expression. Interestingly, another intracellular target of S1P has been identified as prohibitin 2 (PHB2), a protein regulating mitochondrial function (Strub et al. 2011). S1P generated by SK2 binds prohibitin 2 and not prohibitin 1 to regulate cytochrome c oxidase complex IV in mitochondrial respiration and function. Also, mitochondria isolated from SK2 knockout mice showed decreased association of PHB2 and cytochrome c oxidase (Strub et al. 2011) (Fig. 3).
Collectively, SK1- and SK2-generated S1P have distinct roles in the context of their subcellular localization and function. Also, SK1 and SK2 might also have overlapping functions in different cancer models for inducing pro-survival and anti-apoptosis, possibly via distinct mechanisms of action at different subcellular compartments.
6 Role of S1P in Autophagy
Autophagy is a cellular process to degrade long-lived proteins and organelles through lysosomes. The autophagic process can be protective or lethal; the protective autophagy pathway directs degradation of damaged organelles and recycling of amino acid to overcome nutrient deprivation, whereas lethal autophagy leads to caspase independent cell death involving Atg proteins (Kondo and Kondo 2006; Levine and Kroemer 2008). Use of SK2 inhibitor ABC294640 induces autophagic cell death in A498 cells (Beljanski et al. 2010). Mechanistically, Beljanski and colleagues reported that increased ceramide and sphingosine in response to ABC294640 treatment resulted in autophagy in vivo. Moreover, protective autophagy was activated in response to SK1 upregulation by lack of beclin 1 and mTOR inhibition (Lavieu et al. 2006). Recently, it was shown that S1P metabolism plays an important role in switching from protective autophagy to apoptosis through the involvement of SPP1 (S1P phosphohydrolase). It was found that doxorubicin-mediated autophagy was greatly decreased by SPP1 ablation with a concomitant increase in apoptosis by ceramide accumulation via de novo synthesis. Ceramide then activates calpain to cleave Atg5 in SPP1-depleted cells to shift from protective autophagy to apoptosis (Lepine et al. 2011).
7 SK1/S1P Signaling in Drug Resistance
SK/S1P protects cancer cells from drug-induced cell death; therefore, levels of bioactive sphingolipids have been shown to modulate drug resistance. For example, PC3 prostate cancer cells, which are androgen insensitive, are resistant to camptothecin treatment by upregulation of SK1 and S1P1/S1P3 signaling, and these cells became sensitive upon inhibition of SK and S1P formation (Bektas et al. 2005). Radiation-resistant prostate cancer cell line LNCaP showed sensitivity upon coadministration of TNF-α and γ-irradiation to increase the generation of sphingosine and decreased S1P formation (Nava et al. 2000). Moreover, treatment of LNCaP cells with SK inhibitor (N,N-dimethyl sphingosine) sensitized them to radiation-induced apoptosis. In addition, SK1 induced resistance of melanoma cells to ceramide and Fas-induced cell death. The role of SK1/S1P in breast cancer progression was also reported to be important. A microarray study conducted with 1,269 breast cancer tumor samples showed a significant increase in SK1 gene expression and showed a strong correlation between SK1 expression and poor prognosis (Ruckhaberle et al. 2008). Additionally SK1 expression was responsible for resistance to tamoxifen-induced cell death in MCF7 cells. Tamoxifen-resistant MCF7 cells showed elevated SK1 levels and inhibition of SK1 by DMS/SKI-II restored sensitivity of the cells to tamoxifen-induced apoptosis (Sukocheva et al. 2009). In erythroleukemia progression, overexpression of SK1 in non tumorigenic proerythroblasts showed increased tumorigenicity and resistance to cell death. Of note, a microarray-based transcriptome profile showed transcriptional upregulation of SK1 in tumorigenic proerythroblasts. These data suggest SK1 expression to be important in erythroleukemic progression (Le Scolan et al. 2005). Moreover, SK1 is overexpressed in glioblastoma multiforme (GBM) and targeted inhibition of SK1 with an isoform-specific drug; SK1-I significantly decreased tumor proliferation and markedly suppressed pro-survival AKT and its downstream targets (p70S6K and GSK3-α) (Kapitonov et al. 2009; Knapp et al. 2010). SK1-I not only inhibited S1P generation but also increased ceramide to induce apoptosis (Kapitonov et al. 2009). In endometrial cancer, compared with healthy endometrial tissues, the cancer tissues had increased SK1 activity (2.6-fold) and elevated S1P (1.6-fold) in serum, indicating a significant change in sphingolipid metabolism in driving cancer progression (Knapp et al. 2010).
In CML, ceramide/S1P rheostat plays a crucial role in conferring drug resistance. K562 CML cells generate endogenous C18-ceramide in response to imatinib treatment, and interestingly K562 cells which show resistance to imatinib treatment have increased SK1 expression. Importantly, siRNA-mediated knockdown of SK1 sensitized K562-imatinib-resistant cells to apoptosis. These studies showed that overexpression of SK1 induces drug resistance in CML cells by altering the ceramide/S1P rheostat towards S1P accumulation (Baran et al. 2007). Recently, we have shown that SK1/S1P and S1PR2 signaling is important for Bcr-Abl1 stability in CML (chronic myeloid leukemia), leading to imatinib resistance. This study showed that SK1/S1P/S1PR2 prevents Bcr-Abl1 dephosphorylation and subsequent degradation by inhibiting PP2A. Inhibition of SK1/S1P/S1PR2 signaling either by pharmacological or molecular approaches restored PP2A-mediated dephosphorylation of Bcr-Abl1 and also enhanced imatinib or nilotinib (drugs used against CML) mediated growth inhibition. Furthermore, allograft tumors derived from 32D cells expressing either wild-type or mutant Bcr-Abl1 genes were more sensitive to nilotinib upon inhibition of SK1/S1PR2 signaling (Fig. 5). These findings suggest that inhibition of SK1/S1P/S1PR2 signaling overcomes drug resistance in CML (Salas et al. 2011). In a separate study, LAMA84 cells which are resistant to imatinib treatment became sensitized when SK1 is inhibited by F-12509a, a SK1 inhibitor, leading to apoptotic cell death (Bonhoure et al. 2008).

Role of SK1/S1P/S1PR-mediated drug resistance in chronic myelogenous leukemia (CML). SK1/S1P-mediated drug resistance in CML is mediated by S1PR2-mediated PP2A modulation which abolishes proteasomal degradation of Bcr-Abl1 by enhancing its stability, resulting in drug resistance. Pharmacologic inhibitor (SKI-II) or use of molecular approaches (RNAi) by inhibiting SK1 can lead to PP2A-mediated dephosphorylation and degradation of Bcr-Abl1, thereby overcoming drug resistance. CML chronic myelogenous leukemia, PP2A protein phosphatase 2A, SK1 sphingosine kinase 1, S1P sphingosine 1-phosphate, S1PR2 S1P2 receptor 2, SHP1 src homology region 2 domain containing phosphatase 1
8 SK2 and Drug Resistance
Although SK1/S1P signaling has been well established in drug resistance, only recently SK2/S1P has been shown to confer chemoresistance in cancers. Targeted inhibition of SK2 by molecular/pharmacological agents sensitized cancer cells to chemotherapy. Recently, Antoon’s laboratory reported that the SK2 inhibitor ABC294640 decreased estrogen receptor (ER)-positive breast cancer tumor growth by 68.4 % compared to vehicle-treated tumors. Mechanistically, SK2 inhibition decreased estrogen-mediated transcription of ER-regulated genes such as SDF1 and the progesterone receptor (Antoon et al. 2010). Subsequently, inhibition of SK2 by ABC294640 at submicromolar concentration was shown to block proliferation of endocrine therapy-resistant MDA-MB-231 and chemoresistant MCF-7TN-R cells (Antoon et al. 2011). Also ABC294640 diminished NF-κB pro-survival signaling by decreasing activation of Ser536 phosphorylation of the p65 subunit (Antoon et al. 2011). Moreover, ABC294640, which is orally bioavailable, resulted in growth inhibition of xenograft-derived tumors of MCF-7TN-R cells at 50 mg/kg in SCID mice.
Schnitzer et al. (2009) elucidated the role of SK2-mediated chemoresistance in A549 lung cancer cells. In this study, it was demonstrated that hypoxia induces SK2 protein/activity leading to S1P secretion via S1PR1/S1PR3 signaling to activate P42/44 MAPK signaling and confer resistance to etoposide-induced apoptosis (Schnitzer et al. 2009). Recently, Xiao et al. (2012) showed that SK2 confers resistance to sodium butyrate-induced apoptosis in HCT116 colon cancer cells. In fact, sodium butyrate treatment induced the phosphorylation of SK2 by PKD leading to nuclear export, leading to chemoresistance (Xiao et al. 2012).
9 S1P/S1PR2 Signaling in Cancer and Drug Resistance
S1P exerts its signaling function in an autocrine or paracrine manner through five G protein-coupled receptors, termed S1PR1–5 (formerly referred as the EDG family of receptors). These receptors involve heterotrimeric G proteins such as Gi, Gq, and G12/13 to elicit various cellular functions. Importantly, roles of S1P receptors in modulating cancer growth have been studied (Spiegel and Milstien 2003). For instance, S1PR2 has been shown to be important for retinal angiogenesis. When a neonatal mouse was subjected to ischemia-driven retinopathy, the neovascularization event was downregulated in S1P2-null mice compared to wild-type mice. Moreover, the retina of the S1P2-null mouse had significantly reduced proinflammatory Cox-2 mRNA and increased eNOS expression. These findings indicate the involvement of S1PR2 in retinal angiogenesis and prompt the use of receptor antagonists to counteract ocular neovascularization. Additionally, S1P2 plays an important role in Wilm’s tumor, a malignant renal cancer condition. In Wilm’s tumor, S1PR2 mRNA was higher in ten cancer specimens compared to noncancerous tissues (Li et al. 2009). Additionally S1PR2 overexpression was shown to induce Cox-2 mRNA and increased prostaglandin E2 synthesis. SiRNA-mediated knockdown or pharmacological inhibition of S1PR2 in WiT49 cells decreased Cox-2 mRNA proving an important role of S1PR2 signaling in driving renal cancer progression (Li et al. 2009). In another study, the migration of WiT49 Wilm’s tumor cells was attributed to S1PR1 signaling wherein S1PR1/Gi coupling was shown to induce a promigratory phenotype which depended upon the ratio of S1PR1/S1PR2 and further identified PI3K and Rac1 as downstream regulators of cell motility. S1PR2 was also shown important in esophageal cancer motility and migration, in which TGFβ enhanced S1P-mediated activation of ERK1/2 through S1PR2 through non-Smad signaling (Miller et al. 2008).
In a separate study using a cohort of 304 ER-positive, tamoxifen-treated breast cancer samples, increased SK1 and S1P1 and S1P3 receptor expression was observed. Interestingly cytoplasmic expression of S1P1 and S1P3 was found to be associated with reduced disease-specific survival in patients with ER positive breast cancer (Watson et al. 2010). SK1 also confers resistance towards synthetic retinoid N-(4-hydroxyphenyl) retinamide in A2780 ovarian cancer cells; the HPR-resistant cells express more SK1 both at the mRNA and the protein level and became sensitized when treated with SK inhibitor (Illuzzi et al. 2010). Recently, SK1 has been shown to protect androgen-independent LNCaP prostate cancer cells; it does so by increasing S1P signaling through S1P2/3 receptors, and the resistance was attenuated by treating with FTY-720, a sphingosine analog that inhibits S1PR signaling (except S1PR2), by inducing proteasome-mediated degradation of SK1 (Tonelli et al. 2010).
These findings suggest that the SK/S1P/S1PR signaling pathway is crucial in sphingolipid-mediated drug resistance in various cancers, and hence targeting this pathway would be an effective anticancer therapeutic strategy to overcome drug resistance.
10 SK/S1P-Mediated Anticancer Therapeutics
Targeting S1P produced by SK1 and SK2 can sensitize cancer cells to therapeutic intervention. Also targeting SK/S1P is attractive because indirect generation of ceramide and sphingosine can have antiproliferative and pro-apoptotic functions. An array of sphingolipid and non-sphingolipid agents has been developed to target SK/S1P and/or S1P receptors (Table 1).
Pan Sphingosine Kinase Inhibitors. N,N-dimethyl sphingosine (DMS), a pan SK inhibitor, was shown to be effective against a panel of cancer cell lines and exhibited antitumor growth properties in nude mice. Also, l-threo dihydrosphingosine (Safingol) inhibits SK and is currently in phase I clinical trials (Schwartz et al. 1997). Both DMS and Safingol inhibit SK competitively and displayed inhibitory effects towards ceramide kinase and PKC and activate sphingosine-mediated targets such as PI3K and casein kinase 2 (Igarashi et al. 1989; Kedderis et al. 1995; King et al. 2000; McDonald et al. 1991; Megidish et al. 1998; Sugiura et al. 2002). Some of the reported off-target effects of DMS include hemolysis and hepatotoxicity (Kedderis et al. 1995). Interestingly, phenoxodiol, a synthetic analog of plant isoflavone genistein, exhibits anticancer and antiangiogenenic functions by inhibiting SK. Phenoxodiol exerts its anticancer function by inhibiting endothelial cell function both in vitro and in an in vivo model of angiogenesis, and currently phenoxodiol is in clinical trials against ovarian and prostate cancers (De Luca et al. 2005; Gamble et al. 2006). French et al. (2003) have demonstrated the use of non-lipid selective inhibitors of SK and showed inhibition against a panel of cancer cell lines, with greater selectivity to SK than other protein kinases; additionally these SK inhibitors were noncompetitive and displayed antiproliferative functions (French et al. 2003). Furthermore, French et al. (2006) showed SKI-II (4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol), a noncompetitive inhibitor at nanomolar to submicromolar concentration in vitro, and displayed excellent oral bioavailability with antitumor functions in vivo (French et al. 2006). Additionally, other compounds such as F12509a, a sesquiterpene quinone-based competitive inhibitor of SK, and B5354c, a noncompetitive inhibitor isolated from a marine bacterium, were found to inhibit SK in some cancers, but their efficacy and specificity are still under investigation (Kono et al. 2000a, b, 2002).
SK1 Selective Inhibitors. SK1-I (2R, 3S, 4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1, 3-diol is a SK1 selective inhibitor and has shown efficacy against orthotopic as well as xenograft glioblastoma or AML xenograft tumors (Kapitonov et al. 2009; Paugh et al. 2008). Recently, Nagahashi et al. (2012) demonstrated decreased breast cancer progression in an 4T1-Luc orthotopic mice and also showed decreased lymph node and lung metastasis upon SK1-I treatment. Interestingly SK1-I decreased serum S1P levels and caused mammary tumor cells to undergo apoptosis (Nagahashi et al. 2012). Moreover, novel SK1-specific inhibitors such as 6ag, 9ab, and12aa were synthesized and tested in vitro, but the in vivo efficacy still needs to be validated (Xiang et al. 2009). There is a need for the development of SK1-specific small molecule inhibitors to be utilized as effective anticancer agents.
SK2 Selective Inhibitors. ABC294640 is a novel SK2-specific inhibitor with excellent oral bioavailability and toxicology properties. ABC294640 displays antiproliferative effects against a variety of cancer cell lines at nanomolar up to submicromolar concentrations. This SK2 selective compound induced autophagic cell death in breast, prostate, and kidney xenograft tumors in vivo (Antoon et al. 2010; Beljanski et al. 2010, 2011; French et al. 2010). ABC294640 is currently in phase I clinical trials against solid tumors. Moreover, sphingoid analogs such as SG12 and SG14 displayed specificity towards SK2, and in particular SG14 did not inhibit PKC (Kim et al. 2005).
S1P Receptor Selective Compounds. FTY-720, a sphingosine analog, phosphorylated by SK2, acts through S1P receptors (S1PR1, 3, 4, 5) to cause receptor endocytosis and rapid lymphocyte egress in multiple sclerosis (Brinkmann et al. 2010, 2002). FTY-720 has been efficacious against refractory multiple sclerosis and approved by FDA as an anti-MS drug (Brinkmann et al. 2010). FTY-720 also inhibited SK1, ceramide synthase, phospolipase A2, and SGPL1, and it was shown to activate tumor suppressor PP2A (Bandhuvula et al. 2005; Berdyshev et al. 2009; Lahiri et al. 2009; Matsuoka et al. 2003; Neviani et al. 2007; Payne et al. 2007; Vessey et al. 2007). FTY-720 has been reported to inhibit colon and breast cancer cell lines in situ through S1P receptor independent effects. In addition, phosphorylated-FTY720 (P = FTY-720) was found to be important for antiangiogenesis in an in vitro spheroid model, indicating a receptor-dependent function, which can also be mimicked by SEW2871, a S1PR1-specific antagonist (Nagaoka et al. 2008; Schmid et al. 2007). Further investigation is required to delineate the receptor-dependent and receptor-independent functions of FTY-720 against cancer growth and/or proliferation. Recently, (R)-FTY-720-OMe, a stereospecific analog of FTY-720, showed SK2-specific inhibition and caused actin rearrangement in MCF-7 cells (Lim et al. 2011), suggesting that this novel enantiomer can be used against breast cancer. Other receptor antagonists such as VPC4416, VPC2309, VPC25239, and W146 against S1PR1/3 and S1PR1, respectively, have shown promising results in situ (Davis et al. 2005; Sanna et al. 2006).
Antibody-Based Therapeutics. An anti-S1P-mAb that specifically targets and neutralizes S1P has been shown to be effective against lung A549, ovarian SKOV3, breast MDA-MB-231, and melanoma F16/B10 cancer models in situ and in vivo. Anti-S1P-mAb functions as a molecular sponge to neutralize S1P signaling and cause tumor regression in both xenograft and allograft models or to inhibit lung metastasis (Ponnusamy et al. 2012). Sphingomab (LT1002) and its humanized form (LT1009) neutralize bFGF- and VEGF-induced angiogenesis and block S1P-induced endothelial cell tube formation and migration in numerous in vitro assays (Visentin et al. 2006). Interestingly, LT1009 (sonepcizumab, Lpath Inc.) is currently in phase I/II clinical trials as an anticancer drug (Sabbadini 2011).
11 Conclusion and Future Perspectives
Recent developments in sphingolipid biology, especially the discovery of the roles of SK/S1P/S1PR2 signaling in the regulation of cell proliferation, angiogenesis, drug resistance, and metastasis, have added to our understanding of these processes in various cancers. Importantly emerging evidence suggests that sphingolipids have various functions based on their subcellular localization, relative distribution in tissues, fatty acyl chain-length specificities, and their direct downstream targets via lipid-protein binding. Importantly, chain length-specific ceramides were found to be altered in cancers; thus—from a therapeutic perspective—reconstitution of ceramide generation by use of small molecule inhibitors or ceramide analogs/mimetics could be effective anticancer techniques. In contrast, SK and S1P were elevated in various cancers and inhibitors; antagonists and monoclonal antibodies against SK/S1P/S1PR signaling might hold promise for decreasing cancer growth, proliferation, and metastasis. Recent identification of SPNS2 as a novel S1P transporter (Kawahara et al. 2009) will help delineate the mechanism of S1P transport from various cell types, altering systemic S1P accumulation, which then plays a role in inducing tumor metastasis (Ponnusamy et al., 2012). Moreover, identification of the S1PR1 crystal structure can point towards more potent antagonists of S1PR-specific compounds to mediate anticancer functions (Hanson et al. 2012). Although recent research in sphingolipid metabolism and biology has yielded significant mechanistic information towards cancer pathogenesis and therapeutics, limitations exist regarding our understanding of their roles in varying cancer subtypes, subcellular compartmentalization, and direct downstream targets, which often yield context-dependent effects in response to changes in their generation/accumulation. Moreover, the inherent obstacle in studying membrane-bound enzymes and their lipid products involved in metabolism must be overcome to identify molecular and structural details of their functions in the regulation of cancer growth, proliferation, metastasis, and for the development of novel anticancer therapeutics.
References
Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, Igarashi Y, Nozawa Y (2006) High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem Biophys Res Commun 342:1284–1290
Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S (2010) Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465:1084–1088
Andersson A, Ritz C, Lindgren D, Eden P, Lassen C, Heldrup J, Olofsson T, Rade J, Fontes M, Porwit-Macdonald A, Behrendtz M, Hoglund M, Johansson B, Fioretos T (2007) Microarray-based classification of a consecutive series of 121 childhood acute leukemias: prediction of leukemic and genetic subtype as well as of minimal residual disease status. Leukemia 21:1198–1203
Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Ashe HB, Wiese TE, Smith CD, Burow ME, Beckman BS (2010) Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology 151:5124–5135
Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, Smith CD, Burow ME, Beckman BS (2011) Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther 11:678–689
Avery K, Avery S, Shepherd J, Heath PR, Moore H (2008) Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Stem Cells Dev 17:1195–1205
Bandhuvula P, Tam YY, Oskouian B, Saba JD (2005) The immune modulator FTY720 inhibits sphingosine-1-phosphate lyase activity. J Biol Chem 280:33697–33700
Baran Y, Salas A, Senkal CE, Gunduz U, Bielawski J, Obeid LM, Ogretmen B (2007) Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J Biol Chem 282:10922–10934
Bektas M, Jolly PS, Muller C, Eberle J, Spiegel S, Geilen CC (2005) Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: role of Bcl-2 expression. Oncogene 24:178–187
Beljanski V, Knaak C, Smith CD (2010) A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J Pharmacol Exp Ther 333:454–464
Beljanski V, Lewis CS, Smith CD (2011) Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in hepatocellular carcinoma xenografts. Cancer Biol Ther 11:524–534
Berdyshev EV, Gorshkova I, Skobeleva A, Bittman R, Lu X, Dudek SM, Mirzapoiazova T, Garcia JG, Natarajan V (2009) FTY720 inhibits ceramide synthases and up-regulates dihydrosphingosine 1-phosphate formation in human lung endothelial cells. J Biol Chem 284:5467–5477
Betito S, Cuvillier O (2006) Regulation by sphingosine 1-phosphate of Bax and Bad activities during apoptosis in a MEK-dependent manner. Biochem Biophys Res Commun 340:1273–1277
Bonhoure E, Lauret A, Barnes DJ, Martin C, Malavaud B, Kohama T, Melo JV, Cuvillier O (2008) Sphingosine kinase-1 is a downstream regulator of imatinib-induced apoptosis in chronic myeloid leukemia cells. Leukemia 22:971–979
Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, Bruns C, Prieschl E, Baumruker T, Hiestand P, Foster CA, Zollinger M, Lynch KR (2002) The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem 277:21453–21457
Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P (2010) Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov 9:883–897
Chan MM, Lu X, Merchant FM, Iglehart JD, Miron PL (2005) Gene expression profiling of NMU-induced rat mammary tumors: cross species comparison with human breast cancer. Carcinogenesis 26:1343–1353
Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148:988–1000
Davis MD, Clemens JJ, Macdonald TL, Lynch KR (2005) Sphingosine 1-phosphate analogs as receptor antagonists. J Biol Chem 280:9833–9841
De Luca T, Morre DM, Zhao H, Morre DJ (2005) NAD+/NADH and/or CoQ/CoQH2 ratios from plasma membrane electron transport may determine ceramide and sphingosine-1-phosphate levels accompanying G1 arrest and apoptosis. Biofactors 25:43–60
Dolgachev V, Farooqui MS, Kulaeva OI, Tainsky MA, Nagy B, Hanada K, Separovic D (2004) De novo ceramide accumulation due to inhibition of its conversion to complex sphingolipids in apoptotic photosensitized cells. J Biol Chem 279:23238–23249
Doll F, Pfeilschifter J, Huwiler A (2007) Prolactin upregulates sphingosine kinase-1 expression and activity in the human breast cancer cell line MCF7 and triggers enhanced proliferation and migration. Endocr Relat Cancer 14:325–335
Fox TE, Houck KL, O’Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M (2007) Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J Biol Chem 282:12450–12457
French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD (2003) Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res 63:5962–5969
French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD (2006) Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther 318:596–603
French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD (2010) Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther 333:129–139
Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K, Ishida J, Uemura A, Kiyonari H, Abe T, Fukamizu A, Hirashima M, Sawa H, Aoki J, Ishii M, Mochizuki N (2012) The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest 122:1416–1426
Futerman AH, Hannun YA (2004) The complex life of simple sphingolipids. EMBO Rep 5:777–782
Gamble JR, Xia P, Hahn CN, Drew JJ, Drogemuller CJ, Brown D, Vadas MA (2006) Phenoxodiol, an experimental anticancer drug, shows potent antiangiogenic properties in addition to its antitumour effects. Int J Cancer 118:2412–2420
Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM (2004) Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res 64:55–63
Guillermet-Guibert J, Davenne L, Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C, Delisle MB, Cuvillier O, Susini C, Bousquet C (2009) Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol Cancer Ther 8:809–820
Hait NC, Sarkar S, Le Stunff H, Mikami A, Maceyka M, Milstien S, Spiegel S (2005) Role of sphingosine kinase 2 in cell migration toward epidermal growth factor. J Biol Chem 280:29462–29469
Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S (2007) Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J Biol Chem 282:12058–12065
Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325:1254–1257
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9:139–150
Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, Stevens RC (2012) Crystal structure of a lipid G protein-coupled receptor. Science 335:851–855
Heinrich M, Wickel M, Winoto-Morbach S, Schneider-Brachert W, Weber T, Brunner J, Saftig P, Peters C, Kronke M, Schutze S (2000) Ceramide as an activator lipid of cathepsin D. Adv Exp Med Biol 477:305–315
Hisano Y, Kobayashi N, Kawahara A, Yamaguchi A, Nishi T (2011) The sphingosine 1-phosphate transporter, SPNS2, functions as a transporter of the phosphorylated form of the immunomodulating agent FTY720. J Biol Chem 286:1758–1766
Igarashi Y, Hakomori S, Toyokuni T, Dean B, Fujita S, Sugimoto M, Ogawa T, el-Ghendy K, Racker E (1989) Effect of chemically well-defined sphingosine and its N-methyl derivatives on protein kinase C and src kinase activities. Biochemistry 28:6796–6800
Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem 278:46832–46839
Ikeda M, Kihara A, Igarashi Y (2004) Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem Biophys Res Commun 325:338–343
Illuzzi G, Bernacchioni C, Aureli M, Prioni S, Frera G, Donati C, Valsecchi M, Chigorno V, Bruni P, Sonnino S, Prinetti A (2010) Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl)retinamide in human ovarian cancer cells. J Biol Chem 285:18594–18602
Johnson KR, Johnson KY, Crellin HG, Ogretmen B, Boylan AM, Harley RA, Obeid LM (2005) Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J Histochem Cytochem 53:1159–1166
Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, Zipkin RE, Dent P, Kordula T, Milstien S, Spiegel S (2009) Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res 69:6915–6923
Kawahara A, Nishi T, Hisano Y, Fukui H, Yamaguchi A, Mochizuki N (2009) The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 323:524–527
Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T, Wargovich MJ, Reddy BS, Hannun YA, Obeid LM, Zhou D (2006) Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J 20:386–388
Kedderis LB, Bozigian HP, Kleeman JM, Hall RL, Palmer TE, Harrison SD Jr, Susick RL Jr (1995) Toxicity of the protein kinase C inhibitor safingol administered alone and in combination with chemotherapeutic agents. Fundam Appl Toxicol 25:201–217
Kim JW, Kim YW, Inagaki Y, Hwang YA, Mitsutake S, Ryu YW, Lee WK, Ha HJ, Park CS, Igarashi Y (2005) Synthesis and evaluation of sphingoid analogs as inhibitors of sphingosine kinases. Bioorg Med Chem 13:3475–3485
King CC, Zenke FT, Dawson PE, Dutil EM, Newton AC, Hemmings BA, Bokoch GM (2000) Sphingosine is a novel activator of 3-phosphoinositide-dependent kinase 1. J Biol Chem 275:18108–18113
Knapp P, Baranowski M, Knapp M, Zabielski P, Blachnio-Zabielska AU, Gorski J (2010) Altered sphingolipid metabolism in human endometrial cancer. Prostaglandins Other Lipid Mediat 92:62–66
Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S (1998) Molecular cloning and functional characterization of murine sphingosine kinase. J Biol Chem 273:23722–23728
Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, Ai Y, Ristimaki AP, Fyrst H, Sano H, Rosenberg D, Saba JD, Proia RL, Hla T (2006) Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol 26:7211–7223
Kondo Y, Kondo S (2006) Autophagy and cancer therapy. Autophagy 2:85–90
Kono K, Tanaka M, Mizuno T, Kodama K, Ogita T, Kohama T (2000a) B-535a, b and c, new sphingosine kinase inhibitors, produced by a marine bacterium; taxonomy, fermentation, isolation, physico-chemical properties and structure determination. J Antibiot (Tokyo) 53:753–758
Kono K, Tanaka M, Ogita T, Kohama T (2000b) Characterization of B-5354c, a new sphingosine kinase inhibitor, produced by a marine bacterium. J Antibiot (Tokyo) 53:759–764
Kono K, Sugiura M, Kohama T (2002) Inhibition of recombinant sphingosine kinases by novel inhibitors of microbial origin, F-12509A and B-5354c. J Antibiot (Tokyo) 55:99–103
Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, Obeid LM, Bielawska A (2007) Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem 282:16718–16728
Lahiri S, Park H, Laviad EL, Lu X, Bittman R, Futerman AH (2009) Ceramide synthesis is modulated by the sphingosine analog FTY720 via a mixture of uncompetitive and noncompetitive inhibition in an Acyl-CoA chain length-dependent manner. J Biol Chem 284:16090–16098
Laviad EL, Kelly S, Merrill AH Jr, Futerman AH (2012) Modulation of ceramide synthase activity via dimerization. J Biol Chem 287(25):21025–21033
Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, Botti J, Codogno P (2006) Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem 281:8518–8527
Le Scolan E, Pchejetski D, Banno Y, Denis N, Mayeux P, Vainchenker W, Levade T, Moreau-Gachelin F (2005) Overexpression of sphingosine kinase 1 is an oncogenic event in erythroleukemic progression. Blood 106:1808–1816
Lepine S, Allegood JC, Edmonds Y, Milstien S, Spiegel S (2011) Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J Biol Chem 286:44380–44390
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Li QF, Wu CT, Guo Q, Wang H, Wang LS (2008) Sphingosine 1-phosphate induces Mcl-1 upregulation and protects multiple myeloma cells against apoptosis. Biochem Biophys Res Commun 371:159–162
Li MH, Sanchez T, Milne GL, Morrow JD, Hla T, Ferrer F (2009) S1P/S1P2 signaling induces cyclooxygenase-2 expression in Wilms tumor. J Urol 181:1347–1352
Lim KG, Sun C, Bittman R, Pyne NJ, Pyne S (2011) (R)-FTY720 methyl ether is a specific sphingosine kinase 2 inhibitor: effect on sphingosine kinase 2 expression in HEK 293 cells and actin rearrangement and survival of MCF-7 breast cancer cells. Cell Signal 23:1590–1595
Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S (2000) Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem 275:19513–19520
Liu H, Chakravarty D, Maceyka M, Milstien S, Spiegel S (2002) Sphingosine kinases: a novel family of lipid kinases. Prog Nucleic Acid Res Mol Biol 71:493–511
Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, Milstien S, Spiegel S (2003) Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem 278:40330–40336
Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Enright E, Stecker K, Estepa-Sabal E, Smith B, Younger J, Balis U, Michaelson J, Bhan A, Habin K, Baer TM, Brugge J, Haber DA, Erlander MG, Sgroi DC (2004) A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell 5:607–616
Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH Jr, Milstien S, Spiegel S (2005) SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem 280:37118–37129
Mandala SM (2001) Sphingosine-1-phosphate phosphatases. Prostaglandins Other Lipid Mediat 64:143–156
Mao C, Saba JD, Obeid LM (1999) The dihydrosphingosine-1-phosphate phosphatases of Saccharomyces cerevisiae are important regulators of cell proliferation and heat stress responses. Biochem J 342(Pt 3):667–675
Matsuoka Y, Nagahara Y, Ikekita M, Shinomiya T (2003) A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Br J Pharmacol 138:1303–1312
McDonald OB, Hannun YA, Reynolds CH, Sahyoun N (1991) Activation of casein kinase II by sphingosine. J Biol Chem 266:21773–21776
Megidish T, Cooper J, Zhang L, Fu H, Hakomori S (1998) A novel sphingosine-dependent protein kinase (SDK1) specifically phosphorylates certain isoforms of 14-3-3 protein. J Biol Chem 273:21834–21845
Meyers-Needham M, Lewis JA, Gencer S, Sentelle RD, Saddoughi SA, Clarke CJ, Hannun YA, Norell H, da Palma TM, Nishimura M, Kraveka JM, Khavandgar Z, Murshed M, Cevik MO, Ogretmen B (2012a) Off-target function of the Sonic hedgehog inhibitor cyclopamine in mediating apoptosis via nitric oxide-dependent neutral sphingomyelinase 2/ceramide induction. Mol Cancer Ther 11:1092–1102
Meyers-Needham M, Ponnusamy S, Gencer S, Jiang W, Thomas RJ, Senkal CE, Ogretmen B (2012b) Concerted functions of HDAC1 and microRNA-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol Med 4:78–92
Michel C, van Echten-Deckert G, Rother J, Sandhoff K, Wang E, Merrill AH Jr (1997) Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide. J Biol Chem 272:22432–22437
Miller AV, Alvarez SE, Spiegel S, Lebman DA (2008) Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol Cell Biol 28:4142–4151
Mitra P, Oskeritzian CA, Payne SG, Beaven MA, Milstien S, Spiegel S (2006) Role of ABCC1 in export of sphingosine-1-phosphate from mast cells. Proc Natl Acad Sci USA 103:16394–16399
Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL (2005) Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol 25:11113–11121
Mukhopadhyay A, Saddoughi SA, Song P, Sultan I, Ponnusamy S, Senkal CE, Snook CF, Arnold HK, Sears RC, Hannun YA, Ogretmen B (2009) Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J 23:751–763
Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Yamada A, Zhao R, Milstien S, Zhou H, Spiegel S, Takabe K (2012) Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res 72:726–735
Nagaoka Y, Otsuki K, Fujita T, Uesato S (2008) Effects of phosphorylation of immunomodulatory agent FTY720 (fingolimod) on antiproliferative activity against breast and colon cancer cells. Biol Pharm Bull 31:1177–1181
Nava VE, Cuvillier O, Edsall LC, Kimura K, Milstien S, Gelmann EP, Spiegel S (2000) Sphingosine enhances apoptosis of radiation-resistant prostate cancer cells. Cancer Res 60:4468–4474
Nava VE, Hobson JP, Murthy S, Milstien S, Spiegel S (2002) Sphingosine kinase type 1 promotes estrogen-dependent tumorigenesis of breast cancer MCF-7 cells. Exp Cell Res 281:115–127
Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, Liu S, Trotta R, Muthusamy N, Gambacorti-Passerini C, Druker BJ, Cortes J, Marcucci G, Chen CS, Verrills NM, Roy DC, Caligiuri MA, Bloomfield CD, Byrd JC, Perrotti D (2007) FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest 117:2408–2421
Nindl I, Dang C, Forschner T, Kuban RJ, Meyer T, Sterry W, Stockfleth E (2006) Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol Cancer 5:30
Ogretmen B, Hannun YA (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4:604–616
Ogretmen B, Kraveka JM, Schady D, Usta J, Hannun YA, Obeid LM (2001) Molecular mechanisms of ceramide-mediated telomerase inhibition in the A549 human lung adenocarcinoma cell line. J Biol Chem 276:32506–32514
Olivera A, Kohama T, Edsall L, Nava V, Cuvillier O, Poulton S, Spiegel S (1999) Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol 147:545–558
Osborne N, Brand-Arzamendi K, Ober EA, Jin SW, Verkade H, Holtzman NG, Yelon D, Stainier DY (2008) The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr Biol 18:1882–1888
Paugh SW, Paugh BS, Rahmani M, Kapitonov D, Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, Spiegel S (2008) A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 112:1382–1391
Payne SG, Oskeritzian CA, Griffiths R, Subramanian P, Barbour SE, Chalfant CE, Milstien S, Spiegel S (2007) The immunosuppressant drug FTY720 inhibits cytosolic phospholipase A2 independently of sphingosine-1-phosphate receptors. Blood 109:1077–1085
Pchejetski D, Golzio M, Bonhoure E, Calvet C, Doumerc N, Garcia V, Mazerolles C, Rischmann P, Teissie J, Malavaud B, Cuvillier O (2005) Sphingosine kinase-1 as a chemotherapy sensor in prostate adenocarcinoma cell and mouse models. Cancer Res 65:11667–11675
Pitson SM (2011) Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci 36:97–107
Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW (2003) Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J 22:5491–5500
Pitson SM, Xia P, Leclercq TM, Moretti PA, Zebol JR, Lynn HE, Wattenberg BW, Vadas MA (2005) Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J Exp Med 201:49–54
Ponnusamy S, Meyers-Needham M, Senkal CE, Saddoughi SA, Sentelle D, Selvam SP, Salas A, Ogretmen B (2010) Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol 6:1603–1624
Ponnusamy S, Selvam SP, Mehrotra S, Kawamori T, Snider AJ, Obeid LM, Shao Y, Sabbadini R, Ogretmen B (2012) Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol Med 4(8):761–775
Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM, Kelsey KT, Turek LP, Ahlquist P (2007) Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res 67:4605–4619
Reynolds CP, Maurer BJ, Kolesnick RN (2004) Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett 206:169–180
Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, Grosch S, Geisslinger G, Holtrich U, Karn T, Kaufmann M (2008) Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat 112:41–52
Sabbadini RA (2011) Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br J Pharmacol 162:1225–1238
Saddoughi SA, Song P, Ogretmen B (2008) Roles of bioactive sphingolipids in cancer biology and therapeutics. Subcell Biochem 49:413–440
Salas A, Ponnusamy S, Senkal CE, Meyers-Needham M, Selvam SP, Saddoughi SA, Apohan E, Sentelle RD, Smith C, Gault CR, Obeid LM, El-Shewy HM, Oaks J, Santhanam R, Marcucci G, Baran Y, Mahajan S, Fernandes D, Stuart R, Perrotti D, Ogretmen B (2011) Sphingosine kinase-1 and sphingosine 1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug resistance by modulation of protein phosphatase 2A. Blood 117:5941–5952
Sankala HM, Hait NC, Paugh SW, Shida D, Lepine S, Elmore LW, Dent P, Milstien S, Spiegel S (2007) Involvement of sphingosine kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res 67:10466–10474
Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, Wei SH, Parker I, Jo E, Cheng WC, Cahalan MD, Wong CH, Rosen H (2006) Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol 2:434–441
Sarkar S, Maceyka M, Hait NC, Paugh SW, Sankala H, Milstien S, Spiegel S (2005) Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett 579:5313–5317
Sato K, Malchinkhuu E, Horiuchi Y, Mogi C, Tomura H, Tosaka M, Yoshimoto Y, Kuwabara A, Okajima F (2007) Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J Neurochem 103:2610–2619
Sauer B, Gonska H, Manggau M, Kim DS, Schraut C, Schafer-Korting M, Kleuser B (2005) Sphingosine 1-phosphate is involved in cytoprotective actions of calcitriol in human fibroblasts and enhances the intracellular Bcl-2/Bax rheostat. Pharmazie 60:298–304
Schmid G, Guba M, Ischenko I, Papyan A, Joka M, Schrepfer S, Bruns CJ, Jauch KW, Heeschen C, Graeb C (2007) The immunosuppressant FTY720 inhibits tumor angiogenesis via the sphingosine 1-phosphate receptor 1. J Cell Biochem 101:259–270
Schnitzer SE, Weigert A, Zhou J, Brune B (2009) Hypoxia enhances sphingosine kinase 2 activity and provokes sphingosine-1-phosphate-mediated chemoresistance in A549 lung cancer cells. Mol Cancer Res 7:393–401
Schwartz GK, Ward D, Saltz L, Casper ES, Spiess T, Mullen E, Woodworth J, Venuti R, Zervos P, Storniolo AM, Kelsen DP (1997) A pilot clinical/pharmacological study of the protein kinase C-specific inhibitor safingol alone and in combination with doxorubicin. Clin Cancer Res 3:537–543
Senkal CE, Ponnusamy S, Manevich Y, Meyers-Needham M, Saddoughi SA, Mukhopadyay A, Dent P, Bielawski J, Ogretmen B (2011) Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J Biol Chem 286(49):42446–42458
Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4:397–407
Stahelin RV, Hwang JH, Kim JH, Park ZY, Johnson KR, Obeid LM, Cho W (2005) The mechanism of membrane targeting of human sphingosine kinase 1. J Biol Chem 280:43030–43038
Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S (2010) Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol 688:141–155
Strub GM, Paillard M, Liang J, Gomez L, Allegood JC, Hait NC, Maceyka M, Price MM, Chen Q, Simpson DC, Kordula T, Milstien S, Lesnefsky EJ, Spiegel S (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J 25:600–612
Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S, Kohama T (2002) Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem 277:23294–23300
Sukocheva O, Wang L, Verrier E, Vadas MA, Xia P (2009) Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology 150:4484–4492
Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, Nagahashi M, Harikumar KB, Hait NC, Milstien S, Spiegel S (2010) Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem 285:10477–10486
Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y (2005) Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res 11:7234–7242
Tonelli F, Lim KG, Loveridge C, Long J, Pitson SM, Tigyi G, Bittman R, Pyne S, Pyne NJ (2010) FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cell Signal 22:1536–1542
Vadas M, Xia P, McCaughan G, Gamble J (2008) The role of sphingosine kinase 1 in cancer: oncogene or non-oncogene addiction? Biochim Biophys Acta 1781:442–447
Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW (2005) Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol 64:695–705
Vessey DA, Kelley M, Zhang J, Li L, Tao R, Karliner JS (2007) Dimethylsphingosine and FTY720 inhibit the SK1 form but activate the SK2 form of sphingosine kinase from rat heart. J Biochem Mol Toxicol 21:273–279
Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, Garland WA, Lu Y, Yu S, Hall HS, Kundra V, Mills GB, Sabbadini RA (2006) Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 9:225–238
Wang G, Silva J, Krishnamurthy K, Tran E, Condie BG, Bieberich E (2005) Direct binding to ceramide activates protein kinase Czeta before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells. J Biol Chem 280:26415–26424
Watson C, Long JS, Orange C, Tannahill CL, Mallon E, McGlynn LM, Pyne S, Pyne NJ, Edwards J (2010) High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am J Pathol 177:2205–2215
Wong YF, Selvanayagam ZE, Wei N, Porter J, Vittal R, Hu R, Lin Y, Liao J, Shih JW, Cheung TH, Lo KW, Yim SF, Yip SK, Ngong DT, Siu N, Chan LK, Chan CS, Kong T, Kutlina E, McKinnon RD, Denhardt DT, Chin KV, Chung TK (2003) Expression genomics of cervical cancer: molecular classification and prediction of radiotherapy response by DNA microarray. Clin Cancer Res 9:5486–5492
Wooten LG, Ogretmen B (2005) Sp1/Sp3-dependent regulation of human telomerase reverse transcriptase promoter activity by the bioactive sphingolipid ceramide. J Biol Chem 280:28867–28876
Wooten-Blanks LG, Song P, Senkal CE, Ogretmen B (2007) Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J 21:3386–3397
Xia P, Gamble JR, Wang L, Pitson SM, Moretti PA, Wattenberg BW, D’Andrea RJ, Vadas MA (2000) An oncogenic role of sphingosine kinase. Curr Biol 10:1527–1530
Xiang Y, Asmussen G, Booker M, Hirth B, Kane JL Jr, Liao J, Noson KD, Yee C (2009) Discovery of novel sphingosine kinase 1 inhibitors. Bioorg Med Chem Lett 19:6119–6121
Xiao M, Liu Y, Zou F (2012) Sensitization of human colon cancer cells to sodium butyrate-induced apoptosis by modulation of sphingosine kinase 2 and protein kinase D. Exp Cell Res 318:43–52
Acknowledgments
We thank the members of the Ogretmen laboratory for their helpful discussions and apologize to those investigators whose important work was not included in this chapter because of space limitations. BO is funded by research grants from the National Institutes of Health. We thank Dr. Jennifer Schnellmann for her editorial review.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Selvam, S.P., Ogretmen, B. (2013). Sphingosine Kinase/Sphingosine 1-Phosphate Signaling in Cancer Therapeutics and Drug Resistance. In: Gulbins, E., Petrache, I. (eds) Sphingolipids in Disease. Handbook of Experimental Pharmacology, vol 216. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1511-4_1
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1511-4_1
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1510-7
Online ISBN: 978-3-7091-1511-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)