
Abstract
Sphingosine-1-phosphate (S1P) can regulate several physiological and pathological processes. S1P signaling via its cell surface receptor S1PR1 has been shown to enhance tumorigenesis and stimulate growth, expansion, angiogenesis, metastasis, and survival of cancer cells. S1PR1-mediated tumorigenesis is supported and amplified by activation of downstream effectors including STAT3, interleukin-6, and NF-κB networks. S1PR1 signaling can also trigger various other signaling pathways involved in carcinogenesis including activation of PI3K/AKT, MAPK/ERK1/2, Rac, and PKC/Ca, as well as suppression of cyclic adenosine monophosphate (cAMP). It also induces immunological tolerance in the tumor microenvironment, while the immunosuppressive function of S1PR1 can also lead to the generation of pre-metastatic niches. Some tumor cells upregulate S1PR1 signaling pathways, which leads to drug resistant cancer cells, mainly through activation of STAT3. This signaling pathway is also implicated in some inflammatory conditions leading to the instigation of inflammation-driven cancers. Furthermore, it can also increase survival via induction of anti-apoptotic pathways, for instance, in breast cancer cells. Therefore, S1PR1 and its signaling pathways can be considered as potential anti-tumor therapeutic targets, alone or in combination therapies. Given the oncogenic nature of S1PR1 and its distribution in a variety of cancer cell types along with its targeting advantages over other molecules of this family, S1PR1 should be considered a favorable target in therapeutic approaches to cancer. This review describes the role of S1PR1 in cancer development and progression, specifically addressing breast cancer, glioma, and hematopoietic malignancies. We also discuss the potential use of S1P signaling modulators as therapeutic targets in cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
S1PR1 expression is dysregulated in various cancer cells. |
Oncogenesis is enhanced by S1PR1 signaling. |
Targeting S1PR1 may provide an effective therapeutic approach in cancer treatment. |
1 Introduction
The sphingosine-1-phosphate (S1P) can regulate several physiological functions, including differentiation, proliferation, migration, and survival of cells. Because of its wide variety of functions, it is considered a potent mediator of tumor growth. Phosphorylation of sphingosine by sphingosine kinases (SPHK1 and 2) leads to generation of S1P at cellular membranes [1]. S1P functions via binding to its receptors, S1PR1–S1PR5 [2]. S1PR1, 2, and 3 are ubiquitously expressed on various cell types, whereas S1PR4 and 5 are mainly present on immune cells and in the nervous system [3]. While S1PR1 and 3 facilitate cell migration, S1PR2 suppresses migration [4,5,6]. However, in a study on oral squamous cell carcinoma, a migratory response through S1PR2 was observed [7]. Moreover, S1PR1 and 3 enhance proliferation and survival of cells, whereas S1PR2 inhibits them [4,5,6]. Although, the concentration of S1P in normal tissues is tightly controlled by S1P lyase or S1P phosphatases to remain in picomolar levels, it is present in high levels in the tumor microenvironment were it enhances cancer progression.
Among the S1PRs, it seems that S1PR1 plays a key role in tumor development [1]. It is also involved in various processes including neovascularization, migration of immune cells, survival of stem cells, and generation of cytokines [8]. It has recently been demonstrated that there is a close relationship between signaling of S1PR1 and persistent activation of STAT3 in tumor cells. There is a positive feedback loop in which these two factors stimulate and activate each other to synergistically enhance tumor growth [9]. In addition to STAT3, S1PR1 signaling can also enhance survival, expansion, and spreading of tumor cells through activation of the extracellular signal–regulated kinase (ERK), Akt, and Rac pathways [10]. Besides using Gi/o signaling molecules, S1PR1 can also instigate or inhibit various signaling pathways, such as stimulating Ras/ERK to enhance cell division, phosphoinositide 3-kinase (PI3K) and PKB/Akt to promote survival, PI3K and Rac to enhance migration, and PKC and phospholipase C (PLC) to upregulate intracellular calcium levels involved in multiple cellular functions, and inhibiting adenylyl cyclase activity to promote tumor spreading [11, 12]. Therefore, it has been shown that S1PR1 signaling can promote all tumor hallmarks [13]. S1PR1-STAT3 signaling also induces intravasation of myeloid cells into distant organs and provides a pre-metastatic microenvironment [14]. Hence, blockade of the S1P/S1PRs axis seems to be effective in cancer therapy (Table 1); however, which one is the more effective target, S1P or S1PR??
The answer can be be identified in studies that address the contrasting effects of various S1PRs’ suppression. While suppression of S1PR1 inhibited angiogenesis, depletion of S1PR2 enhanced tumor development and the angiogenesis process. Therefore, it is evident that suppression of cancer-promoting S1PRs has advantages compared to S1P targeting [1, 15]. S1PR1 targeting not only exerts anti-tumor effects, but also drives higher expression level and activation of S1PR2, which has anti-cancer effects. Association of S1PR1 signaling with inflammation and colitis-associated cancers is another reason to target this receptor in such cancers [16]. Therefore, given the oncogenesis-inducing signaling pathways of S1PR1, the fact it has a high incidence in various cancer cells and its targeting advantages over other molecules of this family, S1PR1 is an interesting target for the development cancer therapies.
2 Structure and Signaling of S1PR1
2.1 Fate of S1P
Generation of S1P from sphingosine is mediated through activation of kinases by various factors, including cytokines, which catalyze SPHK1 phosphorylation [17]. Tumor necrosis factor (TNF)α receptors activate SPHK1, which produces S1P that then binds to TRAF2, and via signaling pathways, NF-κB activation occurs. SPHK2-produced S1P binds to HDAC1/2 in the nucleus and activates various genes (e.g., p21, cfos). S1P produced by SPHK2 in mitochondria binds to PHB2, and together regulate assembly of complex IV of cytochrome C oxidase. It also modulates proteinase activity on the amyloid-β precursor protein by binding to BACE1 at the plasma membrane [18]. Following this, SPHK1 translocates to the cell membrane and produces S1P, which can be secreted to act as a ligand for S1PRs (S1PR1–5) [17]. Based on the cell type, the intracellular generated S1P can be secreted by either spinster homolog 2 (Spns2) or ATP-binding cassette (ABC) transporters. Following secretion, it transduces signaling messages through its five G-protein-coupled receptors (GPCRs) (S1PR1–5) in an autocrine or paracrine manner [19]. This process is called ‘inside-out’ signaling [2].
2.2 Structure of S1PRs
S1PRs are GPCRs [20]. GPCRs use heterotrimeric G-proteins (GTP-binding proteins) composed of α, β, and γ subunits for transducing intracellular signaling [21]. While the βγ subunits are similar in various GPCRs, G-proteins are categorized into four groups based on isoforms of α subunits including Gs, Gi/o, Gq/11, and G12/13 [22].
S1PR1 contains 382 amino acids constituting three general sections: (1) the seven transmembrane α-helices spanning the lipid bilayer (TM1–TM7); (2) three extracellular N-terminus loops (ECL1–ECL3); and (3) three intracellular C terminus loops (ICL1–ICL3). The specific amino acids of the α-helices that are named below participate in hydrogen bonding that, all together, make an interhelical network in the structure of S1PR1: Asn63 from TM1 connects to Asp91 from TM2, Asn86 (TM2) to Ser134 (TM3), Asn86 (TM2) to Trp168 (TM4), Asp91 (TM2) to Ser304 (TM7), Ser131 (TM3) to Ser304 (TM7), and Trp182 (TM4) to His2O1 (TM5). In this interhelical network of hydrogen bonding, TM6 does not have a role [23].
2.3 S1PR1 Signaling
Binding of S1P with S1PR1 can lead to formation of complexes composed of G-protein or β-arrestin which are involved in the modulation of the ERK1/2 signaling pathway and migration [24]. Following binding with S1P, β-arrestin recruits c-Src to S1PR1, which is an important molecule in the S1PR1 signaling process. Subsequently, βγ subunits of G-protein facilitate activation of c-Src, Raf, MEK and ERK1/2 pathways [25, 26]. It has been shown that stimulation of S1PR1 can trigger various signaling pathways, including the RAS/ERK, PI3K/AKT, and RAC pathways. Moreover, it can also stimulate PLC and Ca2+ mobilization pathways [27, 28]. However, in one study, S1PR1 inhibited Ca2+ signaling in some cell lines. In this study, it was observed that co-expression of S1PR1 reversed this inhibitory effect [29]. Stimulation of chemotaxis by S1PR1 is done in part via PI3K and Rac pathways, which are associated with activation of intracellular small GTPases and regulation of the actin cytoskeleton [27, 28]. Thereby, stimulation of S1PR1 can lead to the process of tumor progression, in part through inhibitory G-protein [4].
Among the S1PRs, S1PR 1 and 3 stimulate cell migration via coupling to Gi/o and activating the Rac GTPase pathway. Coupling of S1PR2 and 3 to G12/13 activates the Rho/ROCK pathway, leading to cell migration arrest. Therefore, S1PR1 can enhance the cell migration process based on subsequent activation of Rac in different cell compartments [11].
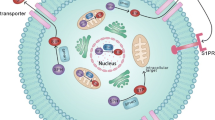
S1PRs are also critical mediators in the maintenance of vascular permeability barriers. While S1PR1 increases the barrier function of endothelium, S1PR2 and 3 decrease it. Coupling of S1PR1 with Gi/o proteins in endothelium activates Rac and rearranges the cytoskeleton, leading to the generation of cortical actin rings and stabilization of cellular shape. This signaling can also enhance cell adherence and tight junctions in order to stabilize cellular connections and enhance barrier function [11]. Following binding to hyaluronan, CD44 transactivates S1PR1, which leads to Rac1 signaling and endothelium barrier enhancement [30]. In addition to CD44, it has been shown that vascular endothelial growth factor (VEGF) signaling in endothelial cells can enhance expression of S1PR1, which can potentially increase endothelial nitric oxide synthase (eNOS) activation and barrier function (Figs. 1, 2) [11, 31].

S1PR1 signaling in tumor cells, leading to activation of various downstream molecules. AC Adenylyl cyclase, AKT Protein kinase B, AP-1 Activator protein 1, cAMP Cyclic adenosine monophosphate, CREB cAMP response element binding protein, DAG diacylglycerol, EGR-1 Early growth response protein 1, ERK Extracellular Signal-regulated Kinase, GDP guanosine diphosphate, GTP Guanosine triphosphate, IL-6 Interleukin 6, IP3 Inositol 1,4,5-triphosphate, JAK2 Janus kinase 2, JNK c-Jun N-terminal kinase, MTOR mechanistic target of rapamycin, MAPK mitogen-activated protein kinase, NOS Nitric oxide synthases, NFAT Nuclear factor of activated T-cells, NF-KB Nuclear Factor kappaB, PGE2 Prostaglandin E2, PIP2 phosphatidylinositol (4,5)-bisphosphate, PI3K Phosphoinositide 3-kinase, PKA protein kinase A, PKC Protein kinase C, PLC Phospholipase C, S1P Sphingosine-1-phosphate, S1PR1 Sphingosine-1-phosphate receptor 1, STAT3 Signal transducer and activator of transcription 3

Fate of sphingosine-1-phosphate (S1P) produced by cancer cells and the impacts of S1PR1 signaling on tumor cells. Signaling pathways activated by S1PR1 stimulate cancer progression processes such as increasing proliferation, survival, angiogenesis, metastasis, inflammation, chemoresistance and decreasing apoptosis. AKT Protein kinase B, AP-1 Activator protein 1, E2/Era Estradiol/estrogen receptor, EGR-1 Early growth response protein 1, ERK Extracellular Signal-regulated Kinase, HIF Hypoxia-inducible factor, IL-6 Interleukin 6, JAK2 Janus kinase 2, NF-KB Nuclear Factor kappaB, NOS Nitric oxide synthases, PGE2 Prostaglandin E2, PI3K Phosphoinositide 3-kinase, S1P Sphingosine-1-phosphate, S1PR1 Sphingosine-1-phosphate receptor 1, SPHK1 Sphingosine Kinase 1, STAT3 Signal transducer and activator of transcription 3, VEGFR Vascular endothelial growth factor receptor
3 S1PR1 Expression and Signaling Role in Tumor Microenvironment
A region of tumor is composed of several cell types such as malignant cancerous cells, normal stromal cells, various lymphocytes, myeloid cells, fibroblasts, mast cells, and endothelial cells. In addition to this complex cellular network, there are multiple non-cell factors such as hypoxia, glucose, cytokines, and adenosine in the tumor microenvironment which provide optimum conditions for expansion of cancerous cells [32, 33].
3.1 Role of S1PR1 in Cell Trafficking
It has been demonstrated that S1PR1 can affect both the trafficking and differentiation of lymphocytes in the tumor microenvironment [27]. It has been reported that S1PR1 signaling not only regulates and suppresses anti-tumor immune responses, but also enhances tumor growth [34]. As immune cells express S1PR1 and response to S1P gradient in different kinds of immune responses and get orchestrated by its signaling in various pathological states, it is important to determine its exact impacts on priming of immune cells and clarify the effects of its downstream signaling pathways such as STAT3 on differentiation and function of immune cells in the tumor microenvironment. Since little is known regarding the role of S1PR1 signaling in cell trafficking in the tumor microenvironment and lymphocyte trafficking is required for immune responses and it is important for recognition of cancer cells by immune cells, we suggest that future studies should determine the effect of S1PR1 signaling on immune cells in the cancer microenvironment much more thoroughly and accurately.
3.2 Induction of Immunological Tolerance by S1PR1
S1PR1 signaling causes an immunological tolerance in the tumor microenvironment and promotes cancer progression by inducing differentiation of regulatory T cells (Tregs) via activating JAK/STAT3 signaling and also by inhibiting recruitment and activation of CD8+ T cells [35]. S1PR1 also enhances translocation of tumor antigen-specific Tregs from bone marrow into tumor tissue in patients with breast cancer [36].
S1P can drive the differentiation of T helper 17 (Th17) cells by inducing the formation of STAT3 and interleukin (IL)-6 via S1PR1 signaling. STAT3 stimulates the expression of RORγt and subsequently induces Th17 differentiation [9, 37, 38].
The S1PR1–STAT3 signaling in several cells of pre-metastatic sites enhances infiltration of myeloid cells into distant organs. This infiltration can be disrupted by targeting STAT3/S1PR1 signaling in these cells [8, 14].
3.3 Role of S1PR1 Signaling Network in Regulation of Angiogenesis
Interestingly, it has been suggested that S1PR1 is an effective factor in the development of the angiogenesis process, partly through S1P [39]. Consistently, small interfering RNA (siRNA)-mediated downregulation of S1PR1 led to suppression of tumor angiogenesis [40]. Similarly, functional inhibition of S1PR1 by FTY20 suppressed tumor growth and angiogenesis [41]. The barrier function of endothelial cells and cell spreading is enhanced in part through S1PR1-Gαi-Rac1 and S1PR1-Gαi-Cdc42 pathways. In contrast, an S1PR2-Gα12/13-RhoA pathway attenuates this function [42, 43]. Moreover, related transcriptional enhancer factor-1 (RTEF-1), another angiogenesis-promoting factor, also upregulates the expression of S1PR1 [44]. Since the hypoxia upregulates the expression of S1PR1, it can lead to the enhancement of endothelial cell migration and neovascularization [45]. In contrast, there is evidence indicating anti-angiogenic effects of S1PR1 in the tumor microenvironment. It has been demonstrated that signaling of S1PR1 prevents angiogenic sprouting and increases cell-to-cell adhesion, which were associated with VEGF-A and VE-cadherin in endothelial cells [46, 47]. It is unknown whether this anti-angiogenic effect of S1PR1 is similarly present in tumor-related angiogenesis as well as normal development processes [2]. However, it has been reported that S1PR1 agonism decreases tumor growth through inhibiting adequate vascularization [48]. Further investigation of its exact effect on angiogenesis and endothelial barrier function is requisite, since there are some controversies regarding this matter.
3.4 S1PR1 Role in Cell migration and Apoptosis
Migration of cancerous cells toward platelet-derived growth factor (PDGF) depends on S1PR1 expression. On the other hand, PDGF enhances the expression of sphingosine kinase and upregulates intracellular S1P. Accordingly, it has been shown that suppressing S1PR1 or sphingosine kinase prevented chemotaxis toward PDGF and inhibited activation of Rac, leading to migration arrest of tumor cells [49]. Also in airway smooth muscle cells, PDGF and S1P via complexes of PDGF beta receptor-S1PR1 act to enhance mitogenic signaling and stimulate p42/p44 MAPK phosphorylation [50]. S1PR1 also forms a signaling complex with vascular endothelial growth factor receptor (VEGFR)-2 that altogether evokes ERK1/2 and PKC-alpha and subsequently regulates cell migration of ML-1 thyroid carcinoma [51]. The crosstalk between growth factor receptors and S1PR1 has yet to be elucidated.
Apoptosis and resistance to apoptosis is one of the main drawbacks in cancer treatment [52]. In a recent study, it was shown that S1P decreases apoptosis and increases proliferation of rat-derived endothelial progenitor cells mainly through S1PR1, via the PI3K/Akt pathway. It has been shown that utilizing VPC23019 (a selective S1PR1/S1PR3 antagonist) or W146 (an S1PR1 antagonist) considerably increased apoptosis by activation of caspase-3 [53].
3.5 S1PR1 Role in Inflammatory Environment
The relationship of tumor progression with S1PR1 function has also been demonstrated using the direct correlation of S1PR1 and STAT3 activation in some cancers, for example adenocarcinoma, lymphoma, prostate cancer, melanoma, and breast cancer [9]. Accordingly, it has been reported that downregulation of S1PR1 suppressed the expression of STAT3 target genes, which was associated with tumor regression [54]. S1PR1 increases STAT3 phosphorylation and activation by physical interaction with JAK2. Interestingly, STAT3 makes a positive feedback loop in which STAT3 also stimulates the expression of S1PR1 [8]. Given there are difficulties in targeting STAT3, it seems that suppression of S1PR1 is an easier way to inhibit the feedback loop between these molecules [54,55,56,57]. It has also been shown that promotion of the SPHK1/S1P/S1PR1 axis can lead to increased survival, resistance to apoptosis, enhancement of metastasis and chemoresistance in various cancers through continuous activation of STAT3 [9, 58]. Therefore, suppression of the S1P/S1PR1 axis can also arrest tumor growth in a STAT3-dependent manner [59, 60].
It has been shown that upregulation of SPHK, S1P, and S1PR is associated with disease progression and endocrine resistance in estrogen receptor (ER)-positive breast cancers [61]. Moreover, SPHK1, a proproliferative oncogene kinase, has been shown to promote human thyroid cancer proliferation [62]. Signaling of S1PR1 and S1PR3 could also inhibit cell death in ovarian cancer in part through activation of the AKT signaling pathway [63]. In addition, binding of S1PR1 to S1P enhances proliferation, migration, and angiogenesis processes in cancer cells by activating ERK1/2, Rac, and PI3K signaling pathways, respectively [64]. Therefore, in addition to STAT3, there are various downstream signaling pathways by which S1PR1 can enhance tumor growth [13].
Another important point is the role of the S1P/S1PR1 axis in the promotion of an inflammatory microenvironment leading to generation of inflammation-derived cancers. It has been shown that upregulation of SPHK1 increases the generation of S1P in colitis, leading to overexpression of NF-κB and IL-6, and the subsequent induction of STAT3 and S1PR1. This positive feedback loop provides a sustained inflammatory condition during colitis, which is a critical trigger to convert chronic inflammation into colon cancer [9, 16, 65]. Therefore, targeting S1P/S1PR1 can also be a potent therapeutic approach in colorectal cancer via inhibition of the NF-κB/IL-6/STAT3 inflammatory axis [16, 66]. It has been demonstrated that FTY720/fingolimod blocks the SPHK1/S1P/S1PR1 axis, leading to blockade of the NF-kB/IL-6/STAT3 amplification loop and colitis-associated cancer [65]. There are additional similar studies in various colitis models using fingolimod or other S1PR1 modulators that further substantiate this claim [67,68,69,70].
The S1P/S1PR1 axis is also involved in another inflammatory condition in the lung through the COX-2/PGE2/IL-6 axis. It has been shown that binding of S1PR1 or 3 to S1P can enhance secretion of prostaglandin E2 (PGE2) and IL-6 in a c-Src-dependent manner in human tracheal smooth muscle cells, which can instigate pulmonary carcinogenesis [71,72,73]. In the next section, we will review the role of the S1P/S1PR1 axis in the promotion of various cancers.
4 The Role of S1PR1 in Different Cancers
4.1 Breast Cancer
It has been demonstrated that there is a close relationship between S1P/S1PR, estrogen, and growth factors in breast cancer cells. First, estrogen activates SPHK1, leading to an enhanced level of S1P, which consequently stimulates S1PRs and transactivates growth factor receptors [74]. There is also a complex relation between SPHK/S1P signaling and the growth factor network, as well as between estrogen signaling and the growth factor receptor network in breast cancer cells. Similarly, epidermal growth factor receptors (EGFRs) can affect both the estrogen pathway and the SPHK1 network [74].
Binding of ER with 7β-estradiol (E2) activates SPHK1, which leads to increased S1P levels [75]. Subsequent activation of S1PR1 with S1P triggers activation of Akt/eNOS, leading to migration of endothelial cells [76]. It has been consistently demonstrated that silencing S1PR1 in endothelial cells could potently suppress the E2-mediated induction of Akt/eNOS and migration of endothelial cells [76]. Therefore, it seems that the SPHK1/S1PR1 axis is an important mediator of the estrogen effect on angiogenesis and metastatic processes [76]. In an in vitro study, the relationship between S1PR1/S1PR3 and estrogen signaling in the proliferation, adhesion, viability and lateral motility of breast cancer cells was confirmed using ER-negative (MDA-MB-231) and ER-positive (MCF-7) cells. Silencing both receptors in the ER-negative cell line decreased proliferation, but this reduction was not observed in the ER-positive cell line. Calis et al. proposed that the difference in treatment result can be related to ERs, but they did not determine the exact mechanism [4].
S1PR1 signaling can also modulate survival of breast cancer cells. S1PR1 increases cancer cell survival via downregulation of Bim (pro-apoptotic protein) and upregulation of Mcl-1 (anti-apoptotic protein) in an ERK- and PKC-dependent manner, respectively. Signaling of S1PR1 in CCL39 lung fibroblasts was also associated with downregulation of Bim and resistance to apoptosis, which could be reversed by blockade of ERK activation [77]. Moreover, treatment of human umbilical vein endothelial cells with the S1PR agonist FTY720-phosphate (FTY720-P) led to the induction of pro-survival signals through upregulation of Mcl-1 and a delayed onset of caspase-3 cleavage upon growth factor withdrawal [77]. The expression of S1PR1 was consistently correlated with cancer cell survival in ER breast cancer patients and was associated with increased activation of ERK [77]. Therefore, it seems that signaling of S1PR1 can stimulate breast cancer cell survival in part via downregulation of pro-apoptotic BH3-only protein (Bim) through the MEK/ERK1/2 pathway, and upregulation of the anti-apoptotic protein Mcl-1 through a PI3K/PKC-mediated pathway [77].
In a recent preclinical study on human breast cancer cells, the S1PR1 antibody showed an augmented cytotoxic effect against carboplatin-treated MDA-MB-231 cells and an anti-proliferative impact on SK-BR-3 (HER2 subtype) cells [78]. In contrast, Lei et al. reported a contradictory result on the impact of the S1P/S1PR1 signaling pathway in cancer development. They found a tumor suppressive effect and survival benefit of S1PR1 signaling in breast cancer patients [79]. In conclusion, the relationship between S1PR1 and different factors such as estrogen has been well documented, and it appears to result in enhanced migration; however, the exact pathway remains to be identified. It is also important to discover the correlation of S1PR1 with molecules like HER2neu and other receptors in breast cancer much more precisely. S1PR1 also promotes survival rate of breast cancer cells via maintaining the balance of Bim and MCL-1. The data suggest that S1PR1 promotes breast cancer; however, in a recent study by Lei et al., a suppressive effect of S1PR1 was reported [79]. This report suggests further work is required to confirm the role of S1PR1 in breast cancer, as alternative pathways may be involved. This result might be due to inappropriate S1P concentration or a short follow-up duration, and it is better to examine blocking S1PR1 with other S1PR1 modulators and antagonists rather than only using short hairpin RNA (shRNA). It is also suggested to test them on various breast cancer (BCA) subtypes and stages. Also, we cannot rely on the results of colony formation assays after the addition of S1P to the cell lines to confirm S1PR1’s effect because of the interactional effects of all S1PRs.
4.2 Glioma
It has been demonstrated that S1P can affect a wide variety of glioblastoma multiforme’s (GBM’s) pathogenic features, including proliferation, survival, migration, invasion, tumor growth, and the development of microvascular networks [80]. Increased levels of S1PR1, S1PR2, and S1PR3 were observed in GBM tissue specimens; however, only signaling of S1PR1 and S1PR2 were markedly correlated with patients’ survival rate [81, 82]. It has been suggested that the higher S1P to ceramide ratio contributes to a nearly 100% recurrence rate, implying the S1P/S1PR1 axis is a potent therapeutic target for the treatment of GBM [83]. GBM cells can express S1PR1, S1PR2, S1PR3, and S1PR5, and their effects have been reported on the proliferation of U87-MG GBM cells [81, 82, 84]. The shape change induced by S1P in rat C6 glioma cells was via S1PR2 and S1PR1/S1PR3 together [85]. On the other hand, it has been reported that stimulation of S1PR1 activates Gi and induces the ERK/Egr-1/FGF-2 signaling pathway in C6 glioma cells [85]. Activation of this pathway can also lead to upregulation of urokinase plasminogen activator (uPA), which is implicated in the invasiveness of cancer cells in human U118 cells [86].
Interestingly, it has been demonstrated that reduced expression of S1PR1 was associated with poor survival in patients with glioblastoma. Silencing of S1PR1 in high-expressing glioma cell lines enhanced cell proliferation. On the other hand, induction of S1PR1 expression in these cell lines led to decreased growth and tumor regression. It is well known that induction of ERK signaling following stimulation of S1PR1 enhances proliferation of glioma cells. However, in this study, it was reported that downregulation of S1PR1 suppressed an increase in cell proliferation. This controversy is related to the role of the Egr-1 transcription factor, which plays an important role in cancer cell proliferation and PTEN in glioblastoma. Expression of PTEN and reduced Akt phosphorylation are also associated with expression of S1PR1. Expression of Egr-1 correlates with S1PR1 and survival rate in glioblastoma patients, and both are downregulated in patients with poor survival. Therefore, it seems that dysregulated expression of Egr-1 by signaling of S1PR1 and PTEN is the mechanism behind the glioma proliferation regardless of ERK inactivation [82]. Accordingly, it has been reported that low expression of S1PR1 is associated with high MIB-1 labeling index (a measure of proliferative activity in astrocytomas) and poor survival in glioblastoma [87]. Thus, it seems that downregulation of S1PR1 in glioblastoma enhances disease progression, and we need to use its agonists in order to attenuate disease progression. However, this issue requires further investigation to determine the exact overall effect of S1PR1 in glioma and precisely identify the details of S1PR1 in various glioblastoma stages and subtypes.
4.3 Hematopoietic Malignancies
Expression of S1PR1 has a pivotal role in the retention of lymphocytes in secondary lymphoid organs and facilitates their exit [88, 89]. High levels of S1PR1 expression are also detected in endothelial cells and pericytes, which is important for tumor angiogenesis and metastasis [40, 90]. It seems that blockade of S1PR1 may lead to retention of lymphoma cells in lymphoid organs, which can lead to the suppression of the invasive potential of these cells [54, 91].
4.3.1 Diffuse Large B-Cell Lymphoma
Overexpression of S1PR1 and STAT3 has been detected in patients with diffuse large B-cell lymphoma (DLBCL), which was associated with poor prognosis, implying S1PR1’s prognostic biomarker and therapeutic target potential in these patients [10]. Similarly, the prognostic and therapeutic target potency of S1PR1 has also been demonstrated in patients with primary testicular diffuse large B-cell lymphoma (PT-DLBCL) [92]. It has been suggested that targeting of S1PR1 can be an effective tool to suppress STAT3 signaling in activated B-cell–like DLBCL, which is resistant to chemotherapy and rituximab [54]. Regarding the successful use of CpG-S1PR1 siRNA in the treatment of B16 or CT26 tumor-bearing mice [9, 93], it is possible to utilize this approach for treatment of DLBCL. This strategy not only targets Toll-like receptor 9-expressing malignant cells, but also improves anti-tumor responses by TLR9-expressing immune cells [54].
4.3.2 Chronic Lymphocytic Leukemia
Increased expression of the homing receptors CXCR4/CCR7 and decreased levels of S1PR1 are observed in both the circulating leukemic cells and secondary lymphoid organs of chronic lymphocytic leukemia (CLL) patients. Treatment of these patients with ibrutinib, a Btk inhibitor, could normalize this imbalance and attenuate disease progress [94]. This study suggests the importance of S1PR1 in the pathogenesis of CLL. It seems that upregulation of S1PR1 can be associated with ameliorative effects in these patients. Therefore, increased expression of S1PR1 on CLL cells can augment leukemic cells’ egress into the bloodstream to decrease the reservoir of leukemic cells within survival niches [95]. While the signaling of S1PR1 enhances B-cell circulation in CLL patients, S1PR4 regulates and S1PR2 suppresses S1PR1-mediated signals. Binding of S1PR1 with S1P facilitates egress of all B-cell subsets from bone marrow and secondary lymphoid tissues. Interestingly, it has been shown that β-arrestin 2 enhances S1PR1-mediated migration and increases dissemination of leukemic cells [96]. Lack of S1PR1 expression enhances the survival time of leukemic B cells via increasing B CLL cells’ residency in the niche of secondary lymphoid organs [97].
Expression of S1PR1 has also been demonstrated in both the normal mantle zone B cells and mantle cell lymphoma (MCL) cells. S1PR1 mutations have recently been shown to be recurrent in MCLs [98]. It is suggested that reduced or dysfunctional expression of S1PR1 leads to malignant cells’ retention in the surrounding tissue, which is responsible for a minimal residual disease reservoir causing disease relapse. There are no reports regarding possible mutations of S1PR1 in DLBCLs; however, it is not expressed in the early stages of DLBCL, which is associated with a better outcome [10, 92]. Although the treatment of MCL patients with ibrutinib leads to the entrance of lymphoma cells from the tumor region into the bloodstream [99], about half of the patients show resistance to ibrutinib [100]. Treatment of CLL patients with ibrutinib was also associated with upregulation of S1PR1 [94]. However, the relevance between the effect of S1PR1 mutations and outcome of ibrutinib therapy needs to be further investigated [98].
4.3.3 Classical Hodgkin Lymphoma
With regard to the expression of S1PR1 in a subset of classical Hodgkin lymphoma (CHL) cases, the modulation of this receptor has also been suggested for treatment of S1PR1-positive, refractory/recurrent CHL [101]. It seems that S1PR1 can modulate the expression profile of transcriptions such as ATF-like 3 (BATF3) in Hodgkin lymphoma (HL) cells. S1P induces PI3K in HL cells, in part, through upregulation of S1PR1 and downregulation of S1PR2. PI3K induces the expression of BATF3, and BATF3 upregulates S1PR1 in an oncogenic feed-forward signaling loop [102].
4.3.4 T-Lymphoblastic Lymphoma Cells
High levels of expression of S1PR1, ICAM1 and BCL2 in T-lymphoblastic lymphoma cells also leads to the suppression of tumor cell intravasation. Accordingly, blockade of S1PR1 reduced homotypic adhesion and enhanced tumor cell intravasation, which is done via PI3K-AKT activation. The increased levels of these molecules were detected by clinical biopsy of specimens. The aim of this study was to determine the distinguishing molecular pattern of T-lymphoblastic lymphoma and acute T-lymphoblastic leukemia. In order to compare the molecular alterations between two forms of the disease, a transgenic zebrafish model was examined [103].
4.4 Cancers with Limited Information
4.4.1 Urothelial Carcinoma
Expression of S1PR1 is highly correlated with expression of p53 in non-muscle invasive urothelial carcinoma. This is due to the fact that SPHK1/S1P signaling is a downstream target of p53 function. Therefore, concomitant expression of S1PR1 and p53 has been considered as a prognostic marker in urothelial carcinomas. Accordingly, overexpression of S1PR1 was associated with poor clinicopathological characteristics, upregulation of survival and proliferation promoting factors, p53 expression and STAT3 activation in urothelial carcinoma. Difficulties in targeting p53 and STAT3 make S1PR1 a worthy therapeutic target in the treatment of urothelial carcinoma [13]. However, a recent paper demonstrated that S1PR1 expression did not have any prognostic significance in upper urinary tract urothelial carcinoma, but elevated expression level of STAT3 contributes to low survival and a higher rate of tumor progression [104]. This discrepancy may be in part related to use of different evaluating techniques, ethnicity, sample size or cancer stage and subtype; therefore further investigation is required.
4.4.2 Ovarian Cancer
High expression levels of S1PR1 have been demonstrated in hypoxic ovarian cancer cells (HOCCs). Silencing of S1PR1 in these cells led to reduced cell survival. While this suppression decreased pSTAT3, JAK1 and JAK2 levels, it did not affect total STAT3, TYK2 and AKT expression, implying that S1PR1 enhances STAT3 persistent activation in a positive feedback loop via the induction of JAK signaling in HOCCs [105]. There is evidence indicating that the SPHK1/S1P/S1PR1 axis and also S1PR2, but not S1PR3, are involved in enhancing the angiogenesis process in ovarian cancer [106]. Therefore, it is shown that suppressing the S1P/S1PR axis in ovarian cancer could be beneficial in the treatment of these patients.
4.4.3 Melanoma
Stimulation of S1PR1 and S1PR2 by S1P activates NF-κB through induction of the Akt/PI3K signaling pathway in melanoma cells that lack filamin A (FLNA). It has been reported that FLNA can inhibit S1P-mediated NF-κB activation in melanoma cells, in part through blockade of Akt [107].
4.4.4 Oral Squamous Cell Carcinoma
Little is known regarding the role of the S1P/S1PR axis in the immunopathogenesis of oral squamous cell carcinoma. In a recent report, S1P signaling was shown to enhance tumor aggressiveness in oral squamous cell carcinoma, and targeting the S1P axis was proposed to have therapeutic potential [7]. Another recent study, in 2017, showed that the expression of S1PR1, in association with inflammatory molecules, is increased in late-stage surgical margin samples [108]. It is evident that further investigations are required to explore the role of S1P/S1PR1 in the development of this disease.
4.4.5 Colorectal Cancer
It has been reported that upregulation of S1PR1 in colorectal cancer is associated with poor survival and metachronous liver metastasis. It has been suggested that expression level of S1PR1 can be considered an independent prognostic factor for this disease [109]. Further investigation is needed before a precise view regarding the role of S1PR1 in colorectal cancer can be postulated.
4.4.6 Wilms Tumor
Activation of S1PR1 signaling by S1P can enhance migration and invasion of cancerous cells in Wilms tumor. Activation of S1PR1 induces induction of PI3K and Rac1 through Gi coupling. Therefore, it has been suggested that targeting S1PR1 may be a potent therapeutic approach in Wilms tumor [93].
4.4.7 Thyroid Carcinoma
Little is known regarding the role of S1PR1 signaling in the immunopathogenesis of thyroid carcinoma. It has been reported that S1PR1 in association with VEGFR-2 generates a complex with ERK1/2 and PKC-alpha molecules to regulate migration of ML-1 thyroid carcinoma cells. Accordingly, it has been shown that while the inhibition of VEGFR-2 can prevent the S1P-induced ERK1/2 phosphorylation, silencing of S1PR1 can suppress VEGF-A–induced ERK1/2 phosphorylation [51].
4.4.8 Non-small Cell Lung Cancers
In a recent study, it was shown that apolipoprotein M (ApoM) as an S1P carrier has a role in non-small cell lung cancer oncogenesis. It increases the invasion and proliferation of cancer cells via upregulation of S1PR1 and activation of ERK1/2 and PI3K/AKT pathways [110]. The inflammation inducing property of ApoM has been documented in various studies, but its cancer-driving effect in an S1PR1-dependent manner has not yet been explored thoroughly; therefore further studies on this issue are recommended.
4.4.9 Pancreatic Cancer
In a recent study, it was demonstrated that FTY720, by suppressing the S1PR1/STAT3 loop, inhibited tumor growth and desmoplasia and suppressed resistance to the chemotherapy drug gemcitabine [111].
5 S1PR1 and Drug Resistance
Increased expression of S1PR1 has been detected in drug-resistant neuroblastoma cells, implying that the S1P/S1PR1 axis can be involved in inducing chemoresistance. The cooperative functioning of S1PR1 and STAT3 in the tumor microenvironment of neuroblastoma plays an important role in induction of chemoresistance. Accordingly, silencing S1PR1 in neuroblastoma cells could markedly sensitize cancerous cells to etoposide chemotherapy [112]. Moreover, induction of tamoxifen resistance and reduced disease-specific survival by stimulation of S1PR1, S1PR3, SPHK1, and ERK1/2 has also been detected in ER-positive breast cancer patients. Therefore, it is suggested that expression levels of SPHK1, S1PRs, and ERK1/2 can be considered as biomarkers to anticipate resistance to tamoxifen in ER-positive breast cancer patients [113]. Similarly, evaluation of drug resistance in camptothecin-resistant PC3 (a human prostate cancer) or sensitive LNCaP cells further substantiated the role of SPHK/S1P/S1PR1 in induction of drug resistance. PC3 cells significantly had higher levels of SPHK1, S1P and S1PR1 and S1PR3 compared to LNCaP cells. Moreover, camptothecin-treated PC3 cells showed increased expression and function of SPHK/S1P/S1PR1/3 in vitro. Consistently, silencing SPHK1 and blockade of S1PR1 significantly inhibited cancer cells’ proliferation [114]. Also, in a recent study, it was demonstrated that targeting the loop of S1PR1/STAT3 helps to sensitize pancreatic cancer to the chemotherapy agent gemcitabine [111]. Therefore, it seems that signaling of S1PR1 plays a crucial role in induction of chemoresistance.
6 Targeting S1PR1 for Cancer Therapy
As discussed in the previous sections on the role of S1PR1 in cancer progression, it seems that modulation of this receptor with other cancer-modifying drugs or chemotherapeutics may be considered as a potent anti-cancer therapeutic approach [2, 115].
Suppression of S1PR1 using RNA-based modulators was associated with blockade of STAT3 persistent activation and tumor regression, in vivo [14, 16]. On the other hand, in another study, it was demonstrated that administration of S1PR1 antagonist enhances lymphocyte egress and the cancer angiogenesis process in part through inducing capillary leakage [43]. Although both S1PR1 antagonists and S1PR1 agonists exert different effects on receptor expression, both induce lymphopenia in vivo in a similar manner [116]. It should be noted that the appearance of lymphopenia following treatment with S1PR1 agonists is mainly due to receptor internalization, and maybe degradation, which is known as functional antagonism. However, there is evidence that may lead to the rejection of this hypothesis: systemic administration of an S1PR1 antagonist, VPC44116 or W146, in vivo inhibited S1P receptor agonist and stimulated lymphopenia by inducing capillary leakage [43, 116, 117]. There is another hypothesis, known as the “stromal gate,” which implies that the endothelium is the key cellular target of S1PR1 agonist-mediated lymphopenia [116]. Therefore, S1PR1 antagonists or S1PR1 structural agonists, which result in internalization and degradation, can be considered as potent therapeutic drugs alone or in combination with other anti-cancer therapeutics (Table 1). In the following section, we will discuss the efficacy of various S1PR1-targeting drugs for cancer therapy.
6.1 Fingolimod
The impressive effects and good tolerability of fingolimod (FTY720, Gilenya®) led to Food and Drug Administration (FDA) approval for its use in the treatment of relapsing-remitting multiple sclerosis [118, 119]. However, its apoptosis-inducing effects on cancer cells and apparent prevention of metastasis led to consideration of this drug as a potent anti-cancer therapeutic agent [120]. Due to its resemblance to sphingosine in chemical structure, fingolimod can act as a sphingosine kinase substrate and it is phosphorylated by these kinases and turned into active fingolimod-P. Evidence shows that only SPHK2 phosphorylates fingolimod in vivo. Fingolimod-P structurally resembles S1P and therefore binds to its receptors [118]. It is a non-selective agonist of S1PRs that pharmacologically acts as a functional antagonist by enhancing receptor internalization and degradation [118]. Although it can bind to various S1PRs, including S1PR1, 2, 3, and 5, its effective functions are mediated mainly through S1PR1 [121, 122]. Therefore, its affinity for S1PR1 is significantly higher than for other receptors, and it mainly antagonizes the effects of S1PR1 [91, 123]. It should be noted that fingolimod’s anti-cancer effects can also be exerted through modulation of other targets such as blocking VEGF- and S1P-induced angiogenesis [17, 41]. It has been reported that it can suppress PDGF-B–mediated migration of vascular smooth muscle cells (VSMCs) via downregulation of the S1PR1 and S1PR3 signaling pathways. The combination of fingolimod with an S1PR1-specific siRNA may potently inhibit the growth of VSMCs [124]. With regard to the cross-talk between S1P signaling with tyrosine kinase pathways, a combination of fingolimod with tyrosine kinase inhibitors may also be a promising anti-cancer therapeutic approach [124].
It has been reported that treatment with fingolimod can induce apoptosis in non-muscle invasive urothelial carcinoma cells [13], suppress development of diffuse large B cell lymphoma (DLBCL) cells [54], and prevent resistance to cetuximab in colorectal carcinoma cells [125]. Administration of fingolimod into a T-cell lymphoma model significantly decreased Graft versus host disease (GVHD), enhanced anti-tumor responses and trapped T cells in lymph nodes, whereas it had no effect on their activation [126]. It also decreased the expression of S1PR1 in hepatocellular carcinoma [127]. Treatment with fingolimod potently suppressed phosphorylation and activation of STAT3 in cholangiocarcinoma cells, which was associated with decreased proliferation of cancerous cells [128]. In addition, fingolimod can also affect the migration of HL cells via antagonization of S1PR1, whereas it was enhanced by an S1PR2-specific antagonist [101]. As mentioned above, it also chemosensitized pancreatic cancer to treatment with gemcitabine by targeting the S1PR1-STAT3 pathway [111].
One of the problems in the treatment of breast cancer patients is the lack of ERα expression, which leads to resistance against conventional hormonal therapies. Interestingly, it has been demonstrated that treatment with fingolimod can induce re-expression of this receptor in murine and human breast cancer cells and enhance response to tamoxifen-therapy in ERα-negative syngeneic breast tumors [129].
In spite of the promising anti-cancer effects of fingolimod, the systemic lymphopenia induced with its use potently confines its utilization in cancer therapy [115]. Treatment of patients with multiple sclerosis with fingolimod is associated with various side effects, which include bradycardia, relapse, basal-cell carcinoma and macular edema [130]. Since the treatment of cancer with fingolimod requires higher doses than those used for multiple sclerosis severe side effects in cancer patients following treatment are expected; however, studies regarding the side effects of fingolimod on cancer are limited to mice. Intriguingly, fingolimod did not exert severe adverse effects at a dosage of ≤ 10 mg/kg/day in mouse renal cancer [131]. Accordingly, fingolimod arrested tumor growth and metastasis in mouse breast [120] and bladder [132] xenografts without any side effects. Therefore, targeted delivery of fingolimod or S1PR1 antagonists may solve this limitation [133]. Biocompatible nanocarriers could be a good option for targeted drug delivery [134, 135]. It has been shown that liposome-encapsulated fingolimod allowed for significantly lower doses of fingolimod in the treatment of CLL in mice [136]. Similarly, treatment of triple-negative breast cancer–bearing mice with fingolimod-docetaxel loaded nanoparticles exhibited enhanced anti-tumor effects of both drugs using significantly lower doses, which reduced docetaxel-related side effects and fingolimod-induced lymphopenia [115].
Therefore, nanoparticle-based targeted delivery of fingolimod in combination with other potent therapeutics may lead to the appearance of promising outcomes together with good safety and non-toxicity, which may trigger its fast translation into the clinic [17].
6.2 Other S1PR1 Modulators
As mentioned, side effects associated with administration of fingolimod have limited its usage in cancer therapy. Therefore, in addition to fingolimod, which can bind with various S1PRs, several other S1PR1-specific agonists and antagonists have been developed and evaluated for cancer therapy [2, 137] (Tables 2, 3). However, the same side effects have been observed following administration of these S1PR1 modulators [2]. It is thought that the macular edema observed as one of these side effects is in part due to vascular leakage induced by S1PR1 binding [2, 138]. There is no difference between various S1PR1 agonists (which are functional antagonists) concerning the downregulation and degradation of the receptor. Almost all the evaluated small-molecule modulators downregulate and degrade S1PR1. With regards to the application of various therapeutic strategies and the use of various chemical modulators in cancer immunotherapy, comparison of the effects of these modulators on S1PR1 is difficult, but the possible effects on other receptors may help in this comparison. Here, we introduce various modulators of S1PR1 evaluated in respected studies [121].
VPC23019 is a competitive antagonist of S1PR1 and S1PR3 [139], that blocks agonistic activity. This blockade influences calcium mobilization and cell migration [139]. This suppression effect was observed in agonist-mediated cell migration in bladder and thyroid cancer cells [139, 140]. It also reversed agonist-induced MAPK activation, migration, and ligand-induced receptor internalization in T cells [141]. In a study performed in 2018, treatment with VPC23019 inhibited proliferation of endothelial progenitor cells and enhanced caspase-3 activation and apoptosis [142]. In another study in 2018, the anti-inflammatory impact of VPC23019 was examined, and reduction of eosinophilic inflammation was exhibited following treatment in bronchial asthma [143]. VPC23019 also inhibited apoM-induced properties, such as phosphorylation of Akt, and its anti-apoptosis effects [144]. According to a report, this suppressed the attenuating effect of S1P on immune cells’ adhesion to endothelial cells during the inflammation process [145].
VPC44116 is a selective S1PR1 antagonist that prevents S1P-mediated migration and suppresses the development of HL and Wilms tumor [93, 101]. Utilizing VPC44116, an SIPR1-specific antagonist, in macrophages also inhibited Lipopolysaccharide-induced secretion of pro-inflammatory cytokines [146].
There are also other S1PR1 antagonists that are being developed and require examination in cancer models, such as W123 [141, 147], W146 [70, 116], chemical lead 2 (CL2) [39], NIBR-0213 [138], KRP203 [148], AUY954 [149], CS-0777 [150], TASP0277308 [151], Syl930 and SYL927 [152, 153], as well as SEW2871, which is a selective S1PR1 agonist that cannot decrease the expression of S1PR1 and activates S1PR1 signaling pathways [141, 147]. Although, SEW2871 internalizes the receptor, it does not induce receptor degradation [141]. The efficacy of this drug has been evaluated in the treatment of inflammatory autoimmune diseases [154]. Since this triggers the same effects exerted by S1P, it seems unlikely that it will be a good candidate for cancer therapy. Intriguingly, Rolin et al. have demonstrated that SEW2871 can act as an S1PR1 antagonist and enhances natural killer cell-mediated lysis of K562 cells and dendritic cells, implying the anti-cancer potential of SEW2871 and maybe S1P [155]. We propose work is needed to the investigate the efficacy and side effects of the S1PR1 modulators that are capable of antagonizing S1PR1 signaling, but which have not yet been examined in cancer treatment. Furthermore, studying the exact function of SEW2871 at the tumor site is highly recommended, as it is an agonist, but has played an effective role in treating cancer [155].
7 Conclusion
S1PR1 signaling appears to play an important role in cancer development via the stimulation of growth, migration, angiogenesis, and anti-apoptotic effects in breast, renal, and bladder cancers; therefore the modulation of this receptor can be a promising and novel approach in cancer therapy. Although several S1PR1 modulators, including FTY720, have been developed, few studies have evaluated the efficacy of these drugs in the treatment of human cancer, as preference has been given to other molecules involved in the S1PR1 cycle. Moreover, it is pivotal to design and develop more effective S1PR1 modulators with higher affinity and specificity. This point should be addressed in the design of new modulators that lack the lymphopenia-inducing effect and angiogenic potential of some of the current therapeutic options, as this is the reason why so little work has been undertaken in this field with these agents. Due to the ability of S1PR1 modulators to sensitize cancer cells to chemotherapeutics, which consequently leads to immediate regression of different types of cancer burden, using a combination of these drugs with chemotherapy might be a potent anti-cancer therapeutic approach for future studies. Moreover, the best combination for each cancer type should be investigated. S1PR1 drugs probably can be considered in combination with other treatment approaches, such as radiotherapy. We recommend more research to evaluate and devise more combination-focused approaches along with S1PR1 blockade. Our suggestion is combined targeting of CD44, growth factor receptors or immune checkpoint blockers as an emerging field of study. The possible potential of this receptor in cancer therapy is just beginning to be elucidated, and as S1PR1 modulators have not yet been used in clinical trials, its capacity as a significant target for human cancer therapy should be explored. It may also be important to compare the potency of each S1PR1 blocking agent, such as siRNA, antibodies, and other chemical modulators, with each other in identical states.
Moreover, nano-based anti-cancer therapy is an emerging treatment approach that can efficiently decrease systemic side effects [134, 156]; therefore it is worth exploring a combination of this method with S1PR1 blockage, as this may surmount one of the drawbacks of S1PR1 modulators, the induction of lymphopenia. In order to inhibit drug resistance to chemotherapeutics in cancers with enhanced expression of S1PR1 or its downstream molecules, researchers could examine blocking this pathway’s effect on chemosensitization in different types and stages of cancer [157].
References
Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J Exp Med. 2017;214(9):2695–713. https://doi.org/10.1084/jem.20160392.
Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12(9):688–702. https://doi.org/10.1038/nrd4099.
Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60(2):181–95. https://doi.org/10.1124/pr.107.07113.
Calis IU, Cosan DT, Mutlu F. Effects of S1P1 and S1P3 in ER(+) and ER(−) breast cancer cells. Anticancer Res. 2017;37(10):5469–75. https://doi.org/10.21873/anticanres.11976.
Brocklyn JR. Regulation of cancer cell migration and invasion by sphingosine-1-phosphate. World J Biol Chem. 2010;1(10):307–12. https://doi.org/10.4331/wjbc.v1.i10.307.
Goparaju SK, Jolly PS, Watterson KR, Bektas M, Alvarez S, Sarkar S, et al. The S1P2 receptor negatively regulates platelet-derived growth factor-induced motility and proliferation. Mol Cell Biol. 2005;25(10):4237–49. https://doi.org/10.1128/mcb.25.10.4237-4249.2005.
Patmanathan SN, Johnson SP, Lai SL, Panja Bernam S, Lopes V, Wei W, et al. Aberrant expression of the S1P regulating enzymes, SPHK1 and SGPL1, contributes to a migratory phenotype in OSCC mediated through S1PR2. Sci Rep. 2016;6:25650. https://doi.org/10.1038/srep25650.
Jin L, Liu WR, Tian MX, Fan J, Shi YH. The SphKs/S1P/S1PR1 axis in immunity and cancer: more ore to be mined. World J Surg Oncol. 2016;14:131. https://doi.org/10.1186/s12957-016-0884-7.
Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16(12):1421–8. https://doi.org/10.1038/nm.2250.
Paik JH, Nam SJ, Kim TM, Heo DS, Kim CW, Jeon YK. Overexpression of sphingosine-1-phosphate receptor 1 and phospho-signal transducer and activator of transcription 3 is associated with poor prognosis in rituximab-treated diffuse large B-cell lymphomas. BMC Cancer. 2014;14:911. https://doi.org/10.1186/1471-2407-14-911.
Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115(1):84–105. https://doi.org/10.1016/j.pharmthera.2007.04.006.
Dong H, Claffey KP, Brocke S, Epstein PM. Inhibition of breast cancer cell migration by activation of cAMP signaling. Breast Cancer Res Treat. 2015;152(1):17–28. https://doi.org/10.1007/s10549-015-3445-9.
Go H, Kim PJ, Jeon YK, Cho YM, Kim K, Park BH, et al. Sphingosine-1-phosphate receptor 1 (S1PR1) expression in non-muscle invasive urothelial carcinoma: association with poor clinical outcome and potential therapeutic target. Eur J Cancer (Oxford, England: 1990). 2015;51(14):1937–45. https://doi.org/10.1016/j.ejca.2015.07.021.
Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21(5):642–54. https://doi.org/10.1016/j.ccr.2012.03.039.
Sabbadini R. Targeting sphingosine-1-phosphate for cancer therapy. Br J Cancer. 2006;95(9):1131.
Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23(1):107–20. https://doi.org/10.1016/j.ccr.2012.11.013.
White C, Alshaker H, Cooper C, Winkler M, Pchejetski D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget. 2016;7(17):23106–27. https://doi.org/10.18632/oncotarget.7145.
Yester JW, Tizazu E, Harikumar KB, Kordula T. Extracellular and intracellular sphingosine-1-phosphate in cancer. Cancer Metastasis Rev. 2011;30(3–4):577–97. https://doi.org/10.1007/s10555-011-9305-0.
Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22(1):50–60. https://doi.org/10.1016/j.tcb.2011.09.003.
Watters RJ, Wang HG, Sung SS, Loughran TP, Liu X. Targeting sphingosine-1-phosphate receptors in cancer. Anticancer Agents Med Chem. 2011;11(9):810–7.
Neves SR, Ram PT, Iyengar R. G protein pathways. Science (New York, NY). 2002;296(5573):1636–9. https://doi.org/10.1126/science.1071550.
Siehler S, Manning DR. Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. Biochem Biophys Acta. 2002;1582(1–3):94–9.
O’Sullivan C, Dev KK. The structure and function of the S1P1 receptor. Trends Pharmacol Sci. 2013;34(7):401–12. https://doi.org/10.1016/j.tips.2013.05.002.
Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–82. https://doi.org/10.1146/annurev.physiol.69.022405.154712.
Waters CM, Connell MC, Pyne S, Pyne NJ. c-Src is involved in regulating signal transmission from PDGFbeta receptor-GPCR(s) complexes in mammalian cells. Cell Signal. 2005;17(2):263–77. https://doi.org/10.1016/j.cellsig.2004.07.011.
Pyne NJ, Pyne S. Sphingosine 1-phosphate receptor 1 signaling in mammalian cells. Molecules (Basel, Switzerland). 2017;22(3):344. https://doi.org/10.3390/molecules22030344.
Aoki M, Aoki H, Ramanathan R, Hait NC, Takabe K. Sphingosine-1-phosphate signaling in immune cells and inflammation: roles and therapeutic potential. Mediat Inflamm. 2016;2016:8606878. https://doi.org/10.1155/2016/8606878.
Takuwa Y. Subtype-specific differential regulation of Rho family G proteins and cell migration by the Edg family sphingosine-1-phosphate receptors. Biochem Biophys Acta. 2002;1582(1–3):112–20.
Zu Heringdorf DM, Vincent ME, Lipinski M, Danneberg K, Stropp U, Wang DA, et al. Inhibition of Ca(2+) signalling by the sphingosine 1-phosphate receptor S1P(1). Cell Signal. 2003;15(7):677–87.
Singleton PA, Dudek SM, Ma SF, Garcia JG. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem. 2006;281(45):34381–93. https://doi.org/10.1074/jbc.m603680200.
Igarashi J, Erwin PA, Dantas AP, Chen H, Michel T. VEGF induces S1P1 receptors in endothelial cells: Implications for cross-talk between sphingolipid and growth factor receptors. Proc Natl Acad Sci USA. 2003;100(19):10664–9. https://doi.org/10.1073/pnas.1934494100.
Rodriguez YI, Campos LE, Castro MG, Aladhami A, Oskeritzian CA, Alvarez SE. Sphingosine-1 phosphate: a new modulator of immune plasticity in the tumor microenvironment. Front Oncol. 2016;6:218. https://doi.org/10.3389/fonc.2016.00218.
Kazemi T, Younesi V, Jadidi-Niaragh F, Yousefi M. Immunotherapeutic approaches for cancer therapy: an updated review. Artif Cells Nanomed Biotechnol. 2016;44(3):769–79.
Xie Z, Liu H, Geng M. Targeting sphingosine-1-phosphate signaling for cancer therapy. Sci China Life Sci. 2017;60(6):585–600. https://doi.org/10.1007/s11427-017-9046-6.
Priceman SJ, Shen S, Wang L, Deng J, Yue C, Kujawski M, et al. S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. Cell Rep. 2014;6(6):992–9. https://doi.org/10.1016/j.celrep.2014.02.016.
Rathinasamy A, Domschke C, Ge Y, Bohm HH, Dettling S, Jansen D, et al. Tumor specific regulatory T cells in the bone marrow of breast cancer patients selectively upregulate the emigration receptor S1P1. Cancer Immunol Immunother CII. 2017;66(5):593–603. https://doi.org/10.1007/s00262-017-1964-4.
Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282(13):9358–63. https://doi.org/10.1074/jbc.C600321200.
Maeda Y, Seki N, Kataoka H, Takemoto K, Utsumi H, Fukunari A, et al. IL-17-producing Vgamma4+ gammadelta T cells require sphingosine 1-phosphate receptor 1 for their egress from the lymph nodes under homeostatic and inflammatory conditions. J Immunol (Baltimore, Md: 1950). 2015;195(4):1408–16. https://doi.org/10.4049/jimmunol.1500599.
Yonesu K, Kawase Y, Inoue T, Takagi N, Tsuchida J, Takuwa Y, et al. Involvement of sphingosine-1-phosphate and S1P1 in angiogenesis: analyses using a new S1P1 antagonist of non-sphingosine-1-phosphate analog. Biochem Pharmacol. 2009;77(6):1011–20. https://doi.org/10.1016/j.bcp.2008.12.007.
Chae SS, Paik JH, Furneaux H, Hla T. Requirement for sphingosine 1-phosphate receptor-1 in tumor angiogenesis demonstrated by in vivo RNA interference. J Clin Investig. 2004;114(8):1082–9. https://doi.org/10.1172/jci22716.
LaMontagne K, Littlewood-Evans A, Schnell C, O’Reilly T, Wyder L, Sanchez T, et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Can Res. 2006;66(1):221–31. https://doi.org/10.1158/0008-5472.can-05-2001.
Reinhard NR, Mastop M, Yin T, Wu Y, Bosma EK, Gadella TWJ Jr, et al. The balance between Galphai-Cdc42/Rac and Galpha12/13-RhoA pathways determines endothelial barrier regulation by sphingosine-1-phosphate. Mol Biol Cell. 2017;28(23):3371–82. https://doi.org/10.1091/mbc.E17-03-0136.
Sanna MG, Wang SK, Gonzalez-Cabrera PJ, Don A, Marsolais D, Matheu MP, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2(8):434–41. https://doi.org/10.1038/nchembio804.
He P, Philbrick MJ, An X, Wu J, Messmer-Blust AF, Li J. Endothelial differentiation gene-1, a new downstream gene is involved in RTEF-1 induced angiogenesis in endothelial cells. PLoS One. 2014;9(2):e88143. https://doi.org/10.1371/journal.pone.0088143.
Williams PA, Stilhano RS, To VP, Tran L, Wong K, Silva EA. Hypoxia augments outgrowth endothelial cell (OEC) sprouting and directed migration in response to sphingosine-1-phosphate (S1P). PLoS One. 2015;10(4):e0123437. https://doi.org/10.1371/journal.pone.0123437.
Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell. 2012;23(3):600–10. https://doi.org/10.1016/j.devcel.2012.07.015.
Gaengel K, Niaudet C, Hagikura K, Lavina B, Muhl L, Hofmann JJ, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell. 2012;23(3):587–99. https://doi.org/10.1016/j.devcel.2012.08.005.
Sarkisyan G, Gay LJ, Nguyen N, Felding BH, Rosen H. Host endothelial S1PR1 regulation of vascular permeability modulates tumor growth. Am J Physiol Cell Physiol. 2014;307(1):C14–24. https://doi.org/10.1152/ajpcell.00043.2014.
Hobson JP, Rosenfeldt HM, Barak LS, Olivera A, Poulton S, Caron MG, et al. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science (New York, NY). 2001;291(5509):1800–3. https://doi.org/10.1126/science.1057559.
Waters C, Sambi B, Kong KC, Thompson D, Pitson SM, Pyne S, et al. Sphingosine 1-phosphate and platelet-derived growth factor (PDGF) act via PDGF beta receptor-sphingosine 1-phosphate receptor complexes in airway smooth muscle cells. J Biol Chem. 2003;278(8):6282–90. https://doi.org/10.1074/jbc.M208560200.
Bergelin N, Lof C, Balthasar S, Kalhori V, Tornquist K. S1P1 and VEGFR-2 form a signaling complex with extracellularly regulated kinase 1/2 and protein kinase C-alpha regulating ML-1 thyroid carcinoma cell migration. Endocrinology. 2010;151(7):2994–3005. https://doi.org/10.1210/en.2009-1387.
Nikkhoo A, Rostami N, Hojjat-Farsangi M, Azizi G, Yousefi B, Ghalamfarsa G, et al. Smac mimetics as novel promising modulators of apoptosis in the treatment of breast cancer. J Cell Biochem. 2018. https://doi.org/10.1002/jcb.28205.
Wang H, Huang H, Ding SF. Sphingosine-1-phosphate promotes the proliferation and attenuates apoptosis of endothelial progenitor cells via S1PR1/S1PR3/PI3K/Akt pathway. Cell Biol Int. 2018. https://doi.org/10.1002/cbin.10991.
Liu Y, Deng J, Wang L, Lee H, Armstrong B, Scuto A, et al. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood. 2012;120(7):1458–65. https://doi.org/10.1182/blood-2011-12-399030.
Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51. https://doi.org/10.1038/nri1995.
Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y, et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood. 2008;111(3):1515–23. https://doi.org/10.1182/blood-2007-04-087734.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. https://doi.org/10.1038/nrc2734.
Selvam SP, Ogretmen B. Sphingosine kinase/sphingosine 1-phosphate signaling in cancer therapeutics and drug resistance. Handb Exp Pharmacol. 2013;216:3–27. https://doi.org/10.1007/978-3-7091-1511-4_1.
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–46. https://doi.org/10.1038/nrc3818.
Cheng N, Wang GH. miR-133b, a microRNA targeting S1PR1, suppresses nasopharyngeal carcinoma cell proliferation. Exp Ther Med. 2016;11(4):1469–74. https://doi.org/10.3892/etm.2016.3043.
Sukocheva OA. Expansion of sphingosine kinase and sphingosine-1-phosphate receptor function in normal and cancer cells: from membrane restructuring to mediation of estrogen signaling and stem cell programming. Int J Mol Sci. 2018;19(2):420. https://doi.org/10.3390/ijms19020420.
Guan H, Liu L, Cai J, Liu J, Ye C, Li M, et al. Sphingosine kinase 1 is overexpressed and promotes proliferation in human thyroid cancer. Mol Endocrinol (Baltimore, Md). 2011;25(11):1858–66. https://doi.org/10.1210/me.2011-1048.
Baudhuin LM, Cristina KL, Lu J, Xu Y. Akt activation induced by lysophosphatidic acid and sphingosine-1-phosphate requires both mitogen-activated protein kinase kinase and p38 mitogen-activated protein kinase and is cell-line specific. Mol Pharmacol. 2002;62(3):660–71.
Huang YL, Huang WP, Lee H. Roles of sphingosine 1-phosphate on tumorigenesis. World J Biol Chem. 2011;2(2):25–34. https://doi.org/10.4331/wjbc.v2.i2.25.
Nagahashi M, Hait NC, Maceyka M, Avni D, Takabe K, Milstien S, et al. Sphingosine-1-phosphate in chronic intestinal inflammation and cancer. Adv Biol Regul. 2014;54:112–20. https://doi.org/10.1016/j.jbior.2013.10.001.
Masjedi A, Hashemi V, Hojjat-Farsangi M, Ghalamfarsa G, Azizi G, Yousefi M, et al. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed Pharmacother. 2018;108:1415–24. https://doi.org/10.1016/j.biopha.2018.09.177.
Sanada Y, Mizushima T, Kai Y, Nishimura J, Hagiya H, Kurata H, et al. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS One. 2011;6(9):e23933. https://doi.org/10.1371/journal.pone.0023933.
Song J, Matsuda C, Kai Y, Nishida T, Nakajima K, Mizushima T, et al. A novel sphingosine 1-phosphate receptor agonist, 2-amino-2-propanediol hydrochloride (KRP-203), regulates chronic colitis in interleukin-10 gene-deficient mice. J Pharmacol Exp Ther. 2008;324(1):276–83. https://doi.org/10.1124/jpet.106.119172.
Danese S, Furfaro F, Vetrano S. Targeting S1P in Inflammatory bowel disease: new avenues for modulating intestinal leukocyte migration. J Crohn’s Colitis. 2017. https://doi.org/10.1093/ecco-jcc/jjx107.
Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55(8):1596–608. https://doi.org/10.1194/jlr.R046300.
Durham AL, Adcock IM. The relationship between COPD and lung cancer. Lung Cancer (Amsterdam, Netherlands). 2015;90(2):121–7. https://doi.org/10.1016/j.lungcan.2015.08.017.
Houghton AM, Mouded M, Shapiro SD. Common origins of lung cancer and COPD. Nat Med. 2008;14(10):1023–4. https://doi.org/10.1038/nm1008-1023.
Hsu CK, Lee IT, Lin CC, Hsiao LD, Yang CM. Sphingosine-1-phosphate mediates COX-2 expression and PGE2 /IL-6 secretion via c-Src-dependent AP-1 activation. J Cell Physiol. 2015;230(3):702–15. https://doi.org/10.1002/jcp.24795.
Sukocheva O, Wadham C, Holmes A, Albanese N, Verrier E, Feng F, et al. Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: the role of sphingosine kinase-1. J Cell Biol. 2006;173(2):301–10. https://doi.org/10.1083/jcb.200506033.
Sukocheva OA, Wang L, Albanese N, Pitson SM, Vadas MA, Xia P. Sphingosine kinase transmits estrogen signaling in human breast cancer cells. Mol Endocrinol (Baltimore, Md). 2003;17(10):2002–12. https://doi.org/10.1210/me.2003-0119.
Sukocheva O, Wadham C, Gamble J, Xia P. Sphingosine-1-phosphate receptor 1 transmits estrogens’ effects in endothelial cells. Steroids. 2015;104:237–45. https://doi.org/10.1016/j.steroids.2015.10.009.
Rutherford C, Childs S, Ohotski J, McGlynn L, Riddick M, MacFarlane S, et al. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013;4:e927. https://doi.org/10.1038/cddis.2013.455.
Xiao S, Yang J. Preclinical study of the antitumor effect of sphingosine-1-phosphate receptor 1 antibody (S1PR1-antibody) against human breast cancer cells. Investig New Drugs. 2018. https://doi.org/10.1007/s10637-018-0618-5.
Lei FJ, Cheng BH, Liao PY, Wang HC, Chang WC, Lai HC, et al. Survival benefit of sphingosin-1-phosphate and receptors expressions in breast cancer patients. Cancer Med. 2018;7(8):3743–54. https://doi.org/10.1002/cam4.1609.
Mahajan-Thakur S, Bien-Moller S, Marx S, Schroeder H, Rauch BH. Sphingosine 1-phosphate (S1P) signaling in glioblastoma multiforme—a systematic review. Int J Mol Sci. 2017;18(11):2448. https://doi.org/10.3390/ijms18112448.
Bien-Moller S, Lange S, Holm T, Bohm A, Paland H, Kupper J, et al. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget. 2016;7(11):13031–46. https://doi.org/10.18632/oncotarget.7366.
Yoshida Y, Nakada M, Sugimoto N, Harada T, Hayashi Y, Kita D, et al. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int J Cancer. 2010;126(10):2341–52. https://doi.org/10.1002/ijc.24933.
Sordillo LA, Sordillo PP, Helson L. Sphingosine kinase inhibitors as maintenance therapy of glioblastoma after ceramide-induced response. Anticancer Res. 2016;36(5):2085–95.
Bernhart E, Damm S, Wintersperger A, Nusshold C, Brunner AM, Plastira I, et al. Interference with distinct steps of sphingolipid synthesis and signaling attenuates proliferation of U87MG glioma cells. Biochem Pharmacol. 2015;96(2):119–30. https://doi.org/10.1016/j.bcp.2015.05.007.
Kim K, Kim YL, Sacket SJ, Kim HL, Han M, Park DS, et al. Sphingosine 1-phosphate (S1P) induces shape change in rat C6 glioma cells through the S1P2 receptor: development of an agonist for S1P receptors. J Pharm Pharmacol. 2007;59(7):1035–41. https://doi.org/10.1211/jpp.59.7.0017.
Young N, Pearl DK, Van Brocklyn JR. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol Cancer Res MCR. 2009;7(1):23–32. https://doi.org/10.1158/1541-7786.mcr-08-0061.
Yoshida Y, Nakada M, Harada T, Tanaka S, Furuta T, Hayashi Y, et al. The expression level of sphingosine-1-phosphate receptor type 1 is related to MIB-1 labeling index and predicts survival of glioblastoma patients. J Neurooncol. 2010;98(1):41–7. https://doi.org/10.1007/s11060-009-0064-5.
Cinamon G, Matloubian M, Lesneski MJ, Xu Y, Low C, Lu T, et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol. 2004;5(7):713–20. https://doi.org/10.1038/ni1083.
Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440(7083):540–4. https://doi.org/10.1038/nature04606.
Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell. 2006;9(3):225–38. https://doi.org/10.1016/j.ccr.2006.02.023.
Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–60. https://doi.org/10.1038/nature02284.
Koresawa R, Yamazaki K, Oka D, Fujiwara H, Nishimura H, Akiyama T, et al. Sphingosine-1-phosphate receptor 1 as a prognostic biomarker and therapeutic target for patients with primary testicular diffuse large B-cell lymphoma. Br J Haematol. 2016;174(2):264–74. https://doi.org/10.1111/bjh.14054.
Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27(10):925–32. https://doi.org/10.1038/nbt.1564.
Patrussi L, Capitani N, Martini V, Pizzi M, Trimarco V, Frezzato F, et al. Enhanced chemokine receptor recycling and impaired S1P1 expression promote leukemic cell infiltration of lymph nodes in chronic lymphocytic leukemia. Can Res. 2015;75(19):4153–63. https://doi.org/10.1158/0008-5472.can-15-0986.
Borge M, Remes Lenicov F, Nannini PR, de los Rios Alicandu MM, Podaza E, Ceballos A, et al. The expression of sphingosine-1 phosphate receptor-1 in chronic lymphocytic leukemia cells is impaired by tumor microenvironmental signals and enhanced by piceatannol and R406. J Immunol (Baltimore, Md: 1950). 2014;193(6):3165–74. https://doi.org/10.4049/jimmunol.1400547.
Sic H, Kraus H, Madl J, Flittner KA, von Munchow AL, Pieper K, et al. Sphingosine-1-phosphate receptors control B-cell migration through signaling components associated with primary immunodeficiencies, chronic lymphocytic leukemia, and multiple sclerosis. J Allergy Clin Immunol. 2014;134(2):420–8. https://doi.org/10.1016/j.jaci.2014.01.037.
Capitani N, Patrussi L, Trentin L, Lucherini OM, Cannizzaro E, Migliaccio E, et al. S1P1 expression is controlled by the pro-oxidant activity of p66Shc and is impaired in B-CLL patients with unfavorable prognosis. Blood. 2012;120(22):4391–9. https://doi.org/10.1182/blood-2012-04-425959.
Wasik AM, Wu C, Mansouri L, Rosenquist R, Pan-Hammarstrom Q, Sander B. Clinical and functional impact of recurrent S1PR1 mutations in mantle cell lymphoma. Blood Adv. 2018;2(6):621–5. https://doi.org/10.1182/bloodadvances.2017014860.
Chang BY, Francesco M, De Rooij MF, Magadala P, Steggerda SM, Huang MM, et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood. 2013;122(14):2412–24. https://doi.org/10.1182/blood-2013-02-482125.
Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun. 2017;8:14920. https://doi.org/10.1038/ncomms14920.
Kluk MJ, Ryan KP, Wang B, Zhang G, Rodig SJ, Sanchez T. Sphingosine-1-phosphate receptor 1 in classical Hodgkin lymphoma: assessment of expression and role in cell migration. Lab Investig. 2013;93(4):462–71. https://doi.org/10.1038/labinvest.2013.7.
Vrzalikova K, Ibrahim M, Vockerodt M, Perry T, Margielewska S, Lupino L, et al. S1PR1 drives a feedforward signalling loop to regulate BATF3 and the transcriptional programme of Hodgkin lymphoma cells. Leukemia. 2018;32(1):214–23. https://doi.org/10.1038/leu.2017.275.
Feng H, Stachura DL, White RM, Gutierrez A, Zhang L, Sanda T, et al. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell. 2010;18(4):353–66. https://doi.org/10.1016/j.ccr.2010.09.009.
Matsuzaki K, Fujita K, Hayashi Y, Matsushita M, Nojima S, Jingushi K, et al. STAT3 expression is a prognostic marker in upper urinary tract urothelial carcinoma. PLoS One. 2018;13(8):e0201256. https://doi.org/10.1371/journal.pone.0201256.
McCann GA, Naidu S, Rath KS, Bid HK, Tierney BJ, Suarez A, et al. Targeting constitutively-activated STAT3 in hypoxic ovarian cancer, using a novel STAT3 inhibitor. Oncoscience. 2014;1(3):216–28. https://doi.org/10.18632/oncoscience.26.
Dai L, Liu Y, Xie L, Wu X, Qiu L, Di W. Sphingosine kinase 1/sphingosine-1-phosphate (S1P)/S1P receptor axis is involved in ovarian cancer angiogenesis. Oncotarget. 2017;8(43):74947–61. https://doi.org/10.18632/oncotarget.20471.
Campos LS, Rodriguez YI, Leopoldino AM, Hait NC, Lopez Bergami P, Castro MG, et al. Filamin A expression negatively regulates sphingosine-1-phosphate-induced NF-kappaB activation in melanoma cells by inhibition of Akt signaling. Mol Cell Biol. 2016;36(2):320–9. https://doi.org/10.1128/mcb.00554-15.
Suwanwela J, Osathanon T. Inflammation related genes are upregulated in surgical margins of advanced stage oral squamous cell carcinoma. J Oral Biol Craniofac Res. 2017;7(3):193–7. https://doi.org/10.1016/j.jobcr.2017.05.003.
Lin Q, Wei Y, Zhong Y, Zhu D, Ren L, Xu P, et al. Aberrant expression of sphingosine-1-phosphate receptor 1 correlates with metachronous liver metastasis and poor prognosis in colorectal cancer. Tumour Biol. 2014;35(10):9743–50. https://doi.org/10.1007/s13277-014-2267-4.
Zhu Y, Luo G, Jiang B, Yu M, Feng Y, Wang M, et al. Apolipoprotein M promotes proliferation and invasion in non-small cell lung cancers via upregulating S1PR1 and activating the ERK1/2 and PI3K/AKT signaling pathways. Biochem Biophys Res Commun. 2018;501(2):520–6. https://doi.org/10.1016/j.bbrc.2018.05.029.
Lankadasari MB, Aparna JS, Mohammed S, James S, Aoki K, Binu VS, et al. Targeting S1PR1/STAT3 loop abrogates desmoplasia and chemosensitizes pancreatic cancer to gemcitabine. Theranostics. 2018;8(14):3824–40. https://doi.org/10.7150/thno.25308.
Lifshitz V, Priceman SJ, Li W, Cherryholmes G, Lee H, Makovski-Silverstein A, et al. Sphingosine-1-phosphate receptor-1 promotes environment-mediated and acquired chemoresistance. Mol Cancer Ther. 2017;16(11):2516–27. https://doi.org/10.1158/1535-7163.mct-17-0379.
Watson C, Long JS, Orange C, Tannahill CL, Mallon E, McGlynn LM, et al. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am J Pathol. 2010;177(5):2205–15. https://doi.org/10.2353/ajpath.2010.100220.
Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, et al. High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem Biophys Res Commun. 2006;342(4):1284–90. https://doi.org/10.1016/j.bbrc.2006.02.070.
Alshaker H, Wang Q, Srivats S, Chao Y, Cooper C, Pchejetski D. New FTY720-docetaxel nanoparticle therapy overcomes FTY720-induced lymphopenia and inhibits metastatic breast tumour growth. Breast Cancer Res Treat. 2017;165(3):531–43. https://doi.org/10.1007/s10549-017-4380-8.
Tarrason G, Auli M, Mustafa S, Dolgachev V, Domenech MT, Prats N, et al. The sphingosine-1-phosphate receptor-1 antagonist, W146, causes early and short-lasting peripheral blood lymphopenia in mice. Int Immunopharmacol. 2011;11(11):1773–9. https://doi.org/10.1016/j.intimp.2011.07.004.
Foss FW, Snyder AH, Davis MD, Rouse M, Okusa MD, Lynch KR, et al. Synthesis and biological evaluation of γ-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg Med Chem. 2007;15(2):663–77.
Huwiler A, Zangemeister-Wittke U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: recent findings and new perspectives. Pharmacol Ther. 2017. https://doi.org/10.1016/j.pharmthera.2017.11.001.
Jadidi-Niaragh F, Mirshafiey A. Therapeutic approach to multiple sclerosis by novel oral drugs. Recent Pat Inflamm Allergy Drug Discov. 2011;5(1):66–80. https://doi.org/10.2174/187221311794474900.
Azuma H, Takahara S, Ichimaru N, Wang JD, Itoh Y, Otsuki Y, et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Can Res. 2002;62(5):1410–9.
Lukas S, Patnaude L, Haxhinasto S, Slavin A, Hill-Drzewi M, Horan J, et al. No differences observed among multiple clinical S1P1 receptor agonists (functional antagonists) in S1P1 receptor down-regulation and degradation. J Biomol Screen. 2014;19(3):407–16. https://doi.org/10.1177/1087057113502234.
Sobel K, Monnier L, Menyhart K, Bolinger M, Studer R, Nayler O, et al. FTY720 phosphate activates sphingosine-1-phosphate receptor 2 and selectively couples to Galpha12/13/Rho/ROCK to induce myofibroblast contraction. Mol Pharmacol. 2015;87(6):916–27. https://doi.org/10.1124/mol.114.097261.
Mehling M, Brinkmann V, Antel J, Bar-Or A, Goebels N, Vedrine C, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology. 2008;71(16):1261–7. https://doi.org/10.1212/01.wnl.0000327609.57688.ea.
Mousseau Y, Mollard S, Richard L, Nizou A, Faucher-Durand K, Cook-Moreau J, et al. Fingolimod inhibits PDGF-B-induced migration of vascular smooth muscle cell by down-regulating the S1PR1/S1PR3 pathway. Biochimie. 2012;94(12):2523–31. https://doi.org/10.1016/j.biochi.2012.07.002.
Rosa R, Marciano R, Malapelle U, Formisano L, Nappi L, D’Amato C, et al. Sphingosine kinase 1 overexpression contributes to cetuximab resistance in human colorectal cancer models. Clin Cancer Res. 2013;19(1):138–47. https://doi.org/10.1158/1078-0432.ccr-12-1050.
Kim YM, Sachs T, Asavaroengchai W, Bronson R, Sykes M. Graft-versus-host disease can be separated from graft-versus-lymphoma effects by control of lymphocyte trafficking with FTY720. J Clin Investig. 2003;111(5):659–69. https://doi.org/10.1172/jci16950.
Ushitora Y, Tashiro H, Ogawa T, Tanimoto Y, Kuroda S, Kobayashi T, et al. Suppression of hepatocellular carcinoma recurrence after rat liver transplantation by FTY720, a sphingosine-1-phosphate analog. Transplantation. 2009;88(8):980–6. https://doi.org/10.1097/TP.0b013e3181b9ca69.
Lu Z, Wang J, Zheng T, Liang Y, Yin D, Song R, et al. FTY720 inhibits proliferation and epithelial-mesenchymal transition in cholangiocarcinoma by inactivating STAT3 signaling. BMC Cancer. 2014;14:783. https://doi.org/10.1186/1471-2407-14-783.
Hait NC, Avni D, Yamada A, Nagahashi M, Aoyagi T, Aoki H, et al. The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERalpha expression and enhances hormonal therapy for breast cancer. Oncogenesis. 2015;4:e156. https://doi.org/10.1038/oncsis.2015.16.
Kappos L, Radue EW, O’Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. https://doi.org/10.1056/NEJMoa0909494.
Ubai T, Azuma H, Kotake Y, Inamoto T, Takahara K, Ito Y, et al. FTY720 induced Bcl-associated and Fas-independent apoptosis in human renal cancer cells in vitro and significantly reduced in vivo tumor growth in mouse xenograft. Anticancer Res. 2007;27(1a):75–88.
Azuma H, Takahara S, Horie S, Muto S, Otsuki Y, Katsuoka Y. Induction of apoptosis in human bladder cancer cells in vitro and in vivo caused by FTY720 treatment. J Urol. 2003;169(6):2372–7. https://doi.org/10.1097/01.ju.0000064938.32318.91.
Bahrami B, Hojjat-Farsangi M, Mohammadi H, Anvari E, Ghalamfarsa G, Yousefi M, et al. Nanoparticles and targeted drug delivery in cancer therapy. Immunol Lett. 2017;190:64–83. https://doi.org/10.1016/j.imlet.2017.07.015.
Jadidi-Niaragh F, Atyabi F, Rastegari A, Kheshtchin N, Arab S, Hassannia H, et al. CD73 specific siRNA loaded chitosan lactate nanoparticles potentiate the antitumor effect of a dendritic cell vaccine in 4T1 breast cancer bearing mice. J Control Release. 2017;246:46–59. https://doi.org/10.1016/j.jconrel.2016.12.012.
Jadidi-Niaragh F, Atyabi F, Rastegari A, Mollarazi E, Kiani M, Razavi A, et al. Downregulation of CD73 in 4T1 breast cancer cells through siRNA-loaded chitosan-lactate nanoparticles. Tumor Biol. 2016;37(6):8403–12. https://doi.org/10.1007/s13277-015-4732-0.
Mao Y, Wang J, Zhao Y, Wu Y, Kwak KJ, Chen CS, et al. A novel liposomal formulation of FTY720 (fingolimod) for promising enhanced targeted delivery. Nanomed Nanotechnol Biol Med. 2014;10(2):393–400. https://doi.org/10.1016/j.nano.2013.08.001.
Peyrin-Biroulet L, Christopher R, Behan D, Lassen C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun Rev. 2017;16(5):495–503. https://doi.org/10.1016/j.autrev.2017.03.007.
Bigaud M, Dincer Z, Bollbuck B, Dawson J, Beckmann N, Beerli C, et al. Pathophysiological consequences of a break in S1P1-Dependent homeostasis of vascular permeability revealed by S1P1 competitive antagonism. PLoS One. 2016;11(12):e0168252.
Davis MD, Clemens JJ, Macdonald TL, Lynch KR. Sphingosine 1-phosphate analogs as receptor antagonists. J Biol Chem. 2005;280(11):9833–41. https://doi.org/10.1074/jbc.M412356200.
Balthasar S, Samulin J, Ahlgren H, Bergelin N, Lundqvist M, Toescu EC, et al. Sphingosine 1-phosphate receptor expression profile and regulation of migration in human thyroid cancer cells. Biochem J. 2006;398(3):547–56. https://doi.org/10.1042/bj20060299.
Wei SH, Rosen H, Matheu MP, Sanna MG, Wang SK, Jo E, et al. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary T cells to lymphatic sinuses. Nat Immunol. 2005;6(12):1228–35. https://doi.org/10.1038/ni1269.
Wang H, Huang H, Ding SF. Sphingosine-1-phosphate promotes the proliferation and attenuates apoptosis of Endothelial progenitor cells via S1PR1/S1PR3/PI3K/Akt pathway. Cell Biol Int. 2018;42(11):1492–502. https://doi.org/10.1002/cbin.10991.
Kawa Y, Nagano T, Yoshizaki A, Dokuni R, Katsurada M, Terashita T, et al. Role of S1P/S1PR3 axis in release of CCL20 from human bronchial epithelial cells. PLoS One. 2018;13(9):e0203211. https://doi.org/10.1371/journal.pone.0203211.
Kurano M, Tsuneyama K, Morimoto Y, Shimizu T, Jona M, Kassai H, et al. Apolipoprotein M protects lipopolysaccharide-treated mice from death and organ injury. Thromb Haemost. 2018;118(6):1021–35. https://doi.org/10.1055/s-0038-1641750.
Imeri F, Blanchard O, Jenni A, Schwalm S, Wunsche C, Zivkovic A, et al. FTY720 and two novel butterfly derivatives exert a general anti-inflammatory potential by reducing immune cell adhesion to endothelial cells through activation of S1P(3) and phosphoinositide 3-kinase. Naunyn-Schmiedeberg’s Arch Pharmacol. 2015;388(12):1283–92. https://doi.org/10.1007/s00210-015-1159-5.
Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res. 2008;102(8):950–8. https://doi.org/10.1161/circresaha.107.170779.
Huwiler A, Pfeilschifter J. New players on the center stage: sphingosine 1-phosphate and its receptors as drug targets. Biochem Pharmacol. 2008;75(10):1893–900. https://doi.org/10.1016/j.bcp.2007.12.018.
Khattar M, Deng R, Kahan BD, Schroder PM, Phan T, Rutzky LP, et al. Novel sphingosine-1-phosphate receptor modulator KRP203 combined with locally delivered regulatory T cells induces permanent acceptance of pancreatic islet allografts. Transplantation. 2013;95(7):919–27. https://doi.org/10.1097/TP.0b013e3182842396.
Pan S, Mi Y, Pally C, Beerli C, Chen A, Guerini D, et al. A monoselective sphingosine-1-phosphate receptor-1 agonist prevents allograft rejection in a stringent rat heart transplantation model. Chem Biol. 2006;13(11):1227–34. https://doi.org/10.1016/j.chembiol.2006.09.017.
Nishi T, Miyazaki S, Takemoto T, Suzuki K, Iio Y, Nakajima K, et al. Discovery of CS-0777: a potent, selective, and orally active S1P1 agonist. ACS Med Chem Lett. 2011;2(5):368–72. https://doi.org/10.1021/ml100301k.
Fujii Y, Hirayama T, Ohtake H, Ono N, Inoue T, Sakurai T, et al. Amelioration of collagen-induced arthritis by a novel S1P1 antagonist with immunomodulatory activities. J Immunol (Baltimore, Md: 1950). 2012;188(1):206–15. https://doi.org/10.4049/jimmunol.1101537.
Jin J, Hu J, Zhou W, Wang X, Xiao Q, Xue N, et al. Development of a selective S1P1 receptor agonist, Syl930, as a potential therapeutic agent for autoimmune encephalitis. Biochem Pharmacol. 2014;90(1):50–61. https://doi.org/10.1016/j.bcp.2014.04.010.
Xiao Q, Jin J, Wang X, Hu J, Xi M, Tian Y, et al. Synthesis, identification, and biological activity of metabolites of two novel selective S1P1 agonists. Bioorg Med Chem. 2016;24(10):2273–9. https://doi.org/10.1016/j.bmc.2016.03.059.
Lien YH, Yong KC, Cho C, Igarashi S, Lai LW. S1P(1)-selective agonist, SEW2871, ameliorates ischemic acute renal failure. Kidney Int. 2006;69(9):1601–8. https://doi.org/10.1038/sj.ki.5000360.
Rolin J, Sand KL, Knudsen E, Maghazachi AA. FTY720 and SEW2871 reverse the inhibitory effect of S1P on natural killer cell mediated lysis of K562 tumor cells and dendritic cells but not on cytokine release. Cancer Immunol Immunother CII. 2010;59(4):575–86. https://doi.org/10.1007/s00262-009-0775-7.
Hosseini M, Haji-Fatahaliha M, Jadidi-Niaragh F, Majidi J, Yousefi M. The use of nanoparticles as a promising therapeutic approach in cancer immunotherapy. Artif Cells Nanomed Biotechnol. 2016;44(4):1051–61.
Siahmansouri H, Somi MH, Babaloo Z, Baradaran B, Jadidi-Niaragh F, Atyabi F, et al. Effects of HMGA 2 si RNA and doxorubicin dual delivery by chitosan nanoparticles on cytotoxicity and gene expression of HT-29 colorectal cancer cell line. J Pharm Pharmacol. 2016;68(9):1119–30.
Li MH, Sanchez T, Yamase H, Hla T, Oo ML, Pappalardo A, et al. S1P/S1P1 signaling stimulates cell migration and invasion in Wilms tumor. Cancer Lett. 2009;276(2):171–9. https://doi.org/10.1016/j.canlet.2008.11.025.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
There was no funding.
Conflict of interest
None of the authors (NR, AN, AA, GA, MHF, GG, BY, MY, and FJN) have any conflict of interest to declare.
Rights and permissions
About this article

Cite this article
Rostami, N., Nikkhoo, A., Ajjoolabady, A. et al. S1PR1 as a Novel Promising Therapeutic Target in Cancer Therapy. Mol Diagn Ther 23, 467–487 (2019). https://doi.org/10.1007/s40291-019-00401-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-019-00401-5