
Abstract
Obesity is a major cause for the initiation and progression of kidney injury resulting in chronic kidney disease (CKD) and end stage renal disease. High body mass index is a major risk factor for new-onset CKD as well. The well recognized kidney disease secondary to obesity is glomerulopathy. Typical histological features of obesity related glomerulopathy include glomerulomegaly and focal segmental glomerulosclerosis. In obese individuals, excess excretory load induces hyperperfusion and hyperfiltration by the kidneys leading to glomerulomegaly. Lipid accumulation in the kidney which accompanies excessive fat deposition in the body is implicated in the development of CKD. Obesity is associated with metabolic abnormalities in the adipose tissue such as increased free fatty acids, hyperinsulinemia, insulin resistance, pro inflammatory conditions, reduced adiponectin, leptin resistance and activation of the renin-angiotensin-aldosterone system, all of which mediate injury to the cells of glomeruli and tubules leading to CKD. Despite much progress in our understanding of the mechanisms of obesity related kidney disease, several questions about the pathogenesis of nephropathy associated with obesity remain to be answered. Delineating obesity linked factors, which lead to adaptive and maladaptive changes in the kidney and predispose patients to renal disease could lead to identification of molecular targets and reno-protective and therapeutic strategies to improve outcomes for obese patients with CKD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Obesity
- Chronic kidney disease
- Glomerulopathy
- Glomerulosclerosis
- Adipose tissue
- Adipokines
- Insulin resistance
- Cytokines
Introduction
Obesity is currently an epidemic in developed countries and a global health challenge. A significant association has been noted between obesity and initiation and progression of chronic kidney disease (CKD) in population-based studies [1,2,3,4,5,6]. The incidence of obesity-related renal disease has increased ten times in recent years. Prevalence of obesity which is estimated to rise by 40% in the next decade is expected to result in a parallel escalation in the incidence of obesity related CKD [7, 8].
High body mass index (BMI) is a major risk factor for new-onset CKD [1, 2, 9]. In a study of 75,000 Norwegians, who were followed up for 21 years, increased BMI was found to correlate with initiation of kidney disease or CKD related mortality [5]. A 600% increase in end stage renal disease (ESRD) was observed in those with a BMI more than 40 kg/m2 in a survey of more than 3 lakhs individuals enrolled in the Kaiser Permanent Health System between 1964 and 1985 [8, 10]. Obese individuals are at a greater risk for acute kidney injury, for nephrolithiasis and kidney cancer as well [7].
Until recently, obesity related kidney disease was thought to be linked to obesity associated hypertension, diabetes and cardiovascular disease, which are all common causes of renal disease. There is increasing evidence that even without other risk factors, obesity can initiate kidney disease and also accelerate progression of preexisting renal disease [5, 11, 12]. Several obesity-induced disorders are nephrotoxic [13].
Obesity Related Glomerulopathy
The first record on the link between obesity and massive proteinuria was by Weisinger et al. [14]. Later, many reports confirmed the association of obesity with proteinuria, glomerulomegaly and frequently, focal segmental glomerulosclerosis (FSGS) [15,16,17,18]. FSGS is not observed in all cases of obesity related kidney disease; its presence may depend on the degree of obesity or renal impairment. Among various sub-types of FSGS, the perihilar variant is more common [17].
In 2001, Kambham and colleagues proposed the term ‘obesity-related glomerulopathy’ (ORG) for the microscopic lesions observed in kidney biopsies from obese individuals [17]. The diagnostic criteria for ORG are: BMI values of 30 kg/m2 or greater and absence of clinical as well as biopsy evidence of other renal diseases [17]. The concept of ORG as a nephropathy does not for its diagnosis, depend on the manifestation of proteinuria. Patients with ORG may also not have edema [17]. Kambham et al. noted that ORG is less likely than idiopathic FSGS to present with edema or the degree of proteinuria as seen in nephrotic syndrome. ORG is however frequently associated with hypertension and dyslipidemia [17].
Isolated proteinuria of unknown onset with or without renal impairment, is the initial symptom in most cases. Even with relatively high excretion of protein in the urine, hypoalbuminemia may not be present [17]. The mechanism for this remains obscure. Typically, the clinical condition is stable. Alternately, the patient may have slowly progressive proteinuria. Significantly, weight loss can strikingly reduce proteinuria associated with obesity [18, 19]. In 10–33% of patients diagnosed with ORG, long-term outcomes include progression to ESRD [17].
Renal Changes in Obesity
Structural Changes
Much is currently known about the structural and hemodynamic changes in the kidneys of the obese (Table 12.1) [20,21,22,23].
Autopsies have revealed an increase in kidney weight in those with a high BMI [22]. Increase in kidney weight may result from hypertrophy of individual nephrons secondary to increased tubular and glomerular functions related to increase in BMI. Intracellular or extracellular accumulation of fluid and lipid components may also contribute to a larger kidney.
Glomerular sizes are larger in the obese, even without obvious renal disease [23,24,25]. There is no unanimity on the definition of glomerulomegaly in ORG. A morphometric study found that in patients with ORG and preserved renal function, when compared to control subjects, the mean glomerular volume is increased about 3-fold [26]. The glomerular capillaries are increased in number. The increase in vascular endothelial growth factor (VEGF) expression in the glomeruli of patients with ORG may contribute to the formation of new micro vessels [27]. Cell proliferation and matrix synthesis could also contribute to glomerular enlargement. Glomerulomegaly is accompanied by a 45% reduction in podocyte density [24]. Thickening of the glomerular basement membrane (GBM) considered as an early manifestation of diabetic nephropathy, is seen with obesity as well. Both glycemic and lipid abnormalities in obesity may contribute to GBM thickening, which may not be as severe as seen in patients with type 2 diabetes.
Obesity-induced glomerular hypertrophy and glomerulomegaly may cause glomerular podocytes to enlarge their foot processes to cover the expanded glomerular surface area. Consistent with this, a relative reduction in the coating area of glomerular podocytes on the glomerular surface is found in patients with ORG [28]. This may cause changes in podocyte function and a consequent loss in protein selectivity, podocyte detachment, and replacement by matrix deposition, leading to FSGS. Incidence of foot process fusion among glomerular podocytes is significantly lower in ORG than in idiopathic FSGS [17].
Focal lipid vacuoles are occasionally seen in the cytoplasm of glomerular mesangial cells and tubular epithelial cells [29]. A study comparing the kidney biopsies from obese patients with proteinuria and biopsies from non-obese patients with proteinuria found a 33% increase in the cross-sectional area of proximal tubular epithelial cells and 54% increase in the lumen of the proximal tubules [30].
Changes in Hemodynamics
Hemodynamics in the kidney is markedly altered by obesity. In obese individuals, there is compensatory hyperfiltration to meet the high metabolic needs of the larger fat mass.
A recent study found that glomerular filtration rate (GFR) is higher in obese adults than in those with normal body weight [30, 31]. Renal plasma flow (RPF) is also increased, though not to the same degree. As a result, the filtration fraction (FF) increases, a hemodynamic adjustment that parallel the degree of BMI and adipose tissue mass. Studies in obese individuals suggest that afferent arteriolar vasodilatation, together with efferent arteriolar vasoconstriction, contribute to the increase in FF [31,32,33,34]. By lowering tubular sodium chloride relative to GFR, obesity-dependent mechanisms disrupt the tubuloglomerular feedback (TGF) response, preventing suppression of GFR [35]. Given the high rate of association of hypertension with obesity, inadequate TGF may result in the transmission of systemic blood pressure to the glomerulus contributing not only to increased GFR but also to structural changes in the kidney [35]. Increase in obesity-induced GFR is not however permanent [36, 37].
The changes in renal hemodynamics found in obesity are closely linked to increased salt sensitivity [38]. Activation of the renin–angiotensin–aldosterone system (RAAS) in the kidney is an important mechanism by which salt sensitivity is increased in obesity [37, 39]. Activation of renal sympathetic nerves may also be involved in the increased salt reabsorption seen with obesity [40, 41]. The glomerulus enlarges in response to increases in GFR, RPF, FF and tubular sodium reabsorption.
Potential Mechanisms of Renal Injury
Kidney disease is initiated with increases in GFR and pressure in glomerular capillaries (PGC), followed by hypertrophy of the glomerulus as well as podocytes [42, 43]. Micro albuminuria ensues and progresses to proteinuria. Mesangial cell proliferation, mesangial matrix expansion, nodular glomerulosclerosis and tubulointerstitial injury contribute to a decrease in GFR culminating in ESRD [44, 45]. The exact mechanisms for obesity associated CKD in humans are not yet completely clear. Current understanding is based upon association studies, cell and animal studies and pharmacological manipulations.
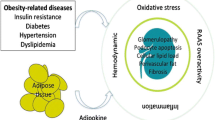
Several obesity-related factors have been implicated in the progression of CKD (Fig. 1). These may act singly or together to cause renal injury. Potential mechanisms are: (i) adverse effects of adaptations to increase in body mass and excretory load, (ii) adverse effects of adaptations to obesity-induced sodium retention [44], (iii) insulin resistance (IR) and progressive hyperglycemia (iv) direct or indirect effects of hyperinsulinemia (v) renal lipotoxicity [46,47,48] (vi) rise in free fatty acids (FFAs) which may aggravate insulin resistance and (vii) fatty acid/triglyceride accumulation within tissues resulting in cellular dysfunction [49].

Mechanisms of renal disease in Obesity. RAAS—Renin-angiotensin-aldosterone system, FFAs—free fatty acids, TNF—tumor necrosis factor, IL—interleukin, CRP—C reactive protein, MCP—monocyte chemo attractive protein, TGF—transforming growth factor, PAI—plasminogen activator inhibitor
The effects of IR, dyslipidemia and oxidative stress are likely to be intensified by dysregulation of adipocytokines such as leptin and adiponectin and proinflammatory cytokines [49]. Resistin, corticosteroids, nutritional status and genetic factors are the other factors implicated.
Excess Excretory Load
Obesity is associated with an excess excretory load resulting from increased body mass and the increased energy intake and tissue turnover required to maintain it. Fat-free body mass also increases in obesity harmonious with functional overload [50]. Organomegaly in obesity includes the kidneys [15]. Chagnac et al. confirmed a 51% increase in renal perperfusion and 31% increase in filtration in severe obesity [33]. The reduced renal resistance with increased FF is compatible with glomerular capillary hypertension [33], a perfect setting for future glomerulosclerosis. Obesity induces single-nephron adaptations typical of the reduced nephron number accompanying CKD.
Adverse Adaptations to Excess Retention of Sodium
Hall et al. proposed that in obesity, there is reduced capacity for sodium excretion, acting at sites proximal to the macula densa through Ang II and sympathetic activation. The reduced sodium chloride delivery to the macula densa site induces afferent vasodilation and renin release to produce compensatory glomerular hyperfiltration, thus restoring normal distal delivery [46]. Thomson et al. considers a mechanism similar for the increase in proximal tubular sodium reabsorption induced by hyperglycemia seen in diabetes [42]. The result is intraglomerular hypertension and proteinuria which form the final common pathway for chronic glomerular and tubular injury, as with excess excretory load induced hyperfiltration.
Hypertension
Hypertension is common in obesity and may be due to a range of factors including sympathetic nervous system activation and angiotensinogen release from adipose tissue [36].
The rise in blood pressure may damage microvasculature within the kidney through intensified RAAS activity. Ang II excess in the kidney can enhance renal injury through proinflammatory mechanisms and can also promote proteinuria-related renal damage [49].
The Role of Adipose Tissue
Adipose tissue is a highly active endocrine organ involved in the clearance and storage of fatty acids, regulation of energy homeostasis and metabolism, insulin function and inflammatory processes. Adipose tissue is involved in progression of diseases associated with metabolic dysfunction [51,52,53,54,55,56]. Increased fat deposition in obesity also correlates with renal injury [29].
Obesity is a chronic low-grade inflammatory condition, in which adipose tissue serves as the source of inflammatory cytokines [57]. Visceral adipose tissue produces less adiponectin and more of pro-inflammatory cytokines, including tumor necrosis factora (TNF-α) and interleukin-6 (IL-6), which can induce IR and promote endothelial dysfunction [58].
There is in obesity, lipid accumulation in the kidney suggesting a role for fat accumulation in the organ for the development of CKD [59,60,61,62]. Visceral fat elaborates bioactive substances which contribute to the abnormal hemodynamic and structural changes leading to obesity-related nephropathy. Visceral adipose tissue mediates obesity-related disease through production of pro-inflammatory cytokines (adipokines) and causing IR [63]. Adipocytes secretes angiogenic factors as well and thus can facilitate vasculogenesis locally and in distal organs [64]. In a study using obese Zucker rats, in parallel with intrarenal inflammation, significant increases in the density of cortical and medullary microvascular was found [65].
Adipocytes contain adipocyte-specific metabolites such as free fatty acids, leptin, and adiponectin and all the components of the RAAS, plasminogen activator inhibitor (PAI), all of which affect renal structure and function. In addition, fat is infiltrated by macrophages that can alter their phenotype and foster a proinflammatory environment which in the kidney, advances pathophysiologic changes associated with obesity. Adipose tissue of patients with ESRD has elevated levels of the pro-inflammatory cytokines TNF-α and monocyte chemoattractant protein-1 (MCP-1) and more infiltration of macrophages [66]. Elevated levels of pro-inflammatory cytokines or chemokines (TNF-α, IL-6, IL-1β, MCP-1) and infiltrated macrophages in the adipose tissue correlate with renal inflammation in obese rodents [67].
Metabolic Factors
Altered fatty acid and cholesterol metabolism is responsible for ipid accumulation, inflammation, oxidative stress and fibrosis in the kidney [68]. BMI seems to determine the degree of accumulation of triglycerides in the human renal cortex [69]. Triglyceride accumulation is seen in both glomerular and tubular cells, more in proximal tubular cells [69].
Lipid disturbances in obesity appears to directly involve in renal damage. Young C57BL/6 mice fed a high fat diet (HFD) have increased body weight, and elevated blood levels of glucose, insulin, triglycerides and cholesterol and lower circulating adiponectin. Proteinuria, glomerulomegaly, expanded mesangial matrix, thickened glomerular basement membrane and podocyte effacement have been observed in these mice [59]. There is evidence that lipid moieties can injure mesangial cells. Low density lipoprotein (LDL), oxidized LDL, and glycated LDL, at levels observed in blood significantly increase synthesis of mesangial matrix components, fibronectin and laminin [70]. Lipid moieties also stimulate production of macrophage migration inhibitory factor in mesangial cells and expression as well as release of inflammatory activators, CD40 and IL-6 [70]. Renal toxicity and proteinuria in mice with hyperlipidemia can be attenuated by treatment with anti-IL-6 monoclonal antibody [71].
Lipids also damage podocytes [28]. Oxidized LDL decreases phosphorylation of AKT, involved in cell survival and can thus contribute to nephrin loss and podocyte apoptosis [72, 73]. In podocytes cultured with palmitate, there is increase in the synthesis of ceramide resulting in reduced insulin-stimulated glucose uptake [74]. Thus, lipid abnormalities may interact with changes in glucose metabolism to actuate nephropathy.
Normal insulin/phosphatidylinositol 3-kinase/AKT and mTOR signalling are critical for podocyte hypertrophy and adaptation. Adipokines and lipid stores in the kidney result in IR in podocytes and maladaptive responses, to cope with the mechanical forces of hyperfiltration [68].
Sterol regulatory element binding protein-1 (SREBP-1) also seem to play a role in the damage to the kidney from lipid accumulation and ensuing inflammatory and fibrotic responses [75, 76]. The effects of HFD on the kidney were not seen in SREBP-1c −/− mutant mice, while SREBP-1a transgenic mice had lipid deposition in the glomeruli, glomerulosclerosis and albuminuria. In patients with ORG, expression of SREBP-1 in the glomeruli is up-regulated [27].
Adipokines
Visceral fat releases into the circulation a large number of adipocytokines with autocrine, paracrine and endocrine activities, which contribute to the pathogenesis of renal injury [77].
The secreted peptides include leptin, adiponectin, resistin, TNF-α, IL-6 and components of the RAAS such as angiotensinogen, ACE and ATII-1R as well as VEGF, MCP-1, RBP-4 and TIMP-1. There is a positive correlation between increase in adipose tissue and elevated inflammatory markers [78]. Axelsson et al. 2004 observed a positive relationship between truncal fat mass and inflammation in patients with ESRD [79]. Adipose tissue also has macrophages and immune cells which secrete pro-inflammatory cytokines. The link between dysfunctional adipose tissue in obesity and raised proinflammatory adipocytokine patterns, systemic inflammation, IR and cellular dysfunction, is well recognized [49]. Be that as it may, role of this link in the pathogenesis of CKD is not well elucidated.
Leptin
Leptin levels rise in response to increase in fat mass. Elevated leptin levels in obese individuals correspond to the fat stores. Central hypothalamic resistance to leptin may also be present in the obese. Leptin has immuno-regulatory and proinflammatory actions in the obese [80].
In patients with obesity, leptin levels rise and there is leptin resistance during the initial stages of development of CKD [81]. When CKD progresses, reduced renal clearance of leptin contributes to hyperleptinemia and is associated with concomitant inflammation [82, 83].
Mice overexpressing leptin have more renal disease than leptin deficient mice [84]. Long-term infusion of recombinant leptin in rats is associated with proteinuria, increased expression of extracellular matrix proteins (collagen type IV), TGF-β and other pro-inflammatory cytokines, macrophage infiltration and glomerulosclerosis [85]. These observations indicate that leptin promotes renal injury in obese subjects. Despite severe obesity, renal dysfunction is not seen in the absence of leptin or mutation in the leptin receptor gene [13].
Adiponectin
Adiponectin (also called Acrp30), one of the most abundant adipokines produced by the adipocytes is down-regulated in obesity and type 2 diabetes [86]. Low adiponectin levels is linked to inflammation, atherosclerosis, IR, and raised blood pressure [87]. Endothelial cell dysfunction, impaired endothelium-dependent vasodilation, enhanced leukocyte-endothelium adhesion and activation of RAAS have been found in both humans and experimental animals with decreased levels of adiponectin.
Adiponectin is also known to aid the normal function of the podocyte [88]. In hypoadiponectinemia, function of podocytes in maintaining glomerular filtration breaks down causing glomerular damage and sclerosis. Glomerulomegaly, collagen deposition in glomeruli, loss of podocyte foot processes, increased TGF-β, and albuminuria have all been observed in adiponectin null mutant mice [89]. Adiponectin restitution reverts podocyte effacement and albuminuria. This benefit is attributed to reduction in oxidant stress [88, 90]. Adiponectin deficiency can also lead to increased NADPH oxidase activity and a rise in the levels of reactive oxygen species.
Adiponectin is an insulin-sensitizing factor as well and has also anti-inflammatory effects. Reduced plasma adiponectin level is inversely correlated with IR in obese patients [88, 89]. Though obese subjects have consistently low circulating adiponectin levels, in patients with CKD due to obesity, adiponectin levels are increased, possibly because of renal dysfunction [90, 91].
The role of adiponectin in obesity-related disease has been extensively investigated using transgenic mice or pharmacological globular Acrp30 compound [92,93,94,95,96]. Adiponectin is an important regulator of lipid and glucose metabolism and a key link among TNF-α, MCP-1, and IR.
Effects of adiponectin are tightly linked to the activation of AMP-activated protein kinase (AMPK) [88, 97]. Obesity is known to modulate the activity of AMPK. Steinberg et al. demonstrated that TNF-α could suppress AMPK activation through the TNF receptor 1 (TNFR1), suppress fatty acid oxidation and promote IR in skeletal muscle [98]. How TNF-α inhibits AMPK activation is unclear. Steinberg et al. showed that this process might involve the upregulation of protein phosphatase 2C (PP2C) by TNF-α. Treatment with TNF-α increases PP2C activity and decreases AMPK activation in WT mice but not in the transgenic ob/ob TNFR−/− mice. This change is also accompanied with a reduction of fatty acid oxidation and an increase of diacylglycerol (DAG) and triacylglycerol (TAG) in skeletal muscle [103]. AMPK activation reduces TNF-α and increases adiponectin levels in human adipose tissue, improving insulin sensitivity [99]. A decrease of adiponectin and AMPK activation is also associated with increase in the levels of MCP-1 in human adipocytes [100].
Recently, a role for AMPK in regulating macrophage infiltration and activation has been proposed [101]. AMPK activation completely reverses HFD induced infiltration of macrophages in the kidney [62]. AMPK activation is also a key regulator of lipid accumulation in vacuolated proximal tubular cells, maintenance of integrity of the brush border, as well as nitrotyrosine and NOX4 levels. These findings suggest that AMPK activation may have a role in tubular dysfunction [62].
Macrophage Infiltration of Adipose Tissue and Phenotype Switch in Macrophages
There are evidences for macrophage influx in adipose tissue of obese humans and animal models of obesity [102,103,104]. Macrophage infiltration of adipose tissue results in inflammation and IR [102, 103]. Obesity induces a phenotype switch in macrophages [105,106,107,108]. In lean rodents, M2 phenotype, involved in the resolution of inflammation and tissue repair are predominant; proinflammatory M1 macrophage population is dominant in obese animals [106]. Macrophages that infiltrate the adipose tissue are a source of a large number of proinflammatory mediators such as TNF-α, IL-6, C-reactive protein (CRP), MCP-1 and macrophage migration inhibitory factor [109]. Inhibition of proinflammatory macrophages reduces kidney injury [110,111,112]. There is indication from animal experiments that macrophage AT1 receptor may mediate macrophage polarization [113].
Adiposity-Driven Proinflammatory Cytokines
Inflammation markers are inversely associated with measures of kidney function and positively with albuminuria [114]. Thus, there is a strong evidence for the contribution of inflammation in obesity associated renal disease.
Rapid expansion of adipose tissue results in an altered synthesis of pro-inflammatory adipokines which leads to a state of low-grade inflammation [115]. Among the large number of pro-inflammatory adipokines, TNF-α is one of the most critical mediators of adipose tissue inflammation and development of IR [116, 117].
Fatty acids released by adipocytes promote TNF-α release by macrophages which, in turn, increases IL-6 production in fat cells and thus an inflammatory milieu in both adipose tissue and kidney [82]. TNF-α is an important mediator of progressive renal fibrosis. Gene expressions of both TNF-α and its receptors, as well as IL-6, a signal transducer are seen increased in glomeruli of patients with ORG and indicates the importance of TNF-α and IL-6 in development of ORG [27].
A rise in TNF-α levels is generally associated with increased production of MCP-1 by both adipocytes and macrophages. MCP-1 is also a key mediator of both adipose tissue inflammation and development of IR [117,118,119]. The mediatory role of MCP-1 and its receptor CCR2 in chronic kidney disease has recently received much attention [120]. In human podocytes, MCP-1 regulates nephrin expression via CCR2 [121]. Studies in mesangial cells have revealed that palmitate stimulates marked secretion of MCP-1 indicating that in obesity, circulating saturated fatty acids, such as palmitate may trigger production of MCP-1 [61].
The major source of pro-inflammatory cytokines that directly contribute to renal injury in obese subjects are the infiltrated macrophages [84]. In addition, renal parenchyma also releases proinflammatory cytokines in response to hyperglycemia or locally active vasoactive peptides such as Ang II [122]. These mediators produce low grade chronic inflammation and participate in the pathogenesis of ORG. TNF-α has been shown to reduce the expression of key components of the slit diaphragm, nephrin and podocin, thus contributing to podocytopathy [78]. IL-6 promotes expression of adhesion molecules and oxidative stress [123].
Insulin Resistance
Insulin resistance (IR) is a common accompaniment of obesity and related metabolic syndrome [124, 125]. In obese rodents and humans, inflamed adipose tissue is known to contribute to development of IR [124, 126]. IR is a salient metabolic risk for CKD [127]. Many studies indicate the association between IR and CKD. This association is seen even before the onset of diabetes [128, 129].
The direct link between adipose tissue dysfunction and associated IR with obesity related kidney disease is presently more evident. The low adiponectin levels in obesity is associated with reduced insulin sensitivity, which leads to a pro-inflammatory state in the kidney [130].
In the captive rhesus monkey with spontaneous obesity, glomerular hypertrophy appears in the prediabetic hyperinsulinemic phase; hyperglycemia, hypertension, renal dysfunction, and increase in mesangial matrix deposition are absent at that stage [131].
Insulin, although a weak vasodilator, augments endothelial-dependent vasodilation. Hyperinsulinemia can contribute to preglomerular vasodilation, glomerular hypertension and increase in glomerular capillary permeability [132, 133]. Structural damage is not manifest during this period. Hyperinsulinemia may induce glomerular hypertrophy either directly or by stimulating the IGF-1 receptor [134]. Hyperinsulinemia can also augment Ang II contraction of glomerular mesangial cells [135]. Abrass et al. demonstrated that high-dose insulin stimulates expression of inflammatory collagens in renal mesangial cells in culture [136]. They also found that the altered gene expression after exposure to high insulin is not reversible by later withdrawal of insulin [137, 138].
Walsh et al. through a study in transgenic mice missing insulin receptors in their podocytes, have elucidated the critical role of insulin signaling in normal podocytes [139]. They showed that these mice have normal glomeruli when they are at their early age (three weeks old), but later starting at the age of 5 weeks, have loss of podocyte foot processes, increased glomerular matrix and albuminuria [139].
Free Fatty Acids (FFA) might also contribute to IR [74]. Increased FFA flux from excess adipose tissue to non-adipose organs results in lipid accumulation in ectopic organs including the kidney. Lipid stores later advances impairment of glucose metabolism and insulin sensitivity in these organs. This adverse effect is associated with an increase of ceramide, a highly lipotoxic molecule, that is related to IR [140]. In addition, a dysregulation of the insulin receptor and the impairment of recruitment of the glucose transporter GLUT4 to the cell surface have been noted with FFA increase [74].
Though there are evidences for considering IR as a driver of the renal disease in obesity, how critical is IR for progression of the disease is still unclear.
There are many similarities between the ‘obese’ and ‘diabetic’ kidney; there are also features unique to obesity sans diabetes. Kidneys of obese individuals frequently have lipid deposits (foam cells) in glomeruli and mesangium. This is an evidence for the concept that renal injury is caused by lipotoxicity. Lipid accumulation in the glomerulus may result in the upregulation of SREBP-1 and 2 and promote podocyte apoptosis, mesangial cell proliferation and cytokine synthesis [140].
Vasoactive Peptides
Several vasoactive peptides have been implicated in the pathogenesis of OGR. There is upregulation in the intrarenal RAAS in obesity [141, 142]. Activation of the RAAS leads to both hemodynamic and cellular effects. Ang II leads to increases in efferent arteriolar vasoconstriction and glomerular pressure, sodium retention and cell proliferation [143,144,145]. In cells, Ang II activates protein kinase C (PKC), MAP kinase (MAPK) and transcription factors such as nuclear factor-κB. Their activation leads to alteration in the expression of genes of a number of growth factors and cytokines. Increase in TGF-β promotes podocyte apoptosis, mesangial cell proliferation and extracellular matrix synthesis, cellular events that are important in the development of obesity- and diabetes-associated glomerulopathy [146].
Renin-Angiotensin-Aldosterone System (RAAS)
Adipose tissue has all the components of the RAAS system. RAAS is activated in the adipose tissue of the obese [147, 148]. In obese adipocytes, production of angiotensinogen, aldosterone, and aldosterone-stimulating factor is increased [149,150,151]. In obese individuals, the RAAS is activated in renal tissue as well, resulting in increase in sodium reabsorption through many mechanisms [152,153,154]. Activation of the RAAS in the kidney, especially of aldosterone and or its receptor, is likely to play a major role in the development of kidney injury and proteinuria associated with obesity. Hyperglycemia and angiotensin II are known to upregulate the expression of sodium glucose co-transporter (SGLT-2) [155, 156]. Thus, in obesity, in which both hyperglycemia and RAAS activation occur, renal tubular reabsorption of glucose may be increased via upregulation of the expression of SGLT-2.
RAAS is a major regulator of vasomotor tone and cellular proliferation and thus regulates renal function and structure. Adipocytes and adipose-infiltrating macrophages are important sources of RAAS [157]. Circulating levels of angiotensinogen (Aog) increase with increasing BMI [158]. Adipose-derived increase in circulating RAAS ligands together with adipose-driven increase in renal AT1 is a potent alliance for efferent arteriolar vasoconstriction, to increase glomerular pressure and FF, as well as cellular proliferation and thus culminate in renal damage [159, 160]. There are also evidences to suggest that AT2 may mediate the substantial adipose inflammatory response associated with increased Aog [161].
Aldosterone blockade reduces renal injury. This benefit is independent of its antihypertensive effects and possibly relates to the blocking effects of aldosterone on PAI-1 and TGF-β̣, reactive oxygen intermediates, inflammatory mediators, and podocyte function [162,163,164]. Adipose tissue produces aldosterone independent of AngII as well. At least one oxidized derivative of linoleic acid is known to stimulate aldosterone synthesis [165]. Complement-C1q TNF-related protein 1 (CTRP1), prominently expressed by adipose tissue may also mediate Ang II-independent aldosterone production [166]. Thus, elevated aldosterone in obesity could be injurious to glomeruli through its indirect action which increases GFR and also through its direct effects on the podocyte.
Plasminogen Activator Inhibitor-1 (PAI-1)
Obesity induces PAI-1 in adipose tissue and glomerular cells. PAI-1 is an independent risk factor for renal damage. PAI-1 can decrease protease-dependent matrix degradation and cellular migration [167]. In a model of podocyte injury-associated glomerulosclerosis, renoprotection by PPAR-γ agonist is partially through reduced PAI-1 [168]. PAI-1 modulates podocyte injury as well. Ablation of the kidney in PAI-1 deficient mice caused less of proteinuria, podocyte damage and glomerular sclerosis [169].
Several mechanisms involved in obesity-related organ dysfunction are concomitant. IR is linked to increased levels of Ang II, which is associated with the progression of renal damage in obesity. Indeed, Angiotensin II is an important mediator in the progression of obesity related kidney disease [170,171,172]. Angiotensin II contributes to hyperfiltration and glomerulosclerosis through both hemodynamic and non-hemodynamic effects [18, 173,174,175]. ANG II produces vasoconstriction while insulin induces vascular relaxation by promoting NO production through the phosphatidylinositol 3-kinase (PIK3-Akt) signaling pathway [176]. The inhibitory effects of ANG II on the insulin action is mediated by production of reactive oxygen species (ROS) [177, 178]. In turn, ROS induces inflammatory cytokines such as MCP-1 or TNFα which can then impair the PI3K-Akt pathway of insulin signaling, resulting in IR [116, 179, 180].
Factors Which Increase Susceptibility for Renal Injury in the Obese
Severity of renal impairment does not always correlate with the severity of obesity. Considerable differences in susceptibility to renal injury have been noted among obese individuals and are possibly related to other predisposing factors (Table 2).
Obesity is a risk factor for type 2 diabetes, hypertension and other components of the metabolic syndrome as well as cardiovascular disease. All of them add to the risk for CKD in obese individuals. Each component of MetS (impaired glucose tolerance, hypertension, and dyslipidemia) can induce kidney injury and may also exacerbate pre-existing kidney disease [181]. Combinations of components synergistically increase the risk for CKD, and the risk of progression of pre-existing CKD [182]. The odds ratio for kidney disease is 1.89 when only one component of MetS is present, leaping to 5.85 when all five components of MetS are present together in an individual [183, 184]. A meta-analysis of eleven studies [185] involving 30,146 subjects revealed that MetS is associated with development of Stage III CKD with an odds ratio (OR) of 1.55 (95% CI: 1.34–1.80) [185]. Global as well as segmental glomerulosclerosis are seen in those with MetS. There is also a higher prevalence of tubular atrophy, interstitial fibrosis, and arterial sclerosis [186].
Interestingly, in comparison to normal subjects, patients with obesity related kidney disease have a significantly lower glomerular density as seen in kidney biopsies [26]. Aging related decreases in the number of glomerular podocytes can also greatly influence susceptibility to renal injury in obese individuals [187].
In obesity, nephron overwork and risk of intraglomerular hypertension would be exaggerated in those with intra uterine growth retardation and those who have had unilateral nephrectomy, as they have already a lower nephron number [188, 189].
Pulmonary hypertension secondary to sleep apnea and non-alcoholic fatty liver disease which commonly accompany obesity may also catalyze renal injury associated with obesity [190, 191].
Conclusions
Obesity is a major cause for the initiation and progression of renal injury resulting in CKD and ESRD. Many factors together cause renal vasodilation, glomerular hyperfiltration and albuminuria, leading to glomerulopathy. In obese individuals, excess excretory load induces hyperperfusion and hyperfiltration by the kidneys leading to glomerulomegaly. Obesity associated metabolic abnormalities such as increased FFA, hyperinsulinemia, IR, pro inflammatory conditions, reduced adiponectin, and leptin resistance mediate injury to glomerular and tubular cells and lead to CKD.
To a large extent, both obesity and related CKD can be prevented by following a healthy lifestyle. Renin-angiotensin-aldosterone blockade is effective in the short-term. SREBP antagonists, PPARα agonists, FXR and TGR5 agonists and LXR agonists directly target lipid metabolism and hence are considered to have therapeutic value. Delineation of obesity linked factors, which lead to adaptive and maladaptive changes in the kidney and predispose patients to renal disease could lead to identification of molecular targets and reno protective and therapeutic strategies to improve outcomes for obese patients with CKD.
Several questions about the pathogenesis of nephropathy associated with obesity remain to be answered. Whether glomerulomegaly is a cause or only an associated feature of proteinuria in ORG is unclear. Whether glomerulomegaly is a precursor of obesity-related FSGS lesion is also obscure. Whether genetic factors have any role in determining the time of onset and the rate of progression of kidney disease in obese subjects is also unknown.
References
Foster MC, Hwang SJ, Larson MG et al (2008) Overweight, obesity, and the development of stage 3 CKD: the Framingham Heart Study. Am J Kidney Dis 52:39–48
Gelber RP, Kurth T, Kausz AT et al (2005) Association between body mass index and CKD in apparently healthy men. Am J Kidney Dis 46:871–880
Kramer H, Luke A, Bidani A et al (2005) Obesity and prevalent and incident CKD: the hypertension detection and follow-up program. Am J Kidney Dis 46:587–594
Lu JL, Kalantar-Zadeh K, Ma JZ et al (2014) Association of body mass index with outcomes in patients with CKD. J Am Soc Nephrol 25:2088–2096
Munkhaugen J, Lydersen S, Wideroe TE, Hallan S (2009) Prehypertension, obesity, and risk of kidney disease: 20-year follow-up of the HUNT I study in Norway. Am J Kidney Dis 54:638–646
Vivante A, Golan E, Tzur D et al (2012) Body mass index in 1.2 million adolescents and risk for end-stage renal disease. Arch Intern Med 172:1644–1650
Kovesdy CP, Furth SL, Zoccali C, On Behalf of the World Kidney Day Steering Committee (2017) Obesity and kidney disease: hidden consequences of the epidemic. Am J Nephrol 45:283–291
Chang A, Kramer H (2012) CKD progression: a risky business. Nephrol Dial Transplant 27:2607–2609
Pinto-Sietsma SJ, Navis G, Janssen WM et al (2003) A central body fat distribution is related to renal function impairment, even in lean subjects. Am J Kidney Dis 41:733–741
Olsen NSC, Iseki K, Kramer H, Liu Z, Sharma K (2017) Kidney disease and obesity: epidemiology, mechanisms and treatment. Nat Rev Nephrol 13:181–190
Goncalves Torres MR, Cardoso LG, de Abreu VG et al (2009) Temporal relation between body mass index and renal function in individuals with hypertension and excess body weight. Nutrition 25:914–919
Nomura I, Kato J, Kitamura K (2009) Association between body mass index and chronic kidney disease: a population-based, cross-sectional study of a Japanese community. Vasc Health Risk Manag 5:315–320
Hunley TE, Ma LJ, Kon V (2010) Scope and mechanisms of obesity-related renal disease. Curr Opin Nephrol Hypertens 19:227–234
Weisinger JR, Kempson RL, Eldridge FL, Swenson RS (1974) The nephrotic syndrome: a complication of massive obesity. Ann Intern Med 81:440–447
Verani RR (1992) Obesity-associated focal segmental glomerulosclerosis: Pathological features of the lesion and relationship with cardiomegaly and hyperlipidemia. Am J Kidney Dis 20:629–634
Adelman RD, Restaino IG, Alon US, Blowey DL (2001) Proteinuria and focal segmental glomerulosclerosis in severely obese adolescents. J Pediatr 138:481–485
Kambham N, Markowitz GS, Valeri AM, Lin J, D’Agati VD (2001) Obesity-related glomerulopathy: an emerging epidemic. Kidney Int 59:1498–1509
Praga M, Hernandez E, Morales E et al (2001) Clinical features and long-term outcome of obesity-associated focal segmental glomerulosclerosis. Nephrol Dial Transplant 16:1790–1798
Morales E, Vlaero A, Leon M et al (2003) Benefical effects of weight loss in overweight patients with chronic proteinuric nephropathies. Am J Kidney Dis 41:319–327
Chen HM, Li SJ, Chen HP et al (2008) Obesity-related glomerulopathy in China: a case series of 90 patients. Am J Kidney Dis 52:58–65
Tsuboi N, Koike K, Hirano K et al (2013) Clinical features and long-term renal outcomes of Japanese patients with obesity-related glomerulopathy. Clin Exp Nephrol 17:379–385
Mandal R, Loeffler AG, Salamat S, Fritsch MK (2012) Organ weight changes associated with body mass index determined from a medical autopsy population. Am J Forensic Med Pathol 33:382–389
Samuel T, Hoy WE, Douglas-Denton R et al (2005) Determinants of glomerular volume in different cortical zones of the human kidney. J Am Soc Nephrol 16:3102–3109
Hoy WE, Hughson MD, Zimanyi M et al (2010) Distribution of volumes of individual glomeruli in kidneys at autopsy: association with age, nephron number, birth weight and body mass index. Clin Nephrol 74(suppl 1):S105–S112
Puelles VG, Zimanyi MA, Samuel T et al (2012) Estimating individual glomerular volume in the human kidney: clinical perspectives. Nephrol Dial Transplant 27:1880–1888
Tsuboi N, Utsunomiya Y, Kanzaki G et al (2012) Low glomerular density with glomerulomegaly in obesity-related glomerulopathy. Clin J Am Soc Nephrol 7:735–741
Wu Y, Liu Z, Xiang Z et al (2006) Obesity-related glomerulopathy: insights from gene expression profiles of the glomeruli derived from renal biopsy samples. Endocrinology 147:44–50
Chen HM, Liu ZH, Zeng CH et al (2006) Podocyte lesions in patients with obesity-related glomerulopathy. Am J Kidney Dis 48:772–779
de Vries AP, Ruggenenti P, Ruan XZ, ERA-EDTA Working Group Diabesity et al (2014) Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diab Endocrinol 2:417–426
Tobar A, Ori Y, Benchetrit S et al (2013) Proximal tubular hypertrophy and enlarged glomerular and proximal tubular urinary space in obese subjects with proteinuria. PLoS ONE 8:e75547
Henegar JR, Bigler SA, Henegar LK et al (2001) Functional and structural changes in the kidney in the early stages of obesity. J Am Soc Nephrol 12:1211–1217
Reisin E, Messerli FG, Ventura HO, Frohlich ED (1987) Renal haemodynamic studies in obesity hypertension. J Hypertens 5:397–400
Chagnac A, Weinstein T, Korzets A et al (2000) Glomerular hemodynamics in severe obesity. Am J Physiol Renal Physiol 278:F817–F822
Chagnac A, Weinstein T, Herman M et al (2003) The effects of weight loss on renal function in patients with severe obesity. J Am Soc Nephrol 14:1480–1486
Vallon V, Richter K, Blantz RC et al (1999) Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10:2569–2576
Hall JE, do Carmo JM, Silva AA et al (2015) Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res 13:991–1006
Kim S, Soltani-Bejnood M, Quignard-Boulange A, et al (2006) The adipose renin-angiotensin system modulates systemic markers of insulin sensitivity and activates the intrarenal renin-angiotensin system. J Biomed Biotechnol 27012. doi:10.1155/JBB/2006/27012
Strazzullo P, Barba G, Cappuccio FP et al (2001) Altered renal sodium handling in men with abdominal obesity: a link to hypertension. J Hypertens 19:2157–2164
Li C, Lin Y, Luo R et al (2016) Intrarenal renin-angiotensin system mediates fatty acid-induced ER stress in the kidney. Am J Physiol Renal Physiol 310:F351–F363
Moss NG (1982) Renal function and renal afferent and efferent nerve activity. Am J Physiol 243:F425–F433
Kassab S, Kato T, Wilkins FC et al (1995) Renal denervation attenuates the sodium retention and hypertension associated with obesity. Hypertension 25:893–897
Thomson SC, Vallon V, Blantz RC (2004) Kidney function in early diabetes: the tubular hypothesis of glomerular filtration. Am J Physiol Renal Physiol 286:F8–F15
Hostetter TH (2003) Hyperfiltration and glomerulosclerosis. Semin Nephrol 23:194–199
Caramori ML, Mauer M (2003) Diabetes and nephropathy. Curr Opin Nephrol Hypertens 12:273–282
Leon CA, Raij L (2005) Interaction of haemodynamic and metabolic pathways in the genesis of diabetic nephropathy. J Hypertens 23:1931–1937
Hall JE, Henegar JR, Dwyer TM et al (2004) Is obesity a major cause of chronic kidney disease? Adv Ren Replace Ther 11:41–54
Adelman RD (2002) Obesity and renal disease. Curr Opin Nephrol Hypertens 11:331–335
Praga M (2002) Obesity—a neglected culprit in renal disease. Nephrol Dial Transplant 17:1157–1159
Slee AD (2012) Exploring metabolic dysfunction in chronic kidney disease. Nutr Metab 9:36
Colman RJ, Roecker EB, Ramsey JJ, Kemnitz JW (1998) The effect of dietary restriction on body composition in adult male and female rhesus macaques. Aging (Milano) 10:83–92
Rajala MW, Scherer PE (2003) Minireview: the adipocyte-at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144:3765–3773
Unger RH (2003) Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 144:5159–5165
Meier U, Gressner AM (2004) Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin Chem 50:1511–1525
Schäffler A, Müller-Ladner U, Schölmerich J, Büchler C (2006) Role of adipose tissue as an inflammatory organ in human diseases. Endocr Rev 27:449–467
Katagiri H, Yamada T, Oka Y (2007) Adiposity and cardiovascular disorders. Circ Res 101:27–39
Musso C, Javor E, Cochran E, Balow JE, Gorden P (2006) Spectrum of renal diseases associated with extreme forms of insulin resistance. Clin J Am Soc Nephrol 1:616–622
Wisse BE (2004) The inflammatory syndrome: the role of adipose tissue cytokines in metabolic disorders linked to obesity. J Am Soc Nephrol 15:2792–2800
Hamdy O, Porramatikul S, Al-Ozairi E (2006) Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev 2:367–373
Deji N, Kume S, Araki S et al (2009) Structural and functional changes in the kidneys of high-fat diet-induced obese mice. Am J Physiol Renal Physiol 296:F118–F126
Kume S, Uzu T, Araki S et al (2007) Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. J Am Soc Nephrol 18:2715–2723
Decleves AE, Mathew AV, Cunard R, Sharma K (2011) AMPK mediates the initiation of kidney disease induced by a high-fat diet. J Am Soc Nephrol 22:1846–1855
Decleves AE, Zolkipil Z, Satriano J et al (2014) Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int 85:611–623
Jensen MD (2006) Adipose tissue as an endocrine organ: implications of its distribution on free fatty acid metabolism. Eur Heart J Suppl 8 (Supplement B):B13–B19
Cao Y (2010) Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nat Rev Drug Discov 9:107–115
Iliescu RI, Chade AR (2010) Progressive renal vascular proliferation and injury in obese Zucker rats. Microcirculation 17:250–258
Roubicek T, BArtlova M, Krajickova J et al (2009) Increased production of proinflammatory cytokines in adipose tissue of patients with end-stage renal disease. Nutrition 25:762–768
Stemmer K, Pervez-Tilve D, Ananthakrishnan G et al (2012) High-fat-diet-induced obesity causes an inflammatory and tumor-promoting microenvironment in the rat kidney. Dis Model Mech 5:627–635
D’Agati VD, Chagnac A, De Vries AP et al (2016) Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol 12:453–471
Bobulescu IA, Lotan Y, Zhang J et al (2014) Triglycerides in the human kidney cortex: relationship with body size. PLoS ONE 9(e101285):44–50
Santini E, Lupi R, Baldi S et al (2008) Effects of different LDL particles on inflammatory molecules in human mesangial cells. Diabetologia 51:2117–2125
Tomiyama-Hanayama M, Rakugi H, Kohara M et al (2009) Effect of interleukin-6 receptor blockage on renal injury in apolipoprotein E-deficient mice. Am J Physiol Renal Physiol 297:F679–F684
Bussolati B, Deregibus MC, Fonsato V et al (2005) Statins prevent oxidized LDL-induced injury of glomerular podocytes by activating the phosphatidyl inositol 3-kinase/AKT signaling pathway. J Am Soc Nephrol 16:1936–1947
Cormack-Aboud FC, Brinkkoetter PT, Pippin JW et al (2009) Rosuvastatin protects against podocyte apoptosis in vitro. Nephrol Dial Transplant 24:404–412
Lennon R, Pons D, Sabin MA et al (2009) Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol Dial Transplant 24:3288–3296
Sun L, Halaihel N, Zhang W et al (2002) Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem 277:18919–18927
Jiang T, Wang Z, Proctor G et al (2005) Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J Biol Chem 280:32317–32325
Tang J, Yan H, Zhuang S (2012) Inflammation and oxidative stress in obesity-related glomerulopathy. Int J Nephrol 608397
Ikezumi Y, Suzuki T, Karasawa T et al (2008) Activated macrophages down-regulate podocyte nephrin and podocin expression via stress-activated protein kinases. Biochem Biophys Res Commun 376:706–711
Axelsson J, Qureshi AR, Suliman ME et al (2004) Truncal fat mass as a contributor to inflammation in end-stage renal disease. Am J Clin Nutr 80:1222–1229
Fernández-Riejos P, Najib S, Santos-Alvarez J et al (2010) Role of leptin in the activation of immune cells. Mediators Inflamm 568343
Kastarinen H, Kesaniemi YA, Ukkola O (2009) Leptin and lipid metabolism in chronic kidney failure. Scand J Clin Lab Invest 69:401–408
Sharma K, Considine RV, Michael B et al (1997) Plasma leptin is partly cleared by the kidney and is elevated in hemodialysis patients. Kidney Int 51:1980–1985
Mak RH, Cheung W (2007) Adipokines and gut hormones in end-stage renal disease. Perit Dial Int 27(Suppl 2):S298–S302
Mathew AV, Okada S, Sharma K (2011) Obesity related kidney disease. Curr Diabetes Rev 7:41–49
Wolf G, Ziyadeh FN (2006) Leptin and renal fibrosis. Contrib Nephrol 151:175–183
Hotta K, Funahashi T, Arita Y et al (2000) Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 20:1595–1599
Kadowaki T, Yamauchi T, Kubota N et al (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116:1784–1792
Sharma K, Ramachandrarao S, Qiu G et al (2008) Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118:1645–1656
Ohashi K, Iwatani H, Kihara S et al (2007) Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arterioscler Thromb Vasc Biol 27:1910–1917
Cammisotto PG, Bendayan M (2008) Adiponectin stimulates phosphorylation of AMP-activated protein kinase alpha in renal glomeruli. J Mol Histol 39:579–584
Guebre-Egziabher F, Bernhard J, Funahashi T et al (2005) Adiponectin in chronic kidney disease is related more to metabolic disturbances than to decline in renal function. Nephrol Dial Transplant 20:129–134
Saraheimo M, Forsblom C, Thorn L et al (2008) Serum adiponectin and progression of diabetic nephropathy in patients with type 1 diabetes. Diabetes Care 31:1165–1169
Berg AH, Combs TP, Du X et al (2001) The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:947–953
Yamauchi T, Kamon J, Waki H et al (2003) Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem 278:2461–2468
Combs TP, Pajvani UB, Berg AH et al (2004) A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 145:367–383
Maeda N, Shimomura I, Kishida K et al (2002) Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 8:731–737
Kubota N, Terauchi Y, Yamauchi T et al (2002) Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem 277(29):25863–25866
Yamauchi T, Kamon J, Minokoshi Y et al (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8:1288–1295
Steinberg GR, Michell BJ, van Denderen BJ et al (2006) Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab 4:465–474
Lihn AS, Jessen N, Pedersen SB et al (2004) AICAR stimulates adiponectin and inhibits cytokines in adipose tissue. Biochem Biophys Res Commun 316:853–858
Sell H, Dietze-Shroeder D, Eckardt K, Eckel J (2006) Cytokine secretion by human adipocytes is differentially regulated by adiponectin, AICAR, and troglitazone. Biochem Biophys Res Commun 343:700–706
O’Neill LA, Hardie DG (2013) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493:346–355
Weisberg SP, Mc Cann D, Desai M et al (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–808
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Stienstra R, Van Diepen JA, Tack CJ et al (2011) Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A 108:15324–15329
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175–184
Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35
Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR (2008) Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57:3239–3246
King GL (2008) The role of inflammatory cytokines in diabetes and its complications. J Periodontol 79(8 Suppl):1527–1534
Duffield JS, Tipping PG, Kipari T et al (2005) Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol 167:1207–1219
Lim AK, Ma FY, Nikolic-Paterson DJ et al (2009) Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia 52:1669–1679
Wang Y, Wang YP, Zheng G et al (2007) Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int 72:290–299
Mal J, Corsa BA, Zhou J et al (2011) Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am J Physiol Renal Physiol 300:F1203–F1213
Gupta J, Mitra N, Kanetsky PA et al (2012) Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc Nephrol 7:1938–1946
Berg AH, Scherer PE (2005) Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96:939–949
Uysal KT, Wiesbrock SM, Hotamisligil GS (1998) Functional analysis of tumor necrosis factor (TNF) receptors in TNF-alpha-mediated insulin resistance in genetic obesity. Endocrinology 139(12):4832–4838
Hivert MF, Sullivan LM, Fox CS et al (2008) Associations of adiponectin, resistin, and tumor necrosis factor-alpha with insulin resistance. J Clin Endocrinol Metab 93:3165–3172
Kanda H, tateya S, Tamori Y et al (2006) MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116:1494–1505
Kamei N, Tobek K, Suzuki R et al (2006) Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 281:26602–26614
Furuichi K, Kaneko S, Wada T (2009) Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin Exp Nephrol 13:9–14
Tarabra E, Giunti S, Barutta F et al (2009) Effect of the monocyte chemoattractant protein-1/CC chemokine receptor 2 system on nephrin expression in streptozotocin-treated mice and human cultured podocytes. Diabetes 58:2109–2118
Ruiz-Ortega M, Ruperez M, Lorenzo O et al (2002) Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl 82:S12–S22
Patel NS, Chatterjee PK, Di Paola R et al (2005) Endogenous interleukin-6 enhances the renal injury, dysfunction, and inflammation caused by ischemia/reperfusion. J Pharmacol Exp Ther 312:1170–1178
Hotamisligil GS (2003) Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord 27(Suppl 3):S53–S55
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444:860–867
Makki K, Froguel P, Wolowczuk I (2013) Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm 139239
Liao MT, Sung CC, Hung KC et al (2012) Insulin resistance in patients with chronic kidney disease. J Biomed Biotechnol 691369
Chen J, Muntner P, Hamm LL et al (2003) Insulin resistance and risk of chronic kidney disease in nondiabetic US adults. J Am Soc Nephrol 14:469–477
Parvanova AI, Trevisan R, Illiev IP et al (2006) Insulin resistance and microalbuminuria: a cross-sectional, case-control study of 158 patients with type 2 diabetes and different degrees of urinary albumin excretion. Diabetes 55(5):1456–1462
Adamczak M, Wiecek A (2013) The adipose tissue as an endocrine organ. Semin Nephrol 33:2–13
Cusumano AM, Bodkin NL, Hansen BC et al (2002) Glomerular hypertrophy is associated with hyperinsulinemia and precedes overt diabetes in aging rhesus monkeys. Am J Kidney Dis 40:1075–1085
Pete G, Walsh M, Hu Y et al (1996) Insulin-like growth factor-I decreases mean blood pressure and selectively increases regional blood flow in normal rats. Proc Soc Exp Biol Med 213:187–192
Hirschberg R, Adler S (1998) Insulin-like growth factor system and the kidney: physiology, pathophysiology, and therapeutic implications. Am J Kidney Dis 31:901–919
Abrass CK, Raugi GJ, Gabourel LS, Lovett DH (1988) Insulin and insulin-like growth factor I binding to cultured rat glomerular mesangial cells. Endocrinology 123:2432–2439
Kreisberg JI (1982) Insulin requirement for contraction of cultured rat glomerular mesangial cells in response to angiotensin II: Possible role for insulin in modulating glomerular hemodynamics. Proc Natl Acad Sci U S A 79:4190–4192
Abrass CK, Spicer D, Raugi GJ (1994) Insulin induces a change in extracellular matrix glycoproteins synthesized by rat mesangial cells in culture. Kidney Int 46:613–620
Abrass CK, Peterson CV, Raugi GJ (1988) Phenotypic expression of collagen types in mesangial matrix of diabetic and nondiabetic rats. Diabetes 37:1695–1702
Abrass CK, Spicer D, Raugi GJ (1995) Induction of nodular sclerosis by insulin in rat mesangial cells in vitro: studies of collagen. Kidney Int 47:25–37
Welsh GI, Hale LJ, Eremina V et al (2010) Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab 12:329–340
Chavez JA, Knotts TA, Wang LP et al (2003) A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem 278:10297–10303
Ahmed SB, Fisher ND, Stevanovic R, Hollenberg NK (2005) Body mass index and angiotensin-dependent control of the renal circulation in healthy humans. Hypertension 46:1316–1320
Kennefick TM, Anderson S (1997) Role of angiotensin II in diabetic nephropathy. Semin Nephrol 17:441–447
Zhuo JL, Li XC (2007) Novel roles of intracrine angiotensin II and signalling mechanisms in kidney cells. J Renin Angiotensin Aldosterone Syst 8:23–33
Griffin KA, Bidani AK (2006) Progression of renal disease: renoprotective specificity of renin-angiotensin system blockade. Clin J Am Soc Nephrol 1:1054–1065
Crowley SD, Gurley SB, Coffman TM (2007) AT(1) receptors and control of blood pressure: the kidney and more. Trends Cardiovasc Med 17:30–34
Ziyadeh FN (2004) Mediators of diabetic renal disease: the case for TGF-beta as the major mediator. J Am Soc Nephrol Suppl 1:S55–S57
Cooper R, McFarlane-Anderson N, Bennett FI et al (1997) ACE, angiotensinogen and obesity: a potential pathway leading to hypertension. J Hum Hypertens 11:107–111
Achard V, Boullu-Ciocca S, Desbriere R et al (2007) Renin receptor expression in human adipose tissue. Am J Physiol Regul Integr Comp Physiol 292:R274–R282
Bochud M, Nussberger J, Bovet P et al (2006) Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension 48:239–245
Rossi GP, Belfiore A, Bernini G et al (2008) Body mass index predicts plasma aldosterone concentrations in overweight-obese primary hypertensive patients. J Clin Endocrinol Metab 93:2566–2571
Ehrhart-Bornstein M, Arakelyan K, Krug AW et al (2004) Fat cells may be the obesity–hypertension link: human adipogenic factors stimulate aldosterone secretion from adrenocortical cells. Endocr Res 30:865–870
Ohsawa M, Tamura K, Wakui H et al (2014) Deletion of the angiotensin II type 1 receptor-associated protein enhances renal sodium reabsorption and exacerbates angiotensin II-mediated hypertension. Kidney Int 86:570–581
Peti-Peterdi J, Warnock DG, Bell PD (2002) Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol 13:1131–1135
Rozansky DJ (2006) The role of aldosterone in renal sodium transport. Semin Nephrol 26:173–181
Rahmoune H, Thompson PW, Ward JM et al (2005) Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 54:3427–3434
Bautista R, Manning R, Martinez F et al (2004) Angiotensin Iidependent increased expression of Naþ-glucose cotransporter in hypertension. Am J Physiol Renal Physiol 286:F127–F133
Frederich RC Jr, Kahn BB, Peach MJ, Flier JS (1992) Tissue-specific nutritional regulation of angiotensinogen in adipose tissue. Hypertension 19:339–344
Schorr U, Blaschke K, Turan S et al (1998) Relationship between angiotensinogen, leptin and blood pressure levels in young normotensive men. J Hypertens 16:1475–1480
Xu ZG, Lanting L, Vaziri ND et al (2005) Upregulation of angiotensin II type 1 receptor, inflammatory mediators, and enzymes of arachidonate metabolism in obese Zucker rat kidney: reversal by angiotensin II type 1 receptor blockade. Circulation 111:1962–1969
Kim S, Soltani-Bejnood M, Quignard-Boulange A et al (2006) The adipose Renin-Angiotensin system modulates systemic markers of insulin sensitivity and activates the intrarenal renin-angiotensin system. J Biomed Biotechnol 27012
Yvan-Charvet L, Massiera F, Lamande N et al (2009) Deficiency of angiotensin type 2 receptor rescues obesity but not hypertension induced by overexpression of angiotensinogen in adipose tissue. Endocrinology 150:1421–1428
Bomback AS, Klemmer PJ (2008) Renal injury in extreme obesity: the important role of aldosterone. Kidney Int 74:1216
Nagase M, Fujita T (2008) Aldosterone and glomerular podocyte injury. Clin Exp Nephrol 12:233–242
de Paula RB, da Silva AA, Hall JE (2004) Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension 43:41–47
Goodfriend TL, Ball DL, Egan BM et al (2004) Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension 43:358–363
Jeon JH, Kim KY, Kim JH et al (2008) A novel adipokine CTRP1 stimulates aldosterone production. FASEB J 22:1502–1511
Eddy AA, Fogo AB (2006) Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J Am Soc Nephrol 17:2999–3012
Yang HC, Ma LJ, Ma J, Fogo AB (2006) Peroxisome proliferator-activated receptor-gamma agonist is protective in podocyte injury-associated sclerosis. Kidney Int 69:1756–1764
Ma L-J, Fogo AB (2009) PAI-1 and kidney fibrosis. Front Biosci 14:2028–2041
Amann K, Benz K (2013) Structural renal changes in obesity and diabetes. Semin Nephrol 33:23–33
Thethi T, Kamiyama M, Kobori H (2012) The link between the renin-angiotensin-aldosterone system and renal injury in obesity and the metabolic syndrome. Curr Hypertens Rep 14:160–169
Leehey DJ, Singh AK, Alavi N, Singh R (2000) Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl 77:S93–S98
Lu H, Boustany-Kari CM, Daugherty A, Cassis LA (2007) Angiotensin II increases adipose angiotensinogen expression. Am J Physiol Endocrinol Metab 292:E1280–E1287
Ogihara T, Asano T, Ando K et al (2002) Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension 40(6):872–879
Olivares-Reyes JA, Arellano-Plancarte A, Castillo-Hernandez JR (2009) Angiotensin II and the development of insulin resistance: implications for diabetes. Mol Cell Endocrinol 302:128–139
Kim JA, Jang HJ, Martinez-Lemus LA, Sowers JR et al (2012) Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab 302:E201–E208
Blendea MC, Jacobs D, Stump CS et al (2005) Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab 288:E353–E359
Sowers JR (2002) Hypertension, angiotensin II, and oxidative stress. N Engl J Med 346:1999–2001
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS et al (1997) Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389:610–614
Taniyama Y, Hitomi H, Shah A et al (2005) Mechanisms of reactive oxygen species-dependent downregulation of insulin receptor substrate-1 by angiotensin II. Arterioscler Thromb Vasc Biol 25:1142–1147
Wahba IM, Mark RH (2007) Obesity and obesity initiated metabolic syndrome; mechanistic links to chronic kidney diseases. Clin J Am Soc Nephrol 2:550–562
Tanak H, Shiohira Y, Uezu Y et al (2006) Metabolic syndrome and chronic kidney disease in Okinawa, Japan. Kidney Int 69:369–374
Garland JS (2014) Elevated body mass index as a risk factor for chronic kidney disease: current perspectives. Diab Metab Syndr Obes Targets Therapy 7:347–355
Nashar K, Megan B (2014) Relationship between chronic kidney disease and metabolic syndrome: current perspectives. Diab Metab Syndr Obes Targets Therapy 7:421–435
Thomas G, Sehgal AR, Kashyap SR et al (2011) Metabolic syndrome and kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol 6:2364–2373
Alexander MP, Patel TV, Farag YM et al (2009) Kidney pathological changes in metabolic syndrome: a cross-sectional study. Am J Kidney Dis 53:751–759
Tsuboi N, Okabayashi Y, Shimizu A, Yokoo T (2017) The renal pathology of obesity. Kidney Int Rep 2:251–260
Lackland DT, Bendall HE, Osmond C et al (2000) Low birth weights contribute to high rates of early-onset chronic renal failure in the Southeastern United States. Arch Intern Med 160:1472–1476
Praga M, Hernandez E, Herrero JC et al (2000) Influence of obesity on the appearance of proteinuria and renal insufficiency after unilateral nephrectomy. Kidney Int 58:2111–2118
Bolignano D, Rastelli S, Agarwal R et al (2013) Pulmonary hypertension in chronic kidney disease. Am J Kidney Dis 61:612–622
Musso G, Gambino R, Tabibian JH et al (2014) Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med 11:e1001680
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Murlidharan, P., Kamaladevan, S., Balan, S., Kartha, C.C. (2020). Mechanisms for Obesity Related Kidney Disease. In: Tappia, P., Ramjiawan, B., Dhalla, N. (eds) Pathophysiology of Obesity-Induced Health Complications. Advances in Biochemistry in Health and Disease, vol 19. Springer, Cham. https://doi.org/10.1007/978-3-030-35358-2_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-35358-2_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-35357-5
Online ISBN: 978-3-030-35358-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)