
Abstract
Obesity is a risk factor for type 2 diabetes mellitus (DM) and is associated with chronic kidney disease. Activation of the renin-angiotensin-aldosterone system (RAAS) is common in obesity. The RAAS is an important mediator of hypertension. Mechanisms involved in activation of the RAAS in obesity include sympathetic stimulation, synthesis of adipokines in the RAAS by visceral fat, and hemodynamic alterations. The RAAS is known for its role in regulating blood pressure and fluid and electrolyte homeostasis. The role of local/tissue RAAS in specific tissues has been a focus of research. Urinary angiotensinogen (UAGT) provides a specific index of the intrarenal RAAS. Investigators have demonstrated that sex steroids can modulate the expression and activity of the different components of the intrarenal RAAS and other tissues. Our data suggest that obese women without DM and hypertension have significantly higher levels of UAGT than their male counterparts. These differences existed without any background difference in the ratio of microalbumin to creatinine in the urine or the estimated glomerular filtration rate, raising a question about the importance of baseline gender differences in the endogenous RAAS in the clinical spectrum of cardiovascular diseases and the potential utility of UAGT as a marker of the intrarenal RAAS. Animal studies have demonstrated that modifying the amount of angiotensin, the biologically active component of the RAAS, directly influences body weight and adiposity. This article reviews the role of the RAAS in renal injury seen in obesity and the metabolic syndrome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic kidney disease (CKD) is widely recognized as a global health problem. Type 2 diabetes mellitus (DM) is the leading cause of end-stage renal disease (ESRD), and obesity is a major risk factor for both DM and kidney disease. Currently, more than 1.6 billion adults worldwide are overweight, and over 400 million are obese [1, 2]. Obesity is associated with insulin resistance, and the exponential increase in obesity is leading to an increased incidence of DM, hypertension, and dyslipidemia [3, 4]. Several studies suggest a link between obesity and CKD [5–9]. Baseline body mass index (BMI) has been suggested as an independent predictor of CKD progression [10]. Abnormal activation of the renin-angiotensin-aldosterone system (RAAS), oxidative stress, and environmental factors (including excessive dietary intake of salt and fat) are all factors that contribute to the pathology of CKD in obesity and metabolic syndrome. This review discusses the link between the RAAS and renal injury in obesity and the metabolic syndrome.
Epidemiology of Obesity, the Metabolic Syndrome, and Chronic Kidney Disease
Several epidemiologic and longitudinal studies [5–11] provide evidence for the association of obesity and the metabolic syndrome with CKD. In the Kidney Early Evaluation Program (KEEP) [11], 16% of the 6,071 screened eligible persons had a reduced estimated glomerular filtration rate (eGFR) and 44% were obese. The Hypertension Detection and Follow-Up Program (HDFP) [10] studied the incidence of CKD (defined by an eGFR less than 60 mL/min or 1+ or greater proteinuria in urine analysis) in 5,897 hypertensive adults during a 5-year follow-up period. The incidence of CKD at the end of year 5 was 34% in the obese group. After excluding subjects with baseline DM, both the overweight and obese categories were significantly associated with the incidence of CKD. A retrospective cohort study [12] of 320,252 healthcare insurance participants in northern California observed the participants for 15 to 35 years. This study revealed a stepwise increase in the rate of ESRD with increasing BMI, even after adjustment for age, sex, and race. In persons with normal weight (BMI, 18.5–24.9), the rate of ESRD per 100,000 person-years was 10; this rate increased to 108 in persons with a BMI greater than 40. Thus, besides being a risk factor for incident CKD, obesity also appears to accelerate the progression of CKD. A recent study by Santos et al. [13] investigated the impact of obesity on abnormalities of systolic and diastolic regional left ventricular function in patients with or without hypertension or hypertrophy and without heart failure. Half of the subjects in this study were women. In the obese groups with hypertension and hypertrophy, the results suggested impaired regional left ventricular relaxation and segmental atrial and systolic dysfunctions.
According to The National Cholesterol Education Program Adult Treatment Panel III Report [14], diagnosis of the metabolic syndrome can be made if three of the following five characteristics are present: (1) Abdominal obesity, given as waist circumference greater than 102 cm (40 in.) for men or 88 cm (35 in.) for women; (2) triglycerides 150 mg/dL or higher; (3) high-density lipoprotein (HDL) cholesterol less than 40 mg/dL for men or 50 mg/dL for women; (4) blood pressure 130/85 mmHg or higher; and (5) fasting glucose 110 mg/dL or higher. Listing abdominal obesity (recognized by increased waist circumference) as the first criterion reflects the priority given to it as a contributor to metabolic syndrome. The hallmark of the metabolic syndrome is insulin resistance. Both obesity and metabolic syndrome are increasingly prevalent and are risk factors for cardiovascular disease and hypertension [15]. Hypertension affects more than 65 million adults in the United States [16, 17]. According to the 2003–2004 National Health and Nutrition Examination Survey (NHANES) [16], non-Hispanic blacks have the highest prevalence of hypertension (39.1%), whereas analysis of data from NHANES II [18] identifies an increased risk of CKD in the morbidly obese group (BMI ≥ 40). A study in Japan by Ninomiya et al. [19] demonstrated that the multivariate-adjusted mean GFR slope decreases significantly in persons with four or more components of the metabolic syndrome, compared with those who have no more than one component. Chen et al. [7] investigated the development of CKD in a cohort of participants of NHANES III study who had normal baseline kidney function and were observed for more than 21 years. The multivariate-adjusted odds ratio (OR) for CKD was 2.6 in participants with the metabolic syndrome, compared with participants without the metabolic syndrome. The OR also increased from 1.89 to 5.85 as the number of components of the metabolic syndrome that were present increased. Prospective studies, such as one done by Kurella et al. [20], have documented a higher rate of CKD in participants with the metabolic syndrome even after adjustment for consequent development of DM and hypertension. This finding suggests that the metabolic syndrome by itself is an independent risk factor for incident CKD, and the onset of kidney disease may long precede the diagnosis of DM and hypertension. A nationwide Danish study [21] examined more than 40,000 patients in a stroke registry (47.9% women) for the prevalence of cardiovascular risk factors. Women more often had hypertension and obesity. Men and women show significant differences in obesity prevalence by age group after adjustment for race/ethnic group [22]; African American women have the highest prevalence of obesity [23]. There are data in the literature about differences between the sexes in cardiovascular risk factors and macrovascular disease, but little about such differences in microvascular disease.
Pathology of Renal Injury in Obesity
The mechanism by which the metabolic syndrome causes CKD and leads to its progression is probably multifactorial. Activation of the RAAS is common in patients with the metabolic syndrome despite sodium retention and clearly increased extracellular fluid volume [24, 25]. Several mechanisms have been postulated to explain the activation of the RAAS [26••]: (1) sympathetic stimulation [27]; (2) synthesis of adipokines in the RAAS by visceral fat [28]; (3) hemodynamic alterations (interference with renal blood flow) [29].
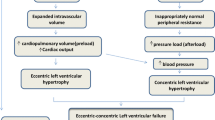
With regards to hemodynamic changes, obesity leads to increased renal plasma flow, GFR, glomerular pressure, and filtration fraction, with resultant net afferent dilatation [30, 31]. These changes could be due to excess excretory load, increased intake of energy, and tissue turnover [32]. Consequences of glomerular hyperperfusion, hyperfiltration, and hypertension are an increase in urinary albumin excretion and glomerulosclerotic damage. Clinically, the first sign of renal injury in patients with obesity is progressive proteinuria, which may precede the decline of the GFR by years [33–35]. Microscopically, the pathology in renal injury in obesity reveals focal and segmental glomerulosclerosis (FSGS) and glomerulomegaly referred to as “obesity-related glomerulopathy.” In obese individuals with normal renal function, renal biopsy features such as increased mesangial matrix, podocyte hypertrophy, mesangial cell proliferation, and glomerulomegaly occur more often than in their nonobese counterparts [36]. Even for a nephron with normal capacity, obesity induces adaptations that are typical of the reduced nephron number accompanying CKD from other causes and resulting eventually in renal failure [32].
Activation of the RAAS and the sympathetic nervous system—as well as physical compression of the kidneys by visceral adiposity—impairs normal pressure natriuresis, increases renal tubular sodium reabsorption, and causes volume expansion and hypertension [29]. Another important mechanism involved in CKD due to obesity is lipotoxicity owing to increased free fatty acid (FFA) levels leading to intracellular shunting of excess FFAs. With heavy proteinuria, the excess albumin-bound FFAs result in lipotoxicity in proximal renal tubular cells, coupled with tubulointerstitial inflammation [37]. Increased release of FFAs from abdominal adipocytes is a characteristic feature of central obesity, contributing to hepatic and peripheral insulin resistance, endothelial dysfunction, and possible blood pressure elevation in persons with abdominal obesity [38, 39].
Insulin Resistance and Salt Sensitivity
In some individuals, increased salt intake leads to a substantial rise in blood pressure that is consistent with enhanced blood-pressure sensitivity to salt. This phenomenon, known as salt sensitivity, has been an area of extensive recent research. Subjects who exhibit salt sensitivity respond with substantial lowering of the blood pressure when dietary sodium is reduced [40•]. Salt sensitivity is multifactorial, involving aspects such as ethnicity, sex steroids, dietary patterns, and adiposity [41]. Salt sensitivity has been associated with hypertension, the metabolic syndrome, type 2 DM, obesity [42], and CKD [43]. In a study of adolescents [42], those who were obese had a significantly greater reduction in mean blood pressure than the nonobese (obese, −12 ± 1 mmHg; nonobese, + 1 ± 2 mmHg). This reduction in blood pressure was seen after the adolescents switched to a low-salt diet (<30 mmol per day) after being on a high-salt diet (>250 mmol per day) for 2 weeks. Weight loss was associated with a reduced rise in blood pressure in response to increased salt intake. Plasma aldosterone and norepinephrine levels were closely related to the responsiveness of blood pressure to salt intake, suggesting an interaction between insulin sensitivity, the RAAS, and the sympathetic nervous system and their influence on the mechanism by which salt affects blood pressure. Both obesity and the metabolic syndrome are associated with insulin resistance. Earlier studies investigating the relationship between insulin resistance and salt sensitivity reported significantly higher levels of plasma insulin concentrations after an oral glucose challenge following a high-salt diet in normotensive and hypertensive individuals who exhibited salt sensitivity, compared with those who were salt resistant [44, 45]. However, not all studies have shown a positive correlation between insulin resistance and salt sensitivity [46]. An acute infusion of angiotensin II (Ang II) has been reported to improve insulin sensitivity in both rats and humans [47–49], whereas chronic infusion of Ang II leads to insulin resistance mediated via oxidative stress [50, 51]. Obesity with excessive production of angiotensinogen (AGT) in adipose tissue probably includes such a pathophysiological state, as mice overexpressing adipose AGT have exhibited reduced insulin sensitivity, as estimated by the homeostasis model assessment index [52•].
Obesity and the Renin-Angiotensin-Aldosterone System
The RAAS is known for its role in regulating blood pressure and fluid and electrolyte homeostasis [53]. The role of the local RAAS in specific tissues has been a focus of research [54]. Evidence suggests the importance of tissue RAAS in the brain, heart, adrenal glands, and vasculature, as well as in the kidney. There is substantial evidence that most Ang II present in renal tissues is delivered to the kidney from the circulation, but it is also generated locally from intrarenal AGT, which is produced locally by the proximal tubule cells. Renin secreted by the juxtaglomerular apparatus cells into the renal interstitium and vascular compartment also provides a pathway for the local generation of angiotensin I. Angiotensin-converting enzyme (ACE) is abundant in the kidney and is present in proximal tubules, distal tubules, and the collecting ducts. Angiotensin I delivered to the kidney thus can also be converted to Ang II, so all the components necessary to generate intrarenal Ang II are present along the nephrons. Ang II acts on the type 1 Ang II receptors (AT1 receptors) in the vasculature, leading to vasoconstriction, and on the zona glomerulosa, where it stimulates the secretion of aldosterone. An increase in adrenocorticotropic hormone (ACTH) and extracellular potassium ion concentrations also stimulate aldosterone secretion by the adrenal glands.
Clinical and experimental studies in patients with primary aldosteronism indicate that excess aldosterone is related to greater cardiac mass, fibrosis, and cardiovascular tissue remodeling than seen in individuals with essential hypertension affected by the same degree of blood pressure elevation [55]. Aldosterone excess results in increased oxidative stress in cardiovascular tissue [56], and this aldosterone excess is associated with impaired insulin metabolic signaling [57]. Increased aldosterone levels are also seen in individuals with obesity and correlate with high blood pressure, high waist circumference, and low HDL cholesterol levels [58]. Adipose tissue results in increased secretion of several cytokines that induce insulin resistance, such as tumor necrosis factor (TNF)-α and interleukin 6 (IL-6) [59]. Some of these cytokines are thought to stimulate aldosterone secretion [60]. Therefore, in obesity, aldosterone production seems to be increased systemically by dietary salt intake and locally in adipose tissue by increased expression of cytokines.
Navaneethan et al. [61] conducted a meta-analysis of 13 studies examining the effect of weight loss on GFR. In smaller, short-duration studies in patients with CKD, nonsurgical weight loss interventions resulted in a decrease in proteinuria and blood pressure. In these individuals, there was no further decline in GFR during a follow-up of about 7.4 months. In morbidly obese individuals with glomerular hyperfiltration, surgical interventions normalized GFR and resulted in decreased blood pressure and microalbuminuria. The limitations of the study are those of a meta-analysis.
Contributions of the Renin-Angiotensin-Aldosterone System to Obesity
AGT is highly expressed in adipose tissue and is constitutively secreted by mature adipocytes in animal models and humans [28]. In rodents, adipose tissue contributes as much as 30% of circulating AGT levels in vivo [62], consistent with the concept that the adipose tissue is an endocrine organ and adipose AGT has a paracrine role. Adipose AGT also may have autocrine effects, as the adipose tissue expresses all the components of RAAS, including renin, ACE, and ACE2. In addition to AT1 and AT2, adipocytes and preadipocytes express receptors for Ang IV and Ang(1-7) [63], allowing for local production of Ang II and other angiotensin peptides in the adipose tissue [62, 64]. Increased local formation of Ang II in adipose tissue was originally seen in rodents with genetic or diet-induced obesity, as well as in humans [65–67]. Conversely, overexpressing AGT in transgenic mice lead to the development of obesity [68]. This observation has been a major contributing factor to the concept that Ang II plays an endocrine role in obesity in vivo. Yvan-Charvet and colleagues showed that mice lacking AGT or AT2 were protected against obesity induced by a high-fat diet; the adipose tissue in these mice demonstrated hypotrophy [69, 70]. Kouyama et al. [71] showed that mice lacking AT1 were also protected from diet-induced obesity, unveiling a synergistic contribution of AT1 and AT2 in mediating the in vivo effects of Ang II on adipose tissue development.
Antagonism of the RAAS at various levels also has been suggested to decrease obesity [72, 73]. Mice lacking the Mas receptor exhibit an increase in abdominal fat mass, associated with higher adipose tissue AGT expression [74]. Studies have demonstrated that fat mass enlargement is prevented in knockout mice lacking renin or ACE with subsequent Ang II production [75, 76]. The reduced fat mass in these RAAS knockout mouse model studies was not shown to be associated with overall food intake. In fact, a higher metabolic rate was seen in RAAS knockout mouse models [69–71, 74–76]. Findings of reduced fat mass also have been shown with RAAS blockade [77–79]. In summary, animal studies have demonstrated that modifying the amount of Ang II, the biologically active component of the RAAS, directly influences body weight and adiposity [69, 71–79].
However, data about the role of Ang II in preadipocyte differentiation in vitro is conflicting [62, 80–82]. Not much is known about the expression and regulation of AT1, AT2, and the Mas receptor in cultured preadipocytes. AT2 mRNA expression is known to be under the control of various growth hormones [83, 84], so it is possible that the use of different culture media may account for the discrepancies between in vitro studies. Yvan-Charvet and colleagues [85] showed that overexpression of AGT in mice led to a slight decrease in the number of adipose cells, suggesting an inhibitory effect of Ang II on preadipocyte differentiation in vivo and pointing to the autocrine role of the RAAS. This effect could be due to the direct action of Ang II on proliferation and differentiation of adipose tissue precursors [64, 86] and the consequent imbalance between AT1 and AT2 [82, 85]. Despite the reduced number of adipose cells, local production of AGT in adipose tissue does promote enlargement of the fat mass. AGT mRNA abundance in adipose tissue may be 60% of that in liver [87], contributing to nearly 30% of circulating AGT levels in rodents [68]. In genetically and diet-induced obesity, adipocytes show increased AGT, an effect that is not observed in liver [65, 66]. Human studies also demonstrate enhanced AGT mRNA expression in visceral and subcutaneous adipose tissue of obese subjects [67, 88].
Urinary Angiotensinogen in Renal Injury and Obesity
We have demonstrated that UAGT is increased in hypertensive subjects [89••] and treatment with RAAS blockers suppresses UAGT [90]. The sandwich enzyme-linked immunosorbent assay (ELISA) [91] for measuring human AGT has been shown to be sensitive and accurate in measuring human AGT without crossreactivity with major proteins (e.g., human immunoglobulins, albumin, or transferrin) in proteinuric urine samples. In subjects with diabetic nephropathy, administration of AT1 receptor blocker (ARB) resulted in greater reduction in urinary albumin excretion in those with high UAGT levels at baseline [90]. The intrarenal RAAS is activated in obesity [92, 93]. A recent study done by Yasue et al. [93] demonstrated that AGT derived from adipose tissue is substantially augmented in obese subjects. The adipose tissue RAAS has been implicated in the pathophysiology of obesity and dysfunction of adipose tissue. The incidence and severity of hypertension [94] and the progression to ESRD [95] is greater in men than in women. Several investigators [96–98] have shown that sex steroids can modulate the expression and activity of the different components of the intrarenal RAAS and other tissues. Thus, a question arises: Are there any gender differences in the baseline endogenous RAAS?
We performed a study [99••] to investigate the difference between obese subjects and nonobese controls in UAGT and the gender differences in UAGT within each group. We selected subjects in a random, stratified manner from an ongoing cross-sectional study of obesity. The inclusion criteria were a BMI of 30 or higher for the obese group and 27 or less for the control group, and ages between 18 and 70 years. Exclusion criteria included pregnancy and diabetes. For the purpose of this substudy, we also excluded subjects with hypertension and eGFR less than 60 mL/min, because the effects on UAGT levels of hypertension [89••, 100] and CKD stage [101–103] have already been reported. Subjects were recruited from outpatient clinics and a community setting. The study protocol was approved by the Institutional Review Board and all the subjects gave informed consent. The subjects underwent a history and physical examination and submitted blood and a random urine sample in a fasting state. Urine was collected and kept in ice until it was aliquotted into cryo-vials. These samples were snap frozen immediately and stored at −70°C for further analysis. Group I was nonobese controls, (BMI ≤ 27) and group II was obese subjects (BMI ≥ 30). Subjects in group I and II did not have DM or hypertension. There were 39 individuals (16 men, 23 women) in group I and 38 (15 men, 23 women) in group II. Baseline characteristics of the members of the two groups are given in Table 1. Urinary albumin, AGT, and creatinine were measured as previously described [54, 89••, 100, 101, 103, 104], and the urinary albumin/creatinine ratio (UALB/UCr) and UAGT/creatinine ratio (UAGT/UCr) were calculated. Analyses were performed using SAS 9.1 for Windows. Variable outcomes were log transformed because they were skewed. T-tests were performed to compare outcomes between the two groups and the two sexes. Because the results of both the original and log scale were either significant or nonsignificant at the same alpha level, only the results of the original scales are reported for ease of interpretation. Multiple linear regression models were fitted to examine variables associated with log-transformed UAGT/UCr.
There was no significant difference for UAGT/UCr between the nonobese controls and the obese group in the original-scale UAGT/UCr (6.3 + 4.3 vs 5.2 + 3.3 μg/g, P = 0.2). Table 2 shows the characteristics of the obese subjects (group II) for both sexes. In group II, there was a significant difference between men and women for UAGT/UCr (3.7 + 2.1 vs 6.3 + 3.6 μg/g, P = 0.01), but there was no significant difference between the sexes in group I for the UAGT/UCr (men, 5.2 + 3.0; women, 7.1 + 4.9 μg/g; P = 0.15). In both groups I and II, there were no significant differences for UALB/UCr. Using stepwise variable selection, for the outcome variable log-transformed UAGT/UCr at 0.10 alpha level, female gender (P = 0.04) was strongly associated with UAGT/UCr. On average, women had higher UAGT/UCr levels than men (Table 2). Systolic blood pressure also had a positive correlation with UAGT/UCr (P = 0.04)
Microalbuminuria has been described as the earliest manifestation of renal injury associated with the metabolic syndrome and diabetic nephropathy, and it is associated with insulin resistance independent of DM [105]. Our data suggest that obese women, even without DM and hypertension, have significantly higher UAGT than obese men with the same characteristics. This is an important finding, as it highlights the existence of UAGT differences even when the urine microalbumin/creatinine ratio is less than 30 mg/g. The small sample size is a limitation, which may be responsible for failure of our data to show any significant difference in UAGT between the healthy and obese groups. Given the exponential increase in the incidence and prevalence of obesity [23], our findings raise an important question about the potential therapeutic value of UAGT in the diagnosis and management of CKD in obesity even before the appearance of DM, hypertension, or both.
Conclusions
Obesity is an independent risk factor for the development and progression of CKD. The RAAS is activated in obesity. AGT plays a role in the control of hypertension [10]. Although most circulating AGT is produced and secreted by the liver, the kidneys also produce AGT [106]. Intrarenal AGT mRNA and protein have also been localized on the proximal tubule cells, indicating that intratubular Ang II could be derived from the AGT that is locally formed and secreted [107, 108]. Urinary excretion of AGT provides a specific index of intrarenal RAAS status. It has been shown to be increased in patients with hypertension [21] and diabetic nephropathy [53, 101, 104], whereas RAAS blockade decreases UAGT [21, 53, 102]. Results from the Kidney Early Evaluation Program (KEEP) [109] demonstrate that in patients with early CKD (stages 1 and 2), the odds of hypertension control (blood pressure < 130/80 mmHg) are greater for African American women (OR, 1.47; 95% CI, 1.14–1.88), white men (OR, 1.85; 95% CI, 1.39–2.46), and white women (OR, 1.69; 95% CI, 1.28–2.22) than for African American men. KEEP was a cross-sectional study that examined 10,827 subjects. It highlights the need to investigate gender and racial differences in those with very early CKD or risk factors for CKD, before progression to late stages of CKD occurs. Data suggest gender differences in obesity and CKD. Small weight loss studies have demonstrated an improvement in CKD or a slowing of its progression.
The RAAS cascade allows for several therapeutic targets for the management of hypertension. These include ACE (via ACE inhibitors), angiotensin type 1 receptors (via ARBs), renin (via direct renin inhibitors), and the aldosterone receptor (via aldosterone receptor blockers). Although effective, neither ACE inhibitors nor ARBs completely block the RAAS, and there is resurgence in efforts to antagonize excess aldosterone by using an aldosterone receptor blocker [98]. Aliskiren, a direct renin inhibitor, has been used to block renin and therefore the RAAS cascade.
Evidence from animal and human data suggests gender differences in the RAAS. Results from our study indicate significant gender differences in UAGT levels in the obese group, with women having significantly higher levels of UAGT than men, though there was no difference in the UALB/UCr or the eGFR. Given the importance of obesity in the pathogenesis of hypertension, the study results point to the importance of gender differences in obese individuals who do not yet have clinical hypertension. With respect to the RAAS, which plays a critical role in renal injury in obesity, these data also establish some baseline gender difference in intrarenal RAAS [98]. This finding places an emphasis on evaluating patients at risk before the clinical diagnosis is overtly present, and for assessing biochemical markers such as UAGT, which is amenable to therapeutic intervention with RAAS blockade. A prospective longitudinal study is needed to assess the utility of UAGT in the paradigm of risk assessment and monitoring of the development and progression of CKD in obesity.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
WHO 2006 Fact Sheet No. 311 Obesity and overweight: World Health Organization. 2006.
Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world—a growing challenge. N Engl J Med. 2007;356(3):213–5.
Ford ES, Mokdad AH, Giles WH. Trends in waist circumference among U.S. adults. Obes Res. 2003;11(10):1223–31.
Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA. 2003;289(1):76–9.
Abrass CK. Overview: obesity: what does it have to do with kidney disease? J Am Soc Nephrol. 2004;15(11):2768–72.
Agnani S, Vachharajani VT, Gupta R, Atray NK, Vachharajani TJ. Does treating obesity stabilize chronic kidney disease? BMC Nephrol. 2005;6:7.
Chen J, Muntner P, Hamm LL, Jones DW, Batuman V, Fonseca V, et al. The metabolic syndrome and chronic kidney disease in U.S. adults. Ann Intern Med. 2004;140(3):167–74.
Ejerblad E, Fored CM, Lindblad P, Fryzek J, McLaughlin JK, Nyrén O. Obesity and risk for chronic renal failure. J Am Soc Nephrol. 2006;17(6):1695–702.
Eknoyan G. Obesity, diabetes, and chronic kidney disease. Curr Diab Rep. 2007;7(6):449–53.
Kramer H, Luke A, Bidani A, Cao G, Cooper R, McGee D. Obesity and prevalent and incident CKD: the Hypertension Detection and Follow-Up Program. Am J Kidney Dis. 2005;46(4):587–94.
Brown WW, Peters RM, Ohmit SE, Keane WF, Collins A, Chen SC, et al. Early detection of kidney disease in community settings: the Kidney Early Evaluation Program (KEEP). Am J Kidney Dis. 2003;42(1):22–35.
Hsu CY, McCulloch CE, Iribarren C, Darbinian J, Go AS. Body mass index and risk for end-stage renal disease. Ann Intern Med. 2006;144(1):21–8.
Santos JL, Salemi VM, Picard MH, Mady C, Coelho OR. Subclinical regional left ventricular dysfunction in obese patients with and without hypertension or hypertrophy. Obesity (Silver Spring). 2011;19(6):1296–303.
Grundy SM, Brewer Jr HB, Cleeman JI, Smith Jr SC, Lenfant C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition 1. Circulation. 2004;109(3):433–8.
Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL, et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA. 2003;289(19):2560–72.
Ong KL, Cheung BM, Man YB, Lau CP, Lam KS. Prevalence, awareness, treatment, and control of hypertension among United States adults 1999–2004. Hypertension. 2007;49(1):69–75.
Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, et al. Heart disease and stroke statistics—2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115(5):e69–e171.
Stengel B, Tarver-Carr ME, Powe NR, Eberhardt MS, Brancati FL. Lifestyle factors, obesity and the risk of chronic kidney disease. Epidemiology. 2003;14(4):479–87.
Ninomiya T, Kiyohara Y, Kubo M, Yonemoto K, Tanizaki Y, Doi Y, et al. Metabolic syndrome and CKD in a general Japanese population: the Hisayama Study. Am J Kidney Dis. 2006;48(3):383–91.
Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol. 2005;16(7):2134–40.
Andersen KK, Andersen ZJ, Olsen TS. Age- and gender-specific prevalence of cardiovascular risk factors in 40,102 patients with first-ever ischemic stroke: a Nationwide Danish Study. Stroke. 2010;41(12):2768–74.
Ogden CL. Disparities in obesity prevalence in the United States: black women at risk. Am J Clin Nutr. 2009;89(4):1001–2.
Centers for Disease Control. Overweight and Obesity: Data and Statistics. Available at http://www.cdc.gov/obesity/data/index.html. Accessed October 29, 2010.
Tuck ML, Sowers J, Dornfeld L, Kledzik G, Maxwell M. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. N Engl J Med. 1981;304(16):930–3.
Engeli S, Böhnke J, Gorzelniak K, Janke J, Schling P, Bader M, et al. Weight loss and the renin-angiotensin-aldosterone system. Hypertension. 2005;45(3):356–62.
•• Nitta K. Possible link between metabolic syndrome and chronic kidney disease in the development of cardiovascular disease. Cardiol Res Pract. 2011. doi:10.4061/2011/963517. Epidemiologic evidence quite often forms the basis of hypotheses that underlie further investigation. This publication summarizes data linking chronic kidney disease to obesity and metabolic syndrome, in the form of epidemiologic and longitudinal studies.
Montani JP, Antic V, Yang Z, Dulloo A. Pathways from obesity to hypertension: from the perspective of a vicious triangle. Int J Obes Relat Metab Disord. 2002;26 Suppl 2:S28–38.
Engeli S, Schling P, Gorzelniak K, Boschmann M, Janke J, Ailhaud G, et al. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003;35(6):807–25.
Hall JE, Henegar JR, Dwyer TM, Liu J, Da Silva AA, Kuo JJ, et al. Is obesity a major cause of chronic kidney disease? Adv Ren Replace Ther. 2004;11(1):41–54.
Chagnac A, Weinstein T, Korzets A, Ramadan E, Hirsch J, Gafter U. Glomerular hemodynamics in severe obesity. Am J Physiol Renal Physiol. 2000;278(5):F817–22.
Henegar JR, Bigler SA, Henegar LK, Tyagi SC, Hall JE. Functional and structural changes in the kidney in the early stages of obesity. J Am Soc Nephrol. 2001;12(6):1211–7.
Bagby SP. Obesity-initiated metabolic syndrome and the kidney: a recipe for chronic kidney disease? J Am Soc Nephrol. 2004;15(11):2775–91.
Kambham N, Markowitz GS, Valeri AM, Lin J, D’Agati VD. Obesity-related glomerulopathy: an emerging epidemic. Kidney Int. 2001;59(4):1498–509.
Praga M, Hernández E, Morales E, Campos AP, Valero MA, Martínez MA, et al. Clinical features and long-term outcome of obesity-associated focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2001;16(9):1790–8.
Adelman RD, Restaino IG, Alon US, Blowey DL. Proteinuria and focal segmental glomerulosclerosis in severely obese adolescents. J Pediatr. 2001;138(4):481–5.
Serra A, Romero R, Lopez D, Navarro M, Esteve A, Perez N, et al. Renal injury in the extremely obese patients with normal renal function. Kidney Int. 2008;73(8):947–55.
Kamijo A, Kimura K, Sugaya T, Yamanouchi M, Hase H, Kaneko T, et al. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney Int. 2002;62(5):1628–37.
Sarafidis PA, Bakris GL. Non-esterified fatty acids and blood pressure elevation: a mechanism for hypertension in subjects with obesity/insulin resistance? J Hum Hypertens. 2007;21(1):12–9.
Sarafidis PA. Obesity, insulin resistance and kidney disease risk: insights into the relationship. Curr Opin Nephrol Hypertens. 2008;17(5):450–6.
• Lastra G, Dhuper S, Johnson MS, Sowers JR. Salt, aldosterone, and insulin resistance: impact on the cardiovascular system. Nat Rev Cardiol. 2010;7(10):577–84. This publication details the genomic and nongenomic effects of aldosterone and the interaction with the mineralocorticoid receptor. It summarizes the basic science and the clinical data linking the RAAS with insulin resistance.
Strazzullo P, Galletti F, Barba G. Altered renal handling of sodium in human hypertension: short review of the evidence. Hypertension. 2003;41(5):1000–5.
Rocchini AP, Key J, Bondie D, Chico R, Moorehead C, Katch V, et al. The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med. 1989;321(9):580–5.
Franco V, Oparil S. Salt sensitivity, a determinant of blood pressure, cardiovascular disease and survival. J Am Coll Nutr. 2006;25(3 Suppl):247S–55S.
Zavaroni I, Coruzzi P, Bonini L, Mossini GL, Musiari L, Gasparini P, et al. Association between salt sensitivity and insulin concentrations in patients with hypertension. Am J Hypertens. 1995;8(8):855–8.
Sharma AM, Ruland K, Spies KP, Distler A. Salt sensitivity in young normotensive subjects is associated with a hyperinsulinemic response to oral glucose. J Hypertens. 1991;9(4):329–35.
Melander O, Groop L, Hulthén UL. Effect of salt on insulin sensitivity differs according to gender and degree of salt sensitivity. Hypertension. 2000;35(3):827–31.
Townsend RR, DiPette DJ. Pressor doses of angiotensin II increase insulin-mediated glucose uptake in normotensive men. Am J Physiol. 1993;265(3 Pt 1):E362–6.
Buchanan TA, Thawani H, Kades W, Modrall JG, Weaver FA, Laurel C, et al. Angiotensin II increases glucose utilization during acute hyperinsulinemia via a hemodynamic mechanism. J Clin Invest. 1993;92(2):720–6.
Juan CC, Chien Y, Wu LY, Yang WM, Chang CL, Lai YH, et al. Angiotensin II enhances insulin sensitivity in vitro and in vivo. Endocrinology. 2005;146(5):2246–54.
Ogihara T, Asano T, Ando K, Chiba Y, Sakoda H, Anai M, et al. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension. 2002;40(6):872–9.
Olivares-Reyes JA, Arellano-Plancarte A, Castillo-Hernandez JR. Angiotensin II and the development of insulin resistance: implications for diabetes. Mol Cell Endocrinol. 2009;302(2):128–39.
• Yvan-Charvet L, Quignard-Boulangé A. Role of adipose tissue renin-angiotensin system in metabolic and inflammatory diseases associated with obesity. Kidney Int. 2011;79(2):162–8. In addition to summarizing the RAAS, this article focuses on the role of adipose tissue on angiotensinogen. It links the animal data on RAAS with clinical studies related to obesity, insulin resistance, and diabetes mellitus.
Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension. 2002;39(2 Pt 2):316–22.
Dzau VJ, Re R. Tissue angiotensin system in cardiovascular medicine. A paradigm shift? Circulation. 1994;89(1):493–8.
Brilla CG, Weber KT. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J Lab Clin Med. 1992;120(6):893–901.
Stas S, Whaley-Connell A, Habibi J, Appesh L, Hayden MR, Karuparthi PR, et al. Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling. Endocrinology. 2007;148(8):3773–80.
Lastra G, Whaley-Connell A, Manrique C, Habibi J, Gutweiler AA, Appesh L, et al. Low-dose spironolactone reduces reactive oxygen species generation and improves insulin-stimulated glucose transport in skeletal muscle in the TG(mRen2)27 rat. Am J Physiol Endocrinol Metab. 2008;295(1):E110–6.
Bochud M, Nussberger J, Bovet P, Maillard MR, Elston RC, Paccaud F, et al. Plasma aldosterone is independently associated with the metabolic syndrome. Hypertension. 2006;48(2):239–45.
Lastra G, Manrique C, Sowers JR. Obesity, cardiometabolic syndrome, and chronic kidney disease: the weight of the evidence. Adv Chronic Kidney Dis. 2006;13(4):365–73.
Fujita T. Mineralocorticoid receptors, salt-sensitive hypertension, and metabolic syndrome. Hypertension. 2010;55(4):813–8.
Navaneethan SD, Yehnert H, Moustarah F, Schreiber MJ, Schauer PR, Beddhu S. Weight loss interventions in chronic kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2009;4(10):1565–74.
Darimont C, Vassaux G, Ailhaud G, Negrel R. Differentiation of preadipose cells: paracrine role of prostacyclin upon stimulation of adipose cells by angiotensin-II. Endocrinology. 1994;135(5):2030–6.
Weiland F, Verspohl EJ. Variety of angiotensin receptors in 3T3-L1 preadipose cells and differentiated adipocytes. Horm Metab Res. 2008;40(11):760–6.
Crandall DL, Armellino DC, Busler DE, McHendry-Rinde B, Kral JG. Angiotensin II receptors in human preadipocytes: role in cell cycle regulation. Endocrinology. 1999;140(1):154–8.
Frederich RC, Kahn BB, Peach MJ, Flier JS. Tissue-specific nutritional regulation of angiotensinogen in adipose tissue. Hypertension. 1992;19(4):339–44.
Hainault I, Nebout G, Turban S, Ardouin B, Ferré P, Quignard-Boulangé A. Adipose tissue-specific increase in angiotensinogen expression and secretion in the obese (fa/fa) Zucker rat. Am J Physiol Endocrinol Metab. 2002;282(1):E59–66.
Van Harmelen V, Ariapart P, Hoffstedt J, Lundkvist I, Bringman S, Arner P. Increased adipose angiotensinogen gene expression in human obesity. Obes Res. 2000;8(4):337–41.
Massiéra F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001;15(14):2727–9.
Massiera F, Seydoux J, Geloen A, Quignard-Boulange A, Turban S, Saint-Marc P, et al. Angiotensinogen-deficient mice exhibit impairment of diet-induced weight gain with alteration in adipose tissue development and increased locomotor activity. Endocrinology. 2001;142(12):5220–5.
Yvan-Charvet L, Even P, Bloch-Faure M, Guerre-Millo M, Moustaid-Moussa N, Ferre P, et al. Deletion of the angiotensin type 2 receptor (AT2R) reduces adipose cell size and protects from diet-induced obesity and insulin resistance. Diabetes. 2005;54(4):991–9.
Kouyama R, Suganami T, Nishida J, Tanaka M, Toyoda T, Kiso M, et al. Attenuation of diet-induced weight gain and adiposity through increased energy expenditure in mice lacking angiotensin II type 1a receptor. Endocrinology. 2005;146(8):3481–9.
Weisinger RS, Begg DP, Jois M. Antagonists of the renin-angiotensin system and the prevention of obesity. Curr Opin Investig Drugs. 2009;10(10):1069–77.
de Kloet AD, Krause EG, Woods SC. The renin angiotensin system and the metabolic syndrome. Physiol Behav. 2010;100(5):525–34.
Santos SH, Fernandes LR, Mario EG, Ferreira AV, Pôrto LC, Alvarez-Leite JI, et al. Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes. 2008;57(2):340–7.
Jayasooriya AP, Mathai ML, Walker LL, Begg DP, Denton DA, Cameron-Smith D, et al. Mice lacking angiotensin-converting enzyme have increased energy expenditure, with reduced fat mass and improved glucose clearance. Proc Natl Acad Sci U S A. 2008;105(18):6531–6.
Takahashi N, Li F, Hua K, Deng J, Wang CH, Bowers RR, et al. Increased energy expenditure, dietary fat wasting, and resistance to diet-induced obesity in mice lacking renin. Cell Metab. 2007;6(6):506–12.
Mathai ML, Naik S, Sinclair AJ, Weisinger HS, Weisinger RS. Selective reduction in body fat mass and plasma leptin induced by angiotensin-converting enzyme inhibition in rats. Int J Obes (Lond). 2008;32(10):1576–84.
de Kloet AD, Krause EG, Kim DH, Sakai RR, Seeley RJ, Woods SC. The effect of angiotensin-converting enzyme inhibition using captopril on energy balance and glucose homeostasis. Endocrinology. 2009;150(9):4114–23.
Lee MH, Song HK, Ko GJ, Kang YS, Han SY, Han KH, et al. Angiotensin receptor blockers improve insulin resistance in type 2 diabetic rats by modulating adipose tissue. Kidney Int. 2008;74(7):890–900.
Janke J, Engeli S, Gorzelniak K, Luft FC, Sharma AM. Mature adipocytes inhibit in vitro differentiation of human preadipocytes via angiotensin type 1 receptors. Diabetes. 2002;51(6):1699–707.
Brücher R, Cifuentes M, Acuña MJ, Albala C, Rojas CV. Larger anti-adipogenic effect of angiotensin II on omental preadipose cells of obese humans. Obesity (Silver Spring). 2007;15(7):1643–6.
Tomono Y, Iwai M, Inaba S, Mogi M, Horiuchi M. Blockade of AT1 receptor improves adipocyte differentiation in atherosclerotic and diabetic models. Am J Hypertens. 2008;21(2):206–12.
Dudley DT, Summerfelt RM. Regulated expression of angiotensin II (AT2) binding sites in R3T3 cells. Regul Pept. 1993;44(2):199–206.
Kambayashi Y, Nagata K, Ichiki T, Inagami T. Insulin and insulin-like growth factors induce expression of angiotensin type-2 receptor in vascular-smooth-muscle cells. Eur J Biochem. 1996;239(3):558–65.
Yvan-Charvet L, Massiéra F, Lamandé N, Ailhaud G, Teboul M, Moustaid-Moussa N, et al. Deficiency of angiotensin type 2 receptor rescues obesity but not hypertension induced by overexpression of angiotensinogen in adipose tissue. Endocrinology. 2009;150(3):1421–8.
Matsushita K, Wu Y, Okamoto Y, Pratt RE, Dzau VJ. Local renin angiotensin expression regulates human mesenchymal stem cell differentiation to adipocytes. Hypertension. 2006;48(6):1095–102.
Lu H, Boustany-Kari CM, Daugherty A, Cassis LA. Angiotensin II increases adipose angiotensinogen expression. Am J Physiol Endocrinol Metab. 2007;292(5):E1280–7.
Umemura S, Nyui N, Tamura K, Hibi K, Yamaguchi S, Nakamaru M, et al. Plasma angiotensinogen concentrations in obese patients. Am J Hypertens. 1997;10(6):629–33.
•• Kobori H, Alper AB, Shenava R, Katsurada A, Saito T, Ohashi N, et al. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension. 2009;53(2):344–50. Given its importance, it is important to have a biochemical marker to assess the intrarenal RAAS. Establishing urinary angiotensinogen as a marker of the status of the intrarenal RAAS was a step forward in forming a bridge between pathophysiology and biochemistry.
Ogawa S, Kobori H, Ohashi N, Urushihara M, Nishiyama A, Mori T, et al. Angiotensin II type 1 receptor blockers reduce urinary angiotensinogen excretion and the levels of urinary markers of oxidative stress and inflammation in patients with type 2 diabetic nephropathy. Biomark Insights. 2009;4:97–102.
Katsurada A, Hagiwara Y, Miyashita K, Satou R, Miyata K, Ohashi N, et al. Novel sandwich ELISA for human angiotensinogen. Am J Physiol Renal Physiol. 2007;293(3):F956–60.
Karlsson C, Lindell K, Ottosson M, Sjöström L, Carlsson B, Carlsson LM. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II. J Clin Endocrinol Metab. 1998;83(11):3925–9.
Yasue S, Masuzaki H, Okada S, Ishii T, Kozuka C, Tanaka T, et al. Adipose tissue-specific regulation of angiotensinogen in obese humans and mice: impact of nutritional status and adipocyte hypertrophy. Am J Hypertens. 2010;23(4):425–31.
Schenck-Gustafsson K. Risk factors for cardiovascular disease in women: assessment and management. Eur Heart J. 1996;17(Suppl D):2–8.
Silbiger SR, Neugarten J. The impact of gender on the progression of chronic renal disease. Am J Kidney Dis. 1995;25(4):515–33.
Ellison KE, Ingelfinger JR, Pivor M, Dzau VJ. Androgen regulation of rat renal angiotensinogen messenger RNA expression. J Clin Invest. 1989;83(6):1941–5.
Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension. 2000;35(1 Pt 2):480–3.
Sartori-Valinotti JC, Iliescu R, Yanes LL, Dorsett-Martin W, Reckelhoff JF. Sex differences in the pressor response to angiotensin II when the endogenous renin-angiotensin system is blocked. Hypertension. 2008;51(4):1170–6.
•• Thethi TK, Kobori H, Fonseca V, Wani J, Parsha K, Leger S, et al. Gender differences in urinary angiotensinogen in individuals with obesity and diabetes mellitus. J Women’s Health. 2009;18(10):1513. Sex differences in urinary angiotensinogen provide valuable information to build on. These data give rise to further investigation into differences in the RAAS that may partly explain differences in clinical outcomes between men and women.
Kobori H, Urushihara M, Xu JH, Berenson GS, Navar LG. Urinary angiotensinogen is correlated with blood pressure in men (Bogalusa Heart Study). J Hypertens. 2010;28(7):1422–8.
Kobori H, Ohashi N, Katsurada A, Miyata K, Satou R, Saito T, et al. Urinary angiotensinogen as a potential biomarker of severity of chronic kidney diseases. J Am Soc Hypertens. 2008;2(5):349–54.
Urushihara M, Kondo S, Kagami S, Kobori H. Urinary angiotensinogen accurately reflects intrarenal Renin-Angiotensin system activity. Am J Nephrol. 2010;31(4):318–25.
Yamamoto T, Nakagawa T, Suzuki H, Ohashi N, Fukasawa H, Fujigaki Y, et al. Urinary angiotensinogen as a marker of intrarenal angiotensin II activity associated with deterioration of renal function in patients with chronic kidney disease. J Am Soc Nephrol. 2007;18(5):1558–65.
Saito T, Urushihara M, Kotani Y, Kagami S, Kobori H. Increased urinary angiotensinogen is precedent to increased urinary albumin in patients with type 1 diabetes. Am J Med Sci. 2009;338(6):478–80.
Lane JT. Microalbuminuria as a marker of cardiovascular and renal risk in type 2 diabetes mellitus: a temporal perspective 1. AmJPhysiol Renal Physiol. 2004;286(3):F442–50.
Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–87.
Darby IA, Sernia C. In situ hybridization and immunohistochemistry of renal angiotensinogen in neonatal and adult rat kidneys. Cell Tissue Res. 1995;281(2):197–206.
Ingelfinger JR, Zuo WM, Fon EA, Ellison KE, Dzau VJ. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. Clin Invest. 1990;85(2):417–23.
Duru OK, Li S, Jurkovitz C, Bakris G, Brown W, Chen SC, et al. Race and sex differences in hypertension control in CKD: results from the Kidney Early Evaluation Program (KEEP). Am J Kidney Dis. 2008;51(2):192–8.
Acknowledgments
Funding for this study is provided by the Tulane University Phase II Research Enhancement Fund and by NIH grants R01D072408 and P20RR017659. This study is also supported in part by the Tulane University and Louisiana State University Health Sciences Center’s Clinical and Translational Research Center as part of the Louisiana Board of Regents RC/EEP Fund.
Dr. Thethi’s work was supported by Award Number K12HD043451 from the Eunice Kennedy Shriver National Institute Of Child Health & Human Development. Dr. Kobori’s work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01D072408), and the National Center for Research Resources (P20RR017659). The content is solely the responsibility of the authors and does not necessarily represent the official views of these agencies. Diabetes research at Tulane University Health Sciences Center is supported in part by the Susan Harling Robinson Fellowship in Diabetes Research and the Tullis-Tulane Alumni Chair in Diabetes. The authors acknowledge excellent technical assistance from Akemi Katsurada, MS, and Jessica L. Mucci, MS (Tulane University).
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thethi, T., Kamiyama, M. & Kobori, H. The Link Between the Renin-Angiotensin-Aldosterone System and Renal Injury in Obesity and the Metabolic Syndrome. Curr Hypertens Rep 14, 160–169 (2012). https://doi.org/10.1007/s11906-012-0245-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-012-0245-z