
Abstract
Body mass index has been found to be the second most important contributor to relative risk for developing end state renal disease (ESRD), after proteinuria. The impact of obesity on the kidney includes a wide spectrum, from characteristic pathologic lesions to increment in urinary albumin excretion (UAE) and proteinuria/or decrease in glomerular filtration rate (GFR). The cause of renal disease associated to obesity is not well understood, but two relevant elements emerge. The first is the presence of obesity-related glomerulopathy, and the second is the fat deposit in the kidney with impact on renal haemodynamics and intrarenal regulation. The mechanisms linking obesity and renal damage are complex and include haemodynamic changes, inflammation, oxidative stress, apoptosis, and finally renal scarring. The protection of kidney damage needs to combine weight reduction with the proper control of the cardiometabolic risk factors associated, hypertension, metabolic syndrome, diabetes and dyslipidaemia. The search for specific treatments merits future research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Overweight [body mass index (BMI) = 25.0–29.9 kg/m2] and obesity (BMI ≥ 30.0 kg/m2), which have reached epidemic levels worldwide, are a widespread condition with an ever-increasing prevalence. In Westernised societies, it is currently estimated that 35 % of the population, and 50 % of those over 50 years of age, are overweight or obese [1•]. These figures are of great medical concern since obesity is a risk factor for a wide range of conditions and it is associated with comorbid diseases. In adults without clinical cardiovascular disease (CVD), increased BMI constitutes a risk factor attributable to the clustering of factors such as hypertension, diabetes, and dyslipidaemia [2]. In the absence of previous CVD, however, many studies have shown that BMI has less predictive value for CVD compared with anthropometric indices of abdominal adiposity, such as waist-to-hip ratio (WHipR) or waist circumference (WC) [3–13]. Likewise, the clustering of metabolic abnormalities around insulin resistance and/or abdominal obesity, the so-called metabolic syndrome, is associated with high risk of CVD, which results from the impact of each of the components [14].
Over the last years, besides the importance of obesity in the prevalence of CV risk factors and in the risk for cardiovascular disease, the impact on the kidney has received attention. This was in part due to the recognition that the presence of chronic kidney disease (CKD), in which obesity seems to play an important role, increases morbidity and mortality beyond the traditional CV risk factors [15, 16]. The impact of obesity on the kidney includes a wide spectrum, from characteristic pathologic lesions to increment in urinary albumin excretion (UAE) and proteinuria/or decrease in glomerular filtration rate (GFR), what today is considered chronic kidney disease (CKD). The epidemiology, pathologic lesions, mechanisms, and consequences of overweight and obesity in the kidney are reviewed.
Epidemiology
The relationship between renal damage and obesity needs to be considered in the frame of the increased relevance of CKD as a mortality and cardiovascular risk condition [15]. A progressive increment of CKD in recent years has been recognised, although this has not been the case in all countries. Data from the USA shows that the prevalence of CKD stages 1 to 4 has increased from the 1988–1994 period to the 1999–2004, 10 % and 13.1 %, respectively [17]. A progressive rise in the prevalence of diabetes and hypertension has been claimed as the cause of the increment, in which overweight and obesity play an important role. In contrast, in the UK, the prevalence of eGFR < 60 ml/min/1–73 m2 decreased within age and gender groups, except in men aged 65–74 years between 2003 and 2010 [18]. This reduction occurred despite the increment of obesity and diabetes in the population, although the prevalence and control of hypertension have improved.
Besides the potential impact of these traditional CV risk factors on the increased risk of CKD, recent epidemiological studies emphasise the direct role of overweight and obesity in producing kidney injury. Obesity is not only related to renal damage, but the kind of obesity is also relevant, as well as the presence of metabolic syndrome. The relationship between BMI and CKD has been analysed in several cross-sectional studies. In general, a positive relationship was observed that was more evident in men as compared to women, although a higher prevalence of CKD in very obese women has been identified. Likewise, a higher rate of GFR decline in obese women as compared to men has been described [19]. In longitudinal studies, however, there were more discrepancies in the results. While some studies have shown that a higher baseline BMI can predict future CKD [20, 21], others did not confirm the association [22, 23].
Due to the fact that the incremental renal risk is derived from mechanisms linked to the presence of visceral fat, markers of this can give a better risk relationship than BMI [24]. Waist and hip ratio, and waist circumference and waist-to-height ratio, an index of visceral adiposity [25], have been linked to risk of CKD.
In the clinical spectrum of metabolic abnormalities associated with obesity, the “metabolic healthy obese” (MHO) and the metabolic syndrome (MetS) are the extremes. The association of increased risk for CKD in these two conditions has been analysed. MHO seems to be a condition in which the risk for metabolic, vascular, and renal damage is very low, although it probably represents the first stage of the process. If visceral fat is measured with more precise methods than BMI or waist circumference, the amount of visceral fat is similar to the amount observed in subjects with metabolic abnormalities, and many of these subjects will progress to overt metabolic abnormalities. Only the presence of some adipokines or hepatokines allows differentiating those MHO with more risk.
MetS, the other extreme of the clinical spectrum, as an independent risk factor for CKD, as has been demonstrated in cross-sectional, cohort-based studies and in a meta-analysis. This increment in risk, around 50 %, is present regardless of age, gender, and other potential confounders. A meta-analysis of 11 studies reported a 55 % increment in the risk to have eGFR <60 ml/min/1.73 m2, an increment in risk that was present even in the absence of diabetes [26, 27••, 28, 29].
The relevance of obesity in renal damage is further reinforced when the impact on end-stage renal disease (ESRD) is evaluated. After proteinuria, body mass index was found to be the second most important contributor to relative risk for developing ESRD among all subjects who were registered as patients and followed for 27–36 years in a large epidemiological study [30].
Finally it is worth commenting on the potential overestimation of CKD when Cystatin C, and the derived formulas are used compared to the creatinine-derived equations in obese subjects. BMI may influence the estimated prevalence of stages 3 and 4 CKD, overestimating them when the MDRD equation is compared to cystatin C-derived equation [31].
Pathology
Two relevant elements emerge as central in the pathology of kidney in the presence of obesity. The first is the presence of a characteristic lesion in the glomerulus, the obesity-related glomerulopathy, and the second is the relevance of fat deposit in the kidney with impact on the renal haemodynamics and intrarenal regulation. Likewise, lipid loading of tubular cells also produces functional alterations and predispose to renal scarring.
Obesity-Related Glomerulopathy
Described in severely obese subjects with nephrotic syndrome, it is characterised by glomerulomegaly and secondary focal glomeruloesclerosis [32, 33]. Obesity-related glomerulopathy has emerged as a distinct pathologic variant of focal segmental glomerulosclerosis, increasing the number of subjects diagnosed in parallel with the obesity epidemic. In addition to the sclerosed glomeruli, glomerulomegaly with mesangial proliferation, matrix accumulation, and a decreased density of more focally effaced and hypertrophied podocytes with milder foot process fusion are characteristic features [34••]. Clinically, it can be differentiated from the idiopathic form by its slower progression of proteinuria and renal insufficiency and lower incidence of nephrotic syndrome [35].
Other features that can be observed associated to MetS are higher prevalence of tubular atrophy, interstitial fibrosis, and arterial sclerosis [36].
Podocyte Damage
When weight is gained, the renal mass and glomerular diameter increase. This produces the necessity for podocytes to enlarge their processes in order to cover the expanded area. The expansion can cause podocyte detachment and the consequent loss in protein selectivity as well as formation of denuded areas, which trigger matrix deposition and more podocyte damage with the final result of glomerulosclerosis [37, 38].
Perivascular Fat Deposits
The perivascular and intracellular fat deposit in the kidney related to obesity and the metabolic abnormalities have different significance and clinical relevance. Perivascular fat in the renal sinus appears to participate in vascular function, modifying the blood flow in the underlying arteries. Some studies have demonstrated that the accumulation of perivascular fat is associated with exercise-induced albuminuria, independent of visceral fat [39]. In the same sense, the renal data of the Framingham Heart Study identifies a relationship between the renal sinus fat and the risk of CKD [40••].
Intracellular Lipid Load
Renal samples from patients with obesity-related glomerulosclerosis demonstrated the presence of intracellular lipid deposits in mesangial, podocytes, and tubular cells. In the mesangial cells, the lipid load produces structural damage and loss of function in maintaining the integrity of the capillary loops. In the podocytes, lipid loading produces metabolic abnormalities of insulin and apoptosis, while in the tubular cells, accumulation of NEFA-bound albumin leads to atrophy and interstitial fibrosis [41].
Mechanisms
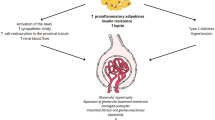
The mechanisms leading to the structural abnormalities described above, the high prevalence of CKD or the increment in the rate of GFR decline in obesity, are complex and are in part related to the effect of obesity-associated cardiometabolic risk factors and their final effector mechanisms. Hypertension, diabetes, dyslipidaemia, and insulin resistance, alone or in clusters, largely contribute to renal damage through multiple effectors, adipokines, lipids, RAAS, SNS, inflammation, oxidative stress, apoptosis, and finally renal scarring [42•] (Fig. 1).

Mechanisms of renal damage in obesity
Haemodynamic Changes
The haemodynamics of kidneys in humans have been analysed indirectly by the assessment of GFR and estimated renal plasma flow (ERPF) and the derived filtration fraction (FF). GFR can be assessed by applying equations or by the calculation of creatinine clearance but overestimates the values in the highest BMI ranges as compared to 125I-iodothalamate, a direct reliable method to assess GFR. Estimated renal plasma flow is calculated using para-amino-hippuric acid or hippuran. Several studies have demonstrated that the FF was increased in proportion to BMI due to the hyperperfusion associated to obesity. The increase in FF is related not only to BMI but also to the body fat distribution, with a higher impact of the waist-hip ratio at the same BMI. Whether the increment in the FF occurs only in the presence of hypertension is a matter of discussion [43]. The increment in FF can be reverted by losing weight, dietary salt restriction, and blockade of the renin-angiotensin system [44–46].
The haemodynamic changes can affect sodium and volume management and contribute to renal damage. The high FF increases tubular sodium reabsorption with the consequent volume expansion and a salt-sensitive state. Whether or not this contributes to accelerate the rate of GFR decline is not well established.
Insulin Resistance
Insulin resistance produces renal damage through changes in renal haemodynamics, releasing inflammatory cytokines and increasing renal endoplasmic reticulum stress. Kidney damage includes glomerular hyperfiltration, endothelial dysfunction, increased vascular permeability, increased glomerular capillary pressure, protein traffic, mesangial hyperplasia, renal hypertrophy, and increased endothelial cell proliferation [47, 48]. Furthermore, the role of insulin in the podocytes has been emphasised in the last years. Recent data strongly suggest that insulin action in podocytes is important for the glomerular function as well as for the morphology, cytoskeleton remodelling, and survival [47]. The constellation of abnormalities related to insulin resistance clustering in the metabolic syndrome (hypertension, dyslipidaemia, diabetes), adipocytokine dysregulation, hyperinsulinaemia, and low-grade inflammation are all involved in worsening kidney function.
Inflammation
A chronic low-grade inflammation is observed in obesity and CKD with the origin in the visceral fat, where a cycle of inflammation between adypocites and recruited proinflammatory monocyte-macrophage M1 cells results in local and remote insulin resistance and inflammation. Secretion of adipokines, which include leptin, adiponectin, TNF-alpha, interleukins 6 and 10, monocyte chemoattracting protein-1, plasminogen activator inhibitor-1, and resistin and lipid mediators of inflammation, impacts on the renal structures contributing to the functional and structural changes associated with obesity [49]. Chronic adipose inflammation, as a result of an imbalance between proinflammatory and anti-inflammatory adipokines, mononuclear cells, and lipid mediators, seems to be a relevant factor in producing the obesity-related glomerulosclerosis [50].
Leptin, which increases the circulating levels in obese subjects, influences kidney function by binding to specific receptors in the mesangial cells, upregulating pro-fibrotic transforming growth factor-beta (TGF-beta) and the TGF-beta receptor II with an increment of type I and type IV collagen fibres in the mesangium [51]. Besides the direct effect, leptin also facilitates renal damage by increasing BP values as a consequence of overactivity of the sympathetic nervous system, through leptin binding to the obRb receptor in the hypothalamus [52]. In contrast, obesity is linked to a reduction of the anti-inflammatory and insulin-sensitising adiponectin. Directly in the kidney, adiponectin influences the function and structure of podocytes through an increment in the AMPK activity, reducing podocyte permeability [53].
Other cytokines have been implicated in the renal damage associated with obesity. Resistin, an adipokine with inflammatory property produced in humans by the monocyte-macrophage cells, appears to increase insulin resistance. Levels of resistin have been found to be related to levels of inflammatory biomarkers and increased in patients with low GFR [54]. Vistafin is an adipokine produced in the adipose tissue and in the kidney with profibrotic and proinflammatory properties. Levels of vistafin are related to the GFR, the lower the GFR, the higher the vistafin [55]. Finally, the role of Fetuin-A should be considered. Fetuin-A [56] is a molecule produced in the liver and in the adipose tissue, which is elevated in obesity and related disorders such as diabetes, metabolic syndrome, and nonalcoholic fatty liver disease. Associated with insulin resistance, it has been implicated in triggering inflammation and fibrosis in the liver and in the kidney. An inverse relationship between fetuin-A levels and adiponectin has been found, since fetuin-A leads to suppression of adiponectin transcription in adipocytes. Both fetuin-A and adiponectin are key proteins contributing to the crosstalk between the liver, adipose tissue, and the kidney.
Overactivity of the Renin-Angiotensin-Aldosterone System
All the major components of the renin-angiotensin-aldosterone system (RAAS) are present in both the kidney and the adipose tissue. In the kidney, it plays a key role in the regulation of blood pressure as well as pressure natriuresis, among others. Adipose tissue produces angiotensinogen, the first piece in the RAAS cascade increasing the activity of the system. Overactivity of the RAAS through the increment in Angiotensin II increases the efferent arteriole tone in the glomerulus and the production of TGF-beta. Likewise, angiotensin II increases fibrosis and nephrin dephosphorilation promoting apoptosis of podocytes [42•].
Overactivity of the RAAS in obesity is not only a consequence of the cascade activation. An increment of aldosterone synthesis has been identified through a direct stimulation in the adrenal gland by adipose derived molecules. Aldosterone levels, by stimulating the mineralocorticoid receptor, promote endothelial dysfunction, inflammation, and fibrosis [57]. Both angiotensin II and aldosterone seem to be key players in the renal damage associated to obesity.
Oxidative Stress and AMPK
Oxidative stress-related mechanisms of kidney damage seem to be linked to the activity of the AMPK, a serine/threonine kinase, through a key role in regulating the NADPH oxydase system, mainly Nox 4 [58]. Other important mechanisms linked to AMPK activity are both the key role in the mitochondrial biogenesis and the role in renal fibrosis. AMPK activation is able to reduce mesangial matrix expansion and reduced TGF-beta 1 excretion [59••]. Likewise, a reduction in glomerular TGF-1beta, collagen, and fibrinonectin accumulation has been observed in several experimental models, increasing AMPK levels. Mitochondrial biogenesis is affected through its effect in the PGC-1alpha, a coactivator of many mitochondrial proteins increasing the mitochondrial content [60].
Recently, the role of AMPK on kidney function has been emphasised. Suppression of AMPK produces cellular hypertrophy, with changes in podocyte morphology, accumulation of matrix molecules, and mesangial expansion [56].
Clinical Consequences
Obesity has been associated with a higher prevalence of urinary albumin excretion, CKD, and higher risk to develop ESRD. Other relevant issues are the impact of losing weight by diet, drugs, or after bariatric procedures in renal function.
Urinary Albumin Excretion and Proteinuria
Microalbuminuria is much more common among obese individuals [61], and several studies have revealed a significant association of microalbuminuria with either obesity or central obesity [62, 63]. The prevalence of microalbuminuria is higher in the presence of central obesity [64]. The presence of a cluster of cardiovascular risk factors such as MetS largely increases the risk.
The increment of UAE associated to obesity is observed early in life. Goknar et al. [65] reported that severe obese children had higher urinary N-acetyl-beta-d-glucosaminidase (NAG) and kidney injury molecule (KIM)-1 levels than healthy controls. In severe obese adolescents, 3 % have proteinuria, 14 % microalbuminuria, and 3 % GFR <60 ml/min/1.73 m2 [66]. In less severe, the prevalence was lower, 2.4 % [67].
Development of heavy proteinuria is not commonly associated to obesity. When present, a focal segmental glomerulosclerosis is observed with relative preservation of foot process morphology and absence of inflammation or traces of immune-mediated mechanisms [68].
Reduced Glomerular Filtration Rate and End-Stage Renal Disease
Obesity has been associated with a higher incidence of CKD defined by the presence of proteinuria and/or a GFR <60 ml/min/1.73 m2 as compared to the nonobese population. There have been several cohort studies of native populations in which the increment of risk induced by obesity has been established [69–71], and in postmenopausal women [72]. Whether or not the risk is only present in those with obesity-induced metabolic abnormalities has been a matter of discussion. While some studies support that the obese metabolically healthy (MHO) do not have increased risk [28, 73–75], or even a reduction in risk [76], other studies are more in favour that the obese MHO is a first stage of obesity and that it is question of time to develop metabolic abnormalities and consequently an increased risk to develop renal dysfunction.
The impact of obesity on dialysis [77] or postransplant patients [78, 79] has also been demonstrated, although limitation of assessment of GFR by using equations should be considered in specific conditions such sarcopenic subjects and postsurgical. Sarcopenia was common in CKD and it may affect the use of BMI, underestimating obesity in people with CKD [80]. Therefore, measurement of body composition beyond BMI should be used whenever possible.
Other Obesity-Associated Conditions and Renal Damage
Two frequent complications of obesity seem to be that it further increases the risk of renal damage in obesity. First, sleep apnea and nocturnal hipoxaemia have been associated with loss of kidney function through activation of the renin-angiotensin system [81] and, second, nonalcoholic fatty liver disease (NAFLD). In a meta-analysis of 33 studies, NAFLD, nonalcoholic steatohepatitis, and advanced fibrosis were associated with an increased risk of prevalence and incidence of CKD with a graded risk from the presence to the severity of NAFLD [82].
It is worthy to comment on the increased risk to develop proteinuria after unilateral nephrectomy or after renal transplantation in obese subjects due to the renal mass reduction [83].
Treatment of Obesity and Renal Damage
The final goal is to reduce the rate of decline of eGFR, delaying the ESRD and in parallel the reduction in cardiovascular morbidity and mortality associated to CKD. The protection of kidney damage needs to combine weight reduction with the proper control of the cardiometabolic risk factors associated, hypertension, metabolic syndrome, diabetes, and dyslipidaemia. Additional salt intake reduction should be implemented if proteinuria is present. Even a small reduction in weight can contribute to achieving control in hypertension and diabetes. Losing weight reduces the UAE or proteinuria [84, 85].
Dietary restriction-induced weight loss reduced proteinuria or microalbuminuria by half, with no significant changes in the GFR, but no data exist about the impact on the rate of GFR decline overtime [86]. After bariatric surgery, reduction in GFR is much higher than in dietary-induced weight loss, reducing the hyperfiltration [86, 87].
The potential role of different classes of drugs in reducing proteinuria and protecting glomerular filtration decline is far from being established. The protective effect of drugs blocking the RAAS, such as angiotensin converting enzyme inhibitors, angiotensin II receptor blockers, or antialdosterone compounds is controversial. Although all of the RAAS blocker agents reduce albuminuria in the short term beyond the BP lowering effect, the long-term impact on reducing the rate of GFR decline is a controversial matter [88, 89].
Experimental studies tried to identify potential drugs that reduce risk of obesity-induced renal dysfunction. They are based on interfering with mechanisms that promote intracellular lipid deposits. Among them are the following: (a) antioxidants, such as lycopene, an antioxidant able to inhibit nuclear factor kappa B and the TNF-alpha production [90], and melatonine, an antioxidant and indoleamine that exerts beneficial effect on mitochondrial morphology and dynamics [91]; (b) compounds which reduce endoplasmic reticulum stress such as scetaminophen reduce the lipid deposits in the tubular cells [92]; and (c) compounds which block the Rac1 activation in mesangial cells [93]. Activation of peroxisome proliferator-activator gamma (PPAR-gamma) and epoxide hydrolase inhibitors, which increases the epoxyeicosatrienoic acid, administrated together have demonstrated the capacity to protect renal injury in hypertensive obese rats [94]. All experimental approaches are still far from having application in humans.
Pharmacologic treatment of obesity has failed to have success due to several reasons, among them the production of secondary effects, mainly cardiovascular. Up to now, no further kidney damage has been described with treatments.
The impact of bariatric surgery on the risk to develop kidney stones has been reported. A decrease in urinary volume and urinary citrate, and increased urinary oxalate and calcium oxalate saturation have been claimed as the mechanism underlying the risk. More restrictive procedures of bariatric surgery reduce the risk [95].
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Prospective Studies Collaboration. Body-mass index and cause specific mortality in 900,000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373:1083–96. The relevance of BMI in mortality is collected based in a large number of prospective studies.
Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9 · 1 million participants. Lancet. 2011;377:557–67.
Lapidus L, Bengtsson C, Larsson B, Pennert K, Rybo E, Sjöström L. Distribution of adipose tissue and risk of cardiovascular disease and death: a 12 year follow-up of participants in the population study of women in Gothenburg Sweden. BMJ. 1984;289:1257–61.
Ducimetière P, Richard J, Cambien F. The pattern of subcutaneous fat distribution in middle-aged men and the risk of coronary heart disease: the Paris prospective study. Int J Obes. 1986;10:229–40.
Rimm EB, Stampfer MJ, Giovannucci E, Ascherio A, Spiegelman D, Colditz GA, et al. Body size and fat distribution as predictors of coronary heart disease among middle-aged and older men. Am J Epidemiol. 1995;141:1117–27.
Rexrode KM, Hennekens CH, Willet WC, et al. A prospective study of body mass index, weight change, and risk of stroke in women. JAMA. 1997;277:1539–45.
Folsom AR, Stevens J, Schreiner PJ, McGovern PG. Body mass index, waist/hip ratio, and coronary heart disease incidence in African Americans and whites. Am J Epidemiol. 1998;148:1187–94.
Molarius A, Seidell JC, Sans S, Tuomilehto J, Kuulasmaa K. Waist and hip circumferences, and waist-hip ratio in 19 populations of the WHO MONICA project. Int J Obes. 1999;23:116–22.
Folsom AR, Kushi LH, Anderson KE, Mink PJ, Olson JE, Hong CP, et al. Associations of general and abdominal obesity with multiple health outcomes in older women. The Iowa women’s health study. Arch Intern Med. 2000;60:2117–28.
Lakka HM, Lakka TA, Tuomilehto J, Salonen JT. Abdominal obesity is associated with increased risk of acute coronary events in men. Eur Heart J. 2002;23:705–13.
Kurth T, Gaziano JM, Berger K, Kase CS, Rexrode KM, Cook NR, et al. Body mass index and the risk of stroke in men. Arch Intern Med. 2002;162:2557–62.
Yusuf S, Hawken S, Ounpuu S, Bautista L, Franzosi MG, Commerford P, et al. Obesity and the risk of myocardial infarction in 27, 000 participants from 52 countries: a case control study. Lancet. 2005;366:1640–9.
Gelber RP, Gaziano JM, Orav EJ, Manson JE, Buring JE, Kurth T. Measures of obesity and cardiovascular risk among men and women. J Am Coll Cardiol. 2008;52:605–15.
Redon J, Cifkova R, Laurent S, Nilsson P, Narkiewicz K, Erdine S, et al. The metabolic syndrome in hypertension: European society of hypertension position statement. J Hypertens. 2008;26:1891–900.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalisation. N Engl J Med. 2004;351:1296–306.
Chronic Kidney Disease Prognosis Consortium, Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375:2073–81.
Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–47.
Aitken GR, Roderick PJ, Fraser S, Mindell JS, O'Donoghue D, Day J, et al. Change in prevalence of chronic kidney disease in England over time: comparison of nationally representative cross-sectional surveys from 2003 to 2010. BMJ Open. 2014;4, e005480.
Komura H, Nomura I, Kitamura K, Kuwasako K, Kato J. Gender difference in relationship between body mass index and development of chronic kidney disease. BMC Res Notes. 2013;6:463.
Iseki K, Ikemiya Y, Kinjo K, Inoue T, Iseki C, Takishita S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int. 2004;65:1870–6.
Stengel B, Tarver-Carr ME, Powe NR, Eberhardt MS, Brancati FL. Lifestyle factors, obesity and the risk of chronic kidney disease. Epidemiology. 2003;14:479–87.
Mohsen A, Brown R, Hoefield R, Kalra PA, O'Donoghue D, Middleton R, et al. Body mass index has no effect on rate of progression of chronic kidney disease in subjects with type 2 diabetes mellitus. J Nephrol. 2012;25:384–93.
Brown RN, Mohsen A, Green D, Hoefield RA, Summers LK, Middleton RJ, et al. Body mass index has no effect on rate of progression of chronic kidney disease in non-diabetic subjects. Nephrol Dial Transplant. 2012;27:2776–80.
França AK, Dos Santos AM, Salgado JV, Hortegal EV, da Silva AA, Salgado FN. Estimated visceral adipose tissue, but not body mass index, is associated with reductions in glomerular filtration rate based on cystatin C in the early stages of chronic kidney disease. Int J Nephrol. 2014;2014:574267.
Odagiri K, Mizuta I, Yamamoto M, Miyazaki Y, Watanabe H, Uehara A. Waist to height ratio is an independent predictor for the incidence of chronic kidney disease. PLoS One. 2014;9:e88873.
Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J Am Soc Nephrol. 2005;16:2134–40.
Thomas G, Sehgal AR, Kashyap SR, Srinivas TR, Kirwan JP, Navaneethan SD. Metabolic syndrome and kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2011;6:2364–73. Systematic review of the association between MetS, its components, and development of microalbuminuria or proteinuria and CKD in 30146 subjects. The study demonstrated the impact of metabolic síndrome in the development of microalbuminuria, proteinuria and CKD.
Song H, Wang X, Cai Q, Ding W, Huang S, Zhuo L. Association of metabolic syndrome with decreased glomerular filtration rate among 75,468 Chinese adults: a cross-sectional study. PLoS One. 2014;9, e113450.
Nashar K, Egan BM. Relationship between chronic kidney disease and metabolic syndrome: current perspectives. Diabetes Metab Syndr Obes. 2014;7:421–35.
Hsu CY, Iribarren C, McCulloch CE, Darbinian J, Go AS. Risk factors for end-stage renal disease: 25-year follow-up. Arch Intern Med. 2009;169:342–50.
Vupputuri S, Fox CS, Coresh J, Woodward M, Muntner P. Differential estimation of CKD using creatinine- versus cystatin C-based estimating equations by category of body mass index. Am J Kidney Dis. 2009;53:993–1001.
Kambham N, Markowitz GS, Valeri AM, Lin J, D'Agati VD. Obesity-related glomerulopathy: an emerging epidemic. Kidney Int. 2001;59:1498–509.
Serra A, Romero R, Lopez D, Navarro M, Esteve A, Perez N, et al. Renal injury in the extremely obese patients with normal renal function. Kidney Int. 2008;73:947–55.
de Vries AP, Ruggenenti P, Ruan XZ, Praga M, Cruzado JM, Bajema IM, et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014;2:417–26. The study collects the evidence of the ectopic lipid accumulation in the kidney and its relationship with obesity related renal disease, including hemodynamic and structural changes.
Tang J, Yan H, Zhuang S. Inflammation and oxidative stress in obesity-related glomerulopathy. Int J Nephrol. 2012;2012:608397.
Alexander MP, Patel TV, Farag YM, Florez A, Rennke HG, Singh AK. Kidney pathological changes in metabolic syndrome: a cross-sectional study. Am J Kidney Dis. 2009;53:751–59.
Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–22.
Matsusaka T, Sandgren E, Shintani A, Kon V, Pastan I, Fogo AB, et al. Podocyte injury damages other podocytes. J Am Soc Nephrol. 2011;22:1275–85.
Wagner R, Machann J, Lehmann R, Rittig K, Schick F, Lenhart J, et al. Exercise-induced albuminuria is associated with perivascular renal sinus fat in individuals at increased risk of type 2 diabetes. Diabetologia. 2012;55:2054–8.
Foster MC, Hwang SJ, Porter SA, Massaro JM, Hoffmann U, Fox CS. Fatty kidney, hypertension, and chronic kidney disease: the Framingham heart study. Hypertension. 2011;58:784–90. In participants from the Framingham Heart Study underwent quantification of renal sinus fat area using computed tomography. With a prevalence of fatty kidney of 30 %, it was a risk for having hypertension independent of potential counfounding factors. Renal sinus fat may play a role in blood pressure regulation and chronic kidney disease.
Stefan N, Artunc F, Heyne N, Machann J, Schleicher ED, Häring HU. Obesity and renal disease: not all fat is created equal and not all obesity is harmful to the kidneys. Nephrol Dial Transplant. 2014. Apr 20.
Felizardo RJ, da Silva MB, Aguiar CF, Câmara NO. Obesity in kidney disease: a heavyweight opponent. World J Nephrol. 2014;3:50–63. The review discussed the consequences of obesity in the context of renal injury.
Ribstein J, du Cailar G, Mimran A. Combined renal effects of overweight and hypertension. Hypertension. 1995;26:610–5.
Krikken JA, Lely AT, Bakker SJ, Navis G. The effect of a shift in sodium intake on renal hemodynamics is determined by body mass index in healthy young men. Kidney Int. 2007;71:260–5.
Chagnac A, Weinstein T, Herman M, Hirsh J, Gafter U, Ori Y. The effects of weight loss on renal function in patients with severe obesity. J Am Soc Nephrol. 2003;14:1480–6.
Ahmed SB, Fisher ND, Stevanovic R, Hollenberg NK. Body mass index and angiotensin-dependent control of the renal circulation in healthy humans. Hypertension. 2005;46:1316–20.
De Cosmo S, Menzaghi C, Prudente S, Trischitta V. Role of insulin resistance in kidney dysfunction: insights into the mechanism and epidemiological evidence. Nephrol Dial Transplant. 2013;28:29–36.
Chen S, Chen Y, Liu X, Li M, Wu B, Li Y, et al. Association of insulin resistance with chronic kidney disease in non-diabetic subjects with normal weight. PLoS One. 2013;8:e74058.
Manabe I. Chronic inflammation links cardiovascular, metabolic and renal diseases. Circ J. 2011;75:2739–48.
Nolan E, O'Meara YM, Godson C. Lipid mediators of inflammation in obesity-related glomerulopathy. Nephrol Dial Transplant. 2013;28(4):iv22–9.
Wolf G, Chen S, Han DC, Ziyadeh FN. Leptin and renal disease. Am J Kidney Dis. 2002;39:1–11.
Nasrallah MP, Ziyadeh FN. Overview of the physiology and pathophysiology of leptin with special emphasis on its role in the kidney. Semin Nephrol. 2013;33:54–65.
Rutkowski JM, Wang ZV, Park AS, Zhang J, Zhang D, Hu MC, et al. Adiponectin promotes functional recovery after podocyte ablation. J Am Soc Nephrol. 2013;24:268–82.
Axelsson J, Bergsten A, Qureshi AR, Heimbürger O, Bárány P, Lönnqvist F, et al. Elevated resistin levels in chronic kidney disease are associated with decreased glomerular filtration rate and inflammation, but not with insulin resistance. Kidney Int. 2006;69:596–604.
Kang YS, Song HK, Lee MH, Ko GJ, Han JY, Han SY, et al. Visfatin is upregulated in type-2 dia- betic rats and targets renal cells. Kidney Int. 2010;78:170–81.
Ix JH, Sharma K. Mechanisms linking obesity, chronic kidney disease, and fatty liver disease: the roles of fetuin-A, adiponectin, and AMPK. J Am Soc Nephrol. 2010;21:406–12.
Nishiyama A, Abe Y. Molecular mechanisms and therapeu- tic strategies of chronic renal injury: renoprotective effects of aldosterone blockade. J Pharmacol Sci. 2006;100:9–16.
Sharma K. Obesity, oxidative stress, and fibrosis in chronic kidney disease. Kidney Int Suppl. 2014;4(4):113–7.
Declèves AE, Zolkipli Z, Satriano J, Wang L, Nakayama T, Rogac M, et al. Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int. 2014;85:611–23. The role of AMP-activated protein kinase, a critical pathway in regulating renal lipid accumulation is reviewed.
Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123:4888–99.
Chen B, Yang D, Chen Y, Xu W, Ye B, Ni Z. The prevalence of microalbuminuria and its relationships with the components of metabolic syndrome in the general population of China. Clin Chim Acta. 2010;411:705–9.
Thoenes M, Reil JC, Khan BV, Bramlage P, Volpe M, Kirch W, et al. Abdominal obesity is associated with microalbuminuria and an elevated cardiovascular risk profile in patients with hypertension. Vasc Health Risk Manag. 2009;55:577–85.
Chandie Shaw PK, Berger SP, Mallat M, Frölich M, Dekker FW, Rabelink TJ. Central obesity is an independent risk factor for albuminuria in nondiabetic south Asian subjects. Diabetes Care. 2007;30:1840–4.
Du N, Peng H, Chao X, Zhang Q, Tian H, Li H. Interaction of obesity and central obesity on elevated urinary albumin-to-creatinine ratio. PLoS One. 2014;9:e98926.
Goknar N, Oktem F, Ozgen IT, Torun E, Kuçukkoc M, Demir AD, et al. Determination of early urinary renal injury markers in obese children. Pediatr Nephrol. 2015;30:139–44.
Xiao N, Jenkins TM, Nehus E, Inge TH, Michalsky MP, Harmon CM, et al. Kidney function in severely obese adolescents undergoing bariatric surgery. Obesity. 2014;22:2319–25.
Lurbe E, Torro MI, Alvarez J, Aguilar F, Fernandez-Formoso JA, Redon J. Prevalence and factors related to urinary albumin excretion in obese youths. J Hypertens. 2013;31:2230–6.
Adelman RD, Restaino IG, Alon US, Blowey DL. Proteinuria and focal segmental glomerulosclerosis in severely obese adolescents. J Pediatr. 2001;138:481–5.
Fox CS, Larson MG, Leip EP, Culleton B, Wilson PW, Levy D. Predictors of new-onset kidney disease in a community- based population. JAMA. 2004;291:844–50.
Ejerblad E, Fored CM, Lindblad P, Fryzek J, McLaughlin JK, Nyrén O. Obesity and risk for chronic renal failure. J Am Soc Nephrol. 2006;17:1695–702.
Tozawa M, Iseki K, Iseki C, Oshiro S, Ikemiya Y, Takishita S. Influence of smoking and obesity on the development of proteinuria. Kidney Int. 2002;62:956–62.
Franceschini N, Gouskova NA, Reiner AP, Bostom A, Howard BV, Pettinger M, Umans JG, Brookhart MA, Winkelmayer WC, Eaton CB, Heiss G, Fine JP. Adiposity patterns and the risk for ESRD in postmenopausal women. Clin J Am Soc Nephrol. 2014; Dec 1.
Hashimoto Y, Tanaka M, Okada H, Senmaru T, Hamaguchi M, Asano M, Yamazaki M, Oda Y, Hasegawa G, Toda H, Nakamura N, Fukui M. Metabolically healthy obesity and risk of incident CKD. Clin J Am Soc Nephrol. 2015; Jan 29.
Song YM, Sung J, Lee K. Longitudinal relationships of metabolic syndrome and obesity with kidney function: healthy twin study. Clin Exp Nephrol. 2015; Jan 30.
Panwar B, Hanks LJ, Tanner RM, Muntner P, Kramer H, McClellan WM, Warnock DG, Judd SE, Gutiérrez OM. Obesity, metabolic health, and the risk of end-stage renal disease. Kidney Int. 2014; Dec 17.
Chen S, Zhou S, Wu B, Zhao Y, Liu X, Liang Y, et al. Association between metabolically unhealthy overweight/obesity and chronic kidney disease: the role of inflammation. Diabetes Metab. 2014;40:423–30.
Hoogeveen EK, Halbesma N, Rothman KJ, Stijnen T, van Dijk S, Dekker FW, et al. Obesity and mortality risk among younger dialysis patients. Clin J Am Soc Nephrol. 2012;7:280–8.
Hoogeveen EK, Aalten J, Rothman KJ, Roodnat JI, Mallat MJ, Borm G, et al. Effect of obesity on the outcome of kidney transplantation: a 20-year follow-up. Transplantation. 2011;91:869–74.
Meier-Kriesche HU, Arndorfer JA, Kaplan B. The impact of body mass index on renal transplant outcomes: a significant independent risk factor for graft failure and patient death. Transplantation. 2002;73:70–4.
Sharma D, Hawkins M, Abramowitz MK. Association of sarcopenia with eGFR and misclassification of obesity in adults with CKD in the United States. Clin J Am Soc Nephrol. 2014;9:2079–88.
Hanly PJ, Ahmed SB. Sleep apnea and the kidney: is sleep apnea a risk factor for chronic kidney disease? Chest. 2014;146:1114–22.
Musso G, Gambino R, Tabibian JH, Ekstedt M, Kechagias S, Hamaguchi M, et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med. 2014;11:e1001680.
Praga M, Hernández E, Herrero JC, Morales E, Revilla Y, Díaz-González R, et al. Influence of obesity on the appearance of proteinuria and renal insufficiency after unilateral nephrectomy. Kidney Int. 2000;58:2111–8.
Morales E, Valero MA, Leo NM, Hernandez E, Praga M. Beneficial effects of weight loss in overweight patients with chronic proteinuric nephropathies. Am J Kidney Dis. 2003;41:319–27.
Saiki A, Nagayama D, Ohhira M, Endoh K, Ohtsuka M, Koide N, et al. Effect of weight loss using formula diet on renal function in obese patients with diabetic nephropathy. Int J Obes. 2005;29:1115–20.
Afshinnia F, Wilt TJ, Duval S, Esmaeili A, Ibrahim HN. Weight loss and proteinuria: systematic review of clinical trials and comparative cohorts. Nephrol Dial Transplant. 2010;25:1173–83.
Navaneethan S, Yehnert H, Moustarah F, Screiber M, Schauer P, Beddhu S. Weight loss interventions in chronic kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2009;10:1565–74.
Pohl MA, Blumenthal S, Cordonnier DJ, De Alvaro F, Deferrari G, Eisner G, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the irbesartan diabetic nephropathy trial: clinical implications and limitations. J Am Soc Nephrol. 2005;16:3027–37.
Pascual JM, Rodilla E, Costa JA, Garcia-Escrich M, Gonzalez C, Redon J. Prognostic value of microalbuminuria during antihypertensive treatment in essential hypertension. Hypertension. 2014;64:1228–34.
Pierine DT, Navarro ME, Minatel IO, Luvizotto RA, Nascimento AF, Ferreira AL, et al. Lycopene supplementation reduces TNF-α via RAGE in the kidney of obese rats. Nutr Diabetes. 2014;4, e142.
Stacchiotti A, Favero G, Giugno L, Lavazza A, Reiter RJ, Rodella LF, et al. Mitochondrial and metabolic dysfunction in renal convoluted tubules of obese mice: protective role of melatonin. PLoS One. 2014;9(10):e111141.
Wang C, Wu M, Arvapalli R, Dai X, Mahmood M, Driscoll H, et al. Acetaminophen attenuates obesity-related renal injury through ER-mediated stress mechanisms. Cell Physiol Biochem. 2014;33:1139–48.
Yoshida S, Ishizawa K, Ayuzawa N, Ueda K, Takeuchi M, Kawarazaki W, et al. Local mineralocorticoid receptor activation and the role of Rac1 in obesity-related diabetic kidney disease. Nephron Exp Nephrol. 2014;126:16–24.
Imig JD, Walsh KA, Hye Khan MA, Nagasawa T, Cherian-Shaw M, Shaw SM, et al. Soluble epoxide hydrolase inhibition and peroxisome proliferator activate receptor γ agonist improve vascular function and decrease renal injury in hypertensive obese rats. Exp Biol Med (Maywood). 2012;237:1402–12.
Abou-Mrad RM, Abu-Alfa AK, Ziyadeh FN. Effects of weight reduction regimens and bariatric surgery on chronic kidney disease in obese patients. Am J Physiol Renal Physiol. 2013;305:F613–7.
Compliance with Ethics Guidelines
Conflict of Interest
Josep Redon and Empar Lurbe declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Hypertension and Obesity
Rights and permissions
About this article

Cite this article
Redon, J., Lurbe, E. The Kidney in Obesity. Curr Hypertens Rep 17, 43 (2015). https://doi.org/10.1007/s11906-015-0555-z
Published:
DOI: https://doi.org/10.1007/s11906-015-0555-z