
Abstract
Plasma catalysis is gaining increasing interest for various applications, as is obvious from the other chapters in this book. However, the underlying mechanisms are very complex and not yet fully understood. A better insight in these mechanisms can be obtained by experiments, but also computer modeling can be very useful for this purpose. This chapter gives an overview and some examples on (i) modeling the effect of plasma on the catalyst and catalytic surface reactions, and (ii) modeling the effect of catalyst on the plasma behavior.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


4.1 Introduction
Plasma catalysis is gaining increasing interest for various applications, as is obvious from the other chapters in this book. However, the underlying mechanisms are very complex and not yet fully understood. A better insight in these mechanisms can be obtained by experiments, but also computer modeling can be very useful for this purpose. Indeed, a model allows us to unravel the individual processes, before combining them into the overall picture, which is not always possible in experiments. This should allow us to obtain detailed insight into the underlying mechanisms.
Although modeling is quite well established for describing the plasma behavior for various kinds of plasmas and diverse applications and computer simulations are also extensively used for describing (thermal) catalytic processes, the modeling attempts reported in literature for describing the combination of both, in plasma catalysis, are very limited up to now. The reason for this is probably the enormous degree of complexity of the entire process.
This chapter gives an overview of the modeling work performed already for plasma catalysis. We start this chapter with a brief overview of plasma-catalyst interactions, to identify the various aspects that need to be considered by modeling.
4.2 Plasma-Catalyst Interactions
The major difference between plasma catalysis and thermal catalysis is that the source gas will already be activated by the plasma, causing among others dissociation, excitation, and ionization of the molecules. Indeed, plasma does not only contain molecules but also various radicals, excited species and ions originating from these molecules, as well as electrons and photons. All these species might interact with the catalyst surface, in addition to electric fields that are also characteristic of a plasma, all contributing to the complexity of plasma catalysis. Vice versa, the catalyst will also affect the plasma behavior in various respects. In the following we will give a brief summary of the effects of plasma on a catalyst, as well as the effects of a catalyst on the plasma.
4.2.1 Effects of the Plasma on a Catalyst
Plasma may affect the catalyst and catalysis mechanisms in various ways. It may cause:
-
(a)
Changes in the physicochemical properties of the catalyst, e.g., a higher adsorption probability [1]; a higher surface area [2, 3]; a change in the oxidation state [4,5,6,7,8,9]; reduced coke formation, thus preventing deactivation of the catalyst [5]; and a change in the work function due to the presence of a voltage and current (or charge accumulation) at the catalyst surface [10, 11], affecting the catalytic activity [12].
-
(b)
The formation of hot spots [13], possibly modifying the local plasma chemistry [14]. These hot spots might even thermally activate the catalyst locally [15], or they can also deactivate the catalyst due to plasma-induced damage [16, 17].
-
(c)
Lower activation barriers, due to the existence of short-lived active species, such as radicals and vibrationally excited species [4].
-
(d)
Activation by photon irradiation [18, 19], although this effect is still under debate, because other studies reported no effect [20, 21]. It is indeed claimed [18, 22, 23] that the UV light originating from the plasma is not intensive enough. Indeed, the UV dose in typical photocatalytic processes should be in the order of several mW/cm2, whereas in typical (air) plasmas it is only in the order of μW/cm2 [22]. However, it is definitely possible that photocatalysts are activated by other (energetic) plasma species, like ions, metastables, or electrons with suitable energy [13, 23, 24].
-
(e)
A change in the reaction pathways, because the plasma contains not only gas molecules, like in thermal catalysis, but also many more species, such as radicals, ions, electrons, and vibrationally and electronically excited species, which can undergo other types of reactions at the catalyst surface. For instance, besides the more common Langmuir-Hinshelwood mechanism, where two adsorbed species undergo a chemical reaction, in plasma catalysis the Eley-Rideal mechanism, where a plasma-produced radical reacts with an adsorbed species, can also occur more readily, while this process occurs in thermal catalysis only at very high temperatures [25].
4.2.2 Effects of the Catalyst on the Plasma
Vice versa, a catalyst may have a profound effect on the plasma. It may cause:
-
(a)
Enhancement of the local electric field in the plasma, because the catalyst is mostly present in a structured packing (e.g., pellets, beads, honeycomb, etc., so-called packed bed reactor), or simply due to the porosity of the catalyst surface [18, 26,27,28]. This enhanced electric field consequently results in an increase of the high energy tail of the electron energy distribution [14, 29, 30].
-
(b)
A change of the discharge type from streamers inside the plasma to streamers along the catalyst surface [6, 31,32,33,34,35,36,37], which might result in more intense plasma around the contact points [13, 38]. The latter will affect the plasma chemistry.
-
(c)
The formation of microdischarges in the catalyst pores [29, 39,40,41,42,43]. This might lead to a strong electric field inside the pores, which will also affect the plasma chemistry.
-
(d)
The adsorption of plasma species on the catalyst surface, affecting the residence time and hence the concentration of species in the plasma [13], while new reactive species might be formed at the catalyst surface. The adsorption will increase with the porosity of the catalyst surface [44].
A schematic overview of some plasma-catalyst interaction processes is presented in Fig. 4.1. More details on these interaction processes can be found in many excellent reviews on this topic (e.g., [20, 22, 25, 32, 45,46,47,48,49,50,51,52,53,54,55,56]) and in Chap. 3 of this book. In the next section, we will illustrate how computer modeling can contribute to obtain a better insight in these interaction mechanisms.

Schematic overview of some plasma-catalyst interactions. (Modified from [46])
4.3 Modeling the Effects of a Plasma on the Catalyst and on the Catalytic Surface Reactions
Modeling the effects of the plasma on the catalyst is quite a difficult task. This is due on one hand to the wide variation in time and length scales in plasma-surface interactions and to the complexity of the interactions on the other hand. While chemical surface reactions of plasma species at the catalyst surface can nowadays be accurately simulated (see examples below), modeling plasma effects, such as plasma-induced morphological changes, surface charging, the effects of photons, etc., is highly challenging. Likewise, simulating adsorption probabilities and coke formation may be feasible, while the enlargment of the surface area or (global) changes in surface oxidation state are much more complicated to simulate.
So far, very little work has been performed in most of these areas. On the other hand, extensive literature exists on simulations of chemical reactions of a source gas at a catalyst surface, without plasma effects. Such simulations can be carried out at the atomistic level, based on either a classical molecular dynamics (MD) or quantum-mechanical approach (mostly density functional theory or DFT); on a mesoscale level, based on a kinetic Monte Carlo (kMC) approach; or on a global level, based on rate equations. Here we give a few examples, being most relevant for the study of plasma catalysis.
In plasma catalysis, as mentioned above, the plasma activates the source gas, creating among others radicals (besides electrons, ions, and excited species). At the molecular level [57], the interaction of radicals with a catalyst surface has been studied by both classical MD simulations (e.g., [58,59,60]) and DFT calculations (e.g., [61,62,63]). Again a few examples will be given in this section. Although the link with plasma catalysis is still weak in most of these studies as they only consider radicals, some simulation works recently appeared in which also other plasma effects, like the effect of electrons [64, 65] and the electric field [66], were addressed.
Some plasma effects, however, were previously considered for another (closely related) application, i.e., carbon nanotube (CNT) growth. The effect of the electric field was investigated in [67], while the effect of ion bombardment was investigated in [68]. The same principle should also apply to plasma catalysis.
The effect of excited species on plasma catalytic processes has also been investigated, more specifically for vibrationally excited CH4 [69]. This, however, is not straightforward, because of the required highly accurate description of the interatomic interactions and forces. This will also be explained below.
To the best of our knowledge, the interaction of photons with catalyst surfaces has not yet been studied in the context of plasma catalysis, and we shall therefore not elaborate on this topic. Finally, the future needs to model plasma-induced effects on a catalyst surface will be discussed.
4.3.1 Changes in the Physicochemical Properties of the Catalyst
The plasma may modify the physicochemical properties of the catalyst, including a change in both the morphological surface properties (e.g., surface structure and surface area) and electronic surface properties (e.g., work function and surface oxidation state). In Sects. 4.3.3 and 4.3.5 we shall discuss plasma-induced changes in catalyst surface reactivity.
So far, however, there are no simulations or model results in the literature available describing the plasma-induced modification of such catalyst physicochemical properties. The effect of such changes, on the other hand, has been described. For instance, Ni et al. [70] developed a heat balance model accounting for temperature gradients resulting from a change in catalyst oxidation state (see Sect. 4.3.2). This model, however, does not elucidate how the plasma may have induced this change in the catalyst oxidation state.
In contrast, there are various reports on models for catalyst restructuring in thermal catalysis, e.g., based on KMC modeling. One example of such a model was developed by Zhdanov et al. [71], to simulate the reconstruction of a Pt(100) surface in the CO-NO reaction. Reconstruction was enabled by considering two types of Pt atoms, i.e., stable and metastable. Although the site density was kept constant, the reconstruction could grow by virtue of accounting for the interactions between the stable and metastable sites. The standard Metropolis algorithm was employed to switch between both states, while accounting for adsorbate-catalyst interactions. An example of the resulting catalyst structure is shown in Fig. 4.2, along with the simulated oscillatory behavior of the reactants.

(a) Simulated oscillatory surface coverage of CO and NO reactants in their reaction on a Pt(100) surface. (b) Snapshot of the reconstructed catalyst structure at the end of the KMC simulation, where the metastable Pt states are shown as black dots. The red, orange, and blue dots denote the CO, NO, and O adsorbates, respectively. The lattice constant is α. (Reproduced with permission from [71])
4.3.2 Hot Spots and Other Thermal Effects
As the plasma contains ions and photons and plasma species which react exo- or endothermically at the catalyst surface, it is natural to imagine that the plasma will affect the thermal balance of the surface. An excellent review paper on this topic can be found in [72]. Several models have been developed to describe the thermal balance of the catalyst in plasma catalysis.
A heat balance model for a packed bed dielectric barrier discharge (DBD) under natural convection was developed by Li et al. [73]. The model was developed in order to understand and explain the experimental observation that the temperature of the plasma phase is significantly higher than the temperature of the pellet phase. The model assumes that no chemical surface reactions (which may be either endothermic or exothermic) occur. Another important limiting feature of the model is that forced convection is not accounted for. Nevertheless, reasonably good agreement with the experimental data is demonstrated. In particular, it can be seen from Fig. 4.3 that the model indeed predicts a gas phase temperature exceeding the pellet phase temperature. Based on the model results, the authors explain that this results from a higher energy consumption in the plasma compared to the pellet phase, while at the same time the heat capacity of the plasma is much less than that of the pellet phase. A second important conclusion of the model from the plasma catalytic point of view is that as the pellet size becomes smaller, the temperature difference between plasma and packed bed increases.

Comparison between the experimental gas phase (Tg) and pellet phase (Tp) temperature and the respective simulated temperatures, as function of position along the length of the DBD. (Reproduced with permission from [73])
In relation to morphological changes in the catalyst, Ni et al. studied oxidative methane coupling as a function of oxygen concentration [70]. It was experimentally found that the formation of ethane and ethylene especially was selectively dependent on the oxygen concentration, which was attributed to temperature inhomogeneities in the catalyst bed due to the microwave heating. In order to investigate this temperature inhomogeneity, a heat transfer model was developed. For a thin catalyst bed of 1 cm length and based on an estimated thermal conductivity of 3 W/m/K, a temperature difference of 85 K exists between the center of the catalyst and its periphery when the gas mixture contains 12.5% of oxygen. In the absence of oxygen, the temperature of the catalyst at the center was calculated to be 168 °C above the catalyst temperature at its periphery. According to the authors, this difference in temperature gradient results from a change in the catalyst properties and in particular a change in catalyst oxidation state from CeO2 to Ce2O3. Therefore, the model succeeds in predicting a temperature gradient as a function of a change in dielectric loss, in agreement with their experiments.
4.3.3 Modification of Catalyst Reaction Kinetic Parameters
It is well known that the reaction rate of any surface reaction is not only determined by the catalyst element but also by the precise geometrical and electronic structure of the active site. Thus, when the plasma modifies, e.g., the catalyst surface morphology, kinetic parameters are likely to be modified as well.
Nozaki et al. carried out a numerical integration of the rate equation for methane steam reforming, in order to obtain the rate coefficient [74]. By calculating the rate coefficient for a plasma-catalyst system and for a regular catalytic system, it was found that while the energy barrier is unchanged, the pre-exponential factor is enhanced by a factor of 50 in the DBD.
Jiang et al. performed a very large number (~18,000) of DFT calculations to explicitly map out a nine-dimensional potential energy surface for the dissociative adsorption of CO2 on Ni(100) [75]. In particular in relation to plasma catalysis, the effect of vibrational excitation on the CO2 dissociation was investigated. From these calculations, the authors found that the vibrational efficacity strongly depends on the translational energy, as can be seen in Fig. 4.4. In particular, below a translational energy of 0.4 eV, the symmetric and asymmetric stretch vibrations show a vibrational efficacity of only 0.3 and 0.2, respectively. In this regime, a small increase in translational energy yields a significant change in dissociation probability. For higher translational energies, however, these efficacities increase to about 2.2, demonstrating the dominant role of vibration in the dissociative chemisorption at these higher translational energies.

Calculated normal incident dissociative sticking probability P0 of CO2 as function of translational energy and for several vibrationally excited modes. (Reproduced with permission from [75])
4.3.4 Reaction Pathways
As the plasma may modify the kinetic parameters, it may of course also affect the actual reaction pathways. For instance, Neyts et al. employed long time scale atomistic simulations based on a classical force field to study the methanol-to-formaldehyde conversion process in the presence and absence of an external electric field [66]. Although the field was not taken into account self-consistently (i.e., the charge distribution at the surface was not modified as a function of the E-field), including the field did demonstrate a significant effect on the surface retention time of the reactants. Moreover, it was found that this residence time is also a function of the polarity of the field: the retention time was decreased by 6% if the field was pointing toward the surface, while it increased by 13% if the field had opposite polarity. However, the time to first conversion (which is representative for the surface reaction time at the pristine surface) did not show any dependence on the electric field polarity nor on its magnitude.
Also employing a classical force field, Somers et al. performed a series of molecular dynamics simulations to investigate the interaction of CHx radicals with various Ni surfaces in a wide temperature range, aiming mainly at methane reforming and H2 formation [58,59,60]. The simulations demonstrate that the consecutive impingement of plasma radicals quickly renders the initially crystalline surface amorphous, altering the surface catalytic properties. This, in turn, is seen to be a result of C diffusion into the (sub-)surface layers, which can be controlled to a certain extent by controlling the catalyst temperature. Thus, reducing the temperature as is typically done in plasma catalysis and allowing the plasma to dissociate CH4 into CH3 and lower radicals enable to maintain a well-controlled catalyst surface and catalyst reactivity.
Often, the accuracy of such studies depends strongly on the accuracy of the available force fields. While various rather accurate force fields have been developed, each new elemental combination requires the fitting of a new parametrization. In order to overcome this issue, DFT calculations may be employed instead.
As an example, Huygh et al. performed extensive DFT calculations to investigate how oxygen vacancies affect the reactivity of the TiO2 anatase (001) surface toward CO2 dissociation [61]. While CO2 was shown not to dissociate at any appreciable rate on the stoichiometric surface, CO2 dissociation does occur on a surface containing oxygen vacancies (see Fig. 4.5). While these calculations do not simulate the plasma directly, they demonstrate that if the plasma can create the required oxygen vacancies, the plasma may indirectly enhance the catalytic CO2 dissociation process.

Minimal CO2 dissociation path on anatase (001) in the presence of a surface oxygen vacancy. (Reproduced with permission from [61])
The same authors also investigated the adsorption of plasma-generated CHx radicals on the same surface, again in relation to the presence of oxygen vacancies. From their DFT calculations, it turns out that the precise location of the vacancies greatly affects the adsorption interaction. This in turn affects the possible surface reactions [62].
Dry reforming of methane on a Ni(111) surface was studied at low temperature, relevant for plasma catalysis, by Shirazi et al. [63], as a function of H coverage. It was found that there is a clear (albeit non-linear) relation between the increase in H coverage and the decrease in energy barriers for methanol and ethane formation. In particular, it was found that the increase in H coverage may sufficiently lower the barriers to make the relevant reactions in the dry reforming process thermally accessible at the low temperatures relevant for plasma catalysis. This is shown in Fig. 4.6, schematically representing the decrease in activation barrier as a function of H coverage for the hydrogenation of CH2OH to methanol.

Influence of H coverage of the Ni(111) surface on the energy profile for hydrogenation of CH2OH to methanol, as obtained from DFT calculations. (Reproduced with permission from [63])
The importance of surface charging for adsorbate/catalyst interactions was recently convincingly demonstrated by Bal et al. using DFT calculations [64, 65]. By adding a fixed proton to the vacuum space above a slab of catalytic material and enforcing charge neutrality in the simulation cell, a negatively charged surface is obtained with a surface charge density that can be tuned by the size of the simulation cell. Additionally, an electric field arises naturally in this setup. The positive counter charge in the vacuum space can then be thought of as the positive space charge in the sheath of the plasma region in contact with the surface.
Using this methodology, Bal et al. investigated the adsorption and splitting of CO2 on charged and neutral surfaces of alumina-supported single atom catalysts (Ti, Ni, and Cu). It was found that the surface charge significantly enhances the reductive power of the catalyst, strongly promoting the adsorption and dissociation of CO2 to CO and O as shown in Fig. 4.7. Furthermore, it was also found that the relative catalytic activity of the investigated materials is modified by the surface charge. The origin of this effect lies in the increase of the reductive power (or the Lewis basicity) of the surface by the negative surface charge. Overall, the CO2 splitting reaction was found to become less endothermic, and furthermore, also the splitting barriers were found to be reduced by the presence of the surface charge. It was therefore concluded that surface charge might play a highly important role in the possible synergism observed in plasma catalysis [64, 65].

Effect of surface charging on CO2 adsorption. (a) and (b) Most favorable adsorption configuration on the support and supported transition metal atom. (c) Adsorption energies on all sites, with and without extra charge. (Reproduced with permission from [64])
Although DFT is sufficiently general enough to model essentially any reactive system relevant in plasma catalysis, it is highly computationally expensive and is limited to small system sizes. Moreover, if dynamics are required, the accessible time scale is typically very limited as well (order of pico- or nanoseconds). To access long time scale dynamics, a novel generic method was recently developed, termed collective variable-driven hyperdynamics (CVHD) [76]. While CVHD was shown to allow the extension of the time scale by up to nine orders of magnitude in classical molecular dynamics, i.e., into the μs-ms-sec region (see, e.g., [66]), it has not been applied in the context of plasma catalysis except for the CO2 splitting process described above [76].
Therefore, various other types of models have been developed and used, which, incidentally, allow for including actual plasma effects somewhat easier than atomistic models. In microkinetic models, a set of elementary reactions is specified. Each elementary reaction is characterized by its rate constant, which can be determined from experiments or from DFT calculations. Solving the set of reactions yields the time evolution of the concentration of each of the species included. Alternatively, other models such as KMC (see also above) can be applied. Finally, simplified kinetic models can be constructed as well, typically to complement experimental data.
As a very recent example of Monte Carlo simulations, Guerra and Marinov developed and compared several dynamical Monte Carlo models to investigate surface reactions [77]. Although it is mentioned that these models were developed to study plasma-surface interactions, the authors acknowledge that only stable neutrals are taken into account, and the surface is considered to be of constant composition and morphology. Thus, ions, photons, electrons, excited species, and electromagnetic fields are not yet considered, and thus the link to actual plasma-surface interactions is still quite weak. Nevertheless, such models hold great promise, as they are much faster than atomistic simulations (for which the link to actual plasmas is also weak), they allow you to study long time scale evolution, and they can be easily coupled with gas phase chemistry. On the other hand, they are strongly dependent on a complete list of possible surface reactions and appropriate model parameters such as sticking coefficients and energy barriers. Very recently, the same authors also provided a description and comparison between KMC models and rate balance equation models [78].
Microkinetic modeling was performed by Blaylock et al. for steam methane reforming over multifaceted nickel catalysts [79, 80]. These authors first performed extensive DFT calculations to determine the kinetic and thermochemistry parameters for a considerable list of possible reactions. Interestingly, from such models the change in reaction pathway can be determined as a function of changing conditions. However, to the best of our knowledge, such models have not yet been performed in the context of plasma catalysis.
In contrast, simplified kinetic models have been developed for plasma catalysis. For instance, Tochikubo developed a combined plasma/catalytic rate equation model to describe the reduction of nitrogen oxides with hydrocarbon admixtures [81]. The catalyst model consists of a set of mass balanced equations for relevant surface reactions. The allowed surface reactions are shown in Fig. 4.8. Although the surface coverage is variable in the model, the number of active sites that are catalytically active is kept fixed in the model. The model is capable of identifying a number of important reactions, and the general trends of the dependency of NOx removal on temperature and hydrocarbon admixture are reproduced. Still, more reliable surface reaction data is required to allow for a more quantitative understanding of the NOx reduction chemistry. In particular, it is assumed that each reaction accounted for is described by the Arrhenius equation and, more importantly, that there are no reactions besides those included in the model. Thus, while atomistic models are limited due to the required interaction model (classical force fields) and the accessible time and length scales, global models are limited due to their inevitably incomplete list of reactions and assumed reaction behavior.

List of surface reactions allowed in the rate equation model of Tochikubo et al. (Reproduced with permission from [81])
Delagrange et al. performed a series of experiments on toluene oxidation in a plasma reactor containing manganese-based catalysts. It was concluded from these experiments that ozone is highly important in the oxidation process. This was confirmed by applying a simplified rate equation model, showing reasonable agreement with the experiments, especially for low amounts of toluene [82]. In particular, the authors concluded from their combined experimental and model results that the increase in toluene oxidation in the presence of the catalyst compared to the plasma alone is probably due to the importance of the surface reactions between plasma-generated ozone and toluene.
Finally, Kim et al. also studied the decomposition of several volatile organic compounds (VOCs) in a plasma-driven setup. Except for styrene, zero-order kinetics was invariably observed. A kinetic model was then constructed to represent these zero-order kinetics with respect to the specific energy input [83]. In this model, the plasma enters through the formation of active surface sites, which are assumed to be proportional to the discharge power density. From the model, a so-called energy constant for each VOC can be defined, whose value is indicative of the energy required to decompose each VOC to a certain degree in the plasma-catalyst setup. Overall, the model results were found to be in good agreement with the experimental results.
4.3.5 Future Needs
From the above, it is quite clear that a lot of work still needs to be done. Currently, there are no models which are capable of describing the influence of the various plasma effects on the surface reactions or on the catalyst properties. The development of such a model is, naturally, complicated by the inherent wide range in time and length scale of the processes and phenomena involved. Therefore, a multi-scale or multi-method approach seems to be most appropriate. In such a multi-method approach, DFT calculations could for instance be used to calculate either reaction probabilities of adsorbates on the catalyst, both for neutral, charged, and excited species, or kinetic parameters for surface reactions including these species. This information can then be used to construct, e.g., a Monte Carlo model, which is capable of handling longer time and length scales. The dynamics of the atomistic processes can be handled through advanced simulation techniques such as collective variable-driven hyperdynamics [76].
4.4 Modeling the Effects of a Catalyst on the Plasma
There are also not many attempts yet to model the effects of a catalyst on the plasma behavior. Here we will discuss the various aspects, more or less following the list presented in Sect. 4.2.2.
4.4.1 Enhancement of the Local Electric Field and Change of the Discharge Type
The most prominent effect of the presence of a catalyst on the plasma characteristics is the change in discharge behavior and the local electric field enhancement. Indeed, most plasma catalysis experiments are carried out in a dielectric barrier discharge, where the catalyst is typically present as pellets, or as a coating on (dielectric/support) beads, in a packed bed DBD reactor. A schematic illustration of such a packed bed DBD reactor is shown in Fig. 4.9.

Schematic illustration of a packed bed DBD reactor. (Reproduced with permission from [84])
The packing beads (or pellets) are typically dielectric materials, and the applied electric field between both electrodes of the DBD reactor thus causes polarization of the dielectric beads. At the contact points between the beads, opposite charges are present, which might cause a strong local electric field enhancement in the plasma. This behavior has been reported based on experiments, as outlined in Sect. 4.2.2 above, but it has also been the subject of modeling.
Chang [27] applied a zero-dimensional (0D) plasma chemistry model for a N2/NF3/O2/H2 mixture in a BaTiO3 packed bed plasma reactor, to predict which by-products were formed. The enhancement factor of the electric field in the voids between the ferroelectric pellets was simply deduced from the ratio of the dielectric constant of the pellets and the gas phase. Takaki et al. [85] developed a simplified time-averaged 1D numerical plasma model for N2, based on solving the transport equations as well as the Poisson equation. They reported that all plasma parameters increased upon increasing the applied potential and dielectric constant of the packing beads. Kang et al. [26] developed a 2D model of a DBD reactor with two stacked ferroelectric beads inside, studying the propagation of the microdischarges during the first 20 ns. The behavior of electrons and ions was described by a set of fluid equations, but no plasma chemical species were taken into account. It was reported that the discharges evolve in three phases, avalanche, streamer, and decay, and also that the streamer discharges tend to be stabilized by the presence of dielectric materials. Russ et al. [86] applied a 2D fluid model to simulate transient microdischarges in a packed bed DBD reactor filled with dry exhaust gas, but only focusing on a short discharge (few tens of nanoseconds). Finally, although not directly applied to a packed bed reactor, Babaeva et al. also showed very interesting modeling results for the effect of dielectric spheres (dust particles) blocking a plasma streamer, using a 2D fluid model in humid air [87]. The effect of particle size, shape, and material properties of the dust particles on the streamer dynamics was investigated, and it was revealed that small dielectric particles (< tens of μm) are enveloped by the streamer, while larger particles can intercept and reinitiate streamers [87].
Kruszelnicki et al. [88] presented 2D fluid model simulations for a packed bed reactor, constructed out of dielectric rods, in humid air, studying in detail the mechanism of discharge propagation. They reported that overall, the discharges in a packed bed reactor can be classified in three modalities: positive restrikes, filamentary microdischarges, and surface ionization waves. Restrikes are formed, following breakdown in regions of high electric field. When the restrikes are confined between two dielectrics, they result in the formation of filamentary microdicharges that bridge the gap between the dielectrics. Eventually, surface charging near the feet of the microdischarges creates electric field components parallel to the dielectric surface, leading to the formation of surface ionization waves. The calculations revealed that the production of reactive species primarily takes place near the surfaces, as a result of restrikes and surface ionization waves. Hence, the production of reactants in a packed bed reactor is not a continuous process, but it rather results from the accumulation of individual, transient events.
In the same paper, the effect of separation between the dielectric rods and the rod orientation in the packed bed reactor was investigated, and it was found that the type of discharge dominating the production of reactive species depends on the dielectric facilitated electric field enhancement, which is a function of the topography and orientation of the dielectric lattice [88]. Whereas filamentary microdischarges and subcritical discharges and their follow-on negative streamers are stable and occupy relatively large volumes, they do not necessarily contribute to a large extent to the plasma chemical processes, because of their lower electron densities and temperatures. On the other hand, restrikes and surface ionization waves are characterized by higher electron densities and temperatures, and thus, in spite of their smaller volume and lifetime, they often produce larger amounts of reactive species. As the packed bed geometry affects the type of discharge that is favored, it will thus also affect the magnitude and reproducibility of reactant production. Finally, the authors also reported that photoionization plays an important role in discharge propagation through the dielectric lattice, as it seeds initial charge in regions of high electric field, which are difficult to access for electrons from the main streamer. This implies that knowledge of the UV spectral distribution is important for the propagation of discharges through packed bed reactors [88].
Figure 4.10 illustrates the calculated time-integrated densities of excited N2 species, as well as experimental data, obtained by fast camera imaging of visible light emission in a packed bed reactor constructed also of dielectric rods, as considered in the model [88]. The formation of a cathode-seeking filamentary microdischarge (FM) between the rods is indicated both in the simulated and experimental results. Lack of plasma near the bottom pole of the top rod is in part due to the direction of the applied electric field, which points upward, toward the cathode. In addition to the filamentary discharges, surface discharges also occur, as is clear from this figure. Ions produced in the positive polarity filamentary microdischarges are accelerated toward the surface of the central rod, positively charging its surface. The latter produces an electric field component parallel to the surface, eventually leading to the development of a surface ionization wave (SIW), also indicated in the figure. This process takes about 4 ns in the model [88].

Calculated time-integrated densities of excited N2 species in a packed bed reactor in humid air, at an applied voltage of −30 kV and bead separation of 0.7 mm, as obtained from 2D fluid simulations by Kushner and coworkers (a), and measured visible light emission, obtained with an ICCD camera at an observation gate width of 0.5 μs, in the packed bed reactor with similar 2D geometry, constructed out of dielectric rods (b). (Reproduced with permission from [88])
Recently, Kang et al. [89] also presented a 2D fluid model to study surface streamer propagation in a simplified packed bed reactor, in comparison with experimental data, obtained from time-resolved intensified charge coupled device (ICCD) imaging.
Within our group PLASMANT, we recently also performed a number of different modeling studies on a packed bed DBD reactor [84, 90,91,92,93,94], and some results of these models will be illustrated below.
-
(a)
Particle-in-Cell-Monte Carlo Collision (PIC-MCC) Model of a Packed Bed DBD Reactor
Zhang et al. [90] developed a 2D particle-in-cell-Monte Carlo collision (PIC-MCC) model to describe the filamentary discharge behavior in a parallel-plate packed bed DBD reactor in air (N2/O2 = 80/20), comparing an unpacked (i.e., empty) and a packed bed DBD reactor, at an applied voltage of −20 kV. The simulations predict that the dielectric packing leads to a transition in discharge behavior from a combination of negative streamers and unlimited surface streamers on the bottom dielectric surface, in case of an empty DBD reactor, to a combination of predominant positive streamers and limited surface streamers on the dielectric surfaces of the beads and plates, in the packed bed DBD reactor. The calculated electron density distribution is plotted in Fig. 4.11, for different moments in time, in a parallel-plate packed bed DBD reactor with five packing beads, assuming a dielectric constant (ε) of 22 for both the packing beads and the upper and lower dielectric barriers.

Electron density (m−3), calculated with the PIC-MCC model, at different times, i.e., (a) 0.1 ns, (b) 0.35 ns, (c) 0.5 ns, and (d) 0.75 ns, in a parallel-plate packed bed DBD reactor with five packing beads, assuming a dielectric constant of the packing beads and the dielectric barriers equal to 22, for an applied voltage of – 20 kV. (Reproduced with permission from [90])
Furthermore, the electric field enhancement inside the dielectric material, as well as in the plasma near the contact points between the beads and the dielectric plates, was demonstrated in this study [90]. Calculations were performed for different dielectric constants of the packing beads and dielectric barriers (i.e., ε = 4, 9, and 22), and the corresponding maximum calculated electric field strengths and electron densities, at different times, for these three different dielectric constants, are listed in Table 4.1.
It is clear that the strength of the electric field and the maximum electron densities generally increase as a function of time, as well as with a rising dielectric constant, because the dielectric materials are more effectively polarized. The electric field values are also much higher than in an empty DBD reactor, resulting in a stronger and faster development of the plasma and thus also higher electron densities. More details about these modeling results can be found in [90].
Gao et al. also applied a PIC-MCC model to study the mode transition from volume to surface discharge in a packed bed DBD in O2/N2 mixtures, upon changing the applied voltage and O2/N2 mixing ratio [91]. A higher voltage yields a mode transition from combined volume + surface discharge to pure surface discharge, because the charged species can escape more easily to the beads due to the strong electric field and thus charge the bead surface. This surface charging will enhance the tangential component of the electric field along the dielectric bead surface, yielding SIWs. The latter give rise to a high concentration of reactive species on the surface, and thus they might enhance the surface activity of the beads, which is of interest for plasma catalysis. The SIWs were found to propagate more slowly with increasing O2 content in the mixture, due to loss of electrons by attachment to O2 molecules.
-
(b)
Fluid Model of a Packed Bed DBD Reactor
Van Laer and Bogaerts developed a 2D fluid model for a packed bed DBD reactor [84]. In principle, such a reactor needs to be modeled in three dimensions, to fully account for the packing geometry, as there is no axial symmetry to reduce the geometry to 2D. However, the mesh size for modeling a packed bed DBD reactor needs to be very small, to account for the regions near the contact points between the beads. More specifically, the distance between two mesh points in the gas gap was around 10 μm, while it was around 1 μm near the contact points and at the material surfaces, yielding up to 100,000 mesh elements in a 2D geometry [84]. Therefore, modeling a packed bed DBD reactor in 3D is not yet feasible within a realistic calculation time. For this reason, Van Laer and Bogaerts have developed two complementary axisymmetric 2D fluid models, in order to approach the 3D geometry. These models are based on a 3D unit cell of a close-packed DBD reactor, i.e., a so-called “contact point” model and a “channel of voids” model; see Fig. 4.12 [84]. The combination of both models allows you to describe the two important features of a packed bed DBD reactor, i.e., (i) the contact between the beads, which is expected to lead to local electric field enhancement in the discharge due to polarization effects, and (ii) the fact that the voids between the beads are connected, allowing the plasma to travel from one side of the discharge gap to the other. The first model considers two packing beads, which are slightly larger than in the real (3D) geometry, to allow them to be in direct contact with each other, while the second model describes three packing beads, with the same size as in reality, with a “channel of voids” in between them (see Fig. 4.12).

3D unit cell of a packed bed DBD reactor (left) and its 2D representations used in the “contact point” model (middle) and “channel of voids” model (right). (Reproduced with permission from [84])
This fluid model was developed in helium, (i) because of the simplified plasma chemistry, thus reducing the calculation time, and (ii) because helium yields a homogeneous discharge in a DBD, which is easier to describe with a fluid model. The model solves conservation equations for the densities of the various plasma species (i.e., He atoms, He+ and He2+ ions, metastable He* atoms, He2* dimers, and the electrons) and for the average electron energy. The other plasma species, i.e., the so-called heavy particles, are assumed to be in thermal equilibrium with the gas, so that no extra energy equation needs to be solved for them. The conservation equations for the species densities are based on source and loss terms, defined by the chemical reactions, while the source of the electron energy is due to heating by the electric field, and the energy loss is again dictated by collisions. In addition, transport is included in the conservation equations, defined by diffusion and by migration in the electric field (for the charged species). These conservation equations are coupled with the Poisson equation for a self-consistent calculation of the electric field distribution from the charged species densities. The packing beads are treated as solid objects in the model, with zero space charge and certain dielectric properties inside the beads, as well as charge accumulation on their surface. The model was developed with the COMSOL Multiphysics Simulation Software. More details can be found in [84].
Figure 4.13 illustrates the calculated time-averaged electric field and electron temperature distributions, for a peak-to-peak voltage of 4 kV and a frequency of 23.5 kHz, in the 2D geometries of both the “contact point” model (a, b) and the “channel of voids” model (c, d). The “contact point” model clearly illustrates the local electric field enhancement near the contact points, due to polarization of the beads, both inside the material and in the gas gap (see Fig. 4.13a). The latter gives rise to more electron heating near the contact points, which is reflected by the higher electron temperature in Fig. 4.13b. The same behavior is also predicted by the “channel of voids” model (see Fig. 4.13c, d), although it is somewhat less pronounced, because the beads are not in direct contact with each other.

Calculated time-averaged 2D profiles of the electric field (Ef) and electron temperature (Te) in a packed bed DBD reactor, as obtained from a 2D fluid model for two complementary 2D geometries, reflecting the specific features of a packed bed DBD reactor, i.e., a “contact point” geometry (a, b) and a “channel of voids” geometry (c, d), at a peak-to-peak voltage of 4 kV and a frequency of 23.5 kHz
At this relatively low applied voltage of 4 kV, the plasma is initiated at the contact points and remains in this region, reflecting the properties of a Townsend discharge. At higher applied voltage, the discharge will spread out more into the bulk of the reactor, from one void space to the other. This is illustrated in Fig. 4.14 for the electron density, which is plotted at four consecutive moments of gas breakdown, at an applied voltage of 7.5 kV (peak-to-peak). The corresponding electrical current profile during one cycle of the applied voltage is plotted in Fig. 4.15. Four current peaks are observed in the first half cycle, which correspond to the electron density profiles illustrated in Fig. 4.14. The first three current peaks correspond to local glow-like discharges, taking place separately and consecutively, and they depend on the time needed for the electric field to reach high enough values to create a breakdown in the gas. The first peak corresponds to a local discharge between the two beads on the left (Fig. 4.14a), the second peak reflects the discharge between the upper left bead and the dielectric layer (Fig. 4.14b), and the third peak represents the simultaneous discharge between the dielectric layer and the top of the right bead and between the bottom of the right bead and the grounded electrode (Fig. 4.14c). Subsequently, a discharge is formed over the whole gas gap, with plasma traveling through the “channel of voids,” from one wall to the other (see Fig. 4.14d and the fourth peak in Fig. 4.15, which is most intense). A similar behavior occurs during the next half cycle, although the timing of the peaks relative to each other can change. Nevertheless, the strongest current peak always corresponds to the discharge travelling through the entire gas gap.

Calculated 2D electron density profiles in the “channel of voids” geometry, at four consecutive moments of gas breakdown, i.e., (a) first, (b) second, (c) third, and (d) fourth breakdown, corresponding to the current peaks of Fig. 4.15, at a peak-to-peak voltage of 7.5 kV and a frequency of 23.5 kHz

Calculated electrical current profile (solid line, left y-axis) during one cycle of the applied voltage (dashed line, right y-axis), for a peak-to-peak voltage of 7.5 kV and a frequency of 23.5 kHz. The current peaks labeled with (a–d) in the first half cycle correspond to the electron density profiles (a–d) in Fig. 4.14. The inset shows where the discharge takes place during peaks (a), (b), and (c)
Hence, this example illustrates that modeling can reveal where exactly the discharge is created and how it expands inside the voids between the packing. This gives some idea on the discharge evolution (along the surface of the beads), but the actual change of discharge type (from streamers inside the plasma to streamers along the catalyst surface) is difficult to capture with this fluid model. More details of the discharge evolution, and how the discharge can travel through the gap, can also be found in [84].
Experimental measurements in a packed bed DBD reactor are not straightforward, among others, due to visual blocking of optical diagnostics by the packing beads. However, Kim and coworkers did measurements in a packed bed DBD reactor, using an ICCD camera [31,32,33,34], and they also observed that at low applied potential the discharge stays local at the contact points, while at higher potential, it spreads across the surface of the packing material [32, 33], in good agreement with the model results of Van Laer and Bogaerts [84]. Similar observations were also made by Tu et al. [35].
Van Laer and Bogaerts also studied the influence of the dielectric constant (ε) of the packing material on the plasma characteristics, for two different gap sizes, i.e., a mm-gap (4.5 mm) and a microgap (0.5 mm) [92]. The calculated time- and space-averaged electric field, electron temperature, and electron and ion densities are plotted as a function of ε in Fig. 4.16, for both gap sizes. Calculations were performed for ε = 5, 9, 25, 100, and 1000, which are representative for silica (SiO2), alumina (Al2O3), zirconia (ZrO2), titania (TiO2), and barium titanate (BaTiO3) materials, respectively.

Space- and time-averaged electric field strength (a), electron temperature (b), and electron and ion density (c), as a function of the dielectric constant of the packing beads, for both a microgap and a mm-gap packed bed DBD reactor
The plasma behavior was found to be significantly different in both cases, even when applying the same voltage. Indeed, the electric field was found to be more enhanced in the microgap reactor, as is obvious from Fig. 4.16a, because the same voltage is applied over a shorter gap. When comparing both y-axes, it is clear that the electric field in the microgap reactor is at least a factor of 5 larger than in the mm-gap reactor, for the same applied voltage and dielectric constant. This stronger electric field results in more current peaks per half cycle, as was illustrated in [91], because the required electric field strength to cause a breakdown is more often reached.
In both the microgap and mm-gap reactor, the calculated electric field increases upon higher dielectric constant of the packing beads, but only up to a certain extent, according to the model. In the mm-gap reactor, the electric field does not increase anymore above ε = 100, while in the microgap reactor, the electric field only increases when ε rises from 5 to 9, but then it stays constant. The reason is that at higher dielectric constants, the model reveals that the electric field enhancement only takes place at the top part of the reactor, where the packing beads are in contact with the dielectric covering the powered electrode, while the electric field near the bottom (grounded) electrode is rather weak, due to less polarization between the lower beads and the grounded electrode. In the mm-gap reactor, this phenomenon only comes into play above ε = 100.
The calculated electron temperature shows a quite similar trend as the electric field strength upon rising dielectric constant; see Fig. 4.16b. In the mm-gap reactor, the electron temperature rises gradually with the dielectric constant, due to the enhanced electric field. In the microgap reactor, the electron temperature only rises from ε = 5 to ε = 9, followed by a slight drop, which is attributed to the fact that the electric field enhancement mainly takes place at the top part of the reactor (see above).
Finally, the calculated electron and ion densities follow more or less the opposite trend as the electric field strength (see Fig. 4.16c). The ion density drops almost one order of magnitude, while the electron density drops by three orders of magnitude between ε = 5 and 9 in the microgap reactor, whereas a more gradual drop is observed between ε = 25 and 1000 for the mm-gap reactor, being again more pronounced for the electrons than for the ions. This drop is attributed to a change in discharge mechanism. For the mm-gap reactor, the plasma loses its ability to travel through the channels between the voids when ε rises from 25 to 1000, resulting in an overall lower electron and ion density. In the microgap reactor, our calculations reveal that the plasma can only travel through the channel between the voids when ε = 5. This was illustrated in detail in [92]. At higher dielectric constants, the electrons and ions get more easily absorbed on the walls and surfaces of the packing beads, due to the small dimensions and enhanced electric field, so the plasma loses its ability to travel through these small channels. This explains the much lower overall electron (and ion) density. The effect is more pronounced for the electrons, because of their smaller mass and thus their higher mobility. Hence, as is clear from Fig. 4.16c, the electron density is much lower than the ion density in the microgap reactor (except for ε = 5), and the same behavior becomes apparent for ε ≥ 100 in the mm-gap reactor, thus resulting in a non-quasineutral plasma at these conditions.
Recently, Van Laer and Bogaerts also applied the above fluid models to study the effect of different bead sizes and dielectric constants of the packing beads, while keeping the interelectrode spacing constant [93]. They demonstrated that the discharge behavior changes considerably as a function of bead size and dielectric constant. At low dielectric constant, the plasma is spread out over the full discharge gap, with a significant density in the voids as well as in the connecting void channels. The electric current profile shows a strong peak during each half cycle. Upon increasing dielectric constant, the plasma becomes localized in the voids, and the current profile consists of many smaller peaks during each half cycle. For large bead sizes, the shift from full gap discharge to localized discharges was found to take place at a higher dielectric constant than for smaller beads. In addition, a higher breakdown voltage seems to be required to cause plasma formation for smaller beads or beads with a lower dielectric constant [93].
Although the above models are developed for helium, we expect a similar behavior in reactive gases that are typically used for environmental applications of plasma catalysis, except that streamer formation will occur in the latter cases. The higher electron temperature will result in more electron impact ionization, excitation, and dissociation of the gas molecules, for the same applied power, and this can explain why a packed bed DBD reactor gives higher pollutant decomposition or CO2 conversion and higher energy efficiencies than an empty reactor, although it is suggested in [92] that this effect might also be (partially) compensated by the lower electron density.
To study in more detail the behavior of streamer propagation in a packed bed DBD, Wang et al. [94] recently developed a 2D fluid model for a packed bed DBD in air, and they investigated the behavior of positive restrikes, filamentary microdischarges, and surface discharges, as well as the transition in discharge modes upon changing the dielectric constant of the packing beads.
Positive restrikes between the dielectrics result in the formation of filamentary microdischarges. Surface charging creates electric field components parallel to the dielectric surface, leading to surface ionization waves. At a low dielectric constant of the packing (e.g., εr = 5), plasma ignition between the beads occurs directly as surface discharges or surface ionization waves, which can connect with the surface of the adjacent bead, as illustrated in Fig. 4.17. On the other hand, at high dielectric constants (e.g., εr = 1000), no surface streamer jumping toward the adjacent bead surface takes place and spatially limited filamentary microdischarges are generated between the beads, as shown in Fig. 4.18. At intermediate dielectric constants, a mixed mode of surface discharges and local discharges is observed [94]. The calculation results were in good qualitative agreement with experiments, as detailed in [94].

Calculated electron number density distribution as a function of time, for a packed bed DBD reactor in dry air, with packing beads of εr = 5. (Adapted from [94] with permission)

Calculated electron number density distribution as a function of time, for a packed bed DBD reactor in dry air, with packing beads of εr = 1000. (Adapted from [94] with permission)
Due to the enhanced electric field, energetic electrons are mainly created in the positive restrikes, local discharges, and surface discharges, causing the production of many reactive species. Our results indicate that a higher dielectric constant constrains the discharge to the contact points of the beads, which may limit the catalyst activation due to the limited catalyst surface area in contact with the discharge. This may have implications for the efficiency of plasma catalysis. Indeed, the best performance is not always reached for packing material with the highest dielectric constant [95, 96].
4.4.2 Formation of Microdischarges in the Catalyst Pores
It is important for plasma catalysis applications to obtain more insight in the occurrence and/or mechanism of microdischarge formation inside catalyst pores, as the presence of plasma species inside catalyst pores might affect the physical and chemical properties of the catalyst, and more importantly, it will increase the active surface area of the catalyst that is available for surface reactions. The formation of microdischarges inside catalyst pores was investigated experimentally by Holzer, Roland, and Kopinke [29, 39, 40], as well as by Hensel et al. [41,42,43], but again, the modeling attempts are more limited.
Bhoj and Kushner have developed a comprehensive multi-scale 2D fluid-surface kinetics model to describe the functionalization of rough polymer (polypropylene) surfaces by an atmospheric pressure dielectric barrier-corona discharge in a He/O2/H2O mixture [97], in humid air [98], and in He/NH3/H2O and He/O2/H2O mixtures [99]. Although it was not a catalyst surface, the study is still relevant for this application, due to the similarities. The multi-scale model accounts for two different spatial scales, i.e., (i) a macroscopic scale (~ mm), defined by the spacing of the corona streamers, and (ii) a microscopic scale (~ μm), defined by the surface roughness, to investigate whether plasma species can penetrate into the microstructure of the surface. It was reported that electrons and ions produced during the corona pulses can only penetrate to a limited extent into the rough surface features for pore sizes of about 1 μm, comparable to the Debye length. Furthermore, the penetration depends on the discharge polarity: in negative discharges, there is limited penetration of electrons, which locally produce reactive species by electron impact, while in positive discharges, a sheath-like region near the surface prevents electrons from penetrating into the surface features, and hence, there is less local producton of radicals.
Figure 4.19 illustrates the calculated density profiles of O, OH, and O3 in the bulk plasma at 50 μs, i.e., halfway during the first interpulse period of a negative corona discharge, as well as the densities of O, OH, O3, and HO2 in the vicinity of the surface at 100 μs, i.e., at the end of the interpulse period. It is clear that the densities of the longer-lived species, which react slowly with the surface, are quite uniform over the scale of the roughness, due to diffusion, and these species are able to penetrate into all surface features over time scales of hundreds of microseconds, while more reactive radicals, such as OH, are more depleted near the surface, because they are consumed before they can diffuse deeper into the surface features. In [99] the treatment of inside surfaces of porous polypropylene microbeads, placed on an electrode, was simulated, and it was revealed that this treatment was sensitive to the view angles to the discharge and the pore size and is controlled by the relative rates of radical transport and surface reactions deep into the pores.

Density profiles of O, OH, and O3 in the bulk plasma at 50 μs, i.e., halfway during the first interpulse period of a negative corona discharge (a), and density profiles of O, OH, O3, and HO2 in the vicinity of the surface at 100 μs, i.e., at the end of the interpulse period (b), as calculated with a multi-scale fluid-surface kinetics model for an atmospheric pressure dielectric barrier-corona discharge in humid air. The contour labels are fractions of the maximum density noted in each figure. (Reproduced with permission from [98])
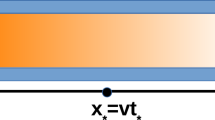
Furthermore, Wang et al. have also studied the propagation of an air plasma through a porous dielectric sheet, with a pore diameter of 100 μm, by means of a fluid model [100]. The model also includes radiation transport and photoionization of O2 by VUV radiation. Figure 4.20 illustrates the electron density inside the pores, at successive times, assuming a photoionization cross section of 10−19 cm2. The maximum values of the electron density are indicated under each frame. The model reveals that less than 1 ns is required for the plasma to penetrate through the porous sheet of 600 μm thickness and that the plasma propagation is controlled by a balance between retarding, due to charging of the internal surfaces of the pores, and photoionization that extends the plasma around corners [100].

Calculated electron density at three moments in time, illustrating plasma propagation through a porous dielectric sheet, assuming a photoionization cross section of 10−19 cm2. (Reproduced with permission from [100])
Recently, within our group PLASMANT, we also developed some models to study the behavior of plasma species inside catalyst pores, and some results will be illustrated below.
-
(a)
Fluid Model for the Formation of Microdischarges in Catalyst Pores
Zhang et al. developed a 2D fluid model to answer the question whether plasma can be formed inside catalyst pores with μm dimensions in a helium DBD [101]. Figure 4.21 illustrates the calculation results for a pore size of 100 μm, at an applied voltage of 20 kV. The total ion density (Fig. 4.21a) increases significantly inside the pore, with a maximum value 7 times higher than in the center of the discharge. The electron density shows a slight increase near the pore, but inside the pore it drops to low values (see Fig. 4.21b). The latter can be explained because the electrons are more easily lost at the walls and also because the electric field pushes the electrons back to the bulk region.

Calculated distributions of the total ion density (a), electron density (b), electric field (c), electron temperature (d), and electron impact ionization rate (e), averaged over one period, for a helium discharge with applied potential of 20 kV, with a 100-μm pore. Note that in (a) and (b) the same color scale is used, to allow comparison, but the values above and below the color scale indicate the maximum and minimum densities in each case
The electric field inside the pore (Fig. 4.21c) is greatly enhanced, yielding a significant rise in electron temperature as well (Fig. 4.21d). As a result, the electron impact ionization is also greatly enhanced inside the pore, as is clear from Fig. 4.21e, giving rise to the formation of the ions and electrons. The same applies to electron impact excitation and the formation of excited plasma species [101]. Hence, the model predicts that for a pore size of 100 μm, the plasma species are effectively generated inside the pore, and in addition, the ions might also migrate into the pore due to the strong electric field.
When investigating the effect of various pore sizes and applied voltages, the modeling results indicate that the microdischarge formation inside the pore occurs more easily at larger pore size and applied voltage [101], which was also observed by Hensel et al. [41, 43]. At a fixed pore size of 30 μm, the presence of the pore seems to have no effect on the plasma properties for an applied voltage of only 2 kV, but above 10 kV, the ionization is significantly enhanced due to the strong electric field and high electron temperature, and the ion density shows a remarkable increase near and in the pore.
Furthermore, at a fixed applied voltage of 20 kV, the model predicts that plasma species can only be created inside catalyst pores with dimensions above 10 μm. Furthermore, while the resulting ion density inside the pores is indeed enhanced for pore sizes of 10 μm and more, the electron density only exhibits a significant increase near and inside the pore for pore sizes above 200 μm, as the electric field pushes the electrons back to the bulk region (see above). Finally, the electric potential shows a strikingly different distribution, depending on the pore size, as illustrated in Fig. 4.22. Inside a 10-μm pore, the potential reaches its maximum at the bottom of the pore, as is clear from Fig. 4.22a. For pore sizes of 20 μm and above, the potential drops gradually from the top of the pore to the bottom; see Fig. 4.22b. Finally, for a 400-μm pore (which may occur in structured catalysts), a clear potential drop in both the vertical and the horizontal direction is predicted, corresponding to the presence of a sheath near the bottom of the pore and the side walls. This is a strong evidence for the presence of a microdischarge inside the pore. The latter is as expected, because the theoretical Debye length is in the order of 40 μm for the typical conditions under study here (i.e., helium plasma with electron temperature and density of 3 eV and 1017 m−3).

Calculated electric potential distribution inside catalyst pores of 10 μm (a), 20 μm (b), and 400 μm (c), for a helium discharge with applied potential of 20 kV
In plasma catalysis, various catalytic support materials can be used, with various dielectric constants. Zhang et al. therefore also investigated the effect of the dielectric constant of the material on the plasma behavior inside the catalyst pores, as well as in the sheath in front of the pores [102]. Figure 4.23 illustrates the calculated electron impact ionization rate, which is characteristic for the plasma generation as explained above, inside and above a pore of 100 μm diameter, for different dielectric constants of the material. At ε ≤ 200, the ionization clearly takes place inside the pore (see Fig. 4.23a–c). The absolute value of the ionization rate depends on the dielectric constant and reaches a maximum at ε = 50 (see Fig. 4.23b). At ε = 300, the ionization still partially takes place inside the pore, but the maximum has shifted to a position in front of the pore (see Fig. 4.23d). At larger dielectric constants, the ionization does not occur inside the pore anymore, but only in the sheath in front of the pore, as illustrated in Fig. 4.23e for ε = 400. At ε = 1000, the ionization is very pronounced in the sheath near the dielectric, but in front of the pore the ionization is reduced (see Fig. 4.23f). The strong ionization in the sheath in front of the dielectric can be explained because the dielectric material is more effectively polarized for larger dielectric constants, and this gives rise to a stronger local electric field in the sheath near the dielectric. The latter yields significant electron heating and thus also enhanced ionization in the sheath. Inside the pore, the electric field is slightly enhanced when ε increases from 4 to 25, but for larger dielectric constants, the polarization of the left sidewall counteracts that of the right sidewall, and as a consequence, the net electric field inside the pore is reduced. Especially for ε = 1000, the electric field inside the pore is very weak, resulting in a low electron temperature, and this significantly limits the plasma generation inside the pore for very high dielectric constants, as is indeed obvious from Fig. 4.23.

Calculated electron impact ionization rate profiles above and inside a pore of 100 μm diameter, averaged over time in 1 AC cycle, for a helium discharge sustained at 20 kV, and for different dielectric constants: (a) ε = 9, (b) ε = 50, (c) ε = 200, (d) ε = 300, (e) ε = 400, (f) ε = 1000
The model calculations reveal that the ionization enhancement inside pores smaller than 100 μm only occurs for materials with smaller dielectric constants, for the same reason as explained above (i.e., the polarization of the left sidewall counteracts that of the right sidewall). More specifically, for pore sizes of 50 μm, 30 μm, and 10 μm, only materials with dielectric constants below 200, 150, and 50, respectively, seem to yield enhanced ionization inside the pores. Note that ε = 300 is a typical value for SrTiO3, ε = 200 corresponds to CaTiO3, and ε = 50 is a typical value for TiO2 [55].
Although some experiments are performed in discharges with different ferroelectric packed bed materials, to our knowledge, no experiments are available for the different plasma behavior inside catalyst pores with different dielectric constants. This behavior is probably different from the behavior in between the beads of packed bed discharges, due to the different dimensions. Hence, the model predictions still have to be validated by experiments, but they suggest that the most common catalyst supports, i.e., Al2O3 and SiO2, with dielectric constants around ε = 8–11 and 4.2, respectively, should allow more easily that microdischarges can be formed inside catalyst pores, while for ferroelectric materials with dielectric constants above 300, it seems much more unlikely that plasma enhancement inside the pores occurs, even not for 100 μm pore sizes.
Finally, Zhang et al. also studied the plasma production inside catalyst pores with different pore shapes and reported that the electric field is significantly enhanced near tip-like structures [103]. A conical pore with small opening yields the strongest electric field at the opening and bottom corners of the pore, causing significant ionization throughout the pore. A cylindrical pore only yields electric field enhancement at the bottom corners, causing only slight enhancement of the ionization rate inside the pore. Finally, a conical pore with large opening yields a maximum electric field at the bottom. In conclusion, the pore shape seems to greatly affect the electric field enhancement and thus the plasma properties [103].
To summarize, this fluid model reveals that plasma species can only be created inside catalyst pores with dimensions above 10 μm, for materials with dielectric constants below 50. These pore sizes are of interest for structured catalysts, but catalytic supports typically have pores in the nm range, and the latter might thus be too small for microdischarge formation inside the pores, according to the fluid model predictions. However, the above studies were applied to a helium plasma, which yields a homogeneous discharge. In contrast, reactive gases, which are more relevant for plasma catalysis applications, exhibit streamer formation, and the latter might be characterized by much higher electron densities. Hence, it is well possible that in this case, the plasma can even be created in nm-sized catalyst pores. This is too small for fluid simulations, so we developed a PIC-MCC model for an air plasma, accounting for streamer formation, and this will be illustrated in the next section.
-
(b)
PIC-MCC Model for the Formation of Microdischarges in Catalyst Pores
In [104] the formation of microdischarges inside both μm- and nm-sized catalyst pores was investigated by a 2D PIC-MCC model, for a DBD operating in dry air in filamentary mode. The calculations reveal that the streamers can penetrate into the surface features of a porous catalyst, and microdischarges can be formed inside both μm- and nm-sized pores. For the μm-sized pores, the ionization mainly occurs inside the pore, in agreement with the fluid model results presented in previous section, while for the nm-sized pores, the ionization is strongest either inside or just above the pores, depending on the pore size. The smaller pores (~ 4–10 nm) are characterized by a more pronounced surface discharge along the dielectric surface, due to the relatively large surface area, while in the larger pores (~ 100 nm or μm-sized) the ionization rate is more pronounced, due to the larger effective ionization region. This is illustrated in Fig. 4.24 for pore sizes of 4, 10, and 100 nm. In the 4-nm pore, the ionization is located above the pore, while in the 10-nm pore, it is both inside and above the pore, and in the 100-nm pore, the maximum ionization rate is clearly inside the pore. The reason why it is mainly in the upper part of the pore is because the electric field, due to space charge separation inside the pore, pushes the electrons out of the pore, inducing many ionization collisions in the upper part of the pore. It is also clear from Fig. 4.24 that the maximum ionization rate drastically increases with pore size. Indeed, although the maximum local electric field decreases for increasing pore sizes between 10 and 100 nm, leading to a lower electron density, the effective ionization region increases with pore size, so there are more electrons available to generate ionization, resulting in an overall higher ionization rate [104].

Calculated electron impact ionization rate (m−3 s−1) for pore sizes of 4 (a), 10 (b), and 100 nm (c), at a time of 1.2 ps. (Reproduced with permission from [104])
Zhang and Bogaerts [105] applied a PIC-MCC model to study the mechanism of streamer propagation in catalyst pores, for various pore diameters in the nm to μm range. They concluded that the Debye length is an important criterion for plasma penetration into catalyst pores, i.e., plasma streamers can only penetrate into pores with diameter larger than the Debye length. The latter depends on the electron density and temperature in the plasma streamer, but is in the order of a few 100 nm up to 1 μm at typical DBD conditions in air. For pores in the range of ~ 50 nm, the calculations revealed that plasma can only penetrate to some extent and only at the beginning of a microdischarge, before the actual plasma streamer reaches the catalyst surface and a sheath is formed. Upon increasing applied voltage, plasma streamers can penetrate into smaller pores, due to the higher plasma density and thus shorter Debye length.
In [106] it was demonstrated with the same model that surface charging (of the dielectric surface or the catalyst pore sidewalls) plays a crucial role in the plasma distribution along the dielectric surface, as well as in the streamer propagation and discharge enhancement inside catalyst pores. The importance greatly depends on the dielectric constant of the material. At εr ≤ 50, surface charging causes the plasma to spread along the dielectric surface and inside the pores, leading to deeper plasma streamer penetration, while for εr > 50 or for metallic coatings, the discharge was found to be more localized, due to very weak surface charging.
Finally, it is worth to stress again that the PIC-MCC simulations reveal that microdischarges can be formed inside nm-sized pores, while the fluid model in previous section predicted that this was only possible for μm-sized pores. The reason is that the fluid model results were obtained for a helium discharge, operating in glow mode, as explained above, while the results presented here are for an air discharge, operating in filamentary mode, which is characterized by much higher electron densities in the streamers, and in this case, the plasma can thus be created even in nm-sized catalyst pores, as long as they are larger than the Debye length at these conditions.
4.4.3 Adsorption of Plasma Species on the Catalyst Surface, and Desorption of Newly Formed Species, that Might Affect the Plasma Chemistry
Plasma species that adsorb on the catalyst surface can give rise to the formation of new species upon reaction at the surface. When these new species desorb from the catalyst surface, they arrive back in the plasma. Hence, when modeling the plasma chemistry for plasma catalysis application, the desorbed species need to be included in the model as well. There exist several models for describing the plasma chemistry for various environmental applications relevant for plasma catalysis, e.g., NOx destruction [107,108,109,110,111], SO2 removal [112, 113], the oxidation of carbon soot [108, 110, 114], VOC remediation [115,116,117,118,119], and hydrocarbon reforming and/or CO2 conversion [120,121,122,123,124,125,126,127,128,129,130,131,132,133], but they are mostly applied to plasma without catalysis, and thus, the effect of desorbed species from a catalyst surface was typically not yet taken into account. Indeed, most often, 0D chemical kinetics models are used for this purpose, so that surface effects are typically not included at all. There exist also some (1D or 2D) fluid models for this application (e.g., [120, 125, 132, 133]), but they typically consider only simplified boundary conditions, based on sticking coefficients at the walls, thus not accounting for possible chemical reactions at a catalyst surface either.
A few papers, however, report on the plasma chemistry, accounting also for the effect of a catalyst. Kim et al. developed a simple kinetic model for the plasma catalytic decomposition of VOCs, predicting zero-order kinetics with respect to the specific energy input, in good agreement with their experimental observations [83]. Tochikubo simulated NOx removal by plasma catalysis, based on a fluid model for a filamentary DBD, dealing with the plasma chemistry and a limited set of 17 surface reactions [81]. However, the synergy expected for plasma catalysis could not be observed, as the plasma simulations were not directly coupled to the surface reaction modeling. Moreover, the authors stated that the input data (activation barriers, rate constants) will need to be improved for better correspondence with experiments. Istadi and Amin developed an artifical neural network for a catalytic DBD reactor for dry reforming of methane, suggesting some synergism between the plasma and the CaO-MnO/CeO2 catalyst, which affects the selectivity toward hydrocarbons with two or more C atoms [134]. Finally, Jiwu and Lei modeled the flue gas desulfurization process by a corona discharge combined with Mn2+ catalysis [135]. The Mn2+ catalyst was however in liquid phase, and thus no surface reactions were included.
There is still a lot of work to be performed in this field, but the latter can only be done when the surface reaction probabilities are known, pointing out the need of more atomic-scale simulations to obtain information on the surface reactions in case of plasma catalysis (see Sect. 4.3 above). When such reaction coefficients become available, the above plasma chemistry models could be upgraded by including the effects of a catalyst surface, through appropriate boundary conditions for the species continuity equations, so that these models, preferably in 2D, become effectively applicable to plasma catalysis.
4.5 Conclusion and Outlook: Ultimate Goal for Modeling Plasma Catalysis
The ultimate goal for modeling plasma catalysis will be to include all the above effects into one comprehensive, multi-level model. This includes describing the dynamic behavior at the atomic and molecular level, which takes place at very short time scales (order of nanoseconds), up to the slower bulk kinetic behavior, as well as mass and heat transfer, taking place at macroscopic time scales. It is clear that such a model has not yet been realized.
In other application fields, however, like for plasma etching and deposition, surface effects have been incorporated already. More specifically, the effect of surface reactions has been accounted for by updating the plasma chemistry with new species formed at the walls (e.g., [136,137,138,139,140,141]). Also the effect of surface temperature on the temperature in the plasma, which could also be relevant for plasma catalysis in case of hot spots (see Sect. 4.3.2 above), has been described already [142]. Typical codes which account for such effects are 2D hybrid models, like the Hybrid Plasma Equipment Model (HPEM) [141] and non-PDPSIM (e.g., [97,98,99]), both developed by Kushner and coworkers. The non-PDPSIM code has already been applied for multi-scale fluid-surface kinetics modeling of plasma treatment of rough polymer surfaces, which can be considered comparable to a porous catalyst surface (see Sect. 4.4.2 above). Furthermore, the HPEM code also allows to calculate the formation and evolution of trench profiles due to etching and the behavior of plasma species inside trenches, by means of a Monte Carlo simulation [143]. Such features would also be of great value for the simulation of plasma species inside catalyst pores.
It is thus clear that such a hybrid model would also be of great value for plasma catalysis applications. This concept is illustrated in Fig. 4.25. As plasma catalysis applications typically entail a comprehensive plasma chemistry, the most suitable type of model for this purpose is a 0D (or global) model, because of its reasonable computational cost, even when including a large number of species and chemical reactions. However, such a model does not account for geometrical (reactor) effects. Therefore, once the plasma chemistry is built up, it should be transferred to 2D or 3D fluid models, which account for geometrical effects, like in a packed bed DBD reactor, or surface effects, to allow updating the plasma chemistry by new species formed at the catalyst surface. Because such a model is computationally very expensive, the plasma chemistry will have to be based on a reduced chemistry set, which can be developed within the 0D chemical kinetics model, by comparing and benchmarking with the full chemistry model. Such a 2D or 3D model also calculates the electric field, which might affect the plasma-catalyst interactions. Furthermore, it provides the fluxes of the various plasma species arriving at the catalyst surface, which can subsequently be used to describe the behavior of plasma species inside a catalyst pore, e.g., by another fluid model or a Monte Carlo model (cf. Sect. 4.4.2 above). Ideally, the latter should also be able to account for changes in the pore shape, similar to what has been developed already for plasma etching applications (see above).

Concept of an integrated model, which would be the ultimate goal for modeling plasma catalysis
This combined, hybrid model should furthermore be combined with atomic-scale simulations for the interaction of the plasma species with the catalyst surface. Indeed, the fluxes of the various plasma species, as well as the electric field near the catalyst surface, calculated by the plasma model, are useful input for atomic-scale simulations. Combining macro-scale plasma models and micro-scale models inside catalyst pores with atomic-scale simulations for the plasma-catalyst interactions is very challenging, due to the small time and length scales of the atomistic simulations, i.e., typically in the order of nanoseconds and nanometers, respectively [46]. Therefore, it is more realistic to use such simulations as a kind of off-line module to the plasma model, providing rate coefficients for the various surface reactions. The latter can then be used as boundary conditions for the plasma species continuity equations in the plasma model, to update the plasma chemistry. This will also allow adding new (desorbed) species from a catalyst surface to the plasma chemistry. In this way, the atomistic simulations can be integrated in a plasma model, yielding a multi-level model for plasma catalysis (see Fig. 4.25).
It is clear that developing such an integrated model will require a lot of efforts, especially because of the large number of plasma-catalyst interactions that need to be accounted for. Hence, it cannot be realized on a short time scale. Nevertheless, we should aim for such a model, which would certainly contribute to a better understanding of plasma catalysis and can help to improve this highly important and rapidly evolving application field. In the meantime, until such a comprehensive model is being developed, the individual modeling approaches, as described in Sect. 4.3 and 4.4 above, can also contribute already to a better understanding of plasma catalysis.
References
Blin-Simiand, N., Tardivaux, P., Risacher, A., Jorand, F., & Pasquiers, S. (2005). Removal of 2-heptanone by dielectric barrier discharges - the effect of a catalyst support. Plasma Processes and Polymers, 2, 256–262.
Hong, J. P., Chu, W., Chernavskii, P. A., & Khodakov, A. Y. (2010). Cobalt species and cobalt-support interaction in glow discharge plasma-assisted Fischer-Tropsch catalysts. Journal of Catalysis, 273, 9–17.
Liu, C. J., Zou, J., Yu, K., Cheng, D., Han, Y., Zhan, J., Ratanatawante, C., & Jang, B. W. L. (2006). Plasma application for more environmentally friendly catalyst preparation. Pure and Applied Chemistry, 78, 1227–1238.
Demidyuk, V., & Whitehead, J. C. (2007). Influence of temperature on gas-phase toluene decomposition in plasma-catalytic system. Plasma Chemistry and Plasma Processing, 27, 85–94.
Shang, S., Liu, G., Chai, X., Tao, X., Li, X., Bai, M., Chu, W., Dai, X., Zhao, Y., & Yin, Y. (2009). Research on Ni/γ-Al2O3 catalyst for CO2 reforming of CH4 prepared by atmospheric pressure glow discharge plasma jet. Catalysis Today, 148, 268–274.
Tu, X., Gallon, H. J., Twigg, M. V., Gorry, P. A., & Whitehead, J. C. (2011). Dry reforming of methane over a Ni/Al2O3 catalyst in a coaxial dielectric barrier discharge reactor. Journal of Physics D: Applied Physics, 44, 274007.
Pylinina, A. I., & Mikhalenko, I. I. (2013). Activation of Cu-, Ag-, Au/ZrO2 catalysts for dehydrogenation of alcohols by low-temperature oxygen and hydrogen plasma. Theoretical and Experimental Chemistry, 49, 65–69.
Guo, Y.-F., Ye, D.-Q., Chen, K.-F., He, J.-C., & Chen, W.-L. (2006). Toluene decomposition using a wire-plate dielectric barrier discharge reactor with manganese oxide catalyst in situ. Journal of Molecular Catalysis. A, Chemical, 245, 93–100.
Mahammadunnisa, S., Reddy, E. L., Ray, D., Subrahmanyam, C., & Whitehead, J. C. (2013). CO2 reduction to syngas and carbon nanofibres by plasma-assisted in situ decomposition of water. International Journal of Greenhouse Gas Control, 16, 361–363.
Liu, C.-J., Mallison, R., & Lobban, L. (1998). Nonoxidative methane conversion to acetylene over zeolite in a low temperature plasma. Journal of Catalysis, 179, 326–334.
Wu, C. C., Wu, C. I., Sturm, J. C., & Kahn, A. (1997). Surface modification of indium tin oxide by plasma treatment: An effective method to improve the efficiency, brightness, and reliability of organic light emitting devices. Applied Physics Letters, 70, 1348–1350.
Poppe, J., Völkening, S., Schaak, A., Schütz, E., Janek, J., & Imbihl, R. (1999). Electrochemical promotion of catalytic CO oxidation on Pt/YSZ catalysts under low pressure conditions. Physical Chemistry Chemical Physics, 1, 5241–5249.
van Durme, J., Dewulf, J., Leys, C., & Van Langenhove, H. (2008). Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: A review. Applied Catalysis B: Environmental, 78, 324–333.
Liu, C. J., Wang, J. X., Yu, K. L., Eliasson, B., Xia, Q., & Xue, B. (2002). Floating double probe characteristics of non-thermal plasmas in the presence of zeolite. Journal of Electrostatics, 54, 149–158.
Kim, H. H., Ogata, A., & Futamura, S. (2006). Effect of different catalysts on the decomposition of VOCs using flow-type plasma-driven catalysis. IEEE Transactions on Plasma Science, 34, 984–995.
Löfberg, A., Essakhi, A., Paul, S., Swesi, Y., Zanota, M.-L., Meille, V., Pitault, I., Supiot, P., Mutel, B., Le Courtois, V., & Bordes-Richard, E. (2011). Use of catalytic oxidation and dehydrogenation of hydrocarbons reactions to highlight improvement of heat transfer in catalytic metallic foams. Chemical Engineering Journal, 176-177, 49–56.
Essakhi, A., Mutel, B., Supiot, P., Löfberg, A., Paul, S., Le Courtois, V., Meille, V., Pitault, I., & Bordes-Richard, E. (2011). Coating of structured catalytic reactors by plasma assisted polymerization of tetramethyldisiloxane. Polymer Engineering and Science, 51, 940–947.
Guaitella, O., Thevenet, F., Puzenat, E., Guillard, C., & Rousseau, A. (2008). C2H2 oxidation by plasma/TiO2 combination: Influence of the porosity, and photocatalytic mechanisms under plasma exposure. Applied Catalysis B: Environmental, 80, 296–305.
Rousseau, A., Guaitella, O., Gatilova, L., Thevenet, F., Guillard, C., Röpcke, J., & Stancu, G. D. (2005). Photocatalyst activation in a pulsed low pressure discharge. Applied Physics Letters, 87, 221501.
Kim, H. H., Ogata, A., & Futamura, S. (2008). Oxygen partial pressure-dependent behavior of various catalysts for the total oxidation of VOCs using cycled system of adsorption and oxygen plasma. Applied Catalysis. B, Environmental, 79, 356–367.
Kim, H. H., Oh, S. M., Ogata, A., & Futamura, S. (2005). Decomposition of gas-phase benzene using plasma-driven catalyst (PDC) reactor packed with Ag/TiO2 catalyst. Applied Catalysis. B, Environmental, 56, 213–220.
Kim, H. H., & Ogata, A. (2011). Nonthermal plasma activates catalyst: From current understanding and future prospects. European Physical Journal Applied Physics, 55, 13806.
Wallis, A. E., Whitehead, J. C., & Zhang, K. (2007). Plasma-assisted catalysis for the destruction of CFC-12 in atmospheric pressure gas streams using TiO2. Catalysis Letters, 113, 29–33.
Mei, D., Zhu, X., He, Y., Yan, J. D., & Tu, X. (2015). Plasma-assisted conversion of CO2 in a dielectric barrier discharge reactor: Understanding the effect of packing materials. Plasma Sources Science and Technology, 24, 015011.
Whitehead, J. C. (2016). Plasma-catalysis: The known knowns, the known unknowns and the unknown unknowns. Journal of Physics D: Applied Physics, 49, 243001.
Kang, W. S., Park, J. M., Kim, Y., & Hong, S. H. (2003). Numerical study on influences of barrier arrangements on dielectric barrier discharge characteristics. IEEE Transactions on Plasma Science, 31, 504–510.
Chang, J. S., Kostov, K. G., Urashima, K., Yamamoto, T., Okayasu, Y., Kato, T., Iwaizumi, T., & Yoshimura, K. (2000). Removal of NF3 from semiconductor-process flue gases by tandem packed-bed plasma and adsorbent hybrid systems. IEEE Transactions on Industry Applications, 36, 1251–1259.
Takuma, T. (1991). Field behaviour at a triple junction in composite dielectric arrangements. IEEE Transactions on Electrical Insulation, 26, 500–509.
Holzer, F., Kopinke, F. D., & Roland, U. (2005). Influence of ferroelectric materials and catalysts on the performance of non-thermal plasma (NTP) for the removal of air pollutants. Plasma Chemistry and Plasma Processing, 25, 595–611.
Tu, X., Gallon, H. J., & Whitehead, J. C. (2011). Electrical and spectroscopic diagnostics of a single-stage plasma-catalysis system: Effect of packing with TiO2. Journal of Physics D: Applied Physics, 44, 482003.
Kim, H. H., Kim, J.-H., & Ogata, A. (2009). Microscopic observation of discharge plasma on the surface of zeolites supported metal nanoparticles. Journal of Physics D: Applied Physics, 42, 135210.
Kim, H. H., Ogata, A., & Song, Y.-H. (2011). Propagation of surface streamers on the surface of HSY zeolites-supported silver nanoparticles. IEEE Transactions on Plasma Science, 39, 2220–2221.
Kim, H. H., & Ogata, A. (2012). Interaction of nonthermal plasma with catalyst for the air pollution control. International Journal of Plasma Environmental Science and Technology, 6, 43–48.
Kim, H. H., Teramoto, Y., Sano, T., Negishi, N., & Ogata, A. (2015). Effects of Si/Al ratio on the interaction of nonthermal plasma and ag/HY catalysts. Applied Catalysis B: Environmental, 166-167, 9–17.
Tu, X., Gallon, H. J., & Whitehead, J. C. (2011). Transition behavior of packed-bed dielectric barrier discharge in argon. IEEE Transactions on Plasma Science, 39, 2172–2173.
Nozaki, T., Muto, N., Kado, S., & Okazaki, K. (2004). Dissociation of vibrationally excited methane on Ni catalyst: Part 2. Process diagnostics by emission spectroscopy. Catalysis Today, 89, 67–74.
Mizuno, A. (2013). Generation of non-thermal plasma combined with catalysts and their application in environmental technology. Catalysis Today, 211, 2–8.
Malik, M. A., Minamitani, Y., & Schoenbach, K. H. (2005). Comparison of catalytic activity of aluminum oxide and silica gel for decomposition of volatile organic compounds (VOCs) in a plasmacatalytic reactor. IEEE Transactions on Plasma Science, 33, 50–56.
Roland, U., Holzer, F., & Kopinke, F.-D. (2005). Combination of non-thermal plasma and heterogeneous catalysis for oxidation of volatile organic compounds: Part 2. Ozone decomposition and deactivation of γ-Al2O3. Applied Catalysis. B, Environmental, 58, 217–226.
Holzer, F., Roland, U., & Kopinke, F.-D. (2002). Combination of non-thermal plasma and heterogeneous catalysis for oxidation of volatile organic compounds: Part 1. Accessibility of the intra-particle volume. Applied Catalysis. B, Environmental, 38, 163–181.
Hensel, K., Katsura, S., & Mizuno, A. (2005). DC microdischarges inside porous ceramics. IEEE Transactions on Plasma Science, 33, 574–575.
Hensel, K., Martisovits, V., Machala, Z., Janda, M., Lestinsky, M., Tardiveau, P., & Mizuno, A. (2007). Electrical and optical properties of AC microdischarges in porous ceramics. Plasma Processes and Polymers, 4, 682–693.
Hensel, K. (2009). Microdischarges in ceramic foams and honeycombs. European Physical Journal D: Atomic, Molecular, Optical and Plasma Physics, 54, 141–148.
Rousseau, A., Guaitella, O., Röpcke, J., Gatilova, L. V., & Tolmachev, Y. A. (2004). Combination of a pulsed microwave plasma with a catalyst for acetylene oxidation. Applied Physics Letters, 85, 2199–2201.
Vandenbroucke, A. M., Morent, R., De Geyter, N., & Leys, C. (2011). Non-thermal plasmas for non-catalytic and catalytic VOC abatement. Journal of Hazardous Materials, 195, 30–54.
Neyts, E. C., & Bogaerts, A. (2014). Understanding plasma catalysis through modelling and simulation - a review. Journal of Physics D: Applied Physics, 47, 224010.
Chen, H. L., Lee, H. M., Chen, S. H., Chang, M. B., Yu, S. J., & Li, S. N. (2009). Removal of volatile organic compounds by single-stage and two-stage plasma catalysis systems: A review of the performance enhancement mechanisms, current status, and suitable applications. Environmental Science & Technology, 43, 2216–2227.
Chen, H. L., Lee, H. M., Chen, S. H., Chao, Y., & Chang, M. B. (2008). Review of plasma catalysis on hydrocarbon reforming for hydrogen production - interaction, integration, and prospects. Applied Catalysis B: Environmental, 85, 1–9.
Whitehead, J. C. (2010). Plasma catalysis: A solution for environmental problems. Pure and Applied Chemistry, 82, 1329–1336.
Tu, X., Gallon, H. J., & Whitehead, J. C. (2013). Plasma-assisted reduction of a NiO/Al2O3 catalyst in atmospheric pressure H2/Ar dielectric barrier discharge. Catalysis Today, 211, 120–125.
Kim, H. H. (2004). Nonthermal plasma processing for air-pollution control: A historical review, current issues, and fture prospects. Plasma Processes and Polymers, 1, 91–110.
Neyts, E. C., & Ostrikov, K. (2015). Nanoscale thermodynamic aspects of plasma catalysis. Catalysis Today, 256, 23–28.
Neyts, E. C. (2016). Plasma-surface interactions in plasma catalysis. Plasma Chemistry and Plasma Processing, 36, 185–212.
Neyts, E. C., Ostrikov, K., Sunkara, M. K., & Bogaerts, A. (2015). Plasma catalysis: Synergistic effects at the nanoscale. Chemical Reviews, 115, 13408–13446.
Kim, H. H., Teramoto, Y., Negishi, N., & Ogata, A. (2015). A multidisciplinary approach to understand the interactions of nonthermal plasma and catalyst: A review. Catalysis Today, 256, 13–22.
Kim, H. H., Teramoto, Y., Ogata, A., Takagi, H., & Nanba, T. (2016). Plasma catalysis for environmental treatment and energy applications. Plasma Chemistry and Plasma Processing, 36, 45–72.
Neyts, E. (2018). Atomistic simulations of plasma catalytic processes. Frontiers of Chemical Science and Engineering, 12, 145–154.
Somers, W., Bogaerts, A., van Duin, A. C. T., & Neyts, E. C. (2014). Interactions of plasma species on nickel catalysts: A reactive molecular dynamics study on the influence of temperature and surface structure. Applied Catalysis B: Environmental, 154-155, 1–8.
Somers, W., Bogaerts, A., van Duin, A. C. T., & Neyts, E. C. (2012). Plasma species interacting with nickel surfaces: Toward an atomic scale understanding of plasma-catalysis. Journal of Physical Chemistry C, 116, 20958–20965.
Somers, W., Bogaerts, A., van Duin, A. C. T., Huygh, S., Bal, K. M., & Neyts, E. C. (2013). Temperature influence on the reactivity of plasma species on a nickel catalyst surface: An atomic scale model. Catalysis Today, 211, 131–136.
Huygh, S., Bogaerts, A., & Neyts, E. C. (2016). How oxygen vacancies activate CO2 dissociation on TiO2 anatase (001). Journal of Physical Chemistry C, 120, 21659–21669.
Huygh, S., & Neyts, E. C. (2015). Adsorption of C and CHx radicals on anatase (001) and the influence of oxygen vacancies. Journal of Physical Chemistry C, 119, 4908–4921.
Shirazi, M., Neyts, E. C., & Bogaerts, A. (2017). DFT study of Ni-catalyzed plasma dry reforming of methane. Applied Catalysis B: Environmental, 205, 605–614.
Bal, K. M., Huygh, S., Bogaerts, A., & Neyts, E. C. (2018). Effect of plasma-induced surface charging on catalytic processes: Application to CO2 activation. Plasma Sources Science and Technology, 27, 024001.
Bal, K. M., & Neyts, E. C. (2018). Modelling molecular adsorption on charged or polarized surfaces: A critical flaw in common approaches. Physical Chemistry Chemical Physics, 20, 8456–8459.
Neyts, E. C., & Bal, K. M. (2017). Effect of electric fields on plasma catalytic hydrocarbon oxidation from atomistic simulations. Plasma Processes and Polymers, 14, e1600158.
Neyts, E. C., van Duin, A. C. T., & Bogaerts, A. (2012). Insights in the plasma-assisted growth of carbon nanotubes through atomic scale simulations: Effect of electric field. Journal of the American Chemical Society, 134, 1256–1260.
Neyts, E. C., Ostrikov, K., Han, Z. J., Kumar, S., van Duin, A. C. T., & Bogaerts, A. (2013). Defect healing and enhanced nucleation of carbon nanotubes by low-energy ion bombardment. Physical Review Letters, 110, 065501.
Somers, W., (2015). Atomic scale simulations of the interactions of plasma species on nickel catalyst surface. University of Antwerp, PhD-thesis.
Ni, B., Lee, C., Sun, R.-C., & Zhang, X. (2009). Microwave assisted heterogeneous catalysis: Effects of varying oxygen concentrations on the oxidative coupling of methane. Reaction Kinetics and Catalysis Letters, 98, 287–302.
Zhdanov, V. P. (1999). Simulation of surface restructuring and oscillations in CO-NO reaction on Pt(100). The Journal of Chemical Physics, 110, 8748–8753.
Kersten, H., Deutsch, H., Steffen, H., Kroesen, G. M. W., & Hippler, R. (2001). The energy balance at substrate surfaces during plasma processing. Vacuum, 63, 385–431.
Li, S., Zheng, W., Tang, Z., & Gu, F. (2012). Plasma heating and temperature difference between gas pellets in packed bed with dielectric barrier discharge under natural convection condition. Heat Transfer Engineering, 33, 609–617.
Nozaki, T., & Okazaki, K. (2013). Non-thermal plasma catalysis of methane: Principles, energy efficiency, and applications. Catalysis Today, 211, 29–38.
JiangB, G. H. (2016). Enhanced dissociative chemisorption of CO2 via vibrational excitation. The Journal of Chemical Physics, 144, 091101.
Bal, K. M., & Neyts, E. C. (2015). Merging metadynamics into hyperdynamics: Accelerated molecular simulations reaching time scales from microseconds to seconds. Journal of Chemical Theory and Computation, 11, 4545–4554.
Guerra, V., & Marinov, D. (2016). Dynamical Monte Carlo methods for plasma-surface reactions. Plasma Sources Science and Technology, 25, 045001.
Marinov, D., Teixeira, C., & Guerra, V. (2017). Deterministic and Monte Carlo methods for simulation of plasma-surface interactions. Plasma Processes and Polymers, 14, 1600175.
Blaylock, D. W., Ogura, T., Green, W. H., & Beran, G. J. O. (2009). Computational investigation of thermochemistry and kinetics of steam methane reforming on Ni(111) under realistic conditions. Journal of Physical Chemistry C, 113, 4898–4908.
Blaylock, D. W., Zhu, Y.-A., & Green, W. H. (2011). Computational investigation of the thermochemistry and kinetics of steam methane reforming over a multi-faceted nickel catalyst. Topics in Catalysis, 54, 828–844.
Toshikubo, F. (2009). Modeling for plasma-enhanced catalytic reduction of nitrogen oxides. Thin Solid Films, 518, 957–961.
Delagrange, S., Pinard, L., & Tatibouët, J.-M. (2009). Combination of a non-thermal plasma and a catalyst for toluene removal from air: Manganese based oxide catalysts. Applied Catalysis. B, Environmental, 68, 92–98.
Kim, H. H., Ogata, A., & Futamura, S. (2005). Atmospheric plasma-driven catalysis for the low temperature decomposition of dilute aromatic compounds. Journal of Physics D: Applied Physics, 38, 1292–1300.
Van Laer, K., & Bogaerts, A. (2016). Fluid modelling of a packed bed dielectric barrier discharge plasma reactor. Plasma Sources Science and Technology, 25, 015002.
Takaki, K., Chang, J.-S., & Kostov, K. G. (2004). Atmospheric pressure of nitrogen plasmas in a ferroelectric packed bed barrier discharge reactor. Part I. Modeling. IEEE Transactions on Dielectrics and Electrical Insulation, 11, 481–490.
Russ, H., Neiger, M., & Lang, J. E. (1999). Simulation of micro discharges for the optimization of energy requirements for removal of NOx from exhaust gases. IEEE Transactions on Plasma Science, 27, 38–39.
Babaeva, N. Y., Bhoj, A. N., & Kushner, M. J. (2006). Streamer dynamics in gases containing dust particles. Plasma Sources Science and Technology, 15, 591–602.
Kruszelnicki, J., Engeling, K. W., Foster, J. E., Xiong, Z., & Kushner, M. J. (2017). Propagation of negative electric discharges through 2-dimensional packed bed reactors. Journal of Physics D: Applied Physics, 50, 025203.
Kang, W. S., Kim, H. H., Teramoto, Y., Ogata, A., Lee, J. Y., Kim, D. W., Hur, M., & Song, Y. H. (2018). Surface streamer propagations on an alumina bead: Experimental observation and numerical modelling. Plasma Sources Science and Technology, 27, 015018.
Zhang, Y., Wang, H.-Y., Jiang, W., & Bogaerts, A. (2015). Two-dimensional particle-in cell/Monte Carlo simulations of a packed-bed dielectric barrier discharge in air at atmospheric pressure. New Journal of Physics, 17, 083056.
Gao, M.-X., Zhang, Y., Wang, H.-Y., Guo, B., Zhang, Q.-Z., & Bogaerts, A. (2018). Mode transition of filaments in packed-bed dielectric barrier discharges. Catalysts, 8, 248.
Van Laer, K., & Bogaerts, A. (2017). Influence of gap size and dielectric constant of the packing material on the plasma behaviour in a packed bed DBD reactor: A fluid modelling study. Plasma Processes and Polymers, 14, e1600129.
Van Laer, K., & Bogaerts, A. (2017). How bead size and dielectric constant affect the plasma behaviour in a packed bed plasma reactor: A modelling study. Plasma Sources Science & Technology, 26, 085007.
Wang, W., Kim, H.-H., Van Laer, K., & Bogaerts, A. (2018). Streamer propagation in a packed bed plasma reactor for plasma catalysis applications. Chemical Engineering Journal, 334, 2467–2479.
Michielsen, I., Uytdenhouwen, Y., Pype, J., Michielsen, B., Mertens, J., Reniers, F., Meynen, V., & Bogaerts, A. (2017). CO2 dissociation in a packed bed DBD reactor: First steps towards a better understanding of plasma catalysis. Chemical Engineering Journal, 326, 477–488.
Uytdenhouwen, Y., Van Alphen, S., Michielsen, I., Meynen, V., Cool, P., & Bogaerts, A. (2018). A packed-bed DBD micro plasma reactor for CO2 dissociation: Does size matter? Chemical Engineering Journal, 348, 557–568.
Bhoj, A. N., & Kushner, M. J. (2006). Multi-scale simulation of functionalizationof rough polymer surfaces using atmospheric pressure plasmas. Journal of Physics D: Applied Physics, 39, 1594–1598.
Bhoj, A. N., & Kushner, M. J. (2008). Repetitively pulsed atmospheric pressure discharge treatment of rough polymer surfaces: I. Humid air discharges. Plasma Sources Science & Technology, 17, 035024.
Bhoj, A. N., & Kushner, M. J. (2008). Repetitively pulsed atmospheric pressure discharge treatment of rough polymer surfaces: II. Treatment of micro-beads in He/NH3/H2O and He/O2/H2O mixtures. Plasma Sources Science & Technology, 17, 035025.
Wang, X. M., Foster, J. E., & Kushner, M. J. (2011). Plasma propagation through porous dielectric sheets. IEEE Transactions on Plasma Science, 39, 2244–2245.
Zhang, Y.-R., Van Laer, K., Neyts, E. C., & Bogaerts, A. (2016). Can plasma be formed in catalyst pores? A modeling investigation. Applied Catalysis B: Environmental, 185, 56–67.
Zhang, Y.-R., Neyts, E. C., & Bogaerts, A. (2016). Influence of the material dielectric constant on plasma generation inside catalyst pores. Journal of Physical Chemistry C, 120, 25923–25934.
Zhang, Y.-R., Neyts, E. C., & Bogaerts, A. (2018). Enhancement of plasma generation in catalyst pores with different shapes. Plasma Sources Science and Technology, 27, 055008.
Zhang, Y., Wang, H.-Y., Zhang, Y.-R., & Bogaerts, A. (2017). Formation of microdischarges inside a mesoporous catalyst in dielectric barrier discharge plasmas. Plasma Sources Science and Technology, 26, 054002.
Zhang, Q.-Z., & Bogaerts, A. (2018). Propagation of a plasma streamer in catalyst pores. Plasma Sources Science and Technology, 27, 035009.
Zhang, Q.-Z., Wang, W.-Z., & Bogaerts, A. (2018). Importance of surface charging during plasma streamer propagation in catalyst pores. Plasma Sources Science and Technology, 27, 065009.
Gentille, A. C., & Kushner, M. J. (1995). Reaction chemistry and optimization of plasma remediation of NxOy from gas streams. Journal of Applied Physics, 78, 2074–2085.
Dorai, R., & Kushner, M. J. (2000). Consequences of propene and propane on plasma remediation of NOx. Journal of Applied Physics, 88, 3739–3747.
Dorai, R., & Kushner, M. J. (2003). Consequences of unburned hydrocarbons on microstreamer dynamics and chemistry during plasma remediation of NOx using dielectric barrier discharges. Journal of Physics D: Applied Physics, 36, 1075–1083.
Dorai, R., & Kushner, M. J. (2002). Repetitively pulsed plasma remediation of NOx in soot laden exhaust using dielectric barrier discharges. Journal of Physics D: Applied Physics, 35, 2954–2968.
Teodoru, S., Kusano, Y., & Bogaerts, A. (2012). The effect of O2 in a humid O2/N2/NOx gas mixture on NOx and N2O remediation by an atmospheric pressure dielectric barrier discharge. Plasma Processes and Polymers, 9, 652–689.
Chang, M. B., Balbach, J. H., Rood, J. J., & Kushner, M. J. (1991). Removal of SO2 from gas streams using a dielectric barrier discharge and combined plasma photolysis. Journal of Applied Physics, 69, 4409–4417.
Chang, M. B., Kushner, M. J., & Rood, M. J. (1992). Removal of SO2 and the simultaneous removal of SO2 and NO from simulated flue gas streams using dielectric barrier discharge plasmas. Plasma Chemistry and Plasma Processing, 12, 565–580.
Chang, M. B., Kushner, M. J., & Rood, M. J. (1992). Gas-phase removal of NO from gas streams via dielectric barrier discharges. Environmental Science & Technology, 26, 777–781.
Storch, D. G., & Kushner, M. J. (1993). Destruction mechanisms for formaldehyde in atmospheric temperature plasmas. Journal of Applied Physics, 73, 51–55.
Evans, D., Rosocha, L. A., Anderson, G. K., Coogan, J. J., & Kushner, M. J. (1993). Plasma remediation of trichloroethylene in silent discharge plasmas. Journal of Applied Physics, 74, 5378–5386.
Aerts, R., Tu, X., De Bie, C., Whitehead, J. C., & Bogaerts, A. (2012). An investigation into the dominant reactions for ethylene destruction in non-thermal atmospheric plasmas. Plasma Processes and Polymers, 9, 994–1000.
Aerts, R., Tu, X., Van Gaens, W., Whitehead, J. C., & Bogaerts, A. (2013). Gas purification by nonthermal plasma: A case study of ethylene. Environmental Science & Technology, 47, 6478–6485.
Vandenbroucke, A. M., Aerts, R., Van Gaens, W., De Geyter, N., Leys, C., Morent, R., & Bogaerts, A. (2015). Modeling and experimental study of tricholoroethylene abatement with a negative dirrect current corona discharge. Plasma Chemistry and Plasma Processing, 35, 217–230.
De Bie, C., Martens, T., van Dijk, J., Paulussen, S., Verheyde, B., & Bogaerts, A. (2011). Dielectric barrier discharges used for the conversion of greenhouse gases: Modeling the plasma chemistry by fluid simulations. Plasma Sources Science and Technology, 20, 024008.
Yang, Y. (2003). Direct non-oxidative methane conversion by non-thermal plasma: Modeling study. Plasma Chemistry and Plasma Processing, 23, 327–346.
Pringle, K. J., Whitehead, J. C., Wilman, J. J., & Wu, J. H. (2004). The chemistry of methane remediation by a non-thermal atmospheric pressure plasma. Plasma Chemistry and Plasma Processing, 24, 421–434.
Agiral, A., Trionfetti, C., Lefferts, L., Seshan, K., & Gardeniers, J. G. E. (2008). Propane conversion at ambient temperatures C–C and C–H bond activation using cold plasma in a microreactor. Chemical Engineering and Technology, 31, 1116–1123.
Pinhao, N. R., Janeco, A., & Branco, J. B. (2011). Influence of helium on the conversion of methane and carbon dioxide in a dielectric barrier discharge. Plasma Chemistry and Plasma Processing, 31, 427–439.
De Bie, C., Verheyde, B., Martens, T., van Dijk, J., Paulussen, S., & Bogaerts, A. (2011). Fluid modelling of the conversion of methane into higher hydrocarbons in an atmospheric pressure dielectric barrier discharge. Plasma Processes and Polymers, 8, 1033–1058.
Aerts, R., Martens, T., & Bogaerts, A. (2012). Influence of vibrational states on CO2 splitting by dielectric barrier discharges. Journal of Physical Chemistry C, 116, 23257–23273.
Snoeckx, R., Aerts, R., Tu, X., & Bogaerts, A. (2013). Plasma-based dry reforming: A computational study ranging from the nanoseconds to seconds time scale. Journal of Physical Chemistry C, 117, 4957–4970.
Snoeckx, R., Setareh, M., Aerts, R., Simon, P., Maghari, A., & Bogaerts, A. (2013). Influence of N2 concentration in a CH4/N2 dielectric barrier discharge used for CH4 conversion into H2. International Journal of Hydrogen Energy, 38, 16098–16120.
Snoeckx, R., Zeng, Y. X., Tu, X., & Bogaerts, A. (2015). Plasma-based dry reforming: Improving the conversion and energy efficiency in a dielectric barrier discharge. RSC Advances, 5, 29799–29808.
Snoeckx, R., Heijkers, S., Van Wesenbeeck, K., Lenaerts, S., & Bogaerts, A. (2016). CO2 conversion in a dielectric barrier discharge plasma: N2 in the mix as a helping hand or problematic impurity? Energy and Environmental Science, 9, 999–1011.
Heijkers, S., Snoeckx, R., Kozák, T., Silva, T., Godfroid, T., Britun, N., Snyders, R., & Bogaerts, A. (2015). CO2 conversion in a microwave plasma reactor in the presence of N2: Elucidating the role of vibrational levels. Journal of Physical Chemistry C, 119, 12815–12828.
De Bie, C., van Dijk, J., & Bogaerts, A. (2015). The dominant pathways for the conversion of methane into oxygenates and syngas in an atmospheric pressure dielectric barrier discharge. Journal of Physical Chemistry C, 119, 22331–22350.
De Bie, C., van Dijk, J., & Bogaerts, A. (2016). CO2 hydrogenation in a dielectric barrier discharge plasma revealed. J. Phys. Chem, C120, 25210–25224.
Istadi, A., & Amin, N. A. S. (2007). Modelling and optimization of catalytic-dielectric barrier discharge plasma reactor for methane and carbon dioxide conversion using hybrid artificial neural network - genetic algorithm technique. Chemical Engineering Science, 62, 6568–6581.
JiwuL, L. F. (2013). Modeling of corona discharge combined with Mn2+ catalysis for the removal of SO2 from simulated flue gas. Chemosphere, 91, 1374–1379.
Tinck, S., Bogaerts, A., & Shamiryan, D. (2011). Simultaneous etching and deposition processes during the etching of silicon with a Cl2/O2/Ar inductively coupled plasma. Plasma Processes and Polymers, 8, 490–499.
Tinck, S., De Schepper, P., & Bogaerts, A. (2013). Numerical investigation of SiO2 coating deposition in wafer processing reactors with SiCl4/O2/Ar inductively coupled plasmas. Plasma Processes and Polymers, 10, 714–730.
Tinck, S., Boullart, W., & Bogaerts, A. (2011). Modeling Cl2/O2/Ar inductively coupled plasmas used for silicon etching: Effects of SiO2 chamber wall coating. Plasma Sources Science and Technology, 20, 045012.
Kushner, M. J. (1987). A phenomenological model for surface deposition kinetics during plasma and sputter deposition of amorphous hydrogenated silicon. Journal of Applied Physics, 62, 4763–4772.
Zhang, D., & Kushner, M. J. (2000). Mechanisms for CF2 radical generation and loss on surfaces in fluorocarbon plasmas. Journal of Vacuum Science and Technology A, 18, 2661–2668.
Kushner, M. J. (2009). Hybrid modelling of low temperature plasmas for fundamental investigations and equipment design. Journal of Physics D: Applied Physics, 42, 194013.
Tinck, S., Tillocher, T., Dussart, R., & Bogaerts, A. (2015). Cryogenic etching of silicon with SF6 inductively coupled plasmas: A combined modelling and experimental study. Journal of Physics D: Applied Physics, 48, 155204.
Hoekstra, R. J., Grapperhaus, H. J., & Kushner, M. J. (1997). Integrated plasma equipment model for polysilicon etch profilesin an inductively coupled plasma reactor with subwafer and superwafer topography. Journal of Vacuum Science and Technology A, 15, 1913–1921.
Acknowledgments
We gratefully acknowledge K. Van Laer, Y.-R. Zhang, Y. Zhang, Q.-Z. Zhang, W. Wang, S. Huygh, and M. Shirazi (University of Antwerp) and M. Kushner (University of Michigan) for providing some of the figures used as illustrations in this chapter. The authors also acknowledge financial support from the Research Council of the University of Antwerp (TOP-BOF project), the IAP/7 (Interuniversity Attraction Poles) program “Physical Chemistry of Plasma-Surface Interactions (PSI)” by the Belgian Federal Science Policy Office (BELSPO), and the Fund for Scientific Research Flanders (FWO, grant no. G.0217.14 N).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bogaerts, A., Neyts, E. (2019). Plasma Catalysis Modeling. In: Tu, X., Whitehead, J., Nozaki, T. (eds) Plasma Catalysis. Springer Series on Atomic, Optical, and Plasma Physics, vol 106. Springer, Cham. https://doi.org/10.1007/978-3-030-05189-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-05189-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-05188-4
Online ISBN: 978-3-030-05189-1
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)