
Abstract
The development of monoclonal antibodies (mAbs) cetuximab and panitumumab, which target the transmembrane protein epidermal growth factor receptor (EGFR), mark a major step forward in the treatment of metastatic colorectal cancer (mCRC). However, this therapeutic progress proved to be effective only in a very restricted subset of patients. Although several mechanisms of resistance, both primary and acquired, have been identified, the only established predictive tumour biomarker for the treatment of mCRC patients is the RAS mutational status. RAS activating mutations predict a lack of response to these therapies while low levels of primary resistance characterize RAS wild type (WT) patients (only about 15%). However, even WT patients that initially respond to anti-EGFR therapy, eventually undergo tumour progression. In this context, there is still more to be done in the search for effective predictive markers with therapeutic applicability. In this chapter, we provide an overview on the mechanisms that contribute to resistance to EGFR-targeted therapy and highlight what is still missing in our understanding of these molecular mechanisms and approaches to overcome them.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Colorectal cancer
- Epidermal growth factor receptor
- Primary resistance
- Secondary resistance
- Targeted therapy
8.1 Introduction
Cancer is a worldwide health problem whose incidence has been increasing every year, severely threatening human wellbeing. Colorectal cancer (CRC) ranks among the third most frequent cancer type and the fourth leading cause of cancer-related death (Ferlay et al. 2015). Although impressive advances in cancer therapy have been achieved over the last 20 years (better surgical techniques, better screening methods, improved postoperative care, regular follow-ups and more effective adjuvant therapies), CRC is still an illness with undesirably high mortality, mainly associated to the metastatic setting. The prognosis of patients with mCRC has been improving, and correspond now to median overall survival (OS) of over 30 months, depending on the treatment options (Heinemann et al. 2014; Venook et al. 2017).
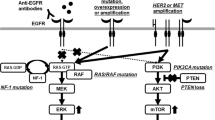
Novel therapies targeting the epidermal growth factor receptor (EGFR) have significantly contributed to the improvement of the OS of mCRC patients. EGFR is a transmembrane receptor belonging to the ErbB tyrosine kinase family which consists of four related proteins: EGFR (ErbB1/HER1), HER2/neu (ErbB2), HER3 (ErbB3) and HER4 (ErbB4) (Hynes and Lane 2005; Wieduwilt and Moasser 2008). All family members contain an extracellular ligand-binding domain with two cysteine-rich regions, a single membrane-spanning region and a cytoplasmic tyrosine kinase domain. In healthy cells, EGFR signalling is tightly regulated by various extracellular ligands, namely EGF, Amphiregulin, Epiregulin and TGFα, which induce homo- and hetero-dimerization with other ErbB members and the subsequent activation of downstream pathways, such as RAS-RAF-MEK-MAPK, PIK3CA-AKT, the SRC family kinases, PLCγ-PKC and JAK/STATs (Fig. 8.1) (Oda et al. 2005). These pathways are involved in several essential cellular processes including proliferation, migration, invasion, survival and angiogenesis. However, in pathologic conditions, constitutive activation of EGFR or downstream effectors (by activating mutations, increased copy number and upregulations) are described as responsible for tumour development and metastasization. EGFR is expressed in various cancers including in CRC with a frequency of 60–80%, where it plays a key role in tumour development and progression (Spano et al. 2005). Therefore, its essential function together with its deregulated activity in cancer justified the rational for the development of EGFR inhibitors . To date, two monoclonal antibodies (mAbs) against EGFR were approved for the treatment of patients with mCRC. Cetuximab is a human-mouse chimeric monoclonal antibody (IgG1 subtype), whereas panitumumab is a fully human anti-EGFR monoclonal antibody (IgG2K subtype). Both antibodies recognize and bind to the extracellular domain of EGFR , not only blocking the ligand-binding region and therefore preventing its ligand-mediated activation, but also causing receptor internalization and degradation, inhibiting further signalling propagation (Ciardiello and Tortora 2008). Furthermore, cetuximab induces antibody-mediated cytotoxicity due to its ability to recruit immune effector cells such as macrophages and monocytes to the tumour, which have specific receptors to bind the antibody’s constant Fc domain (Yang et al. 2013a, b). Cetuximab and panitumumab were proven to give similar benefit in terms of progression-free survival (PFS), overall survival (OS), response rate (RR), as well as quality of life, in several phase II and III clinical trials in combination with chemotherapy or as single agent (Bokemeyer et al. 2009; Van Cutsem et al. 2011; Douillard et al. 2014). Unfortunately however, only a small percentage of mCRC patients are sensitive to anti-EGFR therapy due to primary or innate resistance. And even those that initially respond, eventually acquire resistance and relapse under this therapy (secondary resistance). When used as a single agent in unselected mCRC patients, Cetuximab and panitumumab achieved only a RR of about 10–20% (Cunningham et al. 2004; Van Cutsem et al. 2007). This low RR is caused by the high frequency of genetic or epigenetic alterations in proteins involved in EGFR regulation itself and downstream pathways (such as RAS, BRAF , PI3K and PTEN ) that blunt the response to mAbs targeting EGFR. The subgroup of patients with tumours wild-type for RAS, BRAF, PIK3CA and expressing normal levels of PTEN have the best response to mAbs (De Roock et al. 2010a, b; Karapetis et al. 2014). Nevertheless, still about 10% of these individuals remain resistant to anti-EGFR therapies, suggesting the existence of still unknown alternative mechanisms capable of influencing treatment effectiveness.

EGFR-mediated signalling pathways and clinically available strategies for their inhibition. EGFR ligands bind to the extracellular domain of the receptor leading to its activation and downstream signal propagation, which is essential for tumour cell growth and proliferation. The antibodies cetuximab and panitumumab prevent ligand binding to EGFR, thus blocking receptor signalling. Further targeted agents are available to inhibit EGFR-stimulated downstream pathways and represent potentially valuable tools to overcome resistance to anti-EGFR treatment. Stars indicate oncogenic mutations involved in resistance of tumours to anti-EGFR therapy
To date the molecular mechanisms of response to anti-EGFR mAbs are not yet completely understood. For instance, reports have shown that some patients experience benefit to cetuximab, although having undetectable levels of EGFR (Chung et al. 2005), or occasionally patients that although harbouring RAS activating mutations are able to respond to these therapies (Karapetis et al. 2008; Linardou et al. 2008). Therefore, in this era of targeted medicine, clinical and translational efforts are being made to better understand the molecular complexity of mCRC tumours in order to better adapt the treatment to the molecular characteristics of the specific patient. Furthermore, the identification of clinical relevant predictive biomarkers able to effectively select patients who will likely benefit from these therapies, will not only prevent unnecessary toxicity in resistant patients, but also allow them to receive undelayed alternative treatments.
The aim of this chapter is to provide an overview on the molecular mechanisms that underlie resistance to EGFR -targeted mAbs in mCRC and to discuss possible future directions on how to overcome them.
8.2 Molecular Events That Positively Correlate with Primary Response
8.2.1 Alterations in EGFR Copy Number
Early studies conducted both in heavily pre-treated chemotherapy-refractory patients or in chemotherapy-naïve mCRC patients, have demonstrated that about 80% of unselected mCRC do not benefit from anti-EGFR therapy (Cunningham et al. 2004; Misale et al. 2012). In this context, it was hypothesized that EGFR mutations, levels of expression or levels of its specific ligands could be associated to the lack of response. Soon was realized that, contrarily to non-small-cell lung carcinoma (NSCLC) where mutations in the kinase domains of EGFR correlate with clinical responsiveness to the small molecule tyrosine kinase inhibitors (TKI) gefitinib or erlotinib (Gazdar 2009), point mutations in EGFR are extremely rare in CRC and when they do occur, they are associated with secondary resistance (Barber et al. 2004). Therefore, several studies further assessed whether levels of EGFR expression could correlate with treatment resistance, considering that trastuzumab , a mAb against human epidermal growth factor receptor 2 (HER2/neu) was most effective in patients with metastatic breast tumours overexpressing HER2/neu (Perez et al. 2014). Disappointingly, levels of expression of EGFR were found not to correlate with clinical response to cetuximab or to panitumumab (Chung et al. 2005; Jonker et al. 2007). Curiously, however, alterations in EGFR gene copy number were later confirmed in retrospective analysis of clinical trials to be associated with responses to EGFR-targeted therapy . For example, in a cohort study, about 90% of the patients, who experienced an objective response , had an increase in copy number (three- to five-fold), detected by fluorescence in situ hybridization (FISH) (Moroni et al. 2005). In contrast, only 5% of non-responders showed an increased EGFR copy number. Although only a modest increase in copy number was seen, correlation with response was further confirmed in another large and more homogenous cohort (Sartore-Bianchi et al. 2007). Intriguingly however, is the fact that increased EGFR gene copy number did not seem to correlate with increased expression of this protein (Cappuzzo et al. 2008; Campanella et al. 2010). Therefore, the reason why and how this amplification correlates with response is largely unknown and requires further studies. Furthermore, due to lack of technical standardization and definition of a clear and reproducible cut-off for gene amplification, the assessment of gene copy number by FISH shows high variability, which further makes this biomarker clinically unpractical. Finally, statistical correlation between the increased copy number of EGFR and response to cetuximab and panitumumab is not strong enough to allow the clinical use of this biomarker for the predictive selection of patients (Personeni et al. 2008; Sartore-Bianchi et al. 2012).
8.2.2 Alteration in EGFR-Ligands Expression
Other molecular alterations that positively associate with response are the levels of expression of the EGFR ligands Amphiregulin (AREG) and Epiregulin (EREG). In a prospective clinical trial of 110 patients with mCRC, AREG and EREG levels were higher in pre-treatment tumours from responding patients compared to non-responders (Khambata-Ford et al. 2007). A subsequent in a lager cohort of KRAS WT patients showed similar effects, namely that expression of higher levels of mRNA for either of these ligands was linked to sensitivity to cetuximab monotherapy, improving disease control rate and progression-free survival (Jacobs et al. 2009). Interestingly, patients with KRAS WT or KRAS mutant tumours have similar response rates when ligand expression levels are low and in both cases experience worst responses to cetuximab. It is believed that an autocrine or paracrine loop generated by the increased expression of these ligands is responsible for driving the growth of these tumours. Low levels of expression of AREG and EREG may characterize a tumour that is less dependent on EGFR and, therefore, less sensitive to its inhibition.
Similarly to EGFR gene copy number, the levels of expression of AREG and EREG have so far been difficult to assess (mRNA levels), score and reproduce. Therefore, at present, these markers cannot be used to select patients for cetuximab or panitumumab therapy.
8.3 Primary Resistance to Anti-EGFR Therapy
8.3.1 RAS Mutations
RAS-RAF-MAPK is the signalling pathway mostly studied in cancer given the high frequency of genetic alterations in its components, as well as, its crucial role in cell growth and differentiation. The RAS family is composed of the three genes KRAS , NRAS and HRAS that encode small guanosine-triphosphate (GTP) hydrolases, that act as signal transducers by cycling between a GDP and GTP-bound conformation upon cell surface receptor stimulation (Malumbres and Barbacid 2003). CRC tumours present with about 40% of KRAS mutations, 3–5% of NRAS and less than 1% of HRAS genetic alterations (Bos 1989; Fernández-Medarde and Santos 2011). In tumours, mutations found in RAS family members generally lead to constitutive activation of these proteins and their downstream effector pathway (MAPK pathway), independently of the upstream signalling cascade or growth factor receptor.
A large number of retrospective analyses of data from previous clinical trials over the last decade have led to the discovery that patients with KRAS activating mutations in codons 12 (70–80% of KRAS mutations) or 13 (15–20% of KRAS mutations) of exon 2 do not benefit from cetuximab or panitumumab agents. Clinical trials in which EGFR -targeted mAbs cetuximab or panitumumab were used to treat either chemotherapy-refractory (NCIC trial) or naïve mCRC patients (OPUS, CRYSTAL and PRIME trials), demonstrated that KRAS WT patients had a statistically significant improvement in OS and PFS, whereas KRAS mutant patients did not show any benefit in OS, in PFS or quality of life (Van Cutsem et al. 2011; Douillard et al. 2014; Van Cutsem et al. 2015a, b). The exclusion of patients with RAS mutations has allowed the identification of the subgroup of patients that is more likely to benefit from anti-EGFR therapies. Therefore, in patients with wild-type RAS genotype median OS was 25.8 months versus 20.2 months (HR = 0.77, 95% CI = 0.64–0.94, p = 0.009), in favour of the combination of panitumumab and FOLFOX (infusion of fluorouracil, leucovorin, and oxaliplatin) compared with FOLFOX alone (Douillard et al. 2014). Similar results were presented in the CRYSTAL (28.4 months vs. 20.2 months, HR 0.69, 95% CI = 0.54–0.88) and OPUS trials (ORR 58% vs. 29%; HR 3.33 [95% CI = 1.36–8.17, p = 0.0084), in which randomized patients received first-line cetuximab in combination with FOLFIRI (fluorouracil, leucovorin, and irinotecan) or FOLFOX respectively (Bokemeyer et al. 2011; Van Cutsem et al. 2011).
These results have not only shown that patients harbouring RAS mutations do not experience any benefit from those treatments, but also that in some cases it could even be detrimental for them. PRIME trial shows that the presence of RAS mutations was associated with inferior PFS and OS in patients receiving first line panitumumab plus FOLFOX compared with FOLFOX alone (Douillard et al. 2014). Overall, this information led the American and European health authorities in 2009 to restrict the use of panitumumab and cetuximab only to patients with KRAS exon 2 WT tumours.
However, later retrospective studies revealed that further mutations in KRAS and NRAS genes were also predictive of resistance to anti-EGFR therapies (Heinemann et al. 2014; Van Cutsem et al. 2015a, b). In addition to exon 2, mutations in KRAS exon 3 (codons 59 and 61), exon 4 (codons 117 and 146) and in the homologous codons of NRAS also confer resistance to anti-EGFR therapy, and are altogether called “the expanded RAS mutations” (Heinemann et al. 2014). Thus, a meta-analysis of nine randomized trials confirmed that treatment with mAbs had better efficacy reflected in PFS and OS for RAS WT patients when compared with the expanded RAS mutant group (Sorich et al. 2015). In response to this data, EMA and FDA have updated their recommendations against the use of cetuximab e panitumumab in patients with extended RAS mutations.
Considering that patients with expanded RAS mutations constitute about 53% of all mCRC cases, several attempts have been made to inhibit RAS directly in these patients. Initial approaches have tried to inhibit RAS farnesylation (a necessary step to attach RAS proteins to the cell membrane), which have shown a potent antitumour activity in preclinical studies (Kohl et al. 1995), but this was not confirmed in clinical trials (Macdonald et al. 2005). Another interesting approach was the identification of small-molecule inhibitors that could form a disulfide bond with the cysteine residue in the G12C mutant KRAS protein (about 8% of all KRAS mutations in CRC) (Ostrem et al. 2013). These compounds do not affect WT KRAS but preferentially bind the G12C mutant, inhibiting its activity. Similarly, efforts have been made in identifying compounds that bind and covalently react with the GDP-bound state of KRAS G12C, trapping it in an inactive conformation (Patricelli et al. 2016). However, these studies resulted in only limited demonstration of KRAS inhibition in cells and lack demonstration of in vivo efficacy and specificity. Very recently, Matthew et al. have designed and characterized a promising G12C inhibitor (ARS-1620) with features necessary to achieve in vivo covalent targeting and inhibition of mutant allele-specific G12C cell lines and tumour models (Janes et al. 2018). This could be a promising step towards bringing KRAS mutant specific inhibitors to the clinic. Nevertheless, several other approaches have been used to target RAS: (i) blocking downstream effectors such as MEK (Yoon et al. 2011) and PI3K (Migliardi et al. 2012), (ii) identification of synthetic lethal interactions with mutant KRAS (interactions that when co-occur in a cell result in cellular death) (Costa-Cabral et al. 2016), or (iii) the use of small-molecule inhibitors of KRAS (Welsch et al. 2017). Finally, a combination therapy of inhibitors co-targeting MEK and CDK4/6 with trametinib and palbociclib, respectively, was highly efficacious in KRAS-mutant CRC patient-derived xenografts (Ziemke et al. 2016), but a clinical validations of this strategy is still missing.
Despite these promises, targeting RAS in cancer remains one of the most difficult assignments in cancer therapy. Our incomplete knowledge about RAS-mediated signalling, regulatory feedback loops, pathway redundancy and mechanisms by which RAS activates its downstream effectors, prevents the design of more effective therapies. It is, therefore, essential to fill the gaps of our knowledge regarding RAS-mediated processes in order to develop more effective agents for targeting RAS and its effector pathways in cancer cells.
This is even more relevant given the conflicting data that a number of patients carrying KRAS -mutant tumours are able to respond to either cetuximab or panitumumab. Specifically the role of codon 13 mutation in this mechanism is still controversial. DeRoock et al. studied the role of G13D mutation in response to cetuximab in chemo-refractory patients and their results showed longer OS of 7.6 months compared to 5.7 (P = 0.005) and longer PFS of 4.0 months compared to 1.9 months (P = 0.004) than in G12V mutant patients (De Roock et al. 2010a, b). Although suggesting that patients with G13D-harbouring tumours respond to cetuximab, RR were lower than in KRAS WT patients. The same study further showed in vitro and in mouse models that CRC cells with the G12V mutation were insensitive but with mutation G13D were as sensitive as the KRAS WT to cetuximab. In contrast, a retrospective analysis of three randomized phase III clinical trials showed that patients harbouring KRAS codon 13 mutations did not benefit from receiving panitumumab treatment (Peeters et al. 2013). Explanations for these contradictory results may include differences between cetuximab and panitumumab treatments or in the chemotherapy regimens between the studies. Given that these mutations represent about 19% of the KRAS -mutant tumours, further studies are necessary to unravel the effect of KRAS codon 13 mutation in resistance to anti-EGFR therapy.
8.3.2 BRAF Mutations
Although RAS mutations are effective predictive marker of resistance, not all RAS WT patients respond to cetuximab and panitumumab. Thus, research has turned to the serine-threonine protein kinase BRAF, the main effector of KRAS in EGFR signalling, which is mutated in 5–9% of CRC patients. Importantly, BRAF and KRAS mutations are usually mutually exclusive, therefore, do not tend to coexist in the same tumour. The activating BRAF V600E mutation represents the majority of BRAF mutations and confers poor prognosis to its patients (Di Nicolantonio et al. 2008). OS of mCRC patients harbouring BRAF mutations is about 8.8 months, compared to KRAS mutant of 14.4 months and KRAS WT 20.1 months. Furthermore, De Roock et al. showed that patients with BRAF V600E mutation had a significantly lower response rate to cetuximab than those with WT tumours (8.3% vs. 38.0%, OR = 0.15, P = 0.0012) in chemo-refractory mCRC patients (De Roock et al. 2010a, b). Several multicentre trials and meta-analyses have further confirmed that BRAF V600E mutation resulted in shorter PFS and OS when compared to BRAF WT tumours, indicating its contribution to resistance to anti-EGFR mAbs (Pietrantonio et al. 2015, Tveit et al. 2012, Therkildsen et al. 2014).
Similar to the presence of RAS mutations, BRAF V600E mutation can effectively predict patients that are unlikely to respond to anti-EGFR therapy. It is, therefore, advisable to know both RAS and BRAF status before administering EGFR -targeted therapies .
In this context, diverse strategies have been employed to overcome BRAF-mediated resistance to anti-EGFR therapy . An in vitro study of adding sorafenib (a multi-target small-molecule inhibitor with high affinity for BRAF) to anti-EGFR mAbs showed that even BRAF mutated cells can respond to cetuximab and panitumumab therapy when both inhibitors are used simultaneously (Di Nicolantonio et al. 2008). Based on these results, the combinatory therapy of BRAF and EGFR inhibitors was administered in BRAF-mutant CRC patients and resulted in increased response rates (Al-Marrawi et al. 2013). In addition to sorafenib, other compounds targeting either BRAF (such as vemurafenib) or its downstream effectors are in clinical development and could be exploited in combination with EGFR-targeted mAbs therapy. Thus, monotherapy, doublet and triplet combinations with drugs targeting the MAPK pathway have been tested in BRAF-mutant CRC. Results from vemurafenib monotherapy were disappointing when compared to the clinical activity seen in melanoma, with a median PFS of 2.1 months and only two patients progression-free for more than six months (Kopetz et al. 2015). In contrast to melanoma, CRC express high levels of activated EGFR which reactivate the MAPK pathway after BRAF inhibitor monotherapy (Prahallad et al. 2012, Corcoran et al. 2012). Based on the observed therapy resistance via a feedback activation of EGFR signalling, the BASKET trial was amended to include the assessment of the safety and efficacy of vemurafenib when combined with cetuximab, and showed improved results (median PFS of 3.7 months and OS of 7.1 months) in a heavily pre-treated patient population (Yaeger et al. 2015). Similar results have been seen when combining other BRAF inhibitors , dabrafenib and panitumumab (PFS of 3.5 months) (Atreya et al. 2015, Van Cutsem et al. 2015a, b), as well as encorafenib and cetuximab (RR 23.1%, PFS of 3.7 months) (Gomez-Roca et al. 2014, Van Geel et al. 2017). Phase II results for the latter have been presented with a median PFS of 4.2 months and an ORR of 22% (Tabernero et al. 2016). Chemotherapy was also combined with BRAF and EGFR inhibition in a phase II trial combining irinotecan, cetuximab and vemurafenib. A total of 106 patients were enrolled and results show an increasing PFS to 4.3 months with the addition of vemurafenib compared to the control arm (2.0 months) (Kopetz et al. 2017). Finally, BRAF inhibition can not only also induce EGFR-dependent MAPK reactivation but also PI3K modulation so that triple combinations targeting these pathways have been studied and shown improved results. The MEK116833 trial combining trametinib, panitumumab and dabrafenib included 24 patients which received full dose, with an ORR of 21% and a median PFS of 4.1 months; OS was 9.1 months (Corcoran et al. 2015). A randomised phase II trial which combined encorafenib, cetuximab and alpelisib (a PI3K inhibitor ) revealed a median PFS of 5.4 months in an interim analysis with an ORR of 27% (Tabernero et al. 2016). More recently, the BEACON CRC phase 3 study assessed the safety and efficacy of the combination of the BRAF inhibitor encorafenib, plus MEK inhibitor binimetinib, plus anti-EGFR antibody cetuximab in patients with BRAF mutant CRC after 1 or 2 prior regimens. The confirmed ORR was 41%, with 1 complete and 11 partial responses. In addition, 9 patients had prolonged stable disease up to 9.3 months and CEA/CA19-9 decreased in 96% and 82% of these patients, respectively (Huijberts et al. 2017).
Overall, given that KRAS and BRAF mutations are usually mutually exclusive and highly frequent, together they allow the identification of the majority of non-responder patients, avoiding unnecessary exposure of these patients to ineffective treatments and selecting them for alternative therapeutic options.
8.3.3 Other Putative Players
8.3.3.1 PIK3CA Gene and PTEN Expression
KRAS and BRAF WT status is not enough to define all anti-EGFR-sensitive patients. The EGFR receptor also signals through the PI3K -AKT pathway resulting in tumour cell proliferation and survival (Rommel and Fruman 2014). PIK3CA gene encodes the p110α protein kinase, which is the catalytic subunit of class I PI3Ks. Furthermore, besides direct activation of the PI3K-AKT pathway by EGFR, activated KRAS protein can further bind and directly activate the p110α PI3K protein. Mutations in PIK3CA are reported in approximately 10–18% of mCRC patients and can coexist with either KRAS and BRAF mutations (Nosho et al. 2008). Therefore, several studies have evaluated the predictive value of PIK3CA mutations in resistance to anti-EGFR therapies. Retrospective studies of cetuximab treatments in chemo-refractory mCRC patients have revealed that, in KRAS WT patients, PIK3CA mutations in exon 20 lead to worse outcome shown by lower response rates (0.0% vs. 36.8%; 95% CI 0.00–0.89; P = 0.029) than PIK3CA WT patients (De Roock et al. 2010a, b). Interestingly, mutations in exon 9 (60–65% of PIK3CA mutations) of PIK3CA had no effect on response rates, survival and prognosis. In two further meta-analysis studies on retrospective cohorts, PIK3CA exon 20 mutations, but not exon 9, were associated with absence of response, lower PFS and OS to anti-EGFR mAbs (Sartore-Bianchi et al. 2009; Mao et al. 2012). In vitro studies unravelled different intracellular mechanisms of action: exon 9 mutations release p110α from p85-induced inhibition in a KRAS -GTP dependent way, whereas exon 20 mutations activate the kinase domain, independently of interactions with KRAS. This fact may justify different effects of both mutations in responses to mAbs (Zhao and Vogt 2008; Zhao and Vogt 2010).
Overall, without larger prospective studies, it is still difficult to evaluate the precise role of PIK3CA mutations with respect to the response to EGFR -targeted therapies , especially given that they are mostly found co-occurring with KRAS or BRAF mutations.
PTEN (phosphatase and tensin homologue ) is another potential marker of response to anti-EGFR therapy, given its negative role on the PI3K -AKT signalling pathway. PTEN inhibits the PI3K pathway through its lipid phosphatase activity, behaving in this way as a tumour suppressor protein (Cully et al. 2006). In mCRC, PTEN activity is reduced in about 20–40% of tumours through either PTEN gene silencing ( via promoter hypermethylation or loss of heterozygosity) or mutations (Molinari and Frattini 2014). This loss of PTEN activity resulted in constitutive activation of the PI3K-AKT signalling pathway leading to tumour cell proliferation and survival. Studies on the association between the PTEN status and the response to mAbs are controversial and inconclusive. Frattini et al. have studied a cohort of cetuximab and irinotecan-treated patients and found that lower levels of PTEN were predictive of resistance (Frattini et al. 2007), whereas Laurent-Puig et al. showed no significant differences in terms of RR, PFS and OS in a larger cohort of patients (Laurent-Puig et al. 2009). Moreover, in the NCIC trial, where 572 patients with pretreated mCRC were randomly assigned to receive cetuximab or best supportive care, no statistical significance was found with respect to loss of PTEN and the clinical outcome of patients treated with cetuximab (Karapetis et al. 2014). Nevertheless, two other studies corroborate the fact that loss of PTEN expression (measured by immunohistochemistry) is associated with decreased RR, PFS and OS in mCRC patients treated with anti-EGFR therapy (Loupakis et al. 2009; Sartore-Bianchi et al. 2009). Finally, a recent meta-analysis confirmed that PTEN loss was significantly associated with lack of benefit to mAbs treatment in RAS WT patients. However, this study concluded that the predictive power of BRAF and PIK3CA mutations were stronger than that of PTEN levels (Yang et al. 2013a, b).
Overall, given that technically the assessment of PTEN expression levels by immunohistochemical methods lack standardization, that PTEN alterations co-occur with RAS mutations, and that discordant levels of PTEN expression are seen between primary tumour and metastasis , the loss of PTEN expression cannot be seen at present as a reliable predictive biomarker of response to EGFR -targeted mAbs.
Nevertheless, targeted treatments against PI3K or its downstream effectors such as mTOR and AKT in preclinical models suggest great therapeutic potential when combined with receptor tyrosine kinase inhibitors (Kim et al. 2017). A clinical trial evaluating the combination of mTOR inhibitor everolimus with panitumumab and irinotecan in second-line mCRC patients showed better RR when compared to the treatment without everolimus, in RAS WT patients (Townsend et al. 2018). Another combination that is presently exploited in clinical trials is that of PIK3CA/mTOR inhibitors and MEK inhibitors (Andersen et al. 2015; Temraz et al. 2015).
Despite these promising results, larger prospective studies are needed before the role of PIK3CA mutations and PTEN expression levels in the mechanism of resistance, and their potential predictive value in anti-EGFR therapies can be concluded.
8.3.3.2 JAK/STAT Signalling Pathway
The Janus family of tyrosine kinases (JAK) and the signal transducer and activator of transcription (STAT) family are involved in cytokine receptor signalling as important mediators of cell survival, proliferation, differentiation, and apoptosis (Rawlings 2004). There have been pieces of evidence supporting a role of STAT family member STAT3 in resistance to the EGFR kinase inhibitor gefitinib in cells (Li et al. 2015). Furthermore, this work has shown that inhibition of STAT3 activity by Stattic (STAT3-inhibitor) sensitizes CRC cells to gefitinib treatments. In an independent work, co-treatments of gefitinib with the JAK/STAT3 inhibitor cucurbitacin B led to increased antitumour activity in CRC cells (Yar Saglam et al. 2015). These results indicate that blocking EGFR signalling is more effective in combination with inhibitors of JAK/STAT3, suggesting a putative role of this pathway in the mechanism of resistance to anti-EGFR therapies. However, further studies are required to fully confirm the role of STAT3 in the mechanism of resistance to mAbs targeting EGFR.
8.3.3.3 Others Components
Other mechanisms have been implicated in the resistance to EGFR -targeted therapy in mCRC. Expression of vascular endothelial growth factor 1 (VEGF-1) or its receptor (VEGFR) has been associated to resistance to cetuximab in preclinical models and in patients with mCRC (Bianco et al. 2008). Furthermore, inflammatory markers such as interleukin-8 (IL8) and cyclooxygenases-2 (COX2), as well as the cell cycle regulator cyclin D1 were also shown important for the outcome of patients receiving anti-EGFR therapy. Vallböhmer et al. reported that a combination of low levels of COX2, EGFR and IL8 was a good prognostic marker for patients when compared to high levels of expression of these three genes, with an OS of 13.5 months vs. 2.3 months, respectively (Vallböhmer et al. 2005). Nuclear translocation of EGFR was also identified as a possible marker for resistance dependent on Src family kinases (Li et al. 2009). Nuclear EGFR is associated with transcription of cyclin D1 and consequently proliferation of tumour cells. Expression of the transcription factor nuclear factor κB has also been linked with resistance to cetuximab (Scartozzi et al. 2007). Finally, in in vitro models epithelial-to-mesenchymal transition (EMT) has also been pointed out as a mechanism involved in resistance to anti-EGFR inhibitors , as CRC mesenchymal-like cells were found sevenfold more resistant than epithelial-like cells (Buck et al. 2007). Although interesting, data regarding the previously mentioned proteins is limited and lack further validation. A comprehensive understanding of their contribution to mechanisms involved in cetuximab and panitumumab resistance is desirable and holds the promise for the generation of novel therapeutic opportunities for the treatment of CRC.
8.4 Acquired Resistance to Anti-EGFR Therapy
8.4.1 EGFR Mutations
EGFR mutations are extremely rare in CRC but have been described associated with acquired resistance to mAb treatment (approximately in 20% of patients treated with cetuximab and 1% of patients treated with panitumumab). Montagut et al. have identified EGFR S492R mutation in cell lines that acquired resistance to cetuximab and confirmed these data in patients who relapsed after cetuximab treatment (Montagut et al. 2012). This mutation is located in the extracellular domain of the receptor and prevents binding of the cetuximab antibody, however, does not seem to affect panitumumab binding. Indeed, one patient who had relapsed under cetuximab and harboured the S492R mutation, responded to panitumumab, suggesting a clinical option to overcome cetuximab resistance in these patients. Other mutations occurring in the extracellular domain of EGFR (R451C, S464L, G465R, I491M and K467T) were identified in patients who had relapsed under cetuximab treatment or in cell lines that acquired resistance to cetuximab (Arena et al. 2015). From these mutations, R451C and K467T do not prevent binding of panitumumab to the receptor. This fact resulted in the generation of new EGFR inhibitors consisting of a mixture of more than one mAb that target different epitopes located in the extracellular domain of EGFR. Sym004 (mixture of two different mAbs) and MM-151 (mixture of three fully human IgG1 antibodies) are new treatment options presently under clinical evaluation (Pedersen et al. 2010, Kearns et al. 2015). Phase I clinical trials of both compounds demonstrated their safety. In 42 mCRC patients who had acquired resistance to anti-EGFR therapy, Sym004 treatments induced about 44% of tumour shrinkage and partial response or stable disease for the other patients. In a similar way, MM-151 also showed long-lasting disease control of patients treated with MM-151 in combination with irinotecan.
8.4.2 RAS/RAF Signalling Pathway
As one of the most important signalling pathways downstream of EGFR , the RAS-RAF-MAPK cascade is also one of the most important mechanisms associated with secondary resistance to mAbs (50–80% of cases). Thus, pre-clinical and clinical studies have identified the occurrence of KRAS mutations in metastases that acquired resistance to EGFR inhibitors . Bouchahda et al. reported the first case of CRC liver metastasis harbouring KRAS mutations in a patient who had progressed under cetuximab therapy, although primary and metastatic tumours were KRAS WT before treatment (Bouchahda et al. 2010). In a further study, Misale et al. showed that six out of ten patients that were KRAS WT before the treatment were detected with KRAS mutations in their plasma samples during cetuximab treatment (Misale et al. 2012). The same study, also showed one case of KRAS amplification (an infrequent event in CRC) in a patient who relapsed after cetuximab treatment, showing that either mutations or amplifications could be associated with acquired resistance to mAbs. In addition to the KRAS gene, alterations in NRAS and BRAF were also associated with secondary resistance to EGFR -targeted therapies in pre-clinical models (Misale et al. 2012). Altogether, the occurrence of RAS mutations in relapsed tumours was found to derive from an expansion of pre-existing clones that propagated under the selection pressure of anti-EGFR treatment, rather than from novel spontaneous mutations (Diaz et al. 2012).
8.4.3 HER2/HER3 Expression
Amplification of other receptor tyrosine kinases of the ErbB family has been described as an acquired resistance mechanism to anti-EGFR therapies. Bertotti et al. showed that HER2 gene amplification was correlated with responses to cetuximab in a patient-derived xenografts mouse model (Bertotti et al. 2011). The authors observed that HER2 amplification was only present in 2–3% of KRAS WT patients before treatment, however, in 36% of resistant tumours after cetuximab treatments. They further showed that combination of lapatinib (a small molecule inhibitor of both EGFR and HER2 receptors) with cetuximab or pertuzumab (a monoclonal antibody that inhibits the dimerization of HER2 with other HER receptors) was efficient in a subset of cetuximab-resistant HER2-amplified mCRC xenografts. Based on these findings, the HERACLES phase II was designed to assess the RR of trastuzumab (mAbs targeting HER2) combined with either lapatinib or pertuzumab, in KRAS exon 2 WT and HER2 amplified mCRC patients (Sartore-Bianchi et al. 2016). The initial results concerning the trastuzumab and lapatinib combination showed that 30% of patients achieved an objective response with a median duration of response of 38 weeks. Median PFS was 21 weeks and median OS was 46 weeks. Importantly, these results indicate that HER2 is a good druggable target in mCRC.
It should be noted that HER2 gene amplification was also associated with intrinsic resistance to anti-EGFR therapy, however, given its extremely low frequency in CRC (about 2% of cases), its role in primary resistance seems to be minor (Hynes and Lane 2005).
Additionally to HER2, HER3 has also been described to have a role in the resistance mechanism to EGFR -targeted therapies . In a cohort of mCRC patients treated with irinotecan and cetuximab, HER3 overexpression was associated with lower PFS and OS (Scartozzi et al. 2011). Moreover, HER3 is found mutated in 11% of CRC patients and owns oncogenic activity (Jaiswal et al. 2013). MEHD7945A is a humanized IgG1 mAbs with dual HER3/EGFR activity. This compound has achieved promising results in a phase I trial in patients with pretreated mCRC. However, phase II randomized trial showed no benefit for MEHD7945A plus FOLFIRI when compared to cetuximab plus FOLFIRI in KRAS WT chemo-refractory patients (Van Cutsem et al. 2014).
8.4.4 MET Receptor Expresssion
MET is a tyrosine kinase receptor for the ligand Hepatocyte Growth Factor (HGF), which upon activation leads to several cellular processes such as cell proliferation, invasion, apoptosis and survival (Organ and Tsao 2011). Several pieces of evidence suggest an involvement of the MET pathway (through MET amplification or increased HGF expression) in the mechanism of both primary and secondary resistance to EGFR mAbs in KRAS WT patients (Krumbach et al. 2011). Although in primary samples amplification of MET was only reported in 2% of cases, interaction between EGFR and MET was seen upon activation with TGF-alpha and correlated with acquired resistance to cetuximab in cells (Troiani et al. 2013). Treatment of those cells with MET inhibitor restored cetuximab sensitivity. Furthermore, in vivo studies showed an increased level of MET amplification in cetuximab-resistant patients WT for RAS, BRAF , PIK3CA and HER2, whereas amplification had not been seen in pre-treatment tumours (Bardelli et al. 2013). Finally, in a randomized phase II clinical trial of chemo-refractory KRAS WT anti-EGFR naïve patients, the combination of anti-HGF mAbs and panitumumab led to higher response rates and a trend for better outcome in the patient population with MET overexpression (Van Cutsem et al. 2014). A phase I trial assessing safety of cabozantinib (a small molecule MET inhibitor ) plus panitumumab in chemo-refractory KRAS WT patients is ongoing (Jia et al. 2018).
8.5 Conclusions
The high complexity of mechanisms of resistance to anti-EGFR mAbs, make this therapy only efficient in a restricted CRC patient population. Presently, resistance to EGFR -targeted therapies is known to be mediated by constitutive activation of EGFR signalling cascades through deregulation of the receptor itself or downstream components of the RAS/RAF, PI3K /PTEN and JAK/STAT pathways, as well as, from the activation of alternative tyrosine kinase receptor such HER2 and MET. Despite intensive research done over the last 10 years, RAS mutations are effectively the only approved biomarker of response in clinical practice. More clinical and translational studies are required in order to increase our knowledge on the mechanisms behind anti-EGFR therapy resistance .
Recent efforts to segregate CRC tumours into subgroups based on their biology and gene expression patterns resulted in an unified classification which categorizes the majority of tumours into four groups called consensus molecular subtypes (CMS1–4) (Guinney et al. 2015). CMS1 (immune, 14% of cases) is enriched for microsatellite instable (MSI) tumours that display BRAF mutations, hypermethylation of CpG islands (CIMP), and immune infiltration and activation. CMS2 (canonical, 37% of cases) reflects the classical adenoma-to-carcinoma sequence, encompassing typical WNT/MYC-driven tumours with epithelial characteristics and high somatic copy number alterations (SCNA), whereas CMS3 (metabolic, 13% of cases) is enriched for KRAS -mutated tumours (although KRAS mutations are present in all CMS subtypes) with activation of metabolic pathways. Finally, CMS4 (mesenchymal, 23% of cases) has mesenchymal features, shows high SCNA, stromal content and activation of TGF-β and VEGFR pathways. Clear clinical distinction is also evident with poor prognosis for CMS4 and a relatively good prognosis for CMS1 (Thanki et al. 2017). In this context, Sveen et al. have lately shown that the CMS2 subtype is predicted to respond to EGFR inhibition, whereas tumours with a metabolic and mesenchymal-like phenotype seem strongly resistant, independently of KRAS and BRAF mutation status (Sveen et al. 2018).
Overall, the field is moving towards a more comprehensive picture of the processes involved in therapy resistance , which will certainly lead to the recognition of alternative or combinatory treatments, providing more benefit to patients and sparing unnecessary treatments.
References
Al-Marrawi MY, Saroya BS, Brennan MC, Yang Z, Dykes TM, El-Deiry WS (2013) Off-label use of cetuximab plus sorafenib and panitumumab plus regorafenib to personalize therapy for a patient with V600E BRAF-mutant metastatic colon cancer. Cancer Biol Ther 14:703–710. https://doi.org/10.4161/cbt.25191
Andersen NJ, Boguslawski EB, Kuk CY, Chambers CM, Duesbery NS (2015) Combined inhibition of MEK and mTOR has a synergic effect on angiosarcoma tumorgrafts. Int J Oncol 47:71–80. https://doi.org/10.3892/ijo.2015.2989
Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S, González I, Iglesias M, Gavilan E, Corti G, Hobor S, Crisafulli G, Salido M, Sánchez J, Dalmases A, Bellmunt J, De Fabritiis G, Rovira A, Di Nicolantonio F, Albanell J, Bardelli A, Montagut C (2015) Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res 21:2157–2166. https://doi.org/10.1158/1078-0432.CCR-14-2821
Atreya CE, Van Cutsem E, Bendell JC, Andre T, Schellens JHM, Gordon MS, Jackson McRee A, O’Dwyer PJ, Muro K, Tabernero J, van Geel R, Sidhu R, Greger JG, Rangwala FA, Motw M, Wu Y, Orford KW, Corcoran RB (2015) Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). J Clin Pathol 33(15_suppl):103. https://doi.org/10.1200/jco.2015.33.15_suppl.103
Barber TD, Vogelstein B, Kinzler KW, Velculescu VE (2004) Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med 351:2883. https://doi.org/10.1056/NEJM200412303512724
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, Apicella M, Migliardi G, Galimi F, Lauricella C, Zanon C, Perera T, Veronese S, Corti G, Amatu A, Gambacorta M, Diaz LA, Sausen M, Velculescu VE, Comoglio P, Trusolino L, Di Nicolantonio F, Giordano S, Siena S (2013) Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 3:658–673. https://doi.org/10.1158/2159-8290.CD-12-0558
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, di Nicolantonio F, Buscarino M, Petti C, Ribero D, Russolillo N, Muratore A, Massucco P, Pisacane A, Molinaro L, Valtorta E, Sartore-Bianchi A, Risio M, Capussotti L, Gambacorta M, Siena S, Medico E, Sapino A, Marsoni S, Comoglio PM, Bardelli A, Trusolino L (2011) A molecularly annotated platform of patient- derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1:508–523. https://doi.org/10.1158/2159-8290.CD-11-0109
Bianco R, Rosa R, Damiano V, Daniele G, Gelardi T, Garofalo S, Tarallo V, De Falco S, Melisi D, Benelli R, Albini A, Ryan A, Ciardiello F, Tortora G (2008) Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res 14:5069–5080. https://doi.org/10.1158/1078-0432.CCR-07-4905
Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M, Koralewski P (2011) Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol 22:1535–1546. https://doi.org/10.1093/annonc/mdq632
Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, De Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P (2009) Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 27:663–671. https://doi.org/10.1200/JCO.2008.20.8397
Bos JL (1989) ras oncogenes in human cancer: a review. Cancer Res 49:4682–4689
Bouchahda M, Karaboué A, Saffroy R, Innominato P, Gorden L, Guettier C, Adam R, Lévi F (2010) Acquired KRAS mutations during progression of colorectal cancer metastases: possible implications for therapy and prognosis. Cancer Chemother Pharmacol 66:605–609. https://doi.org/10.1007/s00280-010-1298-9
Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, Iwata KK, Gibson NW, Cagnoni P, Haley JD (2007) Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther 6:532–541. https://doi.org/10.1158/1535-7163.MCT-06-0462
Campanella C, Mottolese M, Cianciulli A, Torsello A, Merola R, Sperduti I, Melucci E, Conti S, Diodoro MG, Zeuli M, Paoletti G, Cognetti F, Garufi C (2010) Epidermal growth factor receptor gene copy number in 101 advanced colorectal cancer patients treated with chemotherapy plus cetuximab. J Transl Med 8:36. https://doi.org/10.1186/1479-5876-8-36
Cappuzzo F, Finocchiaro G, Rossi E, Jänne PA, Carnaghi C, Calandri C, Bencardino K, Ligorio C, Ciardiello F, Pressiani T, Destro A, Roncalli M, Crino L, Franklin WA, Santoro A, Varella-Garcia M (2008) EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol 19:717–723. https://doi.org/10.1093/annonc/mdm492
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, John Simes R, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au H-J, Langer C, Moore MJ (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359:1757–1765. https://doi.org/10.1056/NEJMoa1215817
Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L, Klimstra DS, Fridman D, Kelsen DP, Saltz LB (2005) Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 23:1803–1810. https://doi.org/10.1200/JCO.2005.08.037
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358:1160–1174. https://doi.org/10.1056/NEJMra0707704
Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, Hamid O, Messersmith WA, Daud A, Kurzrock R, Pierobon M, Sun P, Cunningham E, Little S, Orford K, Motwani M, Bai Y, Patel K, Venook AP, Kopetz S (2015) Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-Mutant colorectal cancer. J Clin Oncol 33:4023–4031. https://doi.org/10.1200/JCO.2015.63.2471
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della PP, Dias-Santagata D, Hung KE, Flaherty KT, Piris A, Wargo JA, Settleman J, Mino-Kenudson M, Engelman JA (2012) EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2:227–235. https://doi.org/10.1158/2159-8290.CD-11-0341
Costa-Cabral S, Brough R, Konde A, Aarts M, Campbel J, Marinari E, Riffell J, Bardelli A, Torrance C, Lord CJ, Ashworth A (2016) CDK1 is a synthetic lethal target for KRAS mutant tumours. PLoS One 11:e0149099. https://doi.org/10.1371/journal.pone.0149099
Cully M, You H, Levine AJ, Mak TW (2006) Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 6:184–192. https://doi.org/10.1038/nrc1819
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351:337–345. https://doi.org/10.1056/NEJMoa033025
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010a) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11:753–762. https://doi.org/10.1016/S1470-2045(10)70130-3
De Roock W, Jonker DJ, Di NF, Sartore-bianchi A, Simes J (2010b) Association of KRAS p. G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. J Am Med Assoc 304:1812–1820. https://doi.org/10.1001/jama.2010.1535
Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B (2012) The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486:537–540. https://doi.org/10.1038/nature11219
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2008) Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 26:5705–5712. https://doi.org/10.1200/JCO.2008.18.0786
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Oliner KS, Tian Y, Xu F, Sidhu R (2014) Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol 25:1346–1355. https://doi.org/10.1093/annonc/mdu141
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386. https://doi.org/10.1002/ijc.29210
Fernández-Medarde A, Santos E (2011) Ras in cancer and developmental diseases. Genes and Cancer 2:344–358. https://doi.org/10.1177/1947601911411084
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L (2007) PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 97:1139–1145. https://doi.org/10.1038/sj.bjc.6604009
Gazdar A (2009) Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 28:1–14. https://doi.org/10.1038/onc.2009.198
Gomez-Roca CA, Delord J, Robert C, Hidalgo M, von Moos R, Arance A, Elez E, Michel D, Seroutou A, Demuth T, Tabernero J (2014) 535Pencorafenib (Lgx818), an oral braf inhibitor, in patients (Pts) with Braf V600E metastatic colorectal cancer (Mcrc): results of dose expansion in an open-label, phase 1 study. Ann Oncol 25:iv182–iv183. https://doi.org/10.1093/annonc/mdu333.38
Guinney J, Dienstmann R, Wang X, De Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E, Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 21:1350–1356. https://doi.org/10.1038/nm.3967
Heinemann V, Von Weikersthal LF, Decker T, Kiani A, Vehling-kaiser U, Scholz M, Müller S, Link H, Niederle N, Rost A, Höffk H, Moehler M, Lindig RU, Modest DP, Rossius L, Kirchner T, Jung A, Stintzing S, Jena U (2014) FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol 15:1065–1075. https://doi.org/10.1016/S1470-2045(14)70330-4
Huijberts S, Schellens JHM, Fakih M, Peeters M, Kopetz S, Grothey A, Van Cutsem E, Wasan HS, Desai J, Ciardiello F, Yoshino T, Maharry K, Gollerkeri A, Tabernero J (2017) BEACON CRC (binimetinib [BINI], encorafenib [ENCO], and cetuximab [CTX] combined to treat BRAF-mutant metastatic colorectal cancer [mCRC]): a multicenter, rando.mized, open-label, three-arm phase III study of ENCO plus CTX plus or minus BINI vs irinotecan. J Clin Oncol 35(15 Suppl). https://doi.org/10.1200/JCO.2017.35.15_suppl.TPS3622
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5:341–354. https://doi.org/10.1038/nrc1609
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, Humblet Y, Pénault-Llorca F, De Hertogh G, Laurent-Puig P, Van Cutsem E, Tejpar S (2009) Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 27:5068–5074. https://doi.org/10.1200/JCO.2008.21.3744
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, Janakiraman V, Scholz RP, Bowman KK, Lorenzo M, Li H, Wu J, Yuan W, Peters BA, Kan Z, Stinson J, Mak M, Modrusan Z, Eigenbrot C, Firestein R, Stern HM, Rajalingam K, Schaefer G, Merchant MA, Sliwkowski MX, deSauvage FJ, Seshagiri S (2013) Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23:603–617. https://doi.org/10.1016/j.ccr.2013.04.012
Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, Feng J, Chen JH, Li S, Li S, Long YO, Thach C, Liu Y, Zarieh A, Ely T, Kucharski JM, Kessler LV, Wu T, Yu K, Wang Y, Yao Y, Deng X, Zarrinkar PP, Brehmer D, Dhanak D, Lorenzi MV, Hu-Lowe D, Patricelli MP, Ren P, Liu Y (2018) Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172:578–589.e17. https://doi.org/10.1016/j.cell.2018.01.006
Jia J, Niedzwiecki D, Uronis HE, Morse M, Zafar Y, Hsu SD, Bolch E, Nagy RJ, Lanman RB, Talasaz AA, Haley S, Nixon AB, Strickler JH (2018) A phase I/II trial of cabozantinib (C) with or without panitumumab (P) in patients (pts) with RAS wild-type (WT) metastatic colorectal cancer (mCRC): clinical outcomes in pts with MET amplification (amp) detected in blood. J Clin Oncol 36(15 Suppl):3555
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au H-J, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ (2007) Cetuximab for the treatment of colorectal cancer. N Engl J Med 357:2040–2048. https://doi.org/10.1056/NEJMoa071834
Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O’Callaghan CJ, Marginean C, Zalcberg JR, Simes J, Moore MJ, Tebbutt NC, Price TJ, Shapiro JD, Pavlakis N, Gibbs P, Van Hazel GA, Lee U, Haq R, Virk S, Tu D, Lorimer IAJ (2014) PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer-results from NCIC CTG/AGITG CO.17. Clin Cancer Res 20:744–753. https://doi.org/10.1158/1078-0432.CCR-13-0606
Kearns JD, Bukhalid R, Sevecka M, Tan G, Gerami-Moayed N, Werner SL, Kohli N, Burenkova O, Sloss CM, King AM, Fitzgerald JB, Nielsen UB, Wolf BB (2015) Enhanced targeting of the EGFR network with MM-151, an oligoclonal anti-egfr antibody therapeutic. Mol Cancer Ther 14:1625–1636. https://doi.org/10.1158/1535-7163.MCT-14-0772
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, Burris HA, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ (2007) Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 25:3230–3237. https://doi.org/10.1200/JCO.2006.10.5437
Kim JS, Kim JE, Kim K, Lee J, Park JO, Lim HY, Park YS, Kang WK, Kim ST (2017) The impact of cetuximab plus AKT- or mTOR-inhibitor in a patient-derived colon cancer cell model with wild-type RAS and PIK3CA mutation. J Cancer 8:2713–2719. https://doi.org/10.7150/jca.19458
Kohl NE, Omer CA, Davide JP, Hamilton K, Koblan KS, Kral AM, Miller PJ, Mosser SD, O’neill TJ, Rands E, Schaber MD, Gibbs JB, Oliff A, Conner MW, Anthony NJ, Jane Desolms S, Giuliani EA, Gomez RP, Graham SL, Hartman GD, Anthony NJ (1995) Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med 1:792–797. https://doi.org/10.1038/nm0895-792
Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, Issa JPJ, Gibbs P, James B, Powis G, Nolop KB, Bhattacharya S, Saltz L (2015) Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 33:4032–4038. https://doi.org/10.1200/JCO.2015.63.2497
Kopetz S, McDonough SL, Morris VK, Lenz HJ, Magliocco AM, Atreya CE, Diaz LA, Allegra CJ, Wang SE, Hanyoung Lieu C, Eckhardt SG, Semrad TJ, Kaberle K, Guthrie KA, Hochster HS (2017) Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer. J Clin Oncol 35(4 Suppl):520. https://doi.org/10.1200/JCO.2017.35.4_suppl.520
Krumbach R, Schüler J, Hofmann M, Giesemann T, Fiebig HH, Beckers T (2011) Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer 47:1231–1243. https://doi.org/10.1016/j.ejca.2010.12.019
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, Bouché O, Reid J, Stone S, Penault-Llorca F (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27:5924–5930. https://doi.org/10.1200/JCO.2008.21.6796
Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL (2009) Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 28:3801–3813. https://doi.org/10.1038/onc.2009.234
Li Q, Zhang D, Chen X, He L, Li T, Xu X, Li M (2015) Nuclear PKM2 contributes to gefitinib resistance via upregulation of STAT3 activation in colorectal cancer. Sci Rep 5:1–12. https://doi.org/10.1038/srep16082
Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S (2008) Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol 9:962–972. https://doi.org/10.1016/S1470-2045(08)70206-7
Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, Canestrari E, Funel N, Schiavon G, Petrini I, Magnani M, Tonini G, Campani D, Floriani I, Cascinu S, Falcone A (2009) PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 27:2622–2629. https://doi.org/10.1200/JCO.2008.20.2796
Macdonald JS, McCoy S, Whitehead RP, Iqbal S, Wade JL, Giguere JK, Abbruzzese JL (2005) A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs 23:485–487. https://doi.org/10.1007/s10637-005-2908-y
Malumbres M, Barbacid M (2003) RAS oncogenes: the first 30 years. Nat Rev Cancer 3:459–465. https://doi.org/10.1038/nrc1097
Mao C, Yang ZY, Hu XF, Chen Q, Tang JL (2012) PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol 23:1518–1525. https://doi.org/10.1093/annonc/mdr464
Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A, Risio M, Capussotti L, Marsoni S, Di Nicolantonio F, Bardelli A, Comoglio PM, Trusolino L, Bertotti A (2012) Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res 18:2515–2525. https://doi.org/10.1158/1078-0432.CCR-11-2683
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A (2012) Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 486:532–536. https://doi.org/10.1038/nature11156
Molinari F, Frattini M (2014) Functions and regulation of the PTEN gene in colorectal cancer. Front Oncol 3:1–8. https://doi.org/10.3389/fonc.2013.00326
Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, Minoche A, Somasekar S, Serrano S, Himmelbauer H, Bellmunt J, Rovira A, Settleman J, Bosch F, Albanell J (2012) Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med 18:221–223. https://doi.org/10.1038/nm.2609
Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A (2005) Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 6:279–286. https://doi.org/10.1016/S1470-2045(05)70102-9
Nosho K, Kawasaki T, Longtine JA, Fuchs CS, Ohnishi M, Suemoto Y, Kirkner GJ, Zepf D, Yan L, Ogino S (2008) PIK3CA autation in colorectal cancer: relationship with genetic and epigenetic Alterations. Neoplasia 10:534–541. https://doi.org/10.1593/neo.08336
Oda K, Matsuoka Y, Funahashi A, Kitano H (2005) A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol 1:E1–E17. https://doi.org/10.1038/msb4100014
Organ SL, Tsao MS (2011) An overview of the c-MET signaling pathway. Ther Adv Med Oncol 3:S7–S19. https://doi.org/10.1177/1758834011422556
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503:548–551. https://doi.org/10.1038/nature12796
Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y (2016) Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 6:316–329. https://doi.org/10.1158/2159-8290.CD-15-1105
Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, Kragh M (2010) Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res 70:588–597. https://doi.org/10.1158/0008-5472.CAN-09-1417
Peeters M, Douillard JY, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J (2013) Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol 31:759–765. https://doi.org/10.1200/JCO.2012.45.1492
Perez EA, Hurvitz SA, Amler LC, Mundt KE, Ng V, Guardino E, Gianni L (2014) Relationship between HER2 expression and efficacy with first-line trastuzumab emtansine compared with trastuzumab plus docetaxel in TDM4450g: a randomized phase II study of patients with previously untreated HER2-positive metastatic breast cancer. Breast Cancer Res 16:R50. https://doi.org/10.1186/bcr3661
Personeni N, Fieuws S, Piessevaux H, De Hertogh G, De Schutter J, Biesmans B, De Roock W, Capoen A, Debiec-Rychter M, Van LJL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S (2008) Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin Cancer Res 14:5869–5876. https://doi.org/10.1158/1078-0432.CCR-08-0449
Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, Cabiddu M, Iacovelli R, Bossi I, Lonati V, Ghilardi M, De Braud F, Barni S (2015) Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer 51:587–594. https://doi.org/10.1016/j.ejca.2015.01.054
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483:100–104. https://doi.org/10.1038/nature10868
Rawlings JS (2004) The JAK/STAT signaling pathway. J Cell Sci 117:1281–1283. https://doi.org/10.1242/jcs.00963
Rommel C, Fruman DA (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13:140–156. https://doi.org/10.1038/nrd4204.PI3K
Sartore-Bianchi A, Fieuws S, Veronese S, Moroni M, Personeni N, Frattini M, Torri V, Cappuzzo F, Vander Borght S, Martin V, Skokan M, Santoro A, Gambacorta M, Tejpar S, Varella-Garcia M, Siena S (2012) Standardisation of EGFR FISH in colorectal cancer: results of an international interlaboratory reproducibility ring study. J Clin Pathol 65:218–223. https://doi.org/10.1136/jclinpath-2011-200353
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2009) PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 69:1851–1857. https://doi.org/10.1158/0008-5472.CAN-08-2466
Sartore-Bianchi A, Moroni M, Veronese S, Carnaghi C, Bajetta E, Luppi G, Sobrero A, Barone C, Cascinu S, Colucci G, Cortesi E, Nichelatti M, Gambacorta M, Siena S (2007) Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol 25:3238–3245. https://doi.org/10.1200/JCO.2007.11.5956
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, Troiani T, Ciardiello F, Racca P, Bertotti A, Siravegna G, Torri V, Amatu A, Ghezzi S, Marrapese G, Palmeri L, Valtorta E, Cassingena A, Lauricella C, Vanzulli A, Regge D, Veronese S, Comoglio PM, Bardelli A, Marsoni S, Siena S (2016) Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol 17:738–746. https://doi.org/10.1016/S1470-2045(16)00150-9
Scartozzi M, Bearzi I, Pierantoni C, Mandolesi A, Loupakis F, Zaniboni A, Catalano V, Quadri A, Zorzi F, Berardi R, Biscotti T, Labianca R, Falcone A, Cascinu S (2007) Nuclear factor-kB tumor expression predicts response and survival in irinotecan-refractory metastatic colorectal cancer treated with cetuximab-irinotecan therapy. J Clin Oncol 25:3930–3935. https://doi.org/10.1200/JCO.2007.11.5022
Scartozzi M, Mandolesi A, Giampieri R, Bittoni A, Pierantoni C, Zaniboni A, Galizia E, Giustini L, Silva RR, Bisonni R, Berardi R, Biscotti T, Biagetti S, Bearzi I, Cascinu S (2011) The Role of HER-3 expression in the prediction of clinical outcome for advanced colorectal cancer patients receiving irinotecan and cetuximab. Oncologist 16:53–60. https://doi.org/10.1634/theoncologist.2010-0119
Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS (2015) Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 26:13–21. https://doi.org/10.1093/annonc/mdu378
Spano JP, Lagorce C, Atlan D, Milano G, Domont J, Benamouzig R, Attar A, Benichou J, Martin A, Morere JF, Raphael M, Penault-Llorca F, Breau JL, Fagard R, Khayat D, Wind P (2005) Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann Oncol 16:102–108. https://doi.org/10.1093/annonc/mdi006
Sveen A, Bruun J, Eide PW, Eilertsen IA, Ramirez L, Murumagi A, Arjama M, Danielsen SA, Kryeziu K, Elez E, Tabernero J, Guinney J, Palmer HG, Nesbakken A, Kallioniemi O, Dienstmann R, Lothe RA (2018) Colorectal cancer consensus molecular subtypes translated to preclinical models uncover potentially targetable cancer cell dependencies. Clin Cancer Res 24:794–806. https://doi.org/10.1158/1078-0432.CCR-17-1234
Tabernero J, van Geel R, Guren TK, Yaeger RD, Spreafico A, Faris JE, Yoshino T, Yamada Y, Kim TW, Bendell J, Schuler M, Lenz HJ, Eskens F, Desai J, Hochster H, Avsar E, Demuth T, Sandor V, Elez E, Schellens J (2016) Phase 2 results: encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J Clin Oncol 34(15_suppl):3544. https://doi.org/10.1200/JCO.2016.34.15_suppl.3544
Temraz S, Mukherji D, Shamseddine A (2015) Dual inhibition of MEK and PI3K pathway in KRAS and BRAF mutated colorectal cancers. Int J Mol Sci 16:22976–22988. https://doi.org/10.3390/ijms160922976
Thanki K, Edward Nicholls M, Gomez G, Gajjar A, James Senagore A, Rashidi L, Qiu S, Szabo C, Richard Hellmich M, Chao C (2017) Consensus molecular subtypes of colorectal cancer and their clinical implications. Int Biol Biomed J 3:105–111
Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M (2014) The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol (Madr) 53:852–864. https://doi.org/10.3109/0284186X.2014.895036
Townsend A, Tebbutt N, Karapetis C, Cooper P, Singhal N, Yeend S, Pirc L, Joshi R, Hardingham J, Price T (2018) Phase IB/II study of second-line therapy with panitumumab, irinotecan, and everolimus (PIE) in KRAS wild-type metastatic colorectal cancer. Clin Cancer Res 24:3838–3844. https://doi.org/10.1158/1078-0432.CCR-17-3590
Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A, Sforza V, Nappi A, De Palma R, D’Aiuto E, Berrino L, Bianco R, Ciardiello F (2013) Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res 19:6751–6765. https://doi.org/10.1158/1078-0432.CCR-13-0423
Tveit KM, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, Sigurdsson F, Kure E, Ikdahl T, Skovlund E, Fokstuen T, Hansen F, Hofsli E, Birkemeyer E, Johnsson A, Starkhammar H, Yilmaz MK, Keldsen N, Erdal AB, Dajani O, Dahl O, Christoffersen T (2012) Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol 30:1755–1762. https://doi.org/10.1200/JCO.2011.38.0915
Vallböhmer D, Zhang W, Gordon M, Yang DY, Yun J, Press OA, Rhodes KE, Iqbal S, Danenberg KD, Lenz HJ (2005) Molecular determinants of cetuximab efficacy. J Clin Oncol 23(16 Suppl):3551. https://doi.org/10.1200/jco.2005.23.16_suppl.3551
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon J-L, Van Laethem J-L, Maurel J, Richardson G, Wolf M, Amado RG (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25:1658–1664. https://doi.org/10.1200/JCO.2006.08.1620
Van Cutsem E, Köhne CH, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F (2011) Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 29:2011–2019. https://doi.org/10.1200/JCO.2010.33.5091
Van Cutsem E, Eng C, Nowara E, Swieboda-Sadlej A, Tebbutt NC, Mitchell E, Davidenko I, Stephenson J, Elez E, Prenen H, Deng H, Tang R, McCaffery I, Oliner KS, Chen L, Gansert J, Loh E, Smethurst D, Tabernero J (2014) Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clin Cancer Res 20:4240–4250. https://doi.org/10.1158/1078-0432.CCR-13-2752
Van Cutsem E, Lenz HJ, Köhne CH, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, Han Van Krieken J, Ciardiello F (2015a) Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 33:692–700. https://doi.org/10.1200/JCO.2014.59.4812
Van Cutsem E, Atreya C, André T, Bendell J, Schellens J, Gordon M, McRee A, Yoshino T, Muro K, ODwyer P, Tabernero J, Middleton G, Ducreux M, van Geel R, Sidhu R, Greger J, Rangwala F, Liu Y, Wu Y, Mookerjee B, Corcoran R (2015b) Updated Results of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). Ann Oncol 26(Suppl 4):iv119. https://doi.org/10.1093/annonc/mdv262.07
Van Geel RMJM, Tabernero J, Elez E, Bendell JC, Spreafico A, Schuler M, Yoshino T, Delord JP, Yamada Y, Lolkema MP, Faris JE, Eskens FALM, Sharma S, Yaeger R, Lenz HJ, Wainberg ZA, Avsar E, Chatterjee A, Jaeger S, Tan E, Maharry K, Demuth T, Schellens JHM (2017) A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov 7:610–619. https://doi.org/10.1158/2159-8290.CD-16-0795
Venook AP, Niedzwiecki D, Lenz H-J, Innocenti F, Fruth B, Meyerhardt JA, Schrag D, Greene C, O’Neil BH, Atkins JN, Berry S, Polite BN, O’Reilly EM, Goldberg RM, Hochster HS, Schilsky RL, Bertagnolli MM, El-Khoueiry AB, Watson P, Benson AB, Mulkerin DL, Mayer RJ, Blanke C (2017) Effect of first-line chemotherapy combined with cetuximab or bevacizumab on overall survival in patients with KRAS wild-type advanced or metastatic colorectal cancer. Jama 317:2392. https://doi.org/10.1001/jama.2017.7105
Welsch ME, Kaplan A, Chambers JM, Stokes ME, Bos PH, Zask A, Zhang Y, Sanchez-Martin M, Badgley MA, Huang CS, Tran TH, Akkiraju H, Brown LM, Nandakumar R, Cremers S, Yang WS, Tong L, Olive KP, Ferrando A, Stockwell BR (2017) Multivalent small-molecule pan-RAS inhibitors. Cell 168:878–889.e29. https://doi.org/10.1016/j.cell.2017.02.006
Wieduwilt M, Moasser M (2008) The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci 65:1566–1584. https://doi.org/10.1007/s00018-008-7440-8.The
Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, Lacouture ME, Vakiani E, Saltz LB (2015) Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res 21:1313–1320. https://doi.org/10.1158/1078-0432.CCR-14-2779
Yang X, Zhang X, Mortenson ED, Radkevich-Brown O, Wang Y, Fu YX (2013a) Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Mol Ther 21:91–100. https://doi.org/10.1038/mt.2012.184
Yang Z-Y, Wu X-Y, Huang Y-F, Di M-Y, Zheng D-Y, Chen J-Z, Ding H, Mao C, Tang J-L (2013b) Promising biomarkers for predicting the outcomes of patients with KRAS wild-type metastatic colorectal cancer treated with anti-epidermal growth factor receptor monoclonal antibodies: a systematic review with meta-analysis. Int J Cancer 133:1914–1925. https://doi.org/10.1002/ijc.28153
Yar Saglam AS, Alp E, Elmazoglu Z, Menevse S (2015) Treatment with cucurbitacin B alone and in combination with gefitinib induces cell cycle inhibition and apoptosis via EGFR and JAK/STAT pathway in human colorectal cancer cell lines. Hum Exp Toxicol 35:526–543. https://doi.org/10.1177/0960327115595686
Yoon J, Koo KH, Choi KY (2011) MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res 71:445–453. https://doi.org/10.1158/0008-5472.CAN-10-3058
Zhao L, Vogt PK (2008) Helical domain and kinase domain mutations in p110 of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci USA 105:2652–2657. https://doi.org/10.1073/pnas.0712169105
Zhao L, Vogt PK (2010) Hot-spot mutations in p110α of phosphatidylinositol 3-kinase (pI3K): differential interactions with the regulatory subunit p85 and with RAS. Cell Cycle 9:596–600. https://doi.org/10.4161/cc.9.3.10599
Ziemke EK, Dosch JS, Maust JD, Shettigar A, Sen A, Welling TH, Hardiman KM, Sebolt-Leopold JS (2016) Sensitivity of KRAS-mutant colorectal cancers to combination therapy that cotargets MEK and CDK4/6. Clin Cancer Res 22:405–414. https://doi.org/10.1158/1078-0432.CCR-15-0829
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Martins, M., Mansinho, A., Cruz-Duarte, R., Martins, S.L., Costa, L. (2018). Anti-EGFR Therapy to Treat Metastatic Colorectal Cancer: Not for All. In: Jordan, P. (eds) Targeted Therapy of Colorectal Cancer Subtypes. Advances in Experimental Medicine and Biology, vol 1110. Springer, Cham. https://doi.org/10.1007/978-3-030-02771-1_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-02771-1_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-02770-4
Online ISBN: 978-3-030-02771-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)