
Abstract
The epidermal growth factor receptor (EGFR) and RAS/RAF signaling pathway plays pivotal roles in tumor progression via proliferation, survival, invasion, and immune evasion. Two anti-EGFR monoclonal antibodies, cetuximab and panitumumab, have become essential components in the treatment of patients with metastatic colorectal cancer (mCRC). Treatment with these anti-EGFR antibodies has shown definite benefits when administered in all treatment lines and is strongly recommended as the preferred regimen to prolong survival, especially when administered in the first- and third-lines. Recent efforts have revealed not only mechanisms responsible for resistance to anti-EGFR antibodies, including expanded RAS mutations as a negative predictive biomarker, but also the possibility of continuing anti-EGFR antibody treatment in combination with chemotherapy. Furthermore, the challenges associated with the pharmaceutical development of treatments for patients with mutant-type BRAF mCRC are ongoing. In this review, we provide an overview of the EGFR and RAS/RAF signaling pathway and antitumor activity, focusing on practical aspects such as established treatments including patient selection, treatment strategies, and future perspectives for drug development targeting the EGFR and RAS/RAF signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibodies cetuximab and panitumumab prolong the survival of patients with metastatic colorectal carcinoma (mCRC). |
Small molecule inhibitors to activated BRAF present new treatment options in BRAF mutated mCRC patients. |
Optimizing treatment by integrating these targeted drugs with anti-vascular endothelial growth factor (anti-VEGF) therapy and chemotherapy and finding the best sequencing approaches are current challenges in this area. |
1 Introduction
Colorectal cancer (CRC) is one of the most frequently occurring cancers worldwide, and metastatic CRC (mCRC) continues to be associated with a poor prognosis [1]. Treatment strategies for mCRC patients are limited, but recent efforts to improve survival outcomes among mCRC patients have focused on the combination of conventional chemotherapies with agents targeting biological pathways that are pivotal to cancer pathogenesis. The epidermal growth factor receptor (EGFR) family plays a key role in tumor progression via proliferation, survival, invasion, and immune evasion [2]. Two anti-EGFR monoclonal antibodies, cetuximab and panitumumab, are now approved for the treatment of mCRC worldwide. Furthermore, the benefits of anti-EGFR antibodies have been confirmed to be limited to patients whose tumors do not harbor mutations in retrovirus-associated DNA sequences (RAS) genes, including KRAS and NRAS; these genes encode proteins downstream of EGFR, in the RAS/v-RAF 1 murine leukemia viral oncogene homolog 1 (RAF)/mitogen-activated protein kinase (MAPK) pathway [2,3,4,5,6,7,8]. BRAF and PIK3CA mutations have also been reported as potential predictive biomarkers of the efficacies of these anti-EGFR antibodies [9,10,11,12,13]. Nevertheless, chemotherapy for mCRC patients who harbor BRAF mutant tumors and have a poor prognosis remains insufficient.
In this review, we provide an overview of EGFR and RAS/RAF signaling and antitumor activity of agents that target these pathways, focusing on practical aspects such as established treatments including patient selection, treatment strategies, and future perspectives for drug development targeting the EGFR and RAS/RAF signaling pathway.
2 Clinical Trials Establishing the Use of Anti-EGFR Therapies for mCRC
The anti-EGFR monoclonal antibodies cetuximab and panitumumab were approved for the treatment of mCRC patients in the 2000s. In a phase II study (BOND trial) comparing cetuximab plus irinotecan with cetuximab monotherapy to verify the assumption that cetuximab circumvents irinotecan resistance, cetuximab plus irinotecan improved the response rate (RR) and progression-free survival (PFS) in mCRC patients who were refractory to irinotecan monotherapy [14]. Cetuximab was initially approved based on these results. Subsequently, the NCIC CTG-CO.17 trial comparing cetuximab plus best supportive care (BSC) and BSC alone in EGFR-positive refractory mCRC patients also showed a survival benefit with cetuximab [15].
In the second-line treatment of mCRC, several clinical trials have demonstrated the value of adding cetuximab or panitumumab to conventional chemotherapy in terms of RR and PFS, but unfortunately not in terms of overall survival (OS) [16,17,18,19]. It is discussed that these results could be caused by the impact of a crossover design. Regarding the comparison of anti-EGFR antibodies and bevacizumab treatment beyond progression, subsequent anti-EGFR treatment may affect the lack of improvement in OS [20]. Considering these results, anti-EGFR antibodies only improved the RR compared with bevacizumab beyond progression in the second-line treatment of mCRC patients harboring wild-type KRAS.
In a first-line setting, a phase III trial (CRYSTAL trial) comparing FOLFIRI either alone or in combination with cetuximab in patients with EGFR-positive mCRC demonstrated that combination with cetuximab reduced the risk of mCRC progression (8.9 vs. 8.0 months; hazard ratio [HR] 0.85; 95% confidence interval [CI] 0.72–0.99; P = 0.048) [3]. The value of adding anti-EGFR antibodies to oxaliplatin-based chemotherapy was investigated in three pivotal trials: OPUS, COIN, and PRIME. In the phase II OPUS trial, a significantly higher RR was observed for the FOLFOX4 plus cetuximab arm (46% vs. 36%, P = 0.0064), but PFS or OS did not improve compared with the FOLFOX4 arm [4]. Also, the phase III COIN trial did not confirm a survival benefit for the addition of cetuximab to oxaliplatin-based chemotherapy (FOLFOX or CAPOX) in the first-line treatment of patients with mCRC (17.0 vs. 17.9 months; HR 1.04; 95% CI 0.87–1.23; P = 0.67), even though a higher RR was observed for cetuximab plus oxaliplatin-based chemotherapy (64% vs. 57%, P = 0.049) [21]. The unexpected result of the COIN trial is likely to be attributed to the specific skin toxic effect of the CAPOX regimen when cetuximab was added. Similar results were observed in another phase III trial [22]. Regarding panitumumab, the phase III PRIME trial demonstrated the value of adding panitumumab to FOLFOX4 in terms of PFS (9.6 vs. 8.0 months; HR 0.80; 95% CI 0.66–0.97; P = 0.02) in patients with wild-type KRAS mCRC, but not in terms of OS (23.9 vs. 19.7 months; HR 0.83; 95% CI 0.67–1.02; P = 0.072) [23]. Nowadays, the efficacy of anti-EGFR antibodies is known to differ according to tumor biology. As described below, retrospective subgroup analyses of the CRYSTAL, OPUS, and PRIME trials have revealed that the benefit of anti-EGFR antibodies is only obvious in a molecularly selected population [24,25,26]. A prospective trial that did select for wild-type RAS mCRC patients, the phase III TAILOR trial, demonstrated a survival benefit for adding cetuximab to FOLFOX4 in the first-line treatment of patients with wild-type RAS mCRC (PFS: HR 0.69; 95% CI 0.54–0.89; P = 0.004; OS: HR 0.76; 95% CI 0.61–0.96; P < 0.001), confirming the value of adding anti-EGFR antibodies to oxaliplatin-based chemotherapy [27].
Further, the efficacy of maintenance therapy with anti-EGFR antibodies has been evaluated in several trials. In the phase II MACRO-II trial, single-agent cetuximab following mFOLFOX6 plus cetuximab achieved non-inferiority in terms of PFS at 9 months, with fewer adverse events compared with continuous mFOLFOX6 plus cetuximab (60% vs. 72%, non-inferiority P = 0.25) [28]. A retrospective analysis of the PRIME and PEAK trials also showed that maintenance with panitumumab plus 5-FU/LV after discontinuation of oxaliplatin was well tolerated, and PFS and OS were numerically longer compared to maintenance with panitumumab alone [29]. In the VALENTINO trial, maintenance therapy with single-agent panitumumab following mFOLFOX6 plus panitumumab had a significantly shorter PFS than that with 5-FU/LV plus panitumumab (HR 1.55; 95% CI 1.09–2.20; P = 0.011), and sparing of 5-FU/LV did not contribute to reducing the toxicity [30]. Considering these results, 5-FU/LV plus an anti-EGFR antibody is one of the preferred maintenance regimens using anti-EGFR antibodies, and further investigation comparing it with maintenance therapy with bevacizumab is needed.
3 Development of Predictive Biomarkers in Targeting EGFR
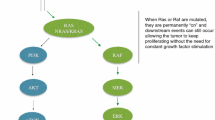
The EGFR and downstream components of the pathway have a key role in tumorigenesis via the regulation of proliferation, angiogenesis and metastasis, and cetuximab and panitumumab bind specifically to both EGFR homodimers and its heterodimers [2, 31,32,33]. In the past few decades, these components of the EGFR and RAS/RAF signaling pathway have been systematically evaluated for their value as predictive biomarkers of EGFR therapies. Although other mechanisms exhibiting resistance to anti-EGFR antibodies, such as HER2/MET amplification and PIK3CA mutation, are important, we will mainly focus on the predictive value of biomarkers involved in the EGFR and RAS/RAF pathway [12, 34] (Fig. 1).

Mechanisms of resistance to anti-EGFR antibodies. Aberrated gene alterations, including EGFR, AREG, EREG, NF-1, RAS, BRAF, PIK3CA, and EGFR S492R mutations, PTEN loss, and HER2/MET amplification, contribute to resistance via the activation of EGFR downstream components, regardless of EGFR blockade. Each mechanism is underlined and noted. EGFR epidermal growth factor receptor
3.1 EGFR and Its Ligands
EGFR overexpression as revealed by immunohistochemistry (IHC) has led to initial clinical trials investigating patient selection for cetuximab or panitumumab with promising results [14, 35]. However, several studies have demonstrated that patients can benefit from the addition of cetuximab in the absence of a positive EGFR IHC result [14, 23, 36,37,38]. Although EGFR alterations including gene overexpression or gene copy number alteration were also expected to be predictive biomarkers for anti-EGFR antibodies, no unified view has been obtained so far [39, 40]. A single point mutation in the ectodomain of EGFR S468R confers acquired or secondary resistance in mCRC treated with cetuximab, although this mechanism is not observed in mCRC treated with panitumumab [41]. This mutation was not detected in wild-type KRAS mCRC before exposure to anti-EGFR antibodies and is not considered to be a mechanism of primary resistance to cetuximab [42]. Regarding EGFR ligands such as epiregulin (EREG) and amphiregulin (AREG), several reports have shown that the gene expression of EGFR ligands in primary tumors significantly predicts the outcomes of patients treated with anti-EGFR antibodies, especially for those with wild-type KRAS mCRC [43,44,45,46]. Another post hoc analysis of the phase III AIO KRK-0207 trial demonstrated the prognostic value of high-expression levels of AREG and EREG in patients undergoing first-line chemotherapy including oxaliplatin, fluoropyrimidine, and bevacizumab [47]. However, further investigation evaluating optimal unified testing methods and cut-offs is needed.
3.2 RAS Mutations
The RAS GTPase is a master signaling protein at the hub of numerous signal transduction pathways (including the EGFR pathway) and interacts with many effector proteins to regulate cell proliferation, survival, migration, and apoptosis [48]. The three major isoforms of RAS (KRAS, NRAS, and HRAS) are mutated in around 45% of mCRC, primarily at the active site at residues G12, G13, and Q61 near the γ-phosphate of the guanosine triphosphate substrate [49,50,51]. The benefit of anti-EGFR antibodies is limited in these cases, with up to 65% of mCRC patients with KRAS mutations in exon 2 in codon 12/13 being resistant to these treatments [2, 7, 8]. Recently, multiple studies have revealed that other mutations in KRAS exons 3 and 4 or NRAS exons 2 to 4 can also predict a lack of benefit from anti-EGFR antibodies, as summarized in Table 1 [26]. Based on these results, in the 2010s, the indications for the use of anti-EGFR antibodies were extended from wild-type KRAS to all wild-type RAS tumors [52,53,54].
This change has led to a reassessment of the value of adding anti-EGFR antibodies in a second-line setting. In a phase III trial (20050181 trial) to evaluate the effect of the addition of panitumumab to FOLFIRI chemotherapy, the HR of the PFS for panitumumab plus FOLFIRI versus FOLFIRI favored panitumumab more strongly in a wild-type RAS population than in a wild-type KRAS exon 2 population (HR 0.70; 95% CI 0.54–0.91; P = 0.007) [55]. Another sub-analysis of a phase II trial comparing panitumumab plus FOLFIRI with bevacizumab plus FOLFIRI in a second-line setting for wild-type KRAS exon 2 mCRC (WJOG6210G trial) demonstrated a tendency toward a better OS for all wild-type RAS tumors in the FOLFIRI plus panitumumab arm (18.9 vs. 16.1 months), resulting in a significant interaction (P for interaction = 0.026) [19]. Furthermore, the RR was 52.5% for FOLFIRI plus panitumumab and 2.6% for FOLFIRI plus bevacizumab (P < 0.001). Recently, the role of EGFR therapies in the second-line treatment of wild-type KRAS mCRC patients after progression on cetuximab has been explored [56, 57]. In the CAPRI-GOIM trial evaluating the possibility of continuing cetuximab treatment, the PFS for treatment with FOLFOX plus cetuximab, compared with FOLFOX alone, was significantly prolonged in all wild-type RAS patients (HR 0.56; 95% CI 0.33–0.94; P = 0.025). This result suggests that continuing cetuximab treatment in combination with chemotherapy might confer therapeutic efficacy in all wild-type RAS patients, leading to the “re-challenge” issue that will be described later (see Sect. 4.1) [58].
The selection of patients on the basis of their RAS status was a notable change in the treatment of mCRC. A remaining issue is whether the antiangiogenic drug bevacizumab or anti-EGFR antibodies are the best option for first-line treatment. Pivotal studies, including two phase III trials, the Cancer and Leukemia Group B (CALGB) 80405 trial and the FIRE-3 trial, and the phase II PEAK trial comparing anti-EGFR antibodies and bevacizumab as a first-line treatment for wild-type KRAS exon 2 mCRC, have tried to resolve this issue. In the overall wild-type RAS population, the FIRE-3 trial and the PEAK trial demonstrated the superiority of anti-EGFR antibodies in terms of OS, RR, depth of response, and early tumor shrinkage [8, 59, 60]. However, the CALGB80405 trial did not show the same superiority of cetuximab in the overall wild-type RAS population [61]. Although this discrepancy might have been caused by differences in post-progression treatment and combination regimens, no unified view has been obtained so far. Several meta-analyses of these three randomized clinical trials have demonstrated the superiority of first-line anti-EGFR antibodies in terms of RR and OS, compared with anti-vascular endothelial growth factor (anti-VEGF) therapy, among wild-type KRAS and the overall wild-type RAS population with mCRC [62, 63]. There seems to be little doubt from these results that EGFR therapies in combination with chemotherapy have a major likelihood of providing an improvement in survival for patients with all wild-type RAS mCRC.
Furthermore, CRC is known to exhibit differences in its incidence, pathogenesis, molecular pathways, and outcome depending on the location of the primary tumor [64, 65]. A recent meta-analysis of over a million CRC patients confirmed the prognostic role of tumor sidedness in all stages of disease. A left-sided primary tumor location was significantly associated with a reduced risk of death, compared with a right-sided primary tumor location (HR 0.82; 95% CI 0.79–0.84; P < 0.001) [66]. Several retrospective analyses have also demonstrated that the primary tumor location may also be a predictive biomarker of anti-EGFR antibodies [67,68,69,70,71,72,73,74]. Several meta-analyses including randomized first-line studies in patients with mCRC support these results, especially in terms of the value of sidedness as a predictive biomarker of the efficacies of anti-EGFR antibodies [62, 75, 76]. These analyses showed a similar worse prognosis for patients with right-sided tumors, compared with those with left-sided tumors, in patients with wild-type RAS mCRC. Moreover, a greater effect of chemotherapy plus anti-EGFR antibody treatment, compared with chemotherapy or chemotherapy plus bevacizumab, was observed in patients with left-sided tumors. For right-sided tumors, there is no significant difference between anti-EGFR and anti-VEGF antibodies in terms of survival in post hoc analyses of the FIRE-3, CALGB80405, and PEAK trials. Nowadays, we understand that chemotherapy plus an anti-EGFR antibody should be the preferred first-line treatment option for patients with mCRC harboring left-sided wild-type RAS tumors [52,53,54].
3.3 BRAF Mutations
BRAF is a serine/threonine kinase that is active directly downstream of KRAS and that activates MEK through its phosphorylation in the RAS signaling pathway [77]. The Cancer Genome Atlas identified BRAF V600E mutations in several malignancies, including CRC [78]. Metastatic CRC harbors BRAF V600E mutations at a frequency of approximately 5–11%, and the BRAF V600E mutation is mutually exclusive with KRAS mutations [51, 79,80,81]. CRC harboring BRAF V600E mutations is known to be associated with right-sided primary tumors, older women, high-grade tumors, and precursor sessile serrated adenomas [82, 83]. Several post hoc analyses of phase III trials have reported the predictive value of BRAF V600E mutations for the efficacy of anti-EGFR antibodies [5, 17, 22, 26, 84,85,86]. However, the predictive values were not in accordance in all the reports and remain controversial. A meta-analysis of phase III trials and a phase II trial in chemorefractory patients reported that the addition of anti-EGFR antibodies did not significantly improve PFS (HR 0.88; 95% CI 0.67–1.14; P = 0.33) or OS (HR 0.91; 95% CI 0.62–1.34, P = 0.63) in the BRAF mutation subgroup [87]. Furthermore, another similar meta-analysis of eight randomized control trials demonstrated that the HR for the OS benefit with anti-EGFR antibodies was 0.97 (95% CI 0.67–1.41) for BRAF mutant tumors and 0.81 (95% CI 0.70–0.95) for BRAF wild-type tumors. However, no statistically significant difference was observed between these tumors (interaction P = 0.43) [88]. On the other hand, in the FIRE-3 trial, early tumor shrinkage as a strong early on-treatment parameter associated with outcome was identified in a certain number of patients with BRAF mutant tumors treated with FOLFIRI plus cetuximab [13]. In the VOLFI trial, the addition of panitumumab to FOLFOXIRI showed a high RR in BRAF mutant mCRC [89]. Considering these results, some patients may receive clinical benefit from treatment with anti-EGFR antibodies even if they are BRAF mutant. Collectively, limited data are available supporting the exclusion of patients with BRAF mutant mCRC from treatment with anti-EGFR antibodies. Nowadays, it may be suspected that the BRAF mutation is not a definitive predictive biomarker for anti-EGFR antibodies but a promising targetable subgroup for combination therapy blocking multiple pathways (see below).
Recently, BRAF mutations that do not result in an amino acid substitution at position 600 (BRAF non-V600E mutations) have been reported. The incidence of BRAF non-V600E mutation is reportedly 1–5% in mCRC, and the mutation’s kinase activity can be classified up to a level of activity similar to that of the BRAF V600E mutation [56, 90,91,92]. Unlike the BRAF V600E mutation, however, BRAF non-V600E mutations are correlated with significantly better survival [93]. A retrospective study reported that the clinical outcomes, including RR, PFS, and OS, were similar between RAS mutation, BRAF V600E mutation, and certain BRAF non-V600E mutations including G469A, L485F, Q524L, L525R, D594G, and V600R; in addition, BRAF non-V600E mutations contributed to a smaller benefit from anti-EGFR antibody [94]. On the other hand, there is evidence that BRAF non-V600E mutant mCRC can respond to panitumumab [95]. BRAF non-V600E mutations including the abovementioned mutations can be classified into three different classes depending on the different extents of dependency on RAS, which may dictate the response to anti-EGFR antibodies [96, 97]. It has been reported that almost half of patients with class 3 BRAF non-V600E mutations responded to anti-EGFR therapy, while response was rare for patients with class 2 BRAF non-V600E mutations [98]. It may be clinically helpful if BRAF tests focused on not only the BRAF V600E mutation but also on non-V600E mutations. Furthermore, the upregulated kinase activity of BRAF and/or alternative signaling through CRAF could lead to the incomplete blockade of the MEK pathway in a preclinical model, and another strategy attempting to inhibit signaling through the MAPK pathway is being developed for these subgroups of BRAF V600E and non-V600E mutations [92, 97, 99].
3.4 NF-1 Alterations
NF-1 is a tumor suppressor gene that encodes a neurofibromin, which functions as a suppressive regulator of the RAS signaling pathway [100]. The frequency of somatic NF-1 mutations in CRC is reportedly 1–6% [101]. In lung cancer, several studies reported that reduced NF-1 expression leading to the activation of the MAPK pathway via NF-1 deletion or mutations was associated with the development of primary and acquired resistance to EGFR tyrosine kinase inhibitors [102, 103]. An analysis of the mutation landscape of 33 Chinese mCRC specimens demonstrated that NF-1 alterations may be candidates for predictive biomarkers for anti-EGFR antibodies [104]. Furthermore, the co-existence of NF-1 mutations and BRAF mutations leads to the development of resistance to BRAF inhibitors in melanoma cells [105]. Therefore, NF-1 alterations are expected to be a potential target of combination therapies inhibiting the EGFR and RAS/RAF signaling pathway in mCRC, and further investigation is needed.
4 Challenges in Targeting EGFR and RAS/RAF Signaling
Although optimizing patient selection according to the RAS mutation status and the primary tumor location have benefited patients with mCRC harboring wild-type RAS tumors, primary or acquired resistance to anti-EGFR antibodies remains an important issue. Over the past decade, many studies have attempted to overcome these resistances by focusing on the EGFR and RAS/RAF signaling pathway.
4.1 Retreatment with Anti-EGFR Antibodies
A recent phase III study has shown that regorafenib and TAS-102 are superior to a placebo in refractory mCRC, although with limited efficacy (RR 1.0% and 1.6%, respectively) [106, 107]. Thus, there is an urgent need to develop more effective later-line treatments for mCRC.
The development of high throughput sequencing technologies has provided not only a highly efficient, rapid, and low-cost DNA sequencing platform, but also the possibility of performing DNA sequencing using “liquid” biopsies. Although tissue biopsy is the standard of care for tumor diagnosis, traditional tumor needle or excisional biopsies from certain metastatic diseases are invasive procedures, and obtaining a sufficient tissue sample can be difficult in patients with mCRC. So far, a liquid biopsy seems to be a promising, minimally invasive technique for diagnosing the current tumor status and monitoring mCRC patients during anti-cancer treatment, with good concordance to tissue specimens [108,109,110,111,112]. Wild-type RAS mCRC tumors are well known to develop resistance to anti-EGFR antibodies by acquiring gene mutations, including RAS, that enable cells to escape from the ongoing treatment, but limited data are available regarding the reversibility of their mutation statuses [34, 113, 114]. There is a hypothesis that the occurrence of disease progression after an initial response in wild-type RAS mCRC may potentially contribute to the progressive prevalence of a mutated clone caused by the acquisition of resistance to the anti-EGFR antibody during therapy. To verify this, several phase II studies have examined re-challenge treatments with anti-EGFR antibodies in patients with wild-type RAS mCRC at the time of diagnosis (Table 2) [115,116,117,118,119,120]. The CRICKET and E-Rechallenge trials demonstrated a tendency toward a higher RR, compared with other trials. A retrospective study analyzing 89 mCRC patients who received cetuximab or cetuximab plus erlotinib also indicated that a response while receiving prior anti-EGFR antibody and a longer interval length between anti-EGFR therapies may be associated with the efficacy of re-challenge treatment with anti-EGFR antibody [121]. An analysis of the post-progression circulating tumor DNA (ctDNA) profiles of patients with wild-type RAS/BRAF mCRC treated with anti-EGFR antibodies who acquired RAS and/or EGFR mutations during therapy supports this hypothesis [122]. This analysis demonstrated that the cumulative half-life of the RAS and EGFR relative mutant allele frequency was 4.4 months, and patients had a higher RR during re-challenge therapies after longer time intervals. Moreover, the CRICKET trial demonstrated that the PFS was longer in patients with wild-type RAS mCRC in ctDNA at the re-challenge baseline (HR 0.44; 95% CI 0.18–0.98; P = 0.026) [119]. Although these results suggest the contribution of the interval length between anti-EGFR antibody therapies in overcoming the acquired resistance to anti-EGFR antibody, an ongoing phase III trial (FIRE-4, NCT02934529) should provide further useful indications. The FIRE-4 trial has a planned enrolment of 550 patients with wild-type RAS mCRC who will receive first-line cetuximab-based therapy and third-line cetuximab as a re-challenge treatment and will include RAS assessment using liquid biopsies to assess progressive disease.
4.2 Development of Combination Therapies Inhibiting the EGFR and RAS/RAF Signaling Pathway
From the very early days of our understanding of the role of BRAF mutation in mCRC, the main issue associated with mutant-type BRAF mCRC has been its dismal prognosis [80]. BRAF V600E mutation is associated with an older age, a female gender, right-sidedness, and a Caucasian ethnicity [123,124,125,126]. Additionally, BRAF V600E mutation leads to diminished DNA mismatch repair via the hypermethylation of the MLH1 gene promoter [127,128,129]. The BRAF V600E mutation is observed in up to 60% of microsatellite instability (MSI)-high tumors and only 5–10% of microsatellite stable (MSS) tumors and is associated with a poor prognosis in both MSS and MSI-high tumors [130]. Regarding BRAF non-V600E mutations, the possible association with a poor prognosis has not been clarified [131].
Vemurafenib, an orally bioavailable, adenosine triphosphate (ATP)-competitive, small-molecule inhibitor of BRAF(V600E) kinase, was shown to inhibit cell proliferation and tumor growth in BRAF V600E mutant CRC cell lines in vivo and in vitro. Based on this rationale, a phase II trial of vemurafenib monotherapy in patients with BRAF V600E mutant mCRC was conducted [132]. However, the efficacy of BRAF inhibition using vemurafenib monotherapy was insufficient, with an RR of 5% and a median PFS of 2.1 months. This lack of efficacy can be explained by the hypothesis that resistance via feedback activation of EGFR may result in the reactivation of the MAPK signaling pathway. Preclinical models have demonstrated a synergistic effect via decreased MAPK signaling with the combined inhibition of BRAF and EGFR [133, 134]. A pilot study of vemurafenib plus panitumumab demonstrated a modest RR (12.5%) [135]. In addition to the rationale behind BRAF and EGFR inhibition, targeting MEK, which is active downstream of BRAF, enabled an improvement in PFS and OS, compared with conventional chemotherapy, in patients with melanoma [136]. In BRAF V600E mutant mCRC, a phase I/II trial of dabrafenib combined with trametinib demonstrated a feasible RR (12%) [137]. Furthermore, a phase II trial (SWOG S1406) revealed that the addition of vemurafenib to cetuximab and irinotecan significantly prolonged PFS (HR 0.42; 95% CI 0.26–0.66; P < 0.001) in patients with BRAF V600E mutant mCRC [138]. Activation of the PI3 K/AKT pathway has been reported to be a mechanism of resistance to BRAF inhibition, and the combined inhibition of BRAF, EGFR, and PIK3A had a synergistic effect in preclinical models [139, 140].
Further synergistic efficacy has been expected for various approaches to MAPK signaling inhibition. Several early phase studies were conducted in patients with BRAF V600E mutant mCRC, and promising results were reported (Table 3) [140,141,142]. Although the addition of alpelisib to encorafenib and cetuximab treatment provided a PFS benefit, it caused additional toxicity and unfortunately did not improve survival (HR 1.21; 95% CI 0.61–2.39) [141]. An ongoing phase III trial (BEACON, NCT02928224) to evaluate encorafenib and cetuximab plus or minus binimetinib versus chemotherapy plus cetuximab in BRAF V600E mutant mCRC patients whose disease had progressed after one or two prior regimens should determine the value of triple inhibition including BRAF and EGFR. This study contains a safety lead-in phase in which the safety and tolerability of the combination of encorafenib, binimetinib, and cetuximab will be assessed prior to the phase III part. The clinical outcomes of the patients enrolled in the safety lead-in phase have been reported, with meaningful clinical activity having been observed. Of the 29 patients with BRAF V600E mutant mCRC, the confirmed objective RR was 41% and the median period of study treatment was 5.3 months [143].
Regarding BRAF non-V600E mutations, BRAF non-V600E mutant cancer cells are reliant on tyrosine kinase receptors for their MAPK activation, and the inhibition of EGFR, BRAF and MEK also inhibits cell proliferation and tumor growth in vivo and in vitro [144]. A phase II study of triple combination chemotherapy with encorafenib, binimetinib, and cetuximab in patients with BRAF non-V600E mutant mCRC (BIGBANG, UMIN000031857) should provide further useful indications.
5 Conclusions
Anti-EGFR antibodies are key drugs in first-line and later-line treatments for patients with wild-type RAS and left-sided mCRC. In second-line treatment, anti-EGFR therapies have been shown to increase the response, but do not confer a survival benefit. Recent efforts to develop non-invasive monitoring techniques, including liquid biopsies, have revealed the potency of monitoring response or resistance to anti-EGFR antibodies and re-challenge treatment with anti-EGFR antibodies. Re-challenge therapy with anti-EGFR antibodies using liquid biopsy test may become a standard of care in mCRC patients who responded to anti-EGFR therapy in the first-line. Furthermore, BRAF-mutant mCRC is not only a potential subgroup exhibiting resistance to anti-EGFR antibodies, but also a potential subgroup that could benefit from combinations of BRAF, MEK, and other pathway inhibitors. Nowadays, immunotherapy has led to clinical benefits in MSI-high CRC. However, only approximately 5% of mCRC display MSI-high tumors, and immunotherapy is still under development in the vast majority of mCRC. Anti-EGFR therapies may become candidates of combination drugs for immunotherapy in the future due to their immunomodulatory functions [145,146,147]. The EGFR and RAS/RAF pathway is a pivotal pathway in mCRC pathogenesis and treatment, and further investigations not only focusing on the direct inhibition of this pathway, but also combination therapy with targeting of the tumor microenvironment are needed to improve the insufficient prognosis in mCRC patients.
References
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358:1160–74.
Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–17.
Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–71.
Bokemeyer C, Van Cutsem E, Rougier P, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–75.
Vaughn CP, Zobell SD, Furtado LV, et al. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50:307–12.
Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–6.
Schwartzberg LS, Rivera F, Karthaus M, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32:2240–7.
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62.
Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12.
Oda K, Okada J, Timmerman L, et al. PIK3CA cooperates with other phosphatidylinositol 3′-kinase pathway mutations to effect oncogenic transformation. Cancer Res. 2008;68:8127–36.
Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–7.
Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–75.
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45.
Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8.
Peeters M, Price TJ, Cervantes A, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:4706–13.
Seymour MT, Brown SR, Middleton G, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14:749–59.
Sobrero AF, Maurel J, Fehrenbacher L, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:2311–9.
Shitara K, Yonesaka K, Denda T, et al. Randomized study of FOLFIRI plus either panitumumab or bevacizumab for wild-type KRAS colorectal cancer-WJOG 6210G. Cancer Sci. 2016;107:1843–50.
Cascinu SLS, Rosati G, et al. A phase III multicenter trial comparing two different sequences of second/third line therapy (cetuximab/irinotecan followed by FOLFOX versus FOLFOX followed by cetuximab/irinotecan) in metastatic KRAS wt colorectal cancer (mCC) patients, refractory to FOLFIRI/bevacizumab. Eur J Cancer. 2015;51(Suppl S3):abstr 2006.
Maughan TS, Adams RA, Smith CG, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–14.
Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30:1755–62.
Douillard JY, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–705.
Van Cutsem E, Kohne CH, Lang I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9.
Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535–46.
Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34.
Qin S, Li J, Wang L, et al. Efficacy and tolerability of first-line cetuximab plus leucovorin, fluorouracil, and oxaliplatin (FOLFOX-4) versus FOLFOX-4 in patients with RAS wild-type metastatic colorectal cancer: the open-label, randomized, phase III TAILOR trial. J Clin Oncol. 2018;36:3031–9 (JCO2018783183).
Aranda E, Garcia-Alfonso P, Benavides M, et al. First-line mFOLFOX plus cetuximab followed by mFOLFOX plus cetuximab or single-agent cetuximab as maintenance therapy in patients with metastatic colorectal cancer: phase II randomised MACRO2 TTD study. Eur J Cancer. 2018;101:263–72.
Modest DP, Rivera F, Bachet JB, et al. Panitumumab-based maintenance after oxaliplatin discontinuation in metastatic colorectal cancer: a retrospective analysis of two randomised trials. Int J Cancer. 2019. https://doi.org/10.1002/ijc.32110.
Pietrantonio FM, De Braud FG, et al. First-line FOLFOX plus panitumumab (Pan) followed by 5FU/LV plus Pan or single-agent Pan as maintenance therapy in patients with RAS wild-type metastatic colorectal cancer (mCRC): the VALENTINO study. J Clin Oncol. 2018;36(15_suppl):3505.
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37.
Yarden Y. The EGFR family and its ligands in human cancer signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3–8.
Ciardiello F, Tortora G. Epidermal growth factor receptor (EGFR) as a target in cancer therapy: understanding the role of receptor expression and other molecular determinants that could influence the response to anti-EGFR drugs. Eur J Cancer. 2003;39:1348–54.
Pietrantonio F, Vernieri C, Siravegna G, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res. 2017;23:2414–22.
Saltz LB, Meropol NJ, Loehrer PJ Sr, et al. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8.
Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64.
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34.
Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803–10.
Laurent-Puig P, Cayre A, Manceau G, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27:5924–30.
Scartozzi M, Bearzi I, Mandolesi A, et al. Epidermal growth factor receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: a fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis. BMC Cancer. 2009;9:303.
Sickmier EA, Kurzeja RJ, Michelsen K, et al. The panitumumab EGFR complex reveals a binding mechanism that overcomes cetuximab induced resistance. PLoS One. 2016;11:e0163366.
Esposito C, Rachiglio AM, La Porta ML, et al. The S492R EGFR ectodomain mutation is never detected in KRAS wild-type colorectal carcinoma before exposure to EGFR monoclonal antibodies. Cancer Biol Ther. 2013;14:1143–6.
Jacobs B, De Roock W, Piessevaux H, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27:5068–74.
Stintzing S, Jung A, Kapaun C, Reiche J, Modest DP, Giessen CA, Vehling-Kaiser U, Stauch M, Hass H, von Weikersthal LF, Kirchner T, Heinemann V. Ligand expression of the EGFR ligands amphiregulin, epiregulin, and amplification of the EGFR gene to predict for treatment efficacy in KRAS wild-type mCRC patients treated with cetuximab plus CAPIRI and CAPOX: analysis of the randomized AIO CRC-0104 trial. J Clin Oncol. 2012;30(15_suppl):3519.
Baker JB, Dutta D, Watson D, et al. Tumour gene expression predicts response to cetuximab in patients with KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2011;104:488–95.
Sunakawa Y, Yang D, Moran M, et al. Combined assessment of EGFR-related molecules to predict outcome of 1st-line cetuximab-containing chemotherapy for metastatic colorectal cancer. Cancer Biol Ther. 2016;17:751–9.
Stintzing S, Ivanova B, Ricard I, et al. Amphiregulin (AREG) and epiregulin (EREG) gene expression as predictor for overall survival (OS) in oxaliplatin/fluoropyrimidine plus bevacizumab treated mCRC patients-analysis of the phase III AIO KRK-0207 trial. Front Oncol. 2018;8:474.
Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1:2–27.
Chang YY, Lin PC, Lin HH, et al. Mutation spectra of RAS gene family in colorectal cancer. Am J Surg. 2016;212(537–544):e533.
Marcus K, Mattos C. Direct Attack on RAS: intramolecular communication and mutation-specific effects. Clin Cancer Res. 2015;21:1810–8.
Yuki STK, Taniguchi H, Hamaguchi T, Akagi K, Denda T, Mizukami T, Oki E, Yamada T, Shiozawa M, Kudo T, Tamura T, Esaki T, Naruge D, Kajiwara T, Nomura S, Fujii S, Shitara K, Ohtsu A, Yoshino T. The nationwide cancer genome screening project in Japan SCRUM-Japan GI-SCREEN: efficient identification of cancer genome alterations in advanced colorectal cancer. Ann Oncol. 2017;28(suppl_5):v158–208.
Benson AB, Venook AP, Al-Hawary MM, et al. NCCN guidelines insights: colon cancer, version 2.2018. J Natl Compr Canc Netw. 2018;16(4):359–69.
Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422.
Yoshino T, Arnold D, Taniguchi H, et al. Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: a JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncol. 2018;29:44–70.
Peeters M, Oliner KS, Price TJ, et al. Analysis of KRAS/NRAS mutations in a phase III study of panitumumab with FOLFIRI compared with FOLFIRI alone as second-line treatment for metastatic colorectal cancer. Clin Cancer Res. 2015;21:5469–79.
Ciardiello F, Normanno N, Maiello E, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol. 2014;25:1756–61.
Ciardiello F, Normanno N, Martinelli E, et al. Cetuximab continuation after first progression in metastatic colorectal cancer (CAPRI-GOIM): a randomized phase II trial of FOLFOX plus cetuximab versus FOLFOX. Ann Oncol. 2016;27:1055–61.
Bennouna J, Hiret S, Bertaut A, et al. Continuation of bevacizumab vs cetuximab plus chemotherapy after first progression in KRAS wild-type metastatic colorectal cancer: the UNICANCER PRODIGE18 randomized clinical trial. JAMA Oncol. 2018;5(1):83–90.
Elez E, Argiles G, Tabernero J. First-line treatment of metastatic colorectal cancer: interpreting FIRE-3, PEAK, and CALGB/SWOG 80405. Curr Treat Options Oncol. 2015;16:52.
Fernando R, Schwartzberg LS, Karthaus M, Fasola G, Canon J-L, Hecht JR, Tian Y, Yu H, Oliner KS, Go WY. Extended RAS analysis and subsequent anti-EGFR and anti-VEGF treatment (tx) in PEAK: a first-line phase 2 study of FOLFOX6 + panitumumab (pmab) or bevacizumab (bev) in metastatic colorectal cancer (mCRC). J Clin Oncol. 2014;32(15_suppl):3629.
Lenz HJND, Lenz HJ, et al. CALGB/SWOG 80405: phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with expanded RAS analyses untreated metastatic adenocarcinoma of the colon or rectum. Proc Am Soc Clin Oncol. 2014;32(abstr LBA3):2014.
Khattak MA, Martin H, Davidson A, Phillips M. Role of first-line anti-epidermal growth factor receptor therapy compared with anti-vascular endothelial growth factor therapy in advanced colorectal cancer: a meta-analysis of randomized clinical trials. Clin Colorectal Cancer. 2015;14:81–90.
Ciliberto D, Staropoli N, Caglioti F, et al. The best strategy for RAS wild-type metastatic colorectal cancer patients in first-line treatment: a classic and Bayesian meta-analysis. Crit Rev Oncol Hematol. 2018;125:69–77.
Lee GH, Malietzis G, Askari A, et al. Is right-sided colon cancer different to left-sided colorectal cancer?—a systematic review. Eur J Surg Oncol. 2015;41:300–8.
Mizukami T, Takahashi M, Sunakawa Y, Yuki S, Kagawa Y, Takashima A, Kato K, Hara H, Denda T, Moriwaki T, Shiozawa M, Oki E, Satoh T, Kawakami H, Esaki T, Nishina T, Okamoto W, Yoshino T, Nakajima TE. Identification of site-specific genome alterations in metastatic colorectal cancer: Sub-study 003 of the SCRUM-Japan GI-SCREEN. J Clin Oncol. 2019;37(suppl 4):abstr 578.
Petrelli F, Tomasello G, Borgonovo K, et al. Prognostic survival associated with left-sided vs right-sided colon cancer: a systematic review and meta-analysis. JAMA Oncol. 2016. https://doi.org/10.1001/jamaoncol.2016.4227.
Boisen MK, Johansen JS, Dehlendorff C, et al. Primary tumor location and bevacizumab effectiveness in patients with metastatic colorectal cancer. Ann Oncol. 2013;24:2554–9.
Venook AP, Ou F-S, Lenz H-J, Kabbarah O, Qu X, Niedzwiecki D, Zemla T, Goldberg RM, Hochster HS, O’Neil BH, Sanoff HK, Mayer RJ, Bertagnolli MM, Blanke CD, Innocenti F. Primary (1°) tumor location as an independent prognostic marker from molecular features for overall survival (OS) in patients (pts) with metastatic colorectal cancer (mCRC): analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol. 2017;35(15_suppl):3503.
Brule SY, Jonker DJ, Karapetis CS, et al. Location of colon cancer (right-sided versus left-sided) as a prognostic factor and a predictor of benefit from cetuximab in NCIC CO.17. Eur J Cancer. 2015;51:1405–14.
Moretto R, Cremolini C, Rossini D, et al. Location of primary tumor and benefit from anti-epidermal growth factor receptor monoclonal antibodies in patients with RAS and BRAF wild-type metastatic colorectal cancer. Oncologist. 2016;21:988–94.
von Einem JC, Heinemann V, von Weikersthal LF, et al. Left-sided primary tumors are associated with favorable prognosis in patients with KRAS codon 12/13 wild-type metastatic colorectal cancer treated with cetuximab plus chemotherapy: an analysis of the AIO KRK-0104 trial. J Cancer Res Clin Oncol. 2014;140:1607–14.
Loupakis F, Yang D, Yau L, et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J Natl Cancer Inst. 2015. https://doi.org/10.1093/jnci/dju427.
Modest DP, Stintzing S, von Weikersthal LF, et al. Exploring the effect of primary tumor sidedness on therapeutic efficacy across treatment lines in patients with metastatic colorectal cancer: analysis of FIRE-3 (AIOKRK0306). Oncotarget. 2017;8:105749–60.
Sunakawa Y, Ichikawa W, Tsuji A, et al. Prognostic impact of primary tumor location on clinical outcomes of metastatic colorectal cancer treated with cetuximab plus oxaliplatin-based chemotherapy: a subgroup analysis of the JACCRO CC-05/06 trials. Clin Colorectal Cancer. 2017;16:e171–80.
Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28:1713–29.
Holch JW, Ricard I, Stintzing S, et al. The relevance of primary tumour location in patients with metastatic colorectal cancer: a meta-analysis of first-line clinical trials. Eur J Cancer. 2017;70:87–98.
Brennan DF, Dar AC, Hertz NT, et al. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472:366–9.
Weinstein JN, Collisson EA, Mills GB, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–20.
Ma BB, Mo F, Tong JH, et al. Elucidating the prognostic significance of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese patients with metastatic colorectal cancer. Asia Pac J Clin Oncol. 2015;11:160–9.
Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–32.
Kawazoe A, Shitara K, Fukuoka S, et al. A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer. 2015;15:258.
Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934.
Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–30.
Karapetis CS, Jonker D, Daneshmand M, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clin Cancer Res. 2014;20:744–53.
Peeters M, Price TJ, Cervantes A, et al. Final results from a randomized phase 3 study of FOLFIRI +/− panitumumab for second-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25:107–16.
Peeters M, Oliner KS, Parker A, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19:1902–12.
Pietrantonio F, Petrelli F, Coinu A, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51:587–94.
Rowland A, Dias MM, Wiese MD, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer. 2015;112:1888–94.
Geissler M, Knorrenschield R, Greeve J, Florschuetz A, Tannapfel A, Wessendorf S, Seufferlein T, Kanzler S, Heinemann V, Held S, Reinacher-Schick A. mFOLFOXIRI + panitumumab versus FOLFOXIRI as first-line treatment in patients with RAS wild-type metastatic colorectal cancer (mCRC): a randomized phase II trial of the AIO (AIO-KRK-0109). Ann Oncol. 2017;28(suppl 5):v158–v208. https://doi.org/10.1093/annonc/mdx393.
Shen Y, Wang J, Han X, et al. Effectors of epidermal growth factor receptor pathway: the genetic profiling of KRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PLoS One. 2013;8:e81628.
Cremolini C, Di Bartolomeo M, Amatu A, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol. 2015;26:2092–7.
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67.
Jones JC, Renfro LA, Al-Shamsi HO, et al. (Non-V600) BRAF mutations define a clinically distinct molecular subtype of metastatic colorectal cancer. J Clin Oncol. 2017;35:2624–30.
Shinozaki E, Yoshino T, Yamazaki K, et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: the biomarker research for anti-EGFR monoclonal antibodies by comprehensive cancer genomics (BREAC) study. Br J Cancer. 2017;117:1450–8.
Wang Y, Jones JC, Kipp BR, Grothey A. Activity of EGFR antibody in non-V600 BRAF mutant metastatic colorectal cancer. Ann Oncol. 2019;30(1):147–9.
Yao Z, Torres NM, Tao A, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–83.
Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548:234–8.
Kotani DSM, Parikh A, Bando H, Seventer EV, Taniguchi H, Yoshino T, Corcoran RB, Yaeger R, Ebi H. Clinicopathological features, efficacy of anti-EGFR therapy, and survival outcomes in patients with BRAF non-V600 mutated metastatic colorectal cancer. J Clin Oncol. 2019;37(suppl 4):abstr 659.
Ursem C, Atreya CE, Van Loon K. Emerging treatment options for BRAF-mutant colorectal cancer. Gastrointest Cancer. 2018;8:13–23.
Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–16.
Philpott C, Tovell H, Frayling IM, et al. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genom. 2017;11:13.
de Bruin EC, Cowell C, Warne PH, et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014;4:606–19.
Pan Y, Yuan C, Cheng C, et al. Frequency and clinical significance of NF1 mutation in lung adenocarcinomas from East Asian patients. Int J Cancer. 2019;144:290–6.
Mei Z, Shao YW, Lin P, et al. SMAD4 and NF1 mutations as potential biomarkers for poor prognosis to cetuximab-based therapy in Chinese metastatic colorectal cancer patients. BMC Cancer. 2018;18:479.
De Raedt T, Brems H, Wolkenstein P, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72:1288–92.
Sunakawa Y, Izawa N, Mizukami T, et al. Profile of trifluridine/tipiracil hydrochloride in the treatment of metastatic colorectal cancer: efficacy, safety, and place in therapy. Onco Targets Ther. 2017;10:4599–605.
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–12.
Taly V, Pekin D, Benhaim L, et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013;59:1722–31.
Danese E, Minicozzi AM, Benati M, et al. Comparison of genetic and epigenetic alterations of primary tumors and matched plasma samples in patients with colorectal cancer. PLoS One. 2015;10:e0126417.
Spindler KL, Pallisgaard N, Vogelius I, Jakobsen A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin Cancer Res. 2012;18:1177–85.
Thierry AR, Mouliere F, El Messaoudi S, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20:430–5.
Grasselli J, Elez E, Caratu G, et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol. 2017;28:1294–301.
Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:827.
Bouchahda M, Karaboue A, Saffroy R, et al. Acquired KRAS mutations during progression of colorectal cancer metastases: possible implications for therapy and prognosis. Cancer Chemother Pharmacol. 2010;66:605–9.
Santini D, Vincenzi B, Addeo R, et al. Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann Oncol. 2012;23:2313–8.
Tsuji A, Eto T, Masuishi T, Satake H, Segawa Y, Tanioka H, Hara H, Kotaka M, Sagawa T, Watanabe T, Nakamura M, Takahashi T, Negoro Y, Manaka D, Fujita H, Suto T, Ichikawa W, Fujii M, Takeuchi M, Nakajima T. Phase II study of third-line cetuximab rechallenge in patients with metastatic wild-type K-RAS colorectal cancer who achieved a clinical benefit in response to first-line cetuximab plus chemotherapy (JACCRO CC-08). Ann Oncol. 2016;27(suppl_6):2016.
Tsuji AMN, Watanabe T, Manaka D, Matsuoka H, Kataoka M, Takeuchi M, Ichikawa W, Fujii M. Phase II study of third-line panitumumab rechallenge in patients with metastatic wild-type KRAS colorectal cancer who achieved a clinical benefit in response to first-line panitumumab plus chemotherapy. Ann Oncol. 2016;27(6):149–206.
Yuki SYK, Muranaka T, Sakata Y, et al. Phase II trial of panitumumab monotherapy for patients with KRAS exon2 wild type colorectal cancer after progression on cetuximab. HGCSG1101. Ann Oncol. 2016;27(suppl_6):497P.
Rossini DCC, Conca E, Santini D, et al. Liquid biopsy to predict benefit from rechallenge with cetuximab (cet) + irinotecan (iri) in RAS/BRAF wild-type metastatic colorectal cancer patients (pts) with acquired resistance to first-line cet + iri: final results and translational analyses of the CRICKET study by GONO. J Clin Oncol. 2018;36(15_suppl):12007.
Osawa HES, Nakamura M, Yamaguchi K et al. Phase II study of cetuximab rechallenge in patients with RAS Wild-type metastatic colorectal cancer: E-Rechallenge trial. Ann Oncol. 2018;29(suppl_8):viii150–viii204.
Liu X, George GC, Tsimberidou AM, et al. Retreatment with anti-EGFR based therapies in metastatic colorectal cancer: impact of intervening time interval and prior anti-EGFR response. BMC Cancer. 2015;15:713.
Parseghian CM, Loree JM, Morris VK, et al. Anti-EGFR resistant clones decay exponentially after progression: implications for anti-EGFR re-challenge. Ann Oncol. 2019;30(2):243–9.
Tie J, Gibbs P, Lipton L, et al. Optimizing targeted therapeutic development: analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer. 2011;128:2075–84.
Loupakis F, Moretto R, Aprile G, et al. Clinico-pathological nomogram for predicting BRAF mutational status of metastatic colorectal cancer. Br J Cancer. 2016;114:30–6.
Yoon HH, Shi Q, Alberts SR, et al. Racial differences in BRAF/KRAS mutation rates and survival in stage III colon cancer patients. J Natl Cancer Inst. 2015. https://doi.org/10.1093/jnci/djv186.
Ghidini M, Petrelli F, Tomasello G. Right versus left colon cancer: resectable and metastatic disease. Curr Treat Options Oncol. 2018;19:31.
Wang L, Cunningham JM, Winters JL, et al. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209–12.
Oliveira C, Pinto M, Duval A, et al. BRAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogene. 2003;22:9192–6.
French AJ, Sargent DJ, Burgart LJ, et al. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin Cancer Res. 2008;14:3408–15.
Clarke CN, Kopetz ES. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J Gastrointest Oncol. 2015;6:660–7.
Zheng G, Tseng LH, Chen G, et al. Clinical detection and categorization of uncommon and concomitant mutations involving BRAF. BMC Cancer. 2015;15:779.
Kopetz S, Desai J, Chan E, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33:4032–8.
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3.
Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–35.
Yaeger R, Cercek A, O’Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–20.
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14.
Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol. 2015;33:4023–31.
Kopetz SSM, Hochster HS, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG 1406). J Clin Oncol. 2017;35(15_suppl):3505.
Mao M, Tian F, Mariadason JM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3 K inhibition or demethylating agents. Clin Cancer Res. 2013;19:657–67.
van Geel R, Tabernero J, Elez E, et al. A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 2017;7:610–9.
Tabernero JRVG, Guren TK, Schellens JHM, et al. Phase 2 results: encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J Clin Oncol. 2016;34(15_suppl):3544.
Atreya CE, Van Cutsem E, Bendell JC, Corcoran RB, et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). J Clin Oncol. 2015;33(15_suppl):103.
Van Cutsem EPC, Huijberts S, Grothey A, et al. BEACON CRC study safety lead-in (SLI) in patients with BRAF V600E metastatic colorectal cancer (mCRC): efficacy and tumor markers. J Clin Oncol. 2018;36(4_suppl):627.
Kotani H, Adachi Y, Kitai H, et al. Distinct dependencies on receptor tyrosine kinases in the regulation of MAPK signaling between BRAF V600E and non-V600E mutant lung cancers. Oncogene. 2018;37:1775–87.
Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27.
Yoshimatsu K, Kato H, Ishibashi K, et al. Second-line chemotherapy with low-dose CPT-11 and cisplatin for colorectal cancer resistant to 5-FU-based chemotherapy. Cancer Chemother Pharmacol. 2003;52:465–8.
Holubec L, Polivka J Jr, Safanda M, et al. The role of cetuximab in the induction of anticancer immune response in colorectal cancer treatment. Anticancer Res. 2016;36:4421–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Takuro Mizukami has received consulting fees from Merck Serono and a speaker honorarium from Merck Serono and Takeda Pharmaceutical Co. Ltd. Takako Eguchi Nakajima has received research grants from Merck Serono, Taiho Pharmaceutical Co. Ltd., Chugai Pharmaceutical Co. Ltd., Takeda Pharmaceutical Co. Ltd., Eli Lilly Japan, and Sanofi KK and a speaker honorarium from Merck Serono, Takeda Pharmaceutical Co. Ltd., Taiho Pharmaceutical Co. Ltd., Chugai Pharmaceutical Co. Ltd., and Eli Lilly Japan. Yu Sunakawa has received consulting fees from Takeda Pharmaceutical Co. Ltd. and a speaker honorarium from Takeda Pharmaceutical Co. Ltd. and Merck Serono. Naoki Izawa declares that he has no conflicts of interest that might be relevant to the contents of this manuscript.
Rights and permissions
About this article

Cite this article
Mizukami, T., Izawa, N., Nakajima, T.E. et al. Targeting EGFR and RAS/RAF Signaling in the Treatment of Metastatic Colorectal Cancer: From Current Treatment Strategies to Future Perspectives. Drugs 79, 633–645 (2019). https://doi.org/10.1007/s40265-019-01113-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01113-0