
Abstract
Colorectal cancer (CRC) remains one of the leading causes of cancer mortality worldwide. Regarded as a heterogeneous disease, a number of biomarkers have been proposed to help in the stratification of CRC patients and to enable the selection of the best therapy for each patient towards personalized therapy. However, although the molecular mechanisms underlying the development of CRC have been elucidated, the therapeutic strategies available for these patients are still quite limited. Thus, over the last few years, a multitude of novel targets and therapeutic strategies have emerged focusing on deregulated molecules and pathways that are implicated in cell growth and survival. Particularly relevant in CRC are the activating mutations in the oncogene PIK3CA that frequently occur in concomitancy with KRAS and BRAF mutations and that lead to deregulation of the major signalling pathways PI3K and MAPK, downstream of EGFR. This review focus on the importance of the PI3K signalling in CRC development, on the current knowledge of PI3K inhibition as a therapeutic approach in CRC and on the implications PI3K signalling molecules may have as potential biomarkers and as new targets for directed therapies in CRC patients.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
4.1 Introduction
Colorectal cancer (CRC) is one of the most common cancer types, and despite intensive research, remains one of the leading causes of cancer mortality worldwide (Torre et al. 2015). It results from the accumulation of genetic and epigenetic alterations in oncogenes and tumour suppressor genes , leading to the transformation of the normal epithelia towards invasive carcinoma (Markowitz and Bertagnolli 2009). Although a large number of molecules have been shown to be altered in these patients, to date, the use of targeted therapies has been limited to anti-epidermal growth factor receptor (EGFR) and anti-vascular endothelial factor (VEGF) agents. Moreover, patients harbouring KRAS and NRAS mutations are currently excluded from anti-EGFR therapies, as these alterations were shown to cause resistance (Allegra et al. 2016). Therefore, it is urgent to unravel novel therapeutic approaches for CRC patients in an attempt to improve patient outcomes and overcome therapy resistance . While several inhibitors are already being tested in preclinical and clinical trials, the interplay of the signalling pathways has proved to be rather complex and no other agents have yet been approved for these patients. In this review, we focus on the key role of the phosphatidylinositol 3-kinase (PI3K) signalling pathway in CRC development, the latest developments in the field of PI3K targeted specific agents as well as on the implications of PI3K inhibition as an alternative therapeutic approach for CRC patients.
4.2 Molecular Aspects of CRC Progression
Colorectal carcinogenesis is characterized by the gradual accumulation of alterations, genetic and epigenetic, in specific oncogenes and tumour suppressor genes in a multistep manner (Markowitz and Bertagnolli 2009).
The classical model of colorectal cancer progression is the adenoma-carcinoma sequence, however this pathway oversimplifies the heterogeneity of CRC and is not able to explain the development of all types of CRC (Fearon and Vogelstein 1990; Walther et al. 2009). Due to the distinct molecular, clinical and pathological characteristics observed in CRC tumours, other mechanisms for CRC development have emerged, namely the serrated pathway (O’Brien et al. 2006). In each of these models, a unique progression pathway is associated with a distinct mutational spectra. While alterations in the genes APC , KRAS and p53 are classic molecular alterations in the Vogelstein pathway, mutations in BRAF are typical of the serrated polyp pathway (Jass 2006; Velho et al. 2010; Vilar and Gruber 2010).
Moreover, these models are often associated with different types of genetic instability. Indeed, according to the type of genetic instability, CRC can be subdivided in different molecular subsets, microsatellite instability (MSI) and microsatellite stability (MSS), the latest characterized by having chromosomal instability (CIN) and observed in the majority of the cases (approximately 85%) (Vilar and Gruber 2010; Cunningham et al. 2010). In contrast, MSI is detected in about 15% of CRC patients and is characterized by a defective mismatch repair system through epigenetic silencing or germline mutations, leading to the accumulation of mutations across the genome mainly in repetitive sequences (microsatellites) (Cunningham et al. 2010). As previously highlighted, MSS and MSI are preferentially observed in the adenoma-carcinoma sequence and the serrated pathway, respectively (Velho et al. 2010; Vilar and Gruber 2010). Notably, while for most patients (about 70%) CRC occurs sporadically (MSI and MSS), in other cases CRC develops in a hereditary context being the most common form the hereditary non-polyposis CRC (HNPCRC) also termed Lynch syndrome (Tops et al. 2009).
Of particular importance in CRC, is the fact that MSI status can be used as a prognostic marker and predictor of therapeutic resistance in CRC patients. More specifically, MSI CRC tumours have been shown to be associated with a better prognosis than MSS tumours (Malesci et al. 2007; Gryfe et al. 2000). In addition, these subsets are known to respond differently to the available therapies and studies indicate that, in contrast to MSS, MSI tumours do not benefit from 5-fluouracil (5-FU) based adjuvant chemotherapies (Ribic et al. 2003).
Overall, to successfully design and develop novel targeted therapies , more studies are needed to clarify the value of specific biomarkers for predictive and prognostic purposes, including MSI status, as well as to better understand the mechanisms underlying the development of CRC in the different molecular subsets.
4.3 The MAPK and PI3K Signalling Pathways and Their Deregulation in CRC
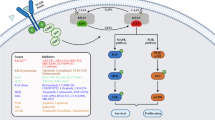
The mitogen activated protein kinase (MAPK) and PI3K are ubiquitous signalling pathways, downstream of EGFR , implicated in a variety of key biological processes including cell proliferation and survival, cell cycle regulation, differentiation, metabolism and apoptosis, among others (Sebolt-Leopold and Herrera 2004; Liu et al. 2009). Figure 4.1 illustrates, in a simplified manner, the classical MAPK and PI3K signalling pathways and their intervenient molecules.

Simplified representation of the MAPK and PI3K signalling pathways. EGF epidermal growth factor, EGFR epidermal growth factor receptor, PI3K phosphatidylinositol 3-kinase, MAPK mitogen activated protein kinase, MEK1/2 MAPK kinase 1/2, ERK1/2 extracellular signal-regulated kinase 1/2, mTOR mammalian target of rapamycin
Overall, these pathways are of major relevance in CRC as activating mutations in genes of these cascades are frequently detected in CRC patients, leading to the constitutive activation of the signalling pathway independently of a stimuli. Indeed, as determined by others and our group, a high frequency of mutations has been observed in KRAS , BRAF and PIK3CA (the gene coding for PI3K p110α, the catalytic subunit of PI3K) (De Roock et al. 2011; Lievre et al. 2010; Oliveira et al. 2004, 2007; Velho et al. 2005, 2008).
Briefly, as part of the RAS-RAF-MAPK cascade, KRAS is a member of the RAS superfamily of GTPases, along with N-RAS and H-RAS, all belonging to the larger class of regulatory GTP hydrolases (Pylayeva-Gupta et al. 2011). By switching between on and off states, GTP- and GDP-bound respectively, KRAS is important in controlling a complex network of signalling pathways by transducing signals from cell surface receptors namely EGFR to specific intracellular effectors (Sebolt-Leopold and Herrera 2004; Samatar and Poulikakos 2014). Upon stimulation, guanine nucleotide exchange factors (GEFs) promote the activation of RAS by stimulating GDP for GTP exchange; conversely, GTPase-activating proteins (GAPs) accelerate RAS-mediated GTP hydrolysis. In their active state, RAS proteins interact and activate their effectors and stimulate downstream signalling pathways (Bos et al. 2007). More specifically, the classical RAS signal transduction pathway comprises sequential phosphorylations of the serine/threonine kinase RAF, MAPK kinase 1/2 (MEK1/2) and extracellular signal-regulated kinase 1/2 (ERK1/2), ultimately modulating other molecules and regulating the distinct biological functions (Sebolt-Leopold and Herrera 2004; Samatar and Poulikakos 2014). Importantly, RAS is also known to activate other molecules and signalling cascades namely the PI3K signalling pathway, with major implications in targeted therapies (Liu et al. 2009; Fernandes et al. 2013; Murillo et al. 2014; Gupta et al. 2007).
The B-RAF serine/threonine kinase, which belongs to the RAF kinase family of protein kinases together with A-RAF and C-RAF, is one of the best characterized RAS effectors. RAF phosphorylates MEK1/2, which in turn phosphorylates and activates ERK1/2 that will modulate downstream effectors (Sebolt-Leopold and Herrera 2004; Samatar and Poulikakos 2014).
On a separate and parallel signalling cascade, PI3Ks are a rather ubiquitous family of lipid kinases activated by receptor tyrosine kinases (RTK) or other molecules as G-proteins (Liu et al. 2009). PI3Ks are able to phosphorylate the 3′-hydroxyl group of phosphatidylinositol and phosphoinositides and these lipid products act as second messengers to trigger a multitude of signalling cascades with impact in key mechanisms as survival, differentiation and metabolism (Liu et al. 2009; Vanhaesebroeck et al. 2012). In terms of classification, PI3Ks are grouped into three classes (IA/IB, II and III), with distinct structures and substrate specificities but class IA have received much attention as they have been implicated in many human cancers. Class IA PI3Ks, able to phosphorylate phosphatidylinositol (4,5)-biphosphate (PIP2), converting it to phosphatidylinositol (3,4,5)-triphosphate (PIP3), are composed of a heterodimer of a p85 regulatory subunit (p85α, p85β, p55γ or splice variants) and a p110 catalytic subunit (p110α, p110β or p110δ) (Liu et al. 2009; Vanhaesebroeck et al. 2012). Notably, the different p110 and p85 isoforms seem to preferentially mediate specific signalling cascades, though with some redundancy as reviewed in (Hennessy et al. 2005). Moreover, while p110α and p110β are ubiquitously expressed, p110δ expression is mostly restricted to the immune system (Engelman et al. 2006; Liu et al. 2009). In CRC, the p110α subunit of PI3K, encoded by the PIK3CA gene, is of particular relevance as it is often mutated in these patients (De Roock et al. 2011). The p110 catalytic isoforms share high homology and have common specific domains namely the p85 binding domain (able to interact with the p85 subunit), a RAS binding domain (to mediate activation by RAS family members) and a kinase catalytic domain (to generate PIP3 and activate downstream targets) (Liu et al. 2009; Thorpe et al. 2015). Similarly, the common p85 subunit domains include a p110-binding domain also termed inter-Src homology 2 (iSH2), and SH2, SH3 and BCR homology (BHD) domains (Liu et al. 2009; Thorpe et al. 2015). Mechanistically, activation (dimerization and autophosphorylation) of the RTK, upon stimulation by growth factors, leads to the recruitment of class IA PI3K to the membrane where the regulatory p85 subunit will bind RTK phosphorylated motifs but also relieve the p85 inhibition of p110; the activated p110 is then able to generate PIP3, a second messenger that provides docking sites for specific proteins, i.e., PIP3 binds to specific domains, as the pleckstrin-homology (PH) domain, of downstream targets including Akt (also termed protein kinase B, PKB) and phosphoinositide-dependent kinase (PDK1); PDK1 is then able to phosphorylate Akt at Thr308 important to activate Akt (Liu et al. 2009; Thorpe et al. 2015). Remarkably, the activation of the serine/threonine-specific protein kinase Akt, one important downstream effector of PI3K, leads to phosphorylation and subsequent activation or inhibition of additional downstream molecules that will ultimately regulate other proteins modulating the many functions of the PI3K signalling cascade (Manning and Cantley 2007). Indeed, a panoply of Akt substrates have been identified including glycogen synthase kinase 3β (GSK-3β), forkhead box O (FoxO) transcription factors, mouse double minute 2 homologue (MDM2), Bcl-2 associated death promoter (Bad), tuberous sclerosis complex 2 (TSC2), proline-rich Akt substrate of 40 kDa (PRAS40) among others, that are involved in cell growth, metabolism , insulin signaling and survival (Manning and Cantley 2007; Manning and Toker 2017). In addition, an important downstream effector of Akt is the mechanistic target of rapamycin (mTOR), a serine/threonine protein kinase. Interestingly, the catalytic subunit mTOR can be found in two distinct complexes, named mTOR complex 1 (mTORC1, composed by mTOR, raptor, mLST8, PRAS40 and DEPTOR) and mTOR complex 2 (mTORC2, composed by mTOR, rictor, mLST8, DEPTOR, Sin1 and Protor) and while mTORC1 is activated by Akt and known to modulate protein synthesis by phosphorylating the key mTOR effectors elF4E Binding Protein (4EBP1) and p70S6 Kinase 1 (S6 K1), mTORC2 is able to phosphorylate Akt at Ser473 and activate it (Sarbassov et al. 2005, 2006; Liu et al. 2009; Manning and Toker 2017; Saxton and Sabatini 2017). Importantly, the phosphorylation of Akt at both Thr308 and Ser473 is required for full Akt activation (Manning and Toker 2017). In contrast to the above mentioned activation mechanisms, negative regulation of the PI3K pathway is also mediated by the tumour suppressor gene phosphatase and tensin homolog (PTEN) that removes the 3′ phosphate from PIP3 hampering the PI3K signalling (Cully et al. 2006). Noteworthy, in addition to the described regulatory mechanisms, the PI3K-Akt-mTOR signaling is also tightly controlled by a number of feedback loops and crosstalk with other signaling pathways as reviewed in (Manning and Toker 2017; Rozengurt et al. 2014). Indeed, an important negative feedback mediated by mTORC1 is known to inhibit the PI3K pathway through distinct mechanisms. In particular, mTORC1 and its downstream effector S6K are able to inhibit the PI3K/Akt signalling through phosphorylation, inhibition and degradation of the insulin receptor substrate I (IRS-I) (Harrington et al. 2004; Manning and Toker 2017). In addition, RTKs are also targets of negative feedback regulation by activated PI3K-Akt-mTOR (Zhang et al. 2007; Chandarlapaty et al. 2011; Manning and Toker 2017).
As previously mentioned, mutations in genes of the MAPK and PI3K pathways are frequent in CRC. Specifically, about 30–40% of CRC patients harbour a mutation in KRAS (mostly affecting codons 12 and 13 of exon 2), whereas mutations in the KRAS effector, BRAF , are detected in about 15% of CRC patients (typically V600E on exon 15) (De Roock et al. 2011; Velho et al. 2010). Notably, KRAS and BRAF oncogenes have been suggested to play different roles in the development and progression of CRC, as KRAS and BRAF mutations are rarely detected in the same tumour and are instead observed as alternative molecular modifications (Rajagopalan et al. 2002; Velho et al. 2010). Moreover, KRAS and BRAF mutation frequencies and patterns are distinct in MSI , MSS, sporadic and hereditary subsets of CRC, with mutations in BRAF mostly found in MSI sporadic CRC and KRAS in MSS and MSI sporadic and hereditary CRC (Oliveira et al. 2004, 2007; Velho et al. 2010; Domingo et al. 2004; Lubomierski et al. 2005). In addition to KRAS and BRAF , PIK3CA mutations are observed in approximately 15% of CRC patients and, in contrast, often arise concomitantly with KRAS or BRAF mutations (De Roock et al. 2011; Velho et al. 2005, 2008). These PIK3CA mutations are of the missense type and are mostly in hotspots involving exon 9 that corresponds to the helical domain of PI3K p100α, and exon 20 that corresponds to the kinase domain of PI3K p100α; two common examples are E545K and H1047R (De Roock et al. 2011; Liu et al. 2009). In contrast to these, alterations in other molecules of the PI3K pathway are rare, except for PTEN for which controversial information has been raised in terms of mutations and loss of expression (De Roock et al. 2011; Nassif et al. 2004).
In any case, aberrant activation of these molecules will have a major impact in cell behaviour with effects on proliferation, survival, invasion and therefore in the initiation and progression of CRC (Thorpe et al. 2015). Of particular importance, and essential to better understand the underlying signalling mechanisms, is the increasing evidence in support of RAS-RAF-MAPK and PI3K-Akt cross-talk resulting in a complex signalling network (Castellano and Downward 2011; Thorpe et al. 2015). However, the extent of such cross-talk and the implications for CRC therapy are still not clear.
4.4 Current Targeted Therapies for CRC and Their Limitations
To date, apart from the conventional therapeutic strategies , CRC is limited to two distinct types of targeted therapy. These include anti-angiogenic and anti-EGFR agents (Ciardiello and Tortora 2008; Welch et al. 2010; Weng et al. 2015). In particular, cetuximab and panitumumab, two anti-EGFR antibodies that bind the extracellular domain of EGFR , have received much attention and are currently approved for the treatment of patients with mCRC (Ciardiello and Tortora 2008). Regrettably, only a subgroup of mCRC patients can benefit from such therapies and only a small percentage will be sensitive (Ciardiello and Tortora 2008; Lievre et al. 2017). More specifically, KRAS mutations have been recognized as predictive biomarkers of resistance to anti-EGFR agents, though some controversy exist as to the type of KRAS mutation (De Roock et al. 2010; Allegra et al. 2016). Nonetheless, CRC patients harbouring somatic KRAS mutations are currently not eligible to cetuximab and panitumumab targeted therapies (Allegra et al. 2009, 2016). Such limitation has a major impact, as about 30–40% of CRC patients do harbour a KRAS mutation (De Roock et al. 2011; Velho et al. 2010). Importantly, not all mCRC patients with wild type KRAS respond to anti-EGFR agents, suggesting that other mechanisms of resistance are involved (Heinemann et al. 2016; Lievre et al. 2017; Price et al. 2016). Indeed, NRAS mutations were also shown to be associated with resistance to anti-EGFR agents, and recommendations are now to exclude these patients from EGFR targeted therapies (Allegra et al. 2016). However, these account for only about 2% of CRC patients, indicating that other molecules are involved (Irahara et al. 2010). Mutations in other genes downstream of EGFR, including BRAF and PIK3CA, have been associated with resistance to EGFR targeted therapies but inconsistent results have been obtained and additional evidence is required to clarify such controversy (De Roock et al. 2011; Mohamed et al. 2018; Lievre et al. 2010; Therkildsen et al. 2014). As above mentioned, mCRC patients can also be offered anti-angiogenic therapy using the anti-VEGF agent bevacizumab, namely in combination with chemotherapy regimens, which has proven some clinical efficacy (Welch et al. 2010).
Altogether, at present, not only the available targeted agents for mCRC patients are limited but also exclude a considerable proportion of patients. Therefore, there is an urgent need to develop novel therapeutic strategies for CRC patients, particularly for those with KRAS mutations.
4.5 Targeting the PI3K Signalling Pathway
The PI3K signalling pathway is a key signalling cascade implicated in many human cancers including CRC (Janku et al. 2018). It is well established that deregulation of the PI3K signalling can occur through activating mutations in the PIK3CA gene, but other important activation mechanisms are known to exist, namely through oncogenic KRAS , with strong implications for CRC patients (Thorpe et al. 2015; Castellano and Downward 2011). Thus, efforts have been made to advance in the development of novel targeted therapies directed to the many molecules of the PI3K signalling cascade. Attractive therapeutic targets include PI3K p110 isoforms, Akt and mTOR, and inhibitors can be isoform specific- or pan-PI3K inhibitors, dual PI3K/mTOR inhibitors, Akt inhibitors and mTORC1 and mTORC2 inhibitors.
A vast number of drugs have already been tested in preclinical assays, however, only a few have reached clinical studies for several cancer types, including CRC (Fig. 4.2). Furthermore, the available data on the clinical effects of PI3K inhibitors is still limited (Rodon et al. 2013; Janku et al. 2018). Notably, despite reports suggesting specific alterations to be predictive of responsiveness, as the presence of PIK3CA mutations for PI3K p110α specific inhibitors, reliable predictive biomarkers of therapeutic response or resistance are still awaited. To date, no PI3K signaling pathway inhibitor has yet been approved for CRC patients and only temsirolimus and everolimus (mTORC1 inhibitors) and copanlisib and idelalisib (PI3K inhibitors ) have been approved for specific types of cancer as subsequently described in more detail (Janku et al. 2018). A brief overview of the current knowledge is described below.

Inhibitors of the PI3K signalling pathway used in clinical trials for several cancer types, including CRC
4.5.1 PI3K Isoforms as Therapeutic Targets
As above mentioned, agents that target the PI3K are classified into pan-class I PI3K inhibitors targeting all class I PI3K isoforms, or into isoform specific-PI3K inhibitors, targeting specifically one p110 isoform (Thorpe et al. 2015; Janku et al. 2018).
Initially, many studies were performed in several cancer type models using the PI3K pan-inhibitors wortmannin and LY294002, but these were only tested in preclinical studies and did not reach clinical trials, in part due to selectivity and toxicity issues (Liu et al. 2009). In particular, the irreversible PI3K inhibitor wortmannin was shown to have antitumour activity in several human tumour cell lines, including a colon carcinoma cell line (Schultz et al. 1995). Moreover, LY294002, a reversible small molecule PI3K inhibitor , demonstrated a remarkable growth-inhibitory and apoptosis-inducing effect in colon cancer cell lines and, experiments using mouse xenografts revealed that LY294002 administration in vivo also resulted in suppression of tumour growth and induction of apoptosis (Semba et al. 2002).
In recent years, novel inhibitors were generated with improved characteristics, namely in terms of specificity, potency and stability while simultaneously minimizing toxicity. In most cases, these inhibitors are ATP competitive agents and many of them are now being evaluated in clinical trials in patients with solid tumours, including CRC, either as monotherapy or in combination with other therapies (Thorpe et al. 2015). The list of inhibitors is vast and includes BKM120, PX-866, XL147, GDC0941, GSK1059615, BYL719, GDC0032, INK1117 (Janku et al. 2018). Some relevant studies focusing on CRC are briefly described.
BKM120 (buparlisib), a pan-PI3K inhibitor , when tested in a panel of 353 cell lines exhibited preferential inhibition of tumour cells harbouring PIK3CA mutations, in contrast to either KRAS or PTEN mutant models (Maira et al. 2012). In addition, BKM120 was shown to reduce cell proliferation in wild type and mutant PI3KCA CRC cells and treatment with cetuximab and BKM120 significantly reduced the growth of xenograft tumours originating from PIK3CA wild type and KRAS mutant cells compared with cetuximab alone (Hong et al. 2016). Noteworthy, the mechanisms underlying resistance to PI3K inhibition are known to involve other molecules. For instance, high nuclear β-catenin concentrations were shown to confer resistance to BKM120 in sphere cultures derived from patients with colon cancer (Tenbaum et al. 2012). Clinical studies have shown that BKM120 was well tolerated and had preliminary antitumour activity (Bendell et al. 2012; Rodon et al. 2014). However, a phase I trial of BKM120 plus mFOLFOX6 (5-FU/LV + oxaliplatin), in patients with refractory solid tumours, including CRC, resulted in increased toxicity compared to either therapy alone (McRee et al. 2015). At present, other clinical trials using BKM120 are under investigation, one in combination with panitumumab in KRAS wild type mCRC patients (NCT01591421) and another in combination with irinotecan in previously treated advanced CRC patients (NCT01304602). PX-866, an irreversible pan-PI3K inhibitor , has been shown to cause prolonged inhibition of PI3K signalling in human tumour xenografts, namely in colon tumour xenografts (Ihle et al. 2004). In clinical trials, it was well tolerated and was associated with prolonged stable disease in patients with advanced solid tumours, namely CRC (Hong et al. 2012). A multicenter phase I study of PX-866 and cetuximab in patients with mCRC or recurrent/metastatic squamous cell carcinoma of the head and neck has shown that PX-866 and cetuximab treatment was tolerated with signs of antitumour activity (Bowles et al. 2014). Subsequently, a randomized phase II study evaluated cetuximab with or without PX-866 in patients with KRAS wild type mCRC; however the addition of PX-866 to cetuximab did not improve progression free survival, objective response rate, or overall survival in patients with mCRC but instead the combination arm had greater toxicity (Bowles et al. 2016). In addition to these, other pan-PI3K inhibitors are being investigated in clinical trials for several tumour types and include XL-147 (pilaralisib), GDC-0941 (pictilisib), CH5132799, GSK1059615, SF1126 and ZSTK474 (Wheler et al. 2017; Sarker et al. 2015; Thorpe et al. 2015; Patnaik et al. 2016; Blagden et al. 2014; Janku et al. 2018). To date, copanlisib (BAY80-6946) is the only pan-PI3K inhibitor , with predominant activity against PI3K p110α and PI3K p110 δ isoforms, approved for relapsed lymphoma (Markham 2017).
In the last few years, isoform specific PI3K inhibitors have also been developed and of particular interest for CRC patients are the PI3K p110α inhibitors, as mutations in the gene that code for the PI3K p110α are frequently observed. BYL719 (alpelisib), a selective inhibitor of the PI3K p110α, was shown to have antitumour activity in preclinical studies and PIK3CA mutation was suggested to be a positive predictor of BYL719 sensitivity (Fritsch et al. 2014). Our group has also demonstrated the potential benefit of targeting PI3K p110α in CRC cells. Notably, not only cells with PIK3CA mutations were sensitive to PI3K p110α inhibition, but also cells with KRAS mutations (Fernandes et al. 2016). In particular, we have shown that the specific inhibition of PI3K p110α, by small interfering RNA (siRNA) or BYL719, had an impact in the viability of SW480 and HCT116 CRC cells harbouring mutations in KRAS and KRAS/PIK3CA, respectively (Fernandes et al. 2016). In addition, PI3K inhibition induced apoptosis in HCT116 cells and cell cycle arrest in SW480 cells suggesting that different mechanisms may be involved (Fernandes et al. 2016). Thus, specific inhibition of the p110α subunit of PI3K could provide an alternative therapeutic approach for CRC patients, particularly those harbouring KRAS mutations, who are currently excluded from EGFR-targeted therapies. In addition to preclinical studies, data from clinical trials is now emerging. The results from the first in-human phase Ia study revealed that BYL719 was tolerable and encouraging preliminary activity was observed in patients with PIK3CA-altered solid tumours (Juric et al. 2018). Moreover, due to the limited efficacy of BRAF inhibitors as single agents in BRAF mutant CRC and since EGFR and PI3K activation have been associated with resistance, a clinical phase Ib study evaluated the selective RAF kinase inhibitor encorafenib plus cetuximab or encorafenib plus cetuximab and BYL719; the results demonstrate that the treatments were tolerable and provided promising clinical activity in BRAF mutant mCRC patients (van Geel et al. 2017). Taselisib (GDC-0032) and MLN1117 (TAK-117, INK1117) are other PI3K p110α inhibitors currently in clinical trials for several cancer types (Janku et al. 2018).
In addition to PI3K p110α inhibitors , other p110 isoforms have been targeted including PI3K p110β and PI3K p110δ, but these have been mostly used for other cancer types. Indeed, idelalisib (CAL-101), a selective PI3K p110δ inhibitor, has already been approved for the treatment of patients with haematological malignancies (Gopal et al. 2014; Yang et al. 2015). In addition, the PI3K p110β inhibitor GSK2636771 and the PI3K p110δ inhibitor INCB050465 are also in clinical trials for various cancer types including CRC.
4.5.2 PI3K/mTOR Axis as a Therapeutic Target
The p110 catalytic domain of PI3K is structurally similar to that of the mTOR and therefore a class of inhibitors has been developed that target both molecules (Takeda et al. 2016). Using this strategy, PI3K-Akt-mTOR activation should be more efficiently inhibited as feedback mechanisms could be prevented. A number of dual inhibitors have been evaluated both in preclinical and clinical settings including BEZ235, XL765, BGT226 and PKI587.
BEZ235 (dactolisib), a potent ATP-competitive dual PI3K-mTOR inhibitor , was shown to have antitumour activity in vitro and in vivo using human tumour cell lines and tumour xenografts (Maira et al. 2008). In addition, BEZ235 was shown to induce tumour regression in genetically engineered mouse models of PIK3CA wild type CRC (Roper et al. 2011). Also in preclinical studies, BEZ235 was able to inhibit the PI3K/mTOR axis and to have antiproliferative and antitumoural activity in cancer cells with both wild type and mutated PIK3CA (Serra et al. 2008). Regarding predictive biomarkers , PIK3CA mutations have been associated with antitumour activity in preclinical and clinical studies, as shown with the association of the PIK3CA mutation H1047R with response to PI3K/AKT/mTOR signalling pathway inhibitors in early-phase clinical trials (Janku et al. 2013). Importantly, other preclinical studies have shown that coexistent mutations in PIK3CA and KRAS in CRC cells conferred resistance to BEZ235 (Kim et al. 2013). In addition, alterations in distinct molecules have also been associated with resistance to PI3K signaling inhibitors as is the case of TRIB2 that was shown to confer in vivo resistance to BEZ235 treatment through activation of Akt (Hill et al. 2017). Nonetheless, additional data is awaited from clinical trials. BEZ235 has also been combined with the mTOR inhibitor everolimus. Indeed, a phase Ib study of BEZ235 combined with everolimus was evaluated in patients with advanced solid malignancies but the combination of BEZ235 and everolimus demonstrated limited efficacy and tolerance (Wise-Draper et al. 2017).
BGT226, another dual PI3K-mTOR inhibitor , has also been evaluated in a clinical trial in patients with advanced solid tumours, including patients with colon cancer. However, BGT226 was shown to have limited preliminary antitumour activity and inconsistent target inhibition (Markman et al. 2012). The first-in-human study of PF05212384 (PKI-587) in patients with advanced cancer demonstrated a manageable safety profile and antitumour activity supporting further clinical development for patients with advanced solid malignancies (Shapiro et al. 2015).
Additional dual PI3K-mTOR inhibitors have been tested in clinical trials for many cancer types, but for some inhibitors, little success or toxicity issues were observed and no further studies were pursued (Janku et al. 2018; LoRusso 2016). More data on CRC is still awaited.
4.5.3 Akt and mTOR as Therapeutic Targets
As a key molecule in the PI3K signalling cascade , Akt has been appointed as a potential therapeutic target. Indeed, MK-2206, a potent allosteric inhibitor of all Akt isoforms, has already been evaluated in preclinical and clinical settings (Brown and Banerji 2017). For instance, in mice with established xenograft tumours, MK-2206 exhibited a significant deceleration of tumour progression and primary patient-derived tumour sphere growth was significantly inhibited by MK-2206 (Malkomes et al. 2016). In the clinic, the first-in-man clinical trial of MK-2206 demonstrated good tolerability with evidence of Akt signalling blockade in patients with advanced solid tumours that included CRC patients (Yap et al. 2011). Further clinical trials using MK-2206, alone or in combination, are ongoing in patients with advanced CRC namely a phase II study in patients with metastatic KRAS wild type and PIK3CA mutant (NCT01186705). Perifosine is another Akt inhibitor that, as MK-2206, targets the PH domain of Akt, thereby preventing its translocation to the plasma membrane and blocking its phosphorylation and activation (Gills and Dennis 2009; Brown and Banerji 2017). In a phase II trial in patients with mCRC, perifosine plus capecitabine showed promising clinical activity when compared with capecitabine alone (Bendell et al. 2011). Other inhibitors , including ATP competitive inhibitors of Akt, are being tested in patients with different cancer types and these include AZD5363, GDC-0068 and GSK2141795 (Janku et al. 2018; LoRusso 2016). Importantly, special attention should be taken when using these inhibitors alone as data indicates that PI3K may signal through both Akt-dependent and Akt-independent mechanisms. Indeed, an Akt-independent signalling downstream of PIK3CA mutations has been described in human cancer cells (Vasudevan et al. 2009).
Inhibitors targeting mTOR are also being evaluated in clinical trials for many cancer types. These inhibitors, which can be rapamycin analogs inhibiting mTORC1, or ATP-competitive inhibiting both mTORC1 and mTORC2, have been investigated in preclinical and clinical studies (Guertin and Sabatini 2009; Papadatos-Pastos et al. 2015). Indeed, temsirolimus, which is an mTORC1 inhibitor, has already been approved for advanced renal cancer and everolimus (RAD001), also an mTORC1 inhibitor, was approved for certain cancer types including advanced renal cancer and particular types of advanced breast cancer (Hudes et al. 2007; Baselga et al. 2012; Motzer et al. 2008; Janku et al. 2018). Temsirolimus and everolimus have also been evaluated in several clinical trials in mCRC patients, either alone or in combination with other therapeutic agents. For instance, in a phase II study in patients with refractory mCRC, the combination of tivozanib (a VEGFR inhibitor ) and everolimus was shown to be well tolerated, with stable disease achieved in 50% of patients (Wolpin et al. 2013). In contrast, in a phase II study in patients with mCRC heavily pretreated, everolimus was well tolerated but did not confer meaningful efficacy (Ng et al. 2013). A phase I trial of everolimus in combination with 5-FU/LV, mFOLFOX6 and mFOLFOX6 plus panitumumab in patients with refractory solid tumours including CRC has shown that the further addition of panitumumab resulted in an unacceptable level of toxicity that cannot be recommended for further study (McRee et al. 2014). A phase II trial of temsirolimus, alone or in combination with irinotecan, in KRAS mutant mCRC revealed that treatment was well tolerated but had limited efficacy in chemotherapy resistant KRAS mutant (Spindler et al. 2013). Nonetheless, plasma KRAS quantification was suggested as a strong predictor of outcome (Spindler et al. 2013). In addition to these, other mTOR inhibitors entered clinical trials including AZD8055, AZD2014 and MLN0128 (LoRusso 2016; Papadatos-Pastos et al. 2015). More studies and data are required evaluating mTOR inhibitors, namely in combination with other regimens.
4.5.4 PI3K Inhibition in Combination with MAPK Targeted Therapies
The available PI3K targeted therapies have shown limited success in CRC patients. Indeed, despite the development of many specific inhibitors , some of which already in clinical trials, none of these have yet been approved for the treatment of patients with CRC. Therefore, in an attempt to improve the response rates of these patients, combined targeted therapies have been proposed and investigated.
Notably, it is well established that the MAPK and PI3K signalling pathways are interconnected and inhibition of one signalling cascade could induce feedback loops and compensatory mechanisms, ultimately leading to resistance (Britten 2013). Moreover, as mutations in KRAS and BRAF are frequently observed in CRC patients, inhibition of both MAPK and PI3K pathways could be a more effective strategy. Thus, several studies have been performed to evaluate the combination of inhibitors targeting molecules of these pathways.
In preclinical models, inhibition of both the MAPK and PI3K signalling pathways have been reported to be synergistic in various cancer types (Temraz et al. 2015). For instance, although treatment with BEZ235 led to marked tumour regression in a mouse model of lung cancer with the PIK3CA H1047R mutation, in KRAS G12D mutant mice only BEZ235 combined with the MEK inhibitor ARRY-142886 induced tumour regression but not BEZ235 alone (Engelman et al. 2008). In CRC, the combination of a PI3K/mTOR (PF-04691502) and a MEK (PD-0325901) inhibitor demonstrated enhanced anti-proliferative effects against CRC cell lines and demonstrated enhanced reduction in tumour growth in patient-derived CRC tumour xenograft models, regardless of KRAS or PI3K mutational status (Pitts et al. 2014). In a panel of CRC cell lines, dual targeting of PI3K (GDC-0941) and MEK (AZD6244) induced synergistic growth inhibition but the combination of specific PI3K inhibitors , rather than dual mTOR/PI3K inhibitors, with MEK inhibitors resulted in greater synergy (Haagensen et al. 2012).The inhibition of MEK and PI3K/mTOR was shown to suppress tumour growth in patient-derived xenografts of RAS-mutant colorectal carcinomas, though it did not cause tumour regression (Migliardi et al. 2012). However, preclinical data also indicates that such therapeutic strategies have limitations namely related to toxicity issues and periodic rather than continuous inhibition has been suggested as an alternative strategy (Will et al. 2014). Indeed, rapid induction of apoptosis by PI3K inhibitors was reported to be dependent of the transient inhibition of RAS–ERK signalling (Will et al. 2014).
In a retrospective analysis, dual targeting of the PI3K and MAPK pathways was evaluated in patients with advanced cancers including CRC and treated with phase I study drugs; the results suggested that dual inhibition may potentially exhibit favourable efficacy compared with inhibition of either pathway, although with greater toxicity (Shimizu et al. 2012). In a biomarker -driven trial, no clinical benefit was observed in CRC patients treated with the Akt inhibitor MK-2206 and the MEK1/2 inhibitor selumetinib; instead, overlapping toxicities limited the ability to dose escalate to achieve exposures likely needed for clinical activity (Do et al. 2015). At present, many clinical trials are ongoing with MAPK and PI3K inhibitors for many cancer types including CRC but data is still awaited (Temraz et al. 2015; Tolcher et al. 2018).
4.6 The Importance of Bioinformatic Tools for Biomarker and Therapy Predictions
The number of studies evaluating specific inhibitors , both in preclinical and clinical settings, is enormous. Whether evaluating single and combination of drugs in cancer cell lines, performing in vivo experiments in animal models, or clinical trials in patients with advanced cancers, the amount of generated information is huge and rather under analysed.
Over the last few years, bioinformatic tools have gained much interest and proved powerful in unravelling some key aspects in cancer research, namely in the discovery of cancer biomarkers and evaluation of therapy responsiveness. Currently, it is well established that despite similarities in the mutational patterns among several cancer types, specific molecular alterations have context specific functional consequences with impact in therapy outcome. For instance, using a computational strategy for integrating (phospho) proteomic and mRNA sequencing data across tumour data sets, it was possible to link the dysregulation of upstream signalling pathways with altered transcriptional programs. More specifically, it was possible to associate PIK3CA activating mutations with altered activities of distinct sets of transcription factors and therefore this model could help to better predict which patients will benefit from targeted and combination therapies (Osmanbeyoglu et al. 2017). In a different study, a model is proposed to integrate oncogene and tumour suppressor activity in CRC cells and used to identify cancer drivers and compute patient-specific gene activity scores (Pavel et al. 2016). In this model, the integrative score improved prediction of drug sensitivity and the gene activity scores were also used to cluster CRC cell lines (Pavel et al. 2016). In addition to these, other studies focusing on bioimaging and bioinformatics have been developed. For instance, a novel method was proposed to characterize E-cadherin signature in gastric cancer cells in order to identify E-cadherin deregulation and functional impairment (Sanches et al. 2015). More specifically, this strategy included a bioimaging pipeline to quantify the expression level and characterize the distribution of the protein from in situ immunofluorescence images (Sanches et al. 2015). As for gastric cancer, bioimaging tools could be used in CRC models with potential implications in biomarker identification and therapy outcome predictions.
4.7 Conclusions and Future Perspectives
The ultimate goal in cancer therapy is to provide patients with treatments that will improve their overall survival and eventually manage cancer as a chronic disease. To achieve successful outcomes, personalized therapy will most probably be needed, i.e., select the best targeted therapy to each patient tumour characteristics. Although many drugs are being developed, there is still the urgent need to develop novel therapeutic strategies.
In recent years, inhibitors to the various molecules of the PI3K-Akt-mTOR cascade have emerged. In most cases, these inhibitors have shown a wide range of adverse effects and limited success. A deeper knowledge of the complex interplay between distinct signalling pathways, as well as a better understanding of feedback-loops disruption and the occurrence of compensatory mechanisms upon PI3K inhibition, will guide and improve the design of novel therapeutic strategies. The available clinical results have shown that dual MAPK and PI3K inhibition is possible, although toxicity has been an issue. Additional combination therapy regimens are being tested and should be considered.
A challenge in the treatment of CRC patients will be the identification of specific biomarkers predictive of therapy responsiveness, for which bioinformatics tools will be essential. Indeed, a proper patient stratification will most probably be a key issue for a successful outcome. Although many studies have been performed to identify such biomarkers, further studies are required. Overall, and despite ongoing clinical trials with some of these drugs, CRC patients are still awaiting alternative therapies to be approved. More data on novel drugs, combination regimens and clinical trials is expected to shed light on CRC best targeted therapies .
This work was financed by FEDER funds through the Operational Programme for Competitiveness Factors (COMPETE), Programa Operacional Regional do Norte (Norte 2020) and by National Funds through the Portuguese Foundation for Science and Technology (FCT), under the projects PTDC/BBB-IMG/0283/2014, PTDC/BIMONC/0171/2012, PTDC/BIM-ONC/0281/2014 and NORTE-01-0145-FEDER-000029.
References
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL (2009) American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 27(12):2091–2096. https://doi.org/10.1200/JCO.2009.21.9170
Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, Schilsky RL (2016) Extended RAS gene mutation testing in metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy: American Society of Clinical Oncology provisional clinical opinion update 2015. J Clin Oncol 34(2):179–185. https://doi.org/10.1200/JCO.2015.63.9674
Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN (2012) Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366(6):520–529. https://doi.org/10.1056/NEJMoa1109653
Bendell JC, Nemunaitis J, Vukelja SJ, Hagenstad C, Campos LT, Hermann RC, Sportelli P, Gardner L, Richards DA (2011) Randomized placebo-controlled phase II trial of perifosine plus capecitabine as second- or third-line therapy in patients with metastatic colorectal cancer. J Clin Oncol 29(33):4394–4400. https://doi.org/10.1200/JCO.2011.36.1980
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J (2012) Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 30(3):282–290. https://doi.org/10.1200/JCO.2011.36.1360
Blagden S, Omlin A, Josephs D, Stavraka C, Zivi A, Pinato DJ, Anthoney A, Decordova S, Swales K, Riisnaes R, Pope L, Noguchi K, Shiokawa R, Inatani M, Prince J, Jones K, Twelves C, Spicer J, Banerji U (2014) First-in-human study of CH5132799, an oral class I PI3K inhibitor, studying toxicity, pharmacokinetics, and pharmacodynamics, in patients with metastatic cancer. Clin Cancer Res 20(23):5908–5917. https://doi.org/10.1158/1078-0432.CCR-14-1315
Bos JL, Rehmann H, Wittinghofer A (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129(5):865–877. https://doi.org/10.1016/j.cell.2007.05.018
Bowles DW, Senzer N, Hausman D, Peterson S, Vo A, Walker L, Cohen RB, Jimeno A (2014) A multicenter phase 1 study of PX-866 and cetuximab in patients with metastatic colorectal carcinoma or recurrent/metastatic squamous cell carcinoma of the head and neck. Investig New Drugs 32(6):1197–1203. https://doi.org/10.1007/s10637-014-0124-3
Bowles DW, Kochenderfer M, Cohn A, Sideris L, Nguyen N, Cline-Burkhardt V, Schnadig I, Choi M, Nabell L, Chaudhry A, Ruxer R, Ucar A, Hausman D, Walker L, Spira A, Jimeno A (2016) A randomized, phase II trial of Cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with metastatic colorectal carcinoma. Clin Colorectal Cancer 15(4):337–344 e332. https://doi.org/10.1016/j.clcc.2016.03.004
Britten CD (2013) PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol 71(6):1395–1409. https://doi.org/10.1007/s00280-013-2121-1
Brown JS, Banerji U (2017) Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol Ther 172:101–115. https://doi.org/10.1016/j.pharmthera.2016.12.001
Castellano E, Downward J (2011) RAS interaction with PI3K: more than just another effector pathway. Genes Cancer 2(3):261–274. https://doi.org/10.1177/1947601911408079
Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N (2011) AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19(1):58–71. https://doi.org/10.1016/j.ccr.2010.10.031
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358(11):1160–1174. https://doi.org/10.1056/NEJMra0707704
Cully M, You H, Levine AJ, Mak TW (2006) Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 6(3):184–192. https://doi.org/10.1038/nrc1819
Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N (2010) Colorectal cancer. Lancet 375(9719):1030–1047. https://doi.org/10.1016/S0140-6736(10)60353-4
De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, Van Cutsem E, O’Callaghan CJ, Khambata-Ford S, Zalcberg JR, Simes J, Karapetis CS, Bardelli A, Tejpar S (2010) Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 304(16):1812–1820. https://doi.org/10.1001/jama.2010.1535
De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S (2011) KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 12(6):594–603. https://doi.org/10.1016/S1470-2045(10)70209-6
Do K, Speranza G, Bishop R, Khin S, Rubinstein L, Kinders RJ, Datiles M, Eugeni M, Lam MH, Doyle LA, Doroshow JH, Kummar S (2015) Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Investig New Drugs 33(3):720–728. https://doi.org/10.1007/s10637-015-0212-z
Domingo E, Espin E, Armengol M, Oliveira C, Pinto M, Duval A, Brennetot C, Seruca R, Hamelin R, Yamamoto H, Schwartz S Jr (2004) Activated BRAF targets proximal colon tumors with mismatch repair deficiency and MLH1 inactivation. Genes Chromosomes Cancer 39(2):138–142. https://doi.org/10.1002/gcc.10310
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7(8):606–619. https://doi.org/10.1038/nrg1879
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14(12):1351–1356. https://doi.org/10.1038/nm.1890
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61(5):759–767
Fernandes MS, Carneiro F, Oliveira C, Seruca R (2013) Colorectal cancer and RASSF family--a special emphasis on RASSF1A. Int J Cancer 132(2):251–258. https://doi.org/10.1002/ijc.27696
Fernandes MS, Melo S, Velho S, Carneiro P, Carneiro F, Seruca R (2016) Specific inhibition of p110alpha subunit of PI3K: putative therapeutic strategy for KRAS mutant colorectal cancers. Oncotarget 7(42):68546–68558. https://doi.org/10.18632/oncotarget.11843
Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, Wang Y, Trappe J, Brachmann SM, Maira SM, Wilson C, Boehm M, Garcia-Echeverria C, Chene P, Wiesmann M, Cozens R, Lehar J, Schlegel R, Caravatti G, Hofmann F, Sellers WR (2014) Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther 13(5):1117–1129. https://doi.org/10.1158/1535-7163.MCT-13-0865
Gills JJ, Dennis PA (2009) Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep 11(2):102–110
Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, Flinn IW, Flowers CR, Martin P, Viardot A, Blum KA, Goy AH, Davies AJ, Zinzani PL, Dreyling M, Johnson D, Miller LL, Holes L, Li D, Dansey RD, Godfrey WR, Salles GA (2014) PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med 370(11):1008–1018. https://doi.org/10.1056/NEJMoa1314583
Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S (2000) Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 342(2):69–77. https://doi.org/10.1056/NEJM200001133420201
Guertin DA, Sabatini DM (2009) The pharmacology of mTOR inhibition. Sci Signal 2(67):pe24. https://doi.org/10.1126/scisignal.267pe24
Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J (2007) Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 129(5):957–968. https://doi.org/10.1016/j.cell.2007.03.051
Haagensen EJ, Kyle S, Beale GS, Maxwell RJ, Newell DR (2012) The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition. Br J Cancer 106(8):1386–1394. https://doi.org/10.1038/bjc.2012.70
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF (2004) The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 166(2):213–223. https://doi.org/10.1083/jcb.200403069
Heinemann V, Rivera F, O’Neil BH, Stintzing S, Koukakis R, Terwey JH, Douillard JY (2016) A study-level meta-analysis of efficacy data from head-to-head first-line trials of epidermal growth factor receptor inhibitors versus bevacizumab in patients with RAS wild-type metastatic colorectal cancer. Eur J Cancer 67:11–20. https://doi.org/10.1016/j.ejca.2016.07.019
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB (2005) Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 4(12):988–1004. https://doi.org/10.1038/nrd1902
Hill R, Madureira PA, Ferreira B, Baptista I, Machado S, Colaco L, Dos Santos M, Liu N, Dopazo A, Ugurel S, Adrienn A, Kiss-Toth E, Isbilen M, Gure AO, Link W (2017) TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat Commun 8:14687. https://doi.org/10.1038/ncomms14687
Hong DS, Bowles DW, Falchook GS, Messersmith WA, George GC, O’Bryant CL, Vo AC, Klucher K, Herbst RS, Eckhardt SG, Peterson S, Hausman DF, Kurzrock R, Jimeno A (2012) A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 18(15):4173–4182. https://doi.org/10.1158/1078-0432.CCR-12-0714
Hong S, Kim S, Kim HY, Kang M, Jang HH, Lee WS (2016) Targeting the PI3K signaling pathway in KRAS mutant colon cancer. Cancer Med 5(2):248–255. https://doi.org/10.1002/cam4.591
Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T, Lustgarten S, Moore L, Motzer RJ, Global AT (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356(22):2271–2281. https://doi.org/10.1056/NEJMoa066838
Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, Kirkpatrick L, Powis G (2004) Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 3(7):763–772
Irahara N, Baba Y, Nosho K, Shima K, Yan L, Dias-Santagata D, Iafrate AJ, Fuchs CS, Haigis KM, Ogino S (2010) NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol 19(3):157–163. https://doi.org/10.1097/PDM.0b013e3181c93fd1
Janku F, Wheler JJ, Naing A, Falchook GS, Hong DS, Stepanek VM, Fu S, Piha-Paul SA, Lee JJ, Luthra R, Tsimberidou AM, Kurzrock R (2013) PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res 73(1):276–284. https://doi.org/10.1158/0008-5472.CAN-12-1726
Janku F, Yap TA, Meric-Bernstam F (2018) Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol 15:273–291. https://doi.org/10.1038/nrclinonc.2018.28
Jass JR (2006) Colorectal cancer: a multipathway disease. Crit Rev Oncog 12(3–4):273–287. https://doi.org/10.1615/CritRevOncog.v12.i3-4.50
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, Middleton MR, Berlin J, Schuler M, Gil-Martin M, Rugo HS, Seggewiss-Bernhardt R, Huang A, Bootle D, Demanse D, Blumenstein L, Coughlin C, Quadt C, Baselga J (2018) Phosphatidylinositol 3-Kinase alpha-selective inhibition with Alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol 36(13):1291-1299. https://doi.org/10.1200/JCO.2017.72.7107
Kim A, Lee JE, Lee SS, Kim C, Lee SJ, Jang WS, Park S (2013) Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int J Cancer 133(4):984–996. https://doi.org/10.1002/ijc.28073
Lievre A, Blons H, Laurent-Puig P (2010) Oncogenic mutations as predictive factors in colorectal cancer. Oncogene 29(21):3033–3043. https://doi.org/10.1038/onc.2010.89
Lievre A, Ouine B, Canet J, Cartier A, Amar Y, Cacheux W, Mariani O, Guimbaud R, Selves J, Lecomte T, Guyetant S, Bieche I, Berger F, de Koning L (2017) Protein biomarkers predictive for response to anti-EGFR treatment in RAS wild-type metastatic colorectal carcinoma. Br J Cancer 117(12):1819–1827. https://doi.org/10.1038/bjc.2017.353
Liu P, Cheng H, Roberts TM, Zhao JJ (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 8(8):627–644. https://doi.org/10.1038/nrd2926
Lorusso PM (2016) Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol 34(31):3803–3815. https://doi.org/10.1200/JCO.2014.59.0018
Lubomierski N, Plotz G, Wormek M, Engels K, Kriener S, Trojan J, Jungling B, Zeuzem S, Raedle J (2005) BRAF mutations in colorectal carcinoma suggest two entities of microsatellite-unstable tumors. Cancer 104(5):952–961. https://doi.org/10.1002/cncr.21266
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7(7):1851–1863. https://doi.org/10.1158/1535-7163.MCT-08-0017
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chene P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, Garcia-Echeverria C, Sellers WR, Voliva CF (2012) Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther 11(2):317–328. https://doi.org/10.1158/1535-7163.MCT-11-0474
Malesci A, Laghi L, Bianchi P, Delconte G, Randolph A, Torri V, Carnaghi C, Doci R, Rosati R, Montorsi M, Roncalli M, Gennari L, Santoro A (2007) Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin Cancer Res 13(13):3831–3839. https://doi.org/10.1158/1078-0432.CCR-07-0366
Malkomes P, Lunger I, Luetticke A, Oppermann E, Haetscher N, Serve H, Holzer K, Bechstein WO, Rieger MA (2016) Selective AKT inhibition by MK-2206 represses colorectal cancer-initiating stem cells. Ann Surg Oncol 23(9):2849–2857. https://doi.org/10.1245/s10434-016-5218-z
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274. https://doi.org/10.1016/j.cell.2007.06.009
Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169(3):381–405. https://doi.org/10.1016/j.cell.2017.04.001
Markham A (2017) Copanlisib: first global approval. Drugs 77(18):2057–2062. https://doi.org/10.1007/s40265-017-0838-6
Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutvoet S, Birle D, Anak O, Hackl W, Baselga J (2012) Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol 23(9):2399–2408. https://doi.org/10.1093/annonc/mds011
Markowitz SD, Bertagnolli MM (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361(25):2449–2460. https://doi.org/10.1056/NEJMra0804588
McRee AJ, Davies JM, Sanoff HG, Goldberg RM, Bernard S, Dees EC, Keller K, Ivanova A, O’Neil BH (2014) A phase I trial of everolimus in combination with 5-FU/LV, mFOLFOX6 and mFOLFOX6 plus panitumumab in patients with refractory solid tumors. Cancer Chemother Pharmacol 74(1):117–123. https://doi.org/10.1007/s00280-014-2474-0
McRee AJ, Sanoff HK, Carlson C, Ivanova A, O’Neil BH (2015) A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Investig New Drugs 33(6):1225–1231. https://doi.org/10.1007/s10637-015-0298-3
Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A, Risio M, Capussotti L, Marsoni S, Di Nicolantonio F, Bardelli A, Comoglio PM, Trusolino L, Bertotti A (2012) Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res 18(9):2515–2525. https://doi.org/10.1158/1078-0432.CCR-11-2683
Mohamed A, Twardy B, AbdAllah N, Akhras A, Ismail H, Zordok M, Schrapp K, Attumi T, Tesfaye A, El-Rayes B (2018) Clinical impact of PI3K/BRAF mutations in RAS wild metastatic colorectal cancer: meta-analysis results. J Gastrointest Cancer. https://doi.org/10.1007/s12029-018-0062-y
Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A, Group R-S (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372(9637):449–456. https://doi.org/10.1016/S0140-6736(08)61039-9
Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, Downward J (2014) RAS interaction with PI3K p110alpha is required for tumor-induced angiogenesis. J Clin Invest 124(8):3601–3611. https://doi.org/10.1172/JCI74134
Nassif NT, Lobo GP, Wu X, Henderson CJ, Morrison CD, Eng C, Jalaludin B, Segelov E (2004) PTEN mutations are common in sporadic microsatellite stable colorectal cancer. Oncogene 23(2):617–628. https://doi.org/10.1038/sj.onc.1207059
Ng K, Tabernero J, Hwang J, Bajetta E, Sharma S, Del Prete SA, Arrowsmith ER, Ryan DP, Sedova M, Jin J, Malek K, Fuchs CS (2013) Phase II study of everolimus in patients with metastatic colorectal adenocarcinoma previously treated with bevacizumab-, fluoropyrimidine-, oxaliplatin-, and irinotecan-based regimens. Clin Cancer Res 19(14):3987–3995. https://doi.org/10.1158/1078-0432.CCR-13-0027
O’Brien MJ, Yang S, Mack C, Xu H, Huang CS, Mulcahy E, Amorosino M, Farraye FA (2006) Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 30(12):1491–1501. https://doi.org/10.1097/01.pas.0000213313.36306.85
Oliveira C, Westra JL, Arango D, Ollikainen M, Domingo E, Ferreira A, Velho S, Niessen R, Lagerstedt K, Alhopuro P, Laiho P, Veiga I, Teixeira MR, Ligtenberg M, Kleibeuker JH, Sijmons RH, Plukker JT, Imai K, Lage P, Hamelin R, Albuquerque C, Schwartz S Jr, Lindblom A, Peltomaki P, Yamamoto H, Aaltonen LA, Seruca R, Hofstra RM (2004) Distinct patterns of KRAS mutations in colorectal carcinomas according to germline mismatch repair defects and hMLH1 methylation status. Hum Mol Genet 13(19):2303–2311. https://doi.org/10.1093/hmg/ddh238
Oliveira C, Velho S, Moutinho C, Ferreira A, Preto A, Domingo E, Capelinha AF, Duval A, Hamelin R, Machado JC, Schwartz S Jr, Carneiro F, Seruca R (2007) KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene 26(1):158–163. https://doi.org/10.1038/sj.onc.1209758
Osmanbeyoglu HU, Toska E, Chan C, Baselga J, Leslie CS (2017) Pancancer modelling predicts the context-specific impact of somatic mutations on transcriptional programs. Nat Commun 8:14249. https://doi.org/10.1038/ncomms14249
Papadatos-Pastos D, Rabbie R, Ross P, Sarker D (2015) The role of the PI3K pathway in colorectal cancer. Crit Rev Oncol Hematol 94(1):18–30. https://doi.org/10.1016/j.critrevonc.2014.12.006
Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Weiss GJ, Sachdev JC, Chadha M, Fulk M, Ejadi S, Mountz JM, Lotze MT, Toledo FG, Chu E, Jeffers M, Pena C, Xia C, Reif S, Genvresse I, Ramanathan RK (2016) First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol 27(10):1928–1940. https://doi.org/10.1093/annonc/mdw282
Pavel AB, Sonkin D, Reddy A (2016) Integrative modeling of multi-omics data to identify cancer drivers and infer patient-specific gene activity. BMC Syst Biol 10:16. https://doi.org/10.1186/s12918-016-0260-9
Pitts TM, Newton TP, Bradshaw-Pierce EL, Addison R, Arcaroli JJ, Klauck PJ, Bagby SM, Hyatt SL, Purkey A, Tentler JJ, Tan AC, Messersmith WA, Eckhardt SG, Leong S (2014) Dual pharmacological targeting of the MAP kinase and PI3K/mTOR pathway in preclinical models of colorectal cancer. PLoS One 9(11):e113037. https://doi.org/10.1371/journal.pone.0113037
Price T, Kim TW, Li J, Cascinu S, Ruff P, Suresh AS, Thomas A, Tjulandin S, Guan X, Peeters M (2016) Final results and outcomes by prior bevacizumab exposure, skin toxicity, and hypomagnesaemia from ASPECCT: randomized phase 3 non-inferiority study of panitumumab versus cetuximab in chemorefractory wild-type KRAS exon 2 metastatic colorectal cancer. Eur J Cancer 68:51–59. https://doi.org/10.1016/j.ejca.2016.08.010
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D (2011) RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11(11):761–774. https://doi.org/10.1038/nrc3106
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE (2002) Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 418(6901):934. https://doi.org/10.1038/418934a
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S (2003) Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 349(3):247–257. https://doi.org/10.1056/NEJMoa022289
Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10(3):143–153. https://doi.org/10.1038/nrclinonc.2013.10
Rodon J, Brana I, Siu LL, De Jonge MJ, Homji N, Mills D, Di Tomaso E, Sarr C, Trandafir L, Massacesi C, Eskens F, Bendell JC (2014) Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig New Drugs 32(4):670–681. https://doi.org/10.1007/s10637-014-0082-9
Roper J, Richardson MP, Wang WV, Richard LG, Chen W, Coffee EM, Sinnamon MJ, Lee L, Chen PC, Bronson RT, Martin ES, Hung KE (2011) The dual PI3K/mTOR inhibitor NVP-BEZ235 induces tumor regression in a genetically engineered mouse model of PIK3CA wild-type colorectal cancer. PLoS One 6(9):e25132. https://doi.org/10.1371/journal.pone.0025132
Rozengurt E, Soares HP, Sinnet-Smith J (2014) Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther 13(11):2477–2488. https://doi.org/10.1158/1535-7163.MCT-14-0330
Samatar AA, Poulikakos PI (2014) Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13(12):928–942. https://doi.org/10.1038/nrd4281
Sanches JM, Figueiredo J, Fonseca M, Duraes C, Melo S, Esmenio S, Seruca R (2015) Quantification of mutant E-cadherin using bioimaging analysis of in situ fluorescence microscopy. A new approach to CDH1 missense variants. Eur J Hum Genet 23(8):1072–1079. https://doi.org/10.1038/ejhg.2014.240
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307(5712):1098–1101. https://doi.org/10.1126/science.1106148
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22(2):159–168. https://doi.org/10.1016/j.molcel.2006.03.029
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, Clarke PA, Raynaud FI, Levy G, Ware JA, Mazina K, Lin R, Wu J, Fredrickson J, Spoerke JM, Lackner MR, Yan Y, Friedman LS, Kaye SB, Derynck MK, Workman P, de Bono JS (2015) First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res 21(1):77–86. https://doi.org/10.1158/1078-0432.CCR-14-0947
Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168(6):960–976. https://doi.org/10.1016/j.cell.2017.02.004
Schultz RM, Merriman RL, Andis SL, Bonjouklian R, Grindey GB, Rutherford PG, Gallegos A, Massey K, Powis G (1995) In vitro and in vivo antitumor activity of the phosphatidylinositol-3-kinase inhibitor, wortmannin. Anticancer Res 15(4):1135–1139
Sebolt-Leopold JS, Herrera R (2004) Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 4(12):937–947. https://doi.org/10.1038/nrc1503
Semba S, Itoh N, Ito M, Harada M, Yamakawa M (2002) The in vitro and in vivo effects of 2-(4-morpholinyl)-8-phenyl-chromone (LY294002), a specific inhibitor of phosphatidylinositol 3′-kinase, in human colon cancer cells. Clin Cancer Res 8(6):1957–1963
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J (2008) NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 68(19):8022–8030. https://doi.org/10.1158/0008-5472.CAN-08-1385
Shapiro GI, Bell-McGuinn KM, Molina JR, Bendell J, Spicer J, Kwak EL, Pandya SS, Millham R, Borzillo G, Pierce KJ, Han L, Houk BE, Gallo JD, Alsina M, Brana I, Tabernero J (2015) First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin Cancer Res 21(8):1888–1895. https://doi.org/10.1158/1078-0432.CCR-14-1306
Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, Wick MJ, Alvarez C, Cavazos A, Mangold GL, Patnaik A (2012) The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 18(8):2316–2325. https://doi.org/10.1158/1078-0432.CCR-11-2381
Spindler KL, Sorensen MM, Pallisgaard N, Andersen RF, Havelund BM, Ploen J, Lassen U, Jakobsen AK (2013) Phase II trial of temsirolimus alone and in combination with irinotecan for KRAS mutant metastatic colorectal cancer: outcome and results of KRAS mutational analysis in plasma. Acta Oncol 52(5):963–970. https://doi.org/10.3109/0284186X.2013.776175
Takeda T, Wang Y, Bryant SH (2016) Structural insights of a PI3K/mTOR dual inhibitor with the morpholino-triazine scaffold. J Comput Aided Mol Des 30(4):323–330. https://doi.org/10.1007/s10822-016-9905-4
Temraz S, Mukherji D, Shamseddine A (2015) Dual inhibition of MEK and PI3K pathway in KRAS and BRAF mutated colorectal cancers. Int J Mol Sci 16(9):22976–22988. https://doi.org/10.3390/ijms160922976
Tenbaum SP, Ordonez-Moran P, Puig I, Chicote I, Arques O, Landolfi S, Fernandez Y, Herance JR, Gispert JD, Mendizabal L, Aguilar S, Ramon y Cajal S, Schwartz S Jr, Vivancos A, Espin E, Rojas S, Baselga J, Tabernero J, Munoz A, Palmer HG (2012) Beta-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med 18(6):892–901. https://doi.org/10.1038/nm.2772
Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M (2014) The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol 53(7):852–864. https://doi.org/10.3109/0284186X.2014.895036
Thorpe LM, Yuzugullu H, Zhao JJ (2015) PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15(1):7–24. https://doi.org/10.1038/nrc3860
Tolcher AW, Peng W, Calvo E (2018) Rational approaches for combination therapy strategies targeting the MAP kinase pathway in solid tumors. Mol Cancer Ther 17(1):3–16. https://doi.org/10.1158/1535-7163.MCT-17-0349
Tops CM, Wijnen JT, Hes FJ (2009) Introduction to molecular and clinical genetics of colorectal cancer syndromes. Best Pract Res Clin Gastroenterol 23(2):127–146. https://doi.org/10.1016/j.bpg.2009.02.002
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA Cancer J Clin 65(2):87–108. https://doi.org/10.3322/caac.21262
van Geel R, Tabernero J, Elez E, Bendell JC, Spreafico A, Schuler M, Yoshino T, Delord JP, Yamada Y, Lolkema MP, Faris JE, Eskens F, Sharma S, Yaeger R, Lenz HJ, Wainberg ZA, Avsar E, Chatterjee A, Jaeger S, Tan E, Maharry K, Demuth T, Schellens JHM (2017) A phase Ib dose-escalation study of Encorafenib and Cetuximab with or without Alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov 7(6):610–619. https://doi.org/10.1158/2159-8290.CD-16-0795
Vanhaesebroeck B, Stephens L, Hawkins P (2012) PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol 13(3):195–203. https://doi.org/10.1038/nrm3290
Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, Boehm JS, Reiling JH, Silver S, Lu Y, Stemke-Hale K, Dutta B, Joy C, Sahin AA, Gonzalez-Angulo AM, Lluch A, Rameh LE, Jacks T, Root DE, Lander ES, Mills GB, Hahn WC, Sellers WR, Garraway LA (2009) AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16(1):21–32. https://doi.org/10.1016/j.ccr.2009.04.012
Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S Jr, Duval A, Carneiro F, Machado JC, Hamelin R, Seruca R (2005) The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer 41(11):1649–1654. https://doi.org/10.1016/j.ejca.2005.04.022
Velho S, Moutinho C, Cirnes L, Albuquerque C, Hamelin R, Schmitt F, Carneiro F, Oliveira C, Seruca R (2008) BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer 8:255. https://doi.org/10.1186/1471-2407-8-255
Velho S, Corso G, Oliveira C, Seruca R (2010) KRAS signaling pathway alterations in microsatellite unstable gastrointestinal cancers. Adv Cancer Res 109:123–143. https://doi.org/10.1016/B978-0-12-380890-5.00004-1
Vilar E, Gruber SB (2010) Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 7(3):153–162. https://doi.org/10.1038/nrclinonc.2009.237
Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D (2009) Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer 9(7):489–499. https://doi.org/10.1038/nrc2645
Welch S, Spithoff K, Rumble RB, Maroun J, Gastrointestinal Cancer Disease Site G (2010) Bevacizumab combined with chemotherapy for patients with advanced colorectal cancer: a systematic review. Ann Oncol 21(6):1152–1162. https://doi.org/10.1093/annonc/mdp533
Weng W, Feng J, Qin H, Ma Y (2015) Molecular therapy of colorectal cancer: progress and future directions. Int J Cancer 136(3):493–502. https://doi.org/10.1002/ijc.28722
Wheler J, Mutch D, Lager J, Castell C, Liu L, Jiang J, Traynor AM (2017) Phase I dose-escalation study of Pilaralisib (SAR245408, XL147) in combination with paclitaxel and carboplatin in patients with solid tumors. Oncologist 22(4):377–e337. https://doi.org/10.1634/theoncologist.2016-0257
Will M, Qin AC, Toy W, Yao Z, Rodrik-Outmezguine V, Schneider C, Huang X, Monian P, Jiang X, de Stanchina E, Baselga J, Liu N, Chandarlapaty S, Rosen N (2014) Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov 4(3):334–347. https://doi.org/10.1158/2159-8290.CD-13-0611
Wise-Draper TM, Moorthy G, Salkeni MA, Karim NA, Thomas HE, Mercer CA, Beg MS, O’Gara S, Olowokure O, Fathallah H, Kozma SC, Thomas G, Rixe O, Desai P, Morris JC (2017) A phase Ib study of the dual PI3K/mTOR inhibitor Dactolisib (BEZ235) combined with Everolimus in patients with advanced solid malignancies. Target Oncol 12(3):323–332. https://doi.org/10.1007/s11523-017-0482-9
Wolpin BM, Ng K, Zhu AX, Abrams T, Enzinger PC, McCleary NJ, Schrag D, Kwak EL, Allen JN, Bhargava P, Chan JA, Goessling W, Blaszkowsky LS, Supko JG, Elliot M, Sato K, Regan E, Meyerhardt JA, Fuchs CS (2013) Multicenter phase II study of tivozanib (AV-951) and everolimus (RAD001) for patients with refractory, metastatic colorectal cancer. Oncologist 18(4):377–378. https://doi.org/10.1634/theoncologist.2012-0378
Yang Q, Modi P, Newcomb T, Queva C, Gandhi V (2015) Idelalisib: first-in-class PI3K felta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res 21(7):1537–1542. https://doi.org/10.1158/1078-0432.CCR-14-2034
Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW (2011) First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 29(35):4688–4695. https://doi.org/10.1200/JCO.2011.35.5263
Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ (2007) PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest 117(3):730–738. https://doi.org/10.1172/JCI28984
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Fernandes, M.S., Sanches, J.M., Seruca, R. (2018). Targeting the PI3K Signalling as a Therapeutic Strategy in Colorectal Cancer. In: Jordan, P. (eds) Targeted Therapy of Colorectal Cancer Subtypes. Advances in Experimental Medicine and Biology, vol 1110. Springer, Cham. https://doi.org/10.1007/978-3-030-02771-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-02771-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-02770-4
Online ISBN: 978-3-030-02771-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)