
Abstract
Purpose
This phase I study investigated the safety, dose-limiting toxicity, and efficacy in three cohorts all treated with the mTOR inhibitor everolimus that was delivered (1) in combination with 5-fluorouracil with leucovorin (5-FU/LV), (2) with mFOLFOX6 (5-FU/LV + oxaliplatin), and (3) with mFOLFOX6 + panitumumab in patients with refractory solid tumors.
Methods
Patients were accrued using a 3-patient cohort design consisting of two sub-trials in which the maximum tolerated combination (MTC) and dose-limiting toxicity (DLT) of everolimus and 5-FU/LV was established in Sub-trial A and of everolimus in combination with mFOLFOX6 and mFOLFOX6 plus panitumumab in Sub-trial B.
Results
Thirty-six patients were evaluable for toxicity, 21 on Sub-trial A and 15 on Sub-trial B. In Sub-trial A, DLT was observed in 1/6 patients enrolled on dose level 1A and 2/3 patients in level 6A. In Sub-trial B, 2/3 patients experienced DLT on level 1B and subsequent patients were enrolled on level 1B-1 without DLT. Three of six patients in cohort 2B-1 experienced grade 3 mucositis, and further study of the combination of everolimus, mFOLFOX6 and panitumumab was aborted. Among the 24 patients enrolled with refractory metastatic colorectal cancer, the median time on treatment was 2.7 months with 45 % of patients remaining on treatment with stable disease for at least 3 months.
Conclusions
While a regimen of everolimus in addition to 5-FU/LV and mFOLFOX6 appears safe and tolerable, the further addition of panitumumab resulted in an unacceptable level of toxicity that cannot be recommended for further study. Further investigation is warranted to better elucidate the role which mTOR inhibitors play in patients with refractory solid tumors, with a specific focus on mCRC as a potential for the combination of this targeted and cytotoxic therapy in future studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer remains the third most common cause of cancer diagnosed in the USA with estimated 50,000 deaths in 2012 [1]. Approximately one-fourth of patients will present with metastatic disease at the time of diagnosis with average life expectancies of about 2 years. The backbone of treatment for patients with metastatic colorectal cancer (mCRC) remains 5-fluorouracil (5-FU), a nucleoside analog that when infused with oxaliplatin (mFOLFOX6) results in median survival rates of approximately 18–20 months. More recently, the repertoire of agents approved to treat mCRC has expanded to include targeted agents that take advantage of key signal transduction pathways involved in colon cancer tumorigenesis. Notable among these is the EGFR pathway, which in tumors expressing wild-type KRAS proteins can be inhibited with antibodies that target the receptor–ligand interaction. EGFR activation results in activation of KRAS, a downstream target of which is the phosphatidylinositide 3-kinase (PI3 K) pathway. The PI3 K pathway plays a key role in cell growth and survival mediated at least in part via activation of mammalian target of rapamycin (mTOR), an intracellular protein kinase implicated in cell cycle control [2].
Everolimus (RAD001) is a novel macrolide derivative of rapamycin that acts by selectively inhibiting mTOR. It is currently approved for the treatment of metastatic renal cell carcinomas and pancreatic neuroendocrine tumors [3]. In vitro studies of everolimus demonstrate inhibition of the proliferation of numerous solid tumor cell lines, including CRC cell lines harboring mutations in KRAS and PIK3CA, the latter of which encodes the active subunit of PI3 K and is altered in 10–30 % of CRC tumors [4]. Everolimus has also been shown to inhibit growth of CRC tumor xenografts in vivo both as a single agent and in combination with chemotherapeutics and additional targeted agents [5, 6].
Studies of single-agent everolimus in refractory solid tumors have not produced a strong signal for activity in colorectal cancer [7]. Three phase II trials have targeted the drug specifically for refractory CRC with the majority of patients achieving stable disease but with disappointing objective response rates [8–10]. Preclinical data on colorectal cancer cell lines and xenografts suggest that mTOR inhibition alone results in increased activation of EGFR and only transient inhibition of the PI3 K pathway [11]. Subsequent cotreatment with the EGFR inhibitor erlotinib has demonstrated more prolonged suppression of the mTOR pathway and resulted in tumor shrinkage. Temsirolimus, an IV administered rapalogue of everolimus, has also been shown to decrease resistance to cetuximab in colon cancer cell lines [12].
With these combinations, however, comes the risk of overlapping toxicity that may limit the dose of everolimus used. An earlier trial of temsirolimus combined with infusional 5-FU in patients with refractory solid tumors reported mucositis as a significant dose-limiting toxicity resulting in two deaths from bowel perforation [13]. Given these concerns, balanced with the potential benefit of inhibiting the PI3 K/AKT/mTOR pathway, we proposed a study investigating the feasibility of everolimus in combination with commonly used chemotherapy backbones for the treatment of mCRC. We developed a phase I trial to determine the dose-limiting toxicities (DLTs) and maximum tolerated combinations (MTC) of everolimus when combined with 5-FU/LV, mFOLFOX6 and mFOLFOX6 plus panitumumab in patients with refractory solid tumors.
Methods
Patient eligibility
Eligible patients for this study had histologically confirmed metastatic solid malignancies with no clearly effective standard therapeutic options available based either on prior therapy or on disease type. Patients with tumor histologies potentially sensitive to EGFR-targeted therapy were recruited preferentially. The study was amended to restrict enrollment of patients with mCRC receiving panitumumab to those with KRAS wild-type tumors after data by Amado et al. [14] were published which reported a requirement of KRAS wild-type status for panitumumab efficacy. Other inclusion criteria included the following: age ≥18 years; Eastern Cooperative Oncology Group (ECOG) performance status of 0–2; evaluable disease by Response Evaluation Criteria in Solid Tumors (RECIST); and a minimum of 3 weeks since major surgery and completion of radiation or completion of all prior systemic anticancer therapy. Patients were required to have adequate organ function, including an absolute neutrophil count (ANC) ≥1,500 cells/mm3, a platelet count ≥100,000/mm3, creatinine clearance ≥60 ml/min as calculated by the Cockcroft–Gault formula, total bilirubin ≤1.2 mg/dl, transaminases ≤2.5 × ULN (or ≤5 × ULN in cases of known liver metastases or primary liver cancer) and magnesium greater than or equal to the lower limits of normal. Patients enrolled on study were required to discontinue all medications that are known inducers or inhibitors of cytochrome P450 3A4 (CYP3A4). Additional exclusion criteria included the presence of grade 2 or greater neuropathy at the time of study entry; pregnancy, breastfeeding or unwilling to use contraception; impairment of gastrointestinal function leading to altered absorption; active bleeding; and any concurrent life-threatening acute medical problem at the discretion of the investigator.
This study received approval from the Institutional Review Board (IRB) of the University of North Carolina at Chapel Hill. This trial was registered with United States National Institutes of Health (trial number: NCT00610948). Written informed consent was obtained from all participants prior to any study-related procedures or treatment.
Study design
The primary objective of this study was to determine the MTC of everolimus in combination with each of the following: 5-FU/LV, mFOLFOX6 and mFOLFOX6 plus panitumumab. Secondary objectives included determining the adverse event profile of the above combinations and evaluating preliminary evidence of anti-tumor activity using RECIST criteria. Toxicity was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 3.0. Any grade 3 or 4 non-hematologic toxicity that occurred during the first cycle of treatment (28 days) was considered a DLT with the exception of grade 3 skin rash or grade 3 nausea/grade 3 diarrhea that could be controlled within 1 week. An ANC of less than 500 cells/mm3 lasting more than 7 days or associated with fever or infection OR a platelet count of less than 25,000/mm3 with major bleeding was considered dose limiting. No intra-patient dose escalation was permitted. Any patient who received at least 1 day of treatment with everolimus was evaluable for toxicity provided they were not replaced. Patients who received therapy for at least one cycle (28 days) or whose treatment was discontinued due to DLT were evaluable for DLT.
This study was conducted serially in two parts: (1) Sub-trial A consisting of everolimus plus 5-FU/LV followed by (2) Sub-trial B consisting of everolimus plus mFOLFOX6 followed by everolimus plus mFOLFOX6 and panitumumab. A separate, parallel trial was conducted with the combination of everolimus and panitumumab. That trial will be reported separately; however, it did help inform the dose escalation of the current trial. Patients in Sub-trial A were treated with 5-FU/LV that was given on Days 1 and 15 of each cycle as a bolus of 5-FU given after LV, followed by a 46-h infusion of 5-FU via ambulatory pump. Everolimus was initially administered as a weekly oral dose, but the study was subsequently amended to daily dosing. Enrollment onto Sub-trial B commenced at one dose lower than the MTC of everolimus and 5-FU/LV as determined in Sub-trial A based on toxicity observed in a prior study of temsirolimus and 5-FU. To start a new treatment of FOLFOX on Day 15 or to initiate a new cycle of therapy, patients were required to have an absolute neutrophil count (ANC) of ≥1,000 cells/mm3 and platelets ≥100,000/mm3 with all toxicities resolved to grade 1 or to entry criteria.
Both sub-trials utilized a standard “3 + 3” trial design. The dose escalation schema is represented in Table 1. If no DLT was observed at the initial dose level after 3 patients completed one cycle of therapy, accrual to the subsequent dose level was permitted. If 1/3 patients in any one dose level experienced DLT, then that cohort was expanded to 6 patients. If a minimum of 2/6 patients in the expanded cohort experienced DLT, the MTC was deemed to be exceeded and the estimated MTC was defined as one dose level lower. If the MTC was surpassed in the first dose level cohort, a dose cohort minus one was utilized.
Baseline assessments prior to treatment consisted of a medical history and physical examination, an assessment of ECOG performance status, chest X-ray, baseline laboratory measurements and an imaging study of evaluable disease. Treatment assessments, including symptom-directed history and physical examination as well as toxicity assessments, were required on a weekly basis during the first 4 weeks of therapy, and then on the first day of each 28 day cycle thereafter. Tumor response was assessed according to RECIST criteria every two cycles (8 weeks) with imaging modalities deemed appropriate by the investigator.
Results
Patient characteristics
A total of thirty-nine patients were treated at a single institution on study from March 2008 until March 2013, with 21 patients enrolled on Sub-trial A and 18 patients on Sub-trial B. Three patients were replaced and hence not evaluable for DLT due to their discontinuation of treatment prior to completing cycle one without DLT (reasons being patient preference, oxaliplatin infusion reaction on day 1 of treatment and insurance denial). The remaining thirty-six received everolimus in combination with 5-FU/LV, mFOLFOX6 or mFOLFOX6 plus panitumumab for at least one cycle (28 days) unless discontinued for DLT and were evaluable for toxicity (Table 2). The median age of all patients enrolled was 57 years (range 35–77). Overall, 24 women and 14 men were treated on study. The majority of patients enrolled on both arms were Caucasian with a higher proportion of African Americans enrolled on Sub-trial B (25 versus 0 %). Twenty-four (67 %) patients enrolled had mCRC. Patients had received a median of two prior therapies.
DLT and determination of MTC
DLT associated with everolimus and chemotherapy is summarized in Table 3. Patients were required to keep a pill diary of their everolimus administration; no issues with compliance were documented. In Sub-trial A, DLT (angioedema) was observed in one of three patients enrolled on dose level 1A. The remaining two patients remained on study for 1 and 6 cycles, respectively. Three additional patients for a total of six were enrolled on this cohort with no additional DLT observed. One patient in cohort 1A experienced a dose delay during cycle 5 secondary to an upper respiratory infection not related to treatment. Subsequent cohorts 2A, 3A, 4A and 5A were completed without DLT. Two out of three patients in cohort 2A had dose delays or reductions for neutropenia and elevated liver enzymes (in cycles 2 and 3, respectively). One patient in cohort 3A experienced a 2-week delay of 5-FU and everolimus in cycle 5 for diarrhea and mucositis. All three patients in cohort 4A had dose reductions (in cycles 2, 3 and 4) for non-dose-limiting toxicity, namely mucositis and hyponatremia. Two of three patients experienced DLT in level 6A, consisting of grade 3 hypophosphatemia, grade 3 hypokalemia and grade 3 hyperglycemia. Therefore, the MTC of Sub-trial A was determined to be 5 mg of everolimus and 5-FU bolus of 400 mg/m2 followed by 5-FU infusion of 2,400 mg/m2. Patients subsequently enrolling on Sub-trial B were initiated on one dose cohort lower (5 mg of everolimus and 5-FU bolus of 400 mg/m2 followed by 5-FU infusion of 1,800 mg).
Within Sub-trial B, one of three patients enrolled on level 1B experienced DLT, specifically grade 3 hypophosphatemia and grade 3 fatigue. The other two patients experienced dose delays in cycles 3 and 5 for neutropenia, thrombocytopenia, fatigue and mucositis. An expansion of three additional patients was planned to this cohort; however, the first patient enrolled in the expansion cohort experienced DLT of grade 3 mucositis and grade 3 dehydration. Thus, subsequent patients were enrolled on level 1B-1 with an oxaliplatin dose of 65 mg/m2. All five patients enrolled at this dose level received treatment without DLT. Two patients in this cohort reported non-dose-limiting toxicity (thrombocytopenia and neutropenia) in cycle 2 that required dose delays or reductions. The final cohort 2B-1 was then initiated with the addition of panitumumab to everolimus and mFOLFOX6. One of three patients in this cohort experienced DLT of grade 3 mucositis. Therefore, the cohort was expanded to six patients. In total, three out of the six patients in cohort 2B-1 experienced grade 3 mucositis. Other adverse events that required dose delays or reductions in this cohort occurred in five out of six patients and included neutropenia, thrombocytopenia, rash, pneumonitis, neuropathy, fatigue and hypokalemia. Therefore, further study of the combination of everolimus, mFOLFOX6 and panitumumab was aborted due to excessive toxicity in spite of reduced doses of all agents.
Adverse events
All patients enrolled on study reported at least one treatment-related adverse event with 33 % of patients in Sub-trial A and 80 % in Sub-trial B reporting at least one grade ≥3 adverse event (Tables 4, 5). The most commonly reported treatment-related toxicity overall in both groups was mucositis. For intolerable grade 2 mucositis, everolimus was held until recovery to ≤grade 1 with reintroduction at the same dose. If grade 2 toxicity recurred, the everolimus dose was reduced. For grades 3 and 4 mucositis, everolimus was held until recovery to entry criteria after which it was restarted at the next lowest dose level and a dose reduction occurred.
In Sub-trial A, other common treatment-related toxicities (all grades) included hypercholesterolemia (62 %), fatigue (52 %), anemia (52 %), leukopenia (48 %), nausea (38 %) and neutropenia (38 %). A total of 12 grade ≥3 treatment-related AEs were reported, including mucositis (n = 3), hypophosphatemia (n = 2), fatigue (n = 1), neutropenia (n = 1), thrombocytopenia (n = 1), anorexia (n = 1), diarrhea (n = 1), hyperglycemia (n = 1), hypokalemia (n = 1) and angioedema (n = 1).
In Sub-trial B, a total of 47 AEs were reported among 15 patients. Across all arms, the most common treatment-related AEs (all grades) were mucositis (67 %), neutropenia (53 %), fatigue (53 %), diarrhea (47 %), anemia (47 %) and thrombocytopenia (47 %). Within Arm 1B, thirteen treatment-related grade ≥3 AEs were reported, including dehydration, diarrhea, fatigue, anemia, mucositis, neutropenia, thrombocytopenia and hypokalemia. In level 1B-1, only four treatment-related grade ≥3 AEs were reported, specifically diarrhea, neutropenia, thrombocytopenia and an oxaliplatin-related infusion reaction. Eight treatment-related grade ≥3 AEs were reported in level 2B-1 and included diarrhea, anemia, mucositis, neutropenia and thrombocytopenia. Only two incidents of acneiform rash were reported among the six patients who received panitumumab, both of which were graded ≤3 in severity.
Clinical benefit
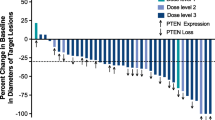
Of the 36 patients evaluated for toxicity on trial, 32 were evaluable for response. Four patients were not evaluable given that they discontinued therapy due to toxicity prior to two cycles of treatment being completed. The median number of cycles received by all evaluable patients was three (range 1–19 in Sub-trial A, range 1–7 in Sub-trial B). Thirteen patients experienced progressive disease at initial evaluation. In Sub-trial B, two patients (diagnosed with cholangiocarcinoma and esophageal adenocarcinoma) had unconfirmed partial responses (PR) and remained on trial for three and seven cycles, respectively. Both of these patients received treatment with everolimus, mFOLFOX6 and panitumumab. Seventeen patients (53 %) achieved a best response of stable disease with 13 of those patients maintaining stable disease for ≥3 months. Among the 24 patients enrolled with mCRC, the median time on treatment was 2.69 months with 11 patients (45 %) remaining on treatment with stable disease for at least 3 months (Fig. 1). Two refractory mCRC patients in Sub-trial A remained on treatment for 21.84 and 10.31 months, respectively, with stable disease.

Time on treatment for all mCRC patients
Discussion
This phase I study was conducted to determine the DLT and MTC of everolimus when combined with 5-FU/LV, mFOLFOX6 and mFOLFOX6 plus panitumumab in patients with refractory solid tumors. It was comprised of two sub-trials that utilized the MTC of everolimus with 5-FU/LV to further evaluate the safety profile of everolimus in combination with doublet chemotherapeutics and an EGFR inhibitor. While a regimen of everolimus in addition to 5-FU/LV and mFOLFOX6 appears safe and tolerable, the further addition of panitumumab resulted in an unacceptable level of toxicity and that combination cannot be recommended for further study.
In reviewing Sub-trial A, in which patients received increasing doses of everolimus combined with 5-FU/LV, the MTC was determined to be 5 mg of daily everolimus combined with standard doses of bolus 5-FU followed LV and a 46 h 5-FU infusion (400 and 2,400 mg/m2, respectively). Dose-limiting toxicities in these patients were primarily metabolic, consisting of hyperglycemia, hypophosphatemia and hypokalemia. Treatment with mTOR inhibitors results in a rise in glucose levels via down-regulation of mRNA translation of glucose transporters, specifically GLUT-5 [15]. Similarly, low phosphate levels are a well-documented side effect of mTOR inhibitors and, while the mechanism is less well understood, can result in skeletal muscle weakness due to depletion of adenosine triphosphate (ATP) if phosphate levels are not properly repleted. The most commonly occurring DLT in Sub-trial B was mucositis. While approximately 80 % of patients in Sub-trial A reported any degree of mucositis, only 3 patients developed ≥grade 3 mucositis. Alternatively, in Sub-trial B, mucositis was reported in 10 out of the 15 patients treated, 30 % of who reported ≥grade 3 mucositis, mostly in those treated with panitumumab. While stomatitis is reportedly a rarer side effect of anti-EGFR antibodies, the possibility of a synergistic effect of everolimus, 5-FU/LV and panitumumab on the mucosa resulting in overlapping toxicity must be considered.
We found the MTC of everolimus to be 5 mg daily in combination with 5-FU/LV (400 mg/m2/2,400 mg/m2) and oxaliplatin (65 mg/m2) administered every 2 weeks. Overall, there were no treatment-related dose delays or reductions during the first cycle of therapy in all cohorts except 2B-1, which included panitumumab. The addition of panitumumab, even at lower than standard doses of 3.6 mg/kg, was not tolerated due to dose-limiting mucositis. In our parallel study of everolimus and panitumumab that will be reported separately, mucositis proved dose limiting leading to an inability to escalate to standardly tolerated doses of each drug when administered as a single agent. For patients with refractory mCRC, however, the combination of 5-FU-based chemotherapy with PI3 K inhibition appears to be safe and well tolerated. Median overall survival (OS) for all evaluable patients enrolled on trial with mCRC was 5.9 months, with patients on Sub-trial A exhibiting a median OS of 6.9 months. These outcomes are similar to those reported for refractory mCRC patients treated with regorafenib, an oral multi-tyrosine kinase inhibitor, which when compared to placebo resulted in an OS of 6.4 months [16]. Thus, further investigation is warranted to better elucidate the role which mTOR inhibitors play in patients with refractory solid tumors, with a specific focus on mCRC as a potential target tumor type for this combination in future studies.
References
Siegel R, Naishadham D, Jemal A (2012) Cancer statistics, 2012. CA Cancer J Clin 62:10–29
Courtney KD, Corcoran RB, Engelman JA (2010) The PI3 K pathway as drug target in human cancer. J Clin Oncol 28:1075–1083
Houghton PJ (2010) Everolimus. Clin Cancer Res 16:1368–1372
Samuels Y, Wang Z, Bardelli A et al (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554
Bradshaw-Pierce EL, Pitts TM, Kulikowski G et al (2013) Utilization of quantitative in vivo pharmacology approaches to assess combination effects of everolimus and irinotecan in mouse xenograft models of colorectal cancer. PLoS ONE 8:e58089
Chu C, Noel-Hudson MS, Boige V et al (2013) Therapeutic efficiency of everolimus and lapatinib in xenograft model of human colorectal carcinoma with KRAS mutation. Fundam Clin Pharmacol 27:434–442
Altomare I, Hurwitz H (2013) Everolimus in colorectal cancer. Expert Opin Pharmacother 14:505–513
Altomare I, Bendell JC, Bullock KE et al (2011) A phase II trial of bevacizumab plus everolimus for patients with refractory metastatic colorectal cancer. Oncologist 16:1131–1137
Ng K, Tabernero J, Hwang J et al (2013) Phase II study of everolimus in patients with metastatic colorectal adenocarcinoma previously treated with bevacizumab-, fluoropyrimidine-, oxaliplatin-, and irinotecan-based regimens. Clin Cancer Res 19:3987–3995
Wolpin BM, Ng K, Zhu AX et al (2013) Multicenter phase II study of tivozanib (AV-951) and everolimus (RAD001) for patients with refractory, metastatic colorectal cancer. Oncologist 18:377–378
Wang Q, Wei F, Li C et al (2013) Combination of mTOR and EGFR kinase inhibitors blocks mTORC1 and mTORC2 kinase activity and suppresses the progression of colorectal carcinoma. PLoS ONE 8:e73175
Wang HW, Yang SH, Huang GD et al (2014) Temsirolimus enhances the efficacy of cetuximab in colon cancer through a CIP2A-dependent mechanism. J Cancer Res Clin Oncol 140:561–571
Punt CJ, Boni J, Bruntsch U, Peters M, Thielert C (2003) Phase I and pharmacokinetic study of CCI-779, a novel cytostatic cell-cycle inhibitor, in combination with 5-fluorouracil and leucovorin in patients with advanced solid tumors. Ann Oncol 14:931–937
Amado RG, Wolf M, Peeters M et al (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26:1626–1634
Soefje SA, Karnad A, Brenner AJ (2011) Common toxicities of mammalian target of rapamycin inhibitors. Target Oncol 6:125–129
Grothey A, Van Cutsem E, Sobrero A et al (2013) Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381:303–312
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
McRee, A.J., Davies, J.M., Sanoff, H.G. et al. A phase I trial of everolimus in combination with 5-FU/LV, mFOLFOX6 and mFOLFOX6 plus panitumumab in patients with refractory solid tumors. Cancer Chemother Pharmacol 74, 117–123 (2014). https://doi.org/10.1007/s00280-014-2474-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-014-2474-0