
Abstract
Phosphoinositide 3-kinase (PI3K) belongs to the intracellular lipid kinases family involved in diverse physiological processes, including proliferation, apoptosis, growth, and metabolism. Recent mutation analysis has shown that the PI3K pathway is one of the most frequently dysregulated pathways in human cancer, including colorectal cancer (CRC). Accordingly, significant effort has been made to develop pharmacological inhibitors targeting PI3K or key nodes in this pathway, such as AKT and mTOR. There are currently more than 20 unique compounds targeting the PI3K pathway being assessed in numerous cancer-related clinical trials. In addition, the mutation status of PI3K pathway in cancers may have predictive and prognostic implications. After 3 decades of the discovery of PI3K, we are now at an exciting intersection in translating our knowledge of the PI3K signaling pathway into developing effective therapeutics for the treatment of cancer. A comprehensive understanding of circuits and regulations of this pathway are essential to the rational development of such therapies. In this chapter, we will discuss recent advances in our understanding of the functions and mutations of PI3K signaling pathway in the pathogenesis of CRC. We will also review current drug-discovery efforts and challenges targeting PI3K signaling for the treatment of CRC.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Vascular Endothelial Growth Factor
- Epidermal Growth Factor Receptor
- Mutant Epidermal Growth Factor Receptor
- mTOR Inhibitor
- PI3K Inhibitor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
6.1 Introduction
Colorectal cancer (CRC) is one of the most common cancers, with an estimated 143,460 new diagnoses and 51,690 deaths in the United States in 2012. CRC remains the third most frequent cancer in the United States, as well as the third most common cause of cancer-related death, accounting for roughly 9 % of US cancer deaths for both men and women. Despite advances in screening, nearly 20 % of patients present with metastatic disease, which carries a poor prognosis, with 5-year survival rates of 12 % (Grothey 2009). While cytotoxic fluorouracil (5-FU)-based regimens have been standard of care as adjuvant and first-line metastatic therapies for CRC treatment, advances have been slow and the efficacy of these agents has reached a plateau (Cook et al. 1969). As such, recent efforts have shifted toward the development of novel therapeutic agents that inhibit specific molecular pathways. Inhibiting the epidermal growth factor receptor (EGFR) with monoclonal antibodies, such as cetuximab and panitumumab, and blocking angiogenesis with antibodies against vascular endothelial growth factor receptor (VEGFR), such as bevacizumab, are successful examples of targeted therapies in CRC. However, the clinical benefits of these targeted therapies in most cases are short-term and often limited to subgroups of patients, indicating the need for evaluating novel biomarkers and identifying new targets for drug therapy of CRC.
The phosphoinositide 3-kinase (PI3K) pathway is one of the most deregulated pathways in human cancer. Several components of this pathway, including PI3K, the v-akt murine thymoma viral oncogene homolog (AKT), and the mammalian target of rapamycin (mTOR), are druggable and plausible targets for cancer therapy. Consequently, the development of therapeutics targeting this pathway has occurred at a rapid pace over the past 10 years, and preclinical and early clinical studies are beginning to suggest strategies to increase efficacy.
In this chapter we provide a comprehensive analysis of genetic alterations in the PI3K pathway detected in CRC and discuss their value as prognostic indicators and potential roles as predictive biomarkers for anti-EGFR therapy. In order to frame the discussion, we begin with a review of the current understanding of the PI3K signaling pathway and the effects that it confers on cellular growth, proliferation, survival, and metabolism. Finally, we discuss some of the current and emerging therapeutic approaches to targeting the PI3K pathway in CRC.
6.2 The Key Players of the PI3K Pathway
6.2.1 PI3Ks
PI3Ks are divided into three classes (I, II, and III) according to their structural characteristics and lipid substrate preferences (Fig. 6.1). Different classes of PI3K also have distinct roles in cellular signaling pathways (Engelman et al. 2006). Class I enzymes are the best characterized of the PI3K classes and are the major class known to be associated with cancer. Therefore, we will mainly focus on class I PI3K throughout this chapter. All PI3K classes catalyze the phosphorylation of inositol-containing lipids, known as phosphatidylinositols (PtdIns), at the 3′-position of the inositol ring. The primary substrate of class I PI3K is phosphatidylinositol (4,5)-bisphosphate (PIP2), which is converted to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 is an important second messenger in cell physiology. Through direct interactions with pleckstrin homology (PH) domains on a wide variety of signaling proteins, including Tec family protein-tyrosine kinases, AKT family kinases, and PDK1 (3-phosphoinositide-dependent protein kinase-1) and with various guanine exchange factors (GEFs) and GTPase-activating proteins (GAPs) of low molecular weight GTP-binding proteins, PIP3 orchestrates a set of events controlling cellular growth, metabolism, proliferation, and survival. In contrast to PIP3, which is produced only from Class I PI3Ks, phosphatidylinositol-3-phosphate (PI3P) is produced by both Class II and Class III PI3Ks. This lipid binds to FYVE domains and PX domains in a variety of proteins to control intracellular membrane trafficking, especially trafficking through early endosomes. Also of interest in cancer is phosphatidylinositol-3,4-bisphosphate (PI3, 4P2), which is produced by Class II PI3Ks via phosphorylation of the 3′ position of phosphatidylinositol-4-phosphate or by SHIP family phosphatases via dephosphorylation of the 5′ position of PIP3. PI3, 4P2, like PIP3, binds to AKT and PDK1 to facilitate AKT activation, but fails to bind to most of the other PIP3 targets.

Classification and domain structure of phosphatidylinositol 3-kinase (PI3K). PI3Ks are divided into three classes (I, II, and III) based on their structural characteristics and lipid substrate preference. Class I PI3Ks are further divided into two subfamilies, IA and IB, according to the receptors with which they interact. Class IA PI3Ks are heterodimers consisting of a p110 catalytic subunit and a p85 regulatory subunit. There are three p110 catalytic isoforms: p110α, p110β, and p110δ, which all have a p85-binding domain (p85BD) at the N-terminus, followed by a Ras-binding domain (RBD), a putative membrane-binding C2 domain (C2), and helical domain (Helical D), and a C-terminal catalytic domain (Catalytic D). The p85 regulatory subunits share a core structure consisting of a p110-binding domain called the iSH2 domain, flanked by SH2 domains. The two longer isoforms, p85α and p85β, have an SH3 domain and BCR homology domain (BHD) located in their extended N-terminal region. Class IB PI3K is a heterodimer composed of the catalytic subunit p110γ and the regulatory subunit p101. p110γ is activated by G protein-coupled receptors (GPCRs). Class II PI3Ks consist of only a p110-like catalytic subunit. There are three class II PI3K isoforms: PI3KC2α, PI3KC2β, and PI3KC2γ, each of which has an extended divergent N-terminus followed by a RBD, a C2 domain, a helical domain, a catalytic domain, PX (Phox homology), and C2 domains at the C-termini. The class III PI3K consists of a heterodimer of the catalytic subunit, VPS34 (homologue of the yeast vacuolar protein 34; also known as PIK3C3 in mammal) and a regulatory subunit, VPS15 (also known as PIK3R4 in mammal). Vps15 consists of a catalytic domain (which is thought to be inactive), HEAT domains (which might mediate protein–protein interactions), and WD repeats, which have structural and functional characteristics similar to a Gβ subunit
6.2.1.1 Class IA PI3Ks and Cancer
Class IA PI3Ks have been most frequently associated with human cancer. These enzymes are heterodimers of p110 family catalytic subunits and p85 family regulatory subunits. PIK3R1 encodes p85α (and its alternative start site variants p55α and p50α). PIK3R2 and PIK3R3 encode the p85β and p55γ isoforms of the p85 regulatory subunit, respectively. This group of subunits is collectively called p85 (Engelman et al. 2006; Bader et al. 2005). The class IA p85 regulatory isoforms have a common structure composed of a p110-binding domain (also called the inter-SH2 domain, iSH2) flanked by two Src-homology 2 (SH2) domains and this core structure is conserved back to worms and flies. The two longer isoforms, p85α and p85β, have an extended N-terminal region containing a Src-homology 3 (SH3) domain and a BCR homology domain (BHD) (Fruman et al. 1998). The SH3 and BHD are speculated to have a negative regulatory role toward the catalytic activity of the p110 subunit. This is consistent with the observation that the p55α and p50α subunits are more effective activators of p110 than is p85α (Vivanco and Sawyers 2002; Inukai et al. 1997; Ueki et al. 2000). Three genes, PIK3CA, PIK3CB, and PIK3CD, encode the highly homologous p110 catalytic subunit isoforms p110α, p110β, and p110δ, respectively. These three p110 subunits are comprised of five domains; an N-terminal adaptor binding domain (ABD) that interacts with the p85 regulatory subunit, a Ras-binding domain (RBD) that mediates activation by the small GTPase Ras, a C2 domain that might be important for membrane anchoring, a helical domain, and a C-terminal catalytic domain. The last four domains have significant sequence homology among all isoforms.
The p85 regulatory subunit is essential for mediating class IA PI3K activation by receptor tyrosine kinases (RTKs), such as EGFR, platelet-derived growth factor receptor (PDGFR), and IGF-1R (insulin-like growth factor-1 receptor). The SH2 domains of p85 bind to phosphotyrosine residues arranged in a pYXXM (in which “pY” indicates a phosphorylated tyrosine) motif on activated RTKs. In some cases, the p85–RTK interactions occur indirectly through adaptor proteins, such as the insulin receptor substrates (IRS1 and IRS2) downstream of IGF-1R (Vivanco and Sawyers 2002; White 1998). Binding of p85 to RTKs or phosphoprotein intermediaries relieves the basal inhibition of p110 by p85 and recruits the p85-p110 heterodimer to the plasma membrane, where it phosphorylates the membrane lipid PIP2 to produce PIP3 (Yu et al. 1998a, b). This leads to activation of various cellular processes, such as proliferation, growth, survival, and metabolism. Interestingly, the p110β isoform is regulated not only by the p85 regulatory subunit but also by binding to Gβγ subunits of heterotrimeric G proteins, suggesting that p110β might integrate signals from GPCRs as well as RTKs (Kurosu et al. 1997; Roche et al. 1998). However, the implication of the p110β activation by GPCRs in cancer remains less well defined. p110α and p110β are expressed ubiquitously, whereas p110δ is predominantly expressed in lymphocytes. Although p110α, p110β, and p110δ have very similar structures and share the p85 regulatory subunits, numerous studies indicated that they may have distinct functions. For example, germline deletion of either p110α or p110β results in embryonic lethality (Bi et al. 1999; Foukas et al. 2006). Mice heterozygous for kinase dead mutation in p110α were viable and fertile, but showed severe defects in the insulin pathway such as hyperinsulinemia, glucose intolerance, and increased adiposity (Foukas et al. 2006). p110δ, although not essential, has an important role in the regulation of the immune compartment, especially B-cell growth (Fruman 2004). As we will be discussed in detail later, both PIK3CA (p110α) and PIK3R1 (p85α) are somatically mutated in various cancers including CRC (Ikenoue et al. 2005; Mizoguchi et al. 2004; Philp et al. 2001; Samuels et al. 2004).
6.2.1.2 Class IB PI3Ks
Similar to class IA PI3Ks, class IB PI3Ks are heterodimers composed of the catalytic subunit p110γ and the regulatory subunit p101 (Fig. 6.1). Although p110γ is highly homologous with the class IA p110 proteins, the p101 regulatory subunit is distinct from the p85 Class IA regulatory subunit. Recently, two additional regulatory subunits, p84 and p87PIKAP (PI3Kγ adaptor protein of 87 kDa), have been identified (Voigt et al. 2006; Suire et al. 2005). The regulatory subunits complexed with p110γ do not have SH2 domains, and thus do not interact with RTKs. Instead, p110γ is activated exclusively by GPCRs through direct interaction with the Gβγ subunit of trimeric G proteins. p110γ is primarily expressed in leukocytes but is also found in the heart, pancreas, liver, and skeletal muscle (Sasaki et al. 2000).
6.2.2 PIP3 Phosphatases
The main consequence of class I PI3K activation is the generation of PIP3 in the plasma membrane. PIP3 functions as a second messenger to activate effector protein kinases such as AKT. Thus, in most tissues, PIP3 has a pivotal role in the actions of insulin, growth factors, and cytokines, thereby mediating effects of diverse physiological processes including proliferation, apoptosis, growth, and metabolism. The cellular levels of PIP3 are hardly detectable in mammalian cells under unstimulated growth conditions and are tightly regulated by the opposing activity of several PIP3 phosphatases (PTEN, SHIP1, and SHIP2). PTEN (phosphatase and tensin homologue, deleted on chromosome ten), also called MMAC1 and TEP1, is an important tumor suppressor and is the PIP3 phosphatase most clearly involved in cancer. Loss of PTEN function occurs through mutations, deletions, or epigenetic silencing in a variety of human cancers at high frequency, making PTEN the second most frequently mutated tumor suppressor gene after p53 (Stokoe 2001). PTEN functionally antagonizes PI3K activity through its intrinsic lipid phosphatase activity that decreases the cellular level of PIP3 by converting PIP3 back into PtdIns (4,5)P2 (PI4,5P2) (Fig. 6.2). Thus, loss of PTEN results in constitutively active signaling through the PI3K pathway, leading to tumor development (Cully et al. 2006).

Structure and generation of phosphatidylinositol-3,4,5-trisphosphate. Phosphatidylinositol-3,4,5-triphosphate (Ptdlns(3,4,5)P3) is an essential second messenger that regulates many cellular processes. Class I PI3Ks phosphorylate the inositol ring of phosphatidylinositol-4,5-triphosphate (PtdIns(4,5)P2) at the 3-position, to generate PtdIns (3,4,5)P3. PTEN (phosphatase and tensin homologues) is a lipid phosphatase that removes phosphate at the 3-position of PtdIns (3,4,5)P3, converting it back to PtdIns (4,5)P2. Additionally, PtdIns (3,4,5)P3 can be dephosphorylated at the 5-position by SHIP1 or SHIP2 to generate PtdIns (3,4)P2
The SHIP phosphatases also act on PIP3, but remove phosphate from the 5-position instead of 3-position, generating PtdIns (3,4)P2 (PI3, 4P2) (Fig. 6.2). PI3, 4P2 can function as a second messenger to recruit certain PH-domain-containing proteins, such as AKT and PDK1 to the plasma membrane. But several PIP3 binding proteins, such as TEC family protein-Tyr kinases, fail to bind to PI3, 4P2. Therefore, although both PTEN and SHIP reduce the level of PIP3 in cells, PTEN terminates all downstream PI3K signaling, while SHIP only terminates a subset of downstream signals. To completely shut off downstream signaling, PI3, 4P2 is dephosphorylated by distinct 4-phosphatases called INPP4A and INPP4B. INPP4B has also been identified as a tumor suppressor in breast and ovarian cancers (Agoulnik et al. 2011). Targeted deletion of PTEN recapitulates many of the ramifications of PTEN loss in human cancers. Homozygous deletion of PTEN causes embryonic lethality, indicating an essential role of PTEN during embryonic development (Di Cristofano et al. 1998). Mice that are heterozygous for PTEN are viable, but have a high incidence of T-cell lymphomas, germline tumors and cancers in several epithelial tissues, including the intestine, endometrium, prostate, and mammary glands (Di Cristofano et al. 1998). Tissue-specific homozygous deletion of PTEN in the prostate epithelium leads to aggressive prostate carcinoma (Wang et al. 2003). Likewise, PTEN deletion in T cells and mammary glands causes aggressive lymphomas and breast tumors, respectively (Kishimoto et al. 2003).
6.2.3 AKT: Direct Effector of PIP3
AKT, also known as protein kinase B (PKB), the human homologue of the retroviral oncogene v-Akt, is the main downstream executor of the PI3K signaling pathway (Fig. 6.3). This serine-threonine protein kinase has three isoforms—AKT1, AKT2, and AKT3. The three isoforms share a similar structure: an amino-terminal pleckstrin homoloy (PH) domain, a central catalytic domain, and a short carboxy-terminal regulatory domain. AKT is activated by a dual regulatory mechanism that requires both translocation to the plasma membrane and phosphorylation at Thr308 and Ser473 (Alessi et al. 1997; Stephens et al. 1998). The generation of PIP3 at the plasma membrane by activated PI3K facilitates the recruitment of both AKT and a second protein kinase, PDK1, to the membrane due to the ability of the PH domains of these proteins to bind to PIP3. PIP3 binding induces a conformational change in AKT, resulting in the exposure of Thr308 for phosphorylation by PDK1. Full activation of AKT requires phosphorylation of Ser473 by TORC2, or in certain situations, another PIK-family protein kinase such as DNA-PK or ATM (Sarbassov et al. 2005). After dual-phosphorylation and activation, AKT phosphorylates an array of target proteins containing the amino acid sequence RxRxxS/T-B (where x represents any amino acid and B is any bulky hydrophobic residue) (Alessi et al. 1996). Currently, more than 100 different AKT substrates have been reported, although it is not clear that all of these are direct substrates in vivo (Manning and Cantley 2007). This variety of substrates indicates broad biological functions mediated by multiple downstream effectors.

Schematic of signaling through the PI3K/AKT pathway. PI3K signaling affects processes related to tumorigenesis such as cell survival, proliferation, growth, and metabolism. Arrows represent activation, while bars reflect inhibition
The three AKT isoforms are very similar in amino acid sequence and might have indistinguishable substrate specificity in vitro (Walker et al. 1998), yet many isoform-specific substrates may exist in vivo (Stambolic and Woodgett 2006). One possible scenario suggesting isoform-specific AKT substrates is that a different cellular localization of the three isoforms could determine their accessibility to a selective group of substrate proteins (Bhaskar and Hay 2007). Furthermore, it is possible that each AKT isoform possesses different functions, as demonstrated by germ line deletions in mice. AKT1 null mice show developmental defects especially in the thymus and testes (Chen et al. 2005; Cho et al. 2001a), AKT2 null mice have defects in glucose homeostasis (Cho et al. 2001b; Garofalo et al. 2003), and AKT3 null mice display defects in brain development (Easton et al. 2005; Tschopp et al. 2005). The relative expression of the three isoforms also differs in mammalian cells in that AKT1 is predominantly expressed in the majority of tissues, AKT2 is the predominant isoform in insulin-responsive tissues such as adipocytes and muscle tissues, while AKT3 is the predominant isoform in the brain and testis (Manning and Cantley 2007). All three AKT isoforms have been found to be mutated or amplified in in subsets of human cancers, albeit at a relatively low frequency.
6.3 Biological Effects of PI3K/AKT Activation
The primary biological effects of AKT activation can be classified into four categories—survival, proliferation (increased cell number), growth (increased cell size), and metabolism (Fig. 6.3). AKT has additional effects on tumor-induced angiogenesis that are mediated, in part, through hypoxia-inducible factor 1, alpha subunit (HIF1A), and vascular endothelial growth factor (VEGF) (Vivanco and Sawyers 2002).
6.3.1 Cell Survival
The balance of proliferation and apoptosis is critical for normal homeostasis. Increased proliferation and/or decreased apoptosis is the basis of tumorigenesis. Even before the relevant substrates were identified, several groups showed a critical role for AKT in promoting cell survival by preventing apoptosis. For example, dominant-negative alleles of AKT induce cell death (Dudek et al. 1997) and constitutively active AKT rescues PTEN-mediated apoptosis (Li et al. 1998). Later, numerous studies led to the discovery that many of the apoptosis-related proteins are directly or indirectly regulated by AKT. AKT protects cells from death by directly phosphorylating several downstream substrates that are involved in apoptosis. AKT negatively regulates the function or expression of several Bcl-2 homology domains (BH3)-only proteins, which exert their pro-apoptotic effects by binding to and inactivating pro-survival Bcl-2 family members. For instance, BAD, a BH3-only protein is a pro-apoptotic member of the Bcl-2 family of proteins that promote cell death by binding to the survival factor BCL-XL, thereby blocking the function of BCL-XL. Phosphorylation of BAD by AKT creates a binding site for 14-3-3 proteins, which triggers release of BAD from BCL-XL (Datta et al. 1997, 2000; del Peso et al. 1997). The consequence is restoration of BCL-XL’s anti-apoptotic function. AKT also inhibits the expression of BH3-only proteins through effects on FOXO transcription factors. Phosphorylation of FOXO proteins by AKT allows 14-3-3 proteins to bind to FOXOs, resulting in their inactivation through sequestration in the cytoplasm (Tran et al. 2003). Through this mechanism, AKT blocks FOXO-mediated transcription of target genes that promote apoptosis, cell-cycle arrest, and metabolic processes (see below). Two major pro-apoptotic targets of FOXO are the BH3-only protein BIM and cytokine FAS ligand (Fas L) (Dijkers et al. 2002; Brunet et al. 1999).
AKT can also influence cell survival through indirect effects on two central regulators of cell death—nuclear factor of kB (NF-kB) and p53. MDM2 is a negative regulator of p53 that targets p53 for degradation by the proteasome through its E3 ubiquitin ligase activity. AKT phosphorylates MDM2, promoting its translocation to the nucleus where it binds to p53 to promote ubiquitination and degradation (Mayo and Donner 2001; Zhou et al. 2001). The BH3-only proteins Puma and Noxa are two transcriptional targets of p53 that seem to be the important targets in p53-induced apoptosis (Villunger et al. 2003). NF-κB is a transcription factor that can promote survival in response to several extracellular stimuli. When it forms a complex with IκB (inhibitor of NF-κB), it remains in the cytoplasm. AKT can have a positive effect on NF-κB function by phosphorylation and activation of IκB kinase (IKK), a kinase that induces degradation of IκB (Ozes et al. 1999; Huang and Chen 2009). Degradation of IκB releases NF-kB from the cytoplasm, and the free NF-κB enters the nucleus to activate its target genes related to increased cell survival.
6.3.2 Cell Proliferation (Cell Cycle)
AKT can stimulate proliferation through multiple downstream targets regulating cell-cycle machinery. The cell cycle is regulated by the coordinated action of cyclin-dependent kinase (CDK) complexes and CDK inhibitors (CKIs). Glycogen synthase kinase-3 (GSK3) phosphorylates cyclin D1 and cyclin E and transcription factors c-Jun and c-Myc, which all play an important role in the G1/S phase cell-cycle transition, targeting them for degradation by the proteasome (Diehl et al. 1998; Wei et al. 2005; Welcker et al. 2003; Yeh et al. 2004). AKT directly phosphorylates GSK3 and blocks its kinase activity, thereby enhancing the stability of these G1/S transition-related proteins. AKT can also negatively regulate the function of the CKI p21 (also known as CIP1 or WAF1). The expression of p21 can also be negatively regulated by activation of MDM2 by AKT (Mayo and Donner 2001; Zhou et al. 2001). Activated MDM2 subsequently down-regulates p53-mediated transcription of p21. Moreover, Akt inhibits p27 expression and retinoblastoma-related protein p130 through phosphorylation and inhibition of the FOXO transcription factors. p27 and p130 are known to cooperate to inhibit the cell cycle at the G1/S transition (Liang and Slingerland 2003).
6.3.3 Cell Growth
One of the well-characterized functions of AKT is its role in promoting cell growth (i.e., an increase in cell size). The major mechanism by which AKT regulates cell mass increase is through activation of the mTOR complex 1 (mTORC1), which is regulated by both nutrients and growth factor signaling. mTOR (mammalian target of Rapamycin), a catalytic subunit of mTORC1, is one of the best-studied downstream responders to AKT activation and belongs to a group of serine-threonine protein kinases of the PI3K superfamily, referred to as class IV PI3Ks, which also includes ATM, ATR, and DNA-PK. mTOR exists in two distinct complexes—mTORC1 and mTORC2. mTORC1 consists of the mTOR catalytic subunit, regulatory associated protein of mTOR (RAPTOR), proline-rich AKT substrate 40 kDa (PRAS40) and a common regulatory subunit called mLST8 (Wullschleger et al. 2006). mTORC2 consists of mTOR, rapamycin-insensitive companion of mTOR (RICTOR), mammalian stress-activated protein kinase interacting protein 1 (SIN1) and mLST8 (Liu et al. 2009). The mTORC1 complex is strongly inhibited by rapamycin treatment, while mTORC2 is not affected by acute treatment and only chronic rapamycin treatment at high concentration inhibits its assembly and signaling capacity (Sarbassov et al. 2006).
mTOR has a dual role in PI3K/AKT signaling; when in the TORC2 complex it participates in activation of AKT via phosphorylation of AKT at Ser473 (as discussed above) and when in the TORC1 complex it is activated downstream of AKT. AKT activates mTORC1 multiple ways. The major mechanism appears to be through phosphorylation and inactivation of TSC2 (tuberous sclerosis complex 2, also called tuberin) (Inoki et al. 2003a; Manning et al. 2002). TSC2 shares homology with GAPs, and its heterodimerization with TSC1 is required to exert a GAP activity toward the small GTPase Rheb (Ras homolog enriched in brain) (Castro et al. 2003; Garami et al. 2003; Inoki et al. 2003b). The GTP-bound form of Rheb strongly activates mTORC1, through direct binding to this complex. Therefore, AKT activates mTORC1 indirectly by phosphorylating and inhibiting the Rheb-GAP activity of TSC2, thereby allowing Rheb-GTP to activate mTORC1 signaling (Manning and Cantley 2007). The most extensively characterized downstream targets of mTORC1 are ribosomal protein S6 kinase (S6K; also known as p70S6K) and eukaryotic translation-initiation factor 4E-binding protein 1 (4EBP1). mTORC1 mediates phosphorylation of S6K at a threonine residue (T381) in a hydrophobic motif at the C terminus of the kinase domain. Phosphorylation at this site allows the recruitment and subsequent phosphorylation and activation of S6K by PDK1 (Pullen and Thomas 1997; Pullen et al. 1998). Active S6K1 appears to play multiple roles in the initiation of protein synthesis through phosphorylation of S6 Ribosomal protein (S6, a component of the ribosome) and other components of the translational machinery, thereby enhancing the translation of mRNAs containing 5′ polypyrimidine tracts (Peterson and Schreiber 1998). Phosphorylation of 4EBP1 by mTORC1 suppresses its ability to bind and inhibit the translation-initiation factor eIF4E (Pause et al. 1994; Gingras et al. 1998). eIF4E that is not inhibited by 4EBP binds an mRNA 5′ cap structure and ultimately bring it to the ribosome, increasing translational efficiency of mRNAs (Pause et al. 1994).
6.3.4 Cellular Metabolism
More than 80 years ago, the biochemist Otto Warburg observed that cancer cells consume glucose fervently and produce more lactate, even in the presence of ample oxygen, as compared with normal cells (Warburg 1956; Vander Heiden et al. 2009). Research over the past few years reinforced his observation—a high rate of glycolysis in cancer, also called “Warburg effect”—and also revealed altered metabolism of lipids, amino acids, and nucleotides in cancer cells (Vander Heiden et al. 2009). Oncogenic events, most notably the uncontrolled activation of the PI3K/AKT pathway, have been found to be directly related to altered metabolisms in cancer. Under normal conditions, AKT2, the primary isoform in insulin-responsive tissues, has been associated with glucose transporter 4 (GLUT4)-containing vesicles upon insulin stimulation, increasing glucose uptake in fat and muscle tissues (Calera et al. 1998; Kohn et al. 1996). GLUT1 is the main glucose transporter in most cell types. Unlike GLUT4, GLUT1 seems to be regulated mainly through alterations in expression levels. Activation of mTORC1, through AKT-mediated phosphorylation of TSC2, regulates both HIF1α-dependent transcription of glycolytic enzymes, including Glut1, and cap-dependent translation of Glut1 mRNA (Taha et al. 1999; Zelzer et al. 1998). Another translational target of mTORC1 is c-Myc, capable of also inducing expression of various glycolytic genes, thus increasing glycolysis (West et al. 1998; Gordan et al. 2007). Furthermore, AKT-mediated phosphorylation and inhibition of GSK3 prevents GSK3 from phosphorylating and inhibiting its substrate glycogen synthase, promoting glycogen synthesis. In the liver, AKT inhibits gluconeogenesis by blocking FOXO-mediated transcription of gluconeogenic enzymes, such as phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase).
In addition to glucose metabolism, lipid synthesis is also regulated by the PI3K/AKT pathway. The PI3K/AKT pathway has been shown to up-regulate lipogenic gene expression through activating SREBP (sterol regulatory element-binding protein) transcription factors, the master regulators of genes involved in cholesterol, fatty acids, triglycerides, and phospholipids synthesis (Gasic 1994; Sundqvist et al. 2005; Porstmann et al. 2005, 2008; Yecies et al. 2011). AKT has also been reported to directly activate ACL (ATP citrate lyase), which functions in an important step in fatty acid biosynthesis (Berwick et al. 2002).
6.4 PI3K Pathway Alterations in CRC
6.4.1 Known PI3K Pathway Mutations in CRC
CRC formation is a multistep process involving cellular transformation proceeding from normal mucosa to microadenomas, to adenomas with increasing dysplasia, to carcinoma. The stepwise CRC model involves many changes in epithelial morphology and is accompanied by characteristic genetic and epigenetic alterations. Both epidemiological (Yoong et al. 2011) and in vitro (Hanahan and Weinberg 2000) studies have shown that cellular transformation requires the accumulation of at least 6–12 mutational events, possibly more (Wood et al. 2007; Sjoblom et al. 2006), a process facilitated by genetic instability (Lengauer et al. 1997; Hirota et al. 1998).
The PI3K pathway is one of the most frequently mutated pathways and has been implicated in driving the progression of pre-invasive adenoma to CRC. Mutations in one or often more than one member of this pathway are found in the majority of CRCs, providing a challenge, and at the same time a potential target, for the treatment of PI3K-addicted CRC tumors with pan-specific or isoform-specific PI3K inhibitors. Activating mutations in PIK3CA, the gene encoding the p110α catalytic subunit of PI3K, were initially detected in approximately one-third of 234 CRCs, but only in 2 of 76 adenomas (Samuels et al. 2004). More recent data evaluating the mutation frequency of PIK3CA in CRC show that it is almost exclusively mutated in established carcinomas and at a lower rate (~13 %, out of n = 9,108 (http://www.sanger.ac.uk/genetics/CGP/cosmic, thereafter COSMIC database) than previously reported, yet still remaining one of the most commonly mutated genes in this tumor type
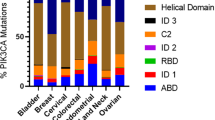
In CRC, there are three mutation hotspot regions within PIK3CA—the H1047R, the E545K, and the E542K mutations. The H1047R mutation is located at the C-terminal portion of the kinase domain, while E545K and E542K mutations are located within the region encoding the helical domain of the protein (Samuels et al. 2004) (Fig. 6.3a). The three aforementioned PIK3CA mutations account for more than 90 % of all the PIK3CA mutations found in CRC. They are all missense (a type of non-synonymous mutation) and confer constitutive lipid kinase activity that promotes cell growth and invasion of human cancer cells (Samuels et al. 2005). Most of the remaining PIK3CA mutations either code for a different amino acid change or target a region adjacent to the hotspot mutations. A list of complete PIK3CA mutations in CRC is summarized in Fig. 6.1.
Aside from PIK3CA activating mutations, the PI3K pathway can be activated by several other mechanisms in CRC, with the most common being loss or mutation of the PTEN tumor suppressor. PTEN is a haploinsufficient tumor suppressor, hence it is more frequently targeted for hemizygous deletions or inactivating mutations of a single allele, thus retaining a normal wild-type allele. Inactivating mutations of PTEN are detected in 6 % of CRCs (out of n = 1,344, COSMIC database), while the overall frequency of PTEN deletion in this tumor type is ~22 % (out of n = 161, COSMIC database) with ~5 % of these being focal, affecting in most cases only PTEN. Another PI3K-related gene that is frequently deleted in CRC (~22 % overall, ~7 % focal) is PIK3R1 (Beroukhim et al. 2010), the gene encoding the p85α inhibitory subunit. PIK3R1 is also the target of point mutations in 4 % (out of n = 560, COSMIC database) of CRCs (Fig. 6.4).

Mutations of PIK3CA identified in colorectal cancer. (a) Histogram displaying the position of somatic mutations in the coding sequence of PIK3CA that are identified in CRC (total no of mutations = 787). (b) Schematic representation of the PIK3CA exons (1–20) and functional domain of the PIK3CA protein based on the SMART database (http://smart.embl-heidelberg.de/). (c) Pie chart showing the proportion of PIK3CA domain mutations in CRC. The frequencies and position of PIK3CA mutations are based on the COSMIC database (http://www.sanger.ac.uk/genetics/CGP/cosmic/)
Given that activation of PI3K signaling begins with the engagement of growth factors by RTKs and recruitment of adaptor proteins, many members of this signaling pathway are altered, although somewhat infrequently, in CRC. Among these, EGFR and the v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERBB2/HER2) are both mutated in 4 % of CRCs (77/2152 for EGFR and 14/365 for ERBB2, COSMIC database), with mutations affecting mostly tumors of the rectum. The ERBB4 gene, on the other hand, has been noted to be mutated in 37 % of CRCs, although the sample size for the mutation detection of this gene is relatively small (out of n = 65, COSMIC database).
The RTK KIT is mutated in 4 % of CRC (out of n = 367, COSMIC database), although activating mutations in this receptor are one of the main forces driving the progression of gastrointestinal stromal tumors (GIST) and occur in >85 % of GISTs (Lengauer et al. 1998). Mutations in the RTK MET and AKT1 are also detected at very low frequencies (1–2 %), occurring in 7out of 310 and 7 out of 917 CRCs, respectively (COSMIC database).
6.4.1.1 Coexistence and Mutual Exclusivity of PI3K Pathway Mutations in CRC
Cancer progression is considered to be a process of Darwinian evolution, where most genetic alterations are products of selective pressure that drive tumor growth and proliferation. The notion of “the survival of the fittest” would therefore prevent mutations in functionally redundant isoforms of the same gene, and/or genes that lie in the same signaling network. In spite of this, almost every known member of the PI3K/Akt signaling network is frequently altered in most cancers, and certain tumor samples carry two or more mutations in this pathway. Furthermore, the mutation frequency of each of the target genes varies dramatically from one tumor type to another, sometimes even among different tumors of the same origin, suggesting that the genetic profiles of all cancers are determined by distinct somatic evolution that drives the accumulation of mutations. In this section we explore the degree of coexistence and/or mutual exclusivity of PI3K pathway alterations in CRC and speculate on mechanisms which drive the unique characteristics of this tumor type.
There is some degree of mutual exclusivity in PIK3CA and PTEN mutations in breast carcinomas and glioblastomas (Suire et al. 2002; Zhou et al. 2002), where the frequency of coexistent mutations falls lower than that expected by chance. While this is also evident for CRCs (Frattini et al. 2007; Yuan and Cantley 2008), the overall frequency of co-mutation varies significantly from one tumor type to another. Endometrial cancers exhibit the highest incidence of co-mutation between PIK3CA and PTEN (35–36 %). Studies have shown that knocking down PTEN expression in HEC-1B cells that carry mutations in both KRAS and PIK3CA further enhances Akt (S473) phosphorylation (Samuels et al. 2004), suggesting that the functional redundancy in the mutation of PTEN and PIK3CA could be the product of selection for combinatorial and additive effects. Furthermore, the PI3K/AKT pathway is complex and often the target of multiple negative feedback loops. Although mutations in either PTEN or PIK3CA can activate the PI3K pathway, tumors bearing both alterations could circumvent negative feedback loops to sustain tumorigenesis.
One of the best understood forms of genetic instability in CRC is deficiency in the DNA mismatch repair (MMR) system. A possible scenario is that concomitant mutation of PIK3CA and KRAS in CRC is the product of a deficiency in the MMR system. Loss of DNA MMR function can be caused by mutations, deletions, or epigenetic silencing of, both copies of one of the major MMR genes, MLH1, MSH2, MSH6, or PMS2. Failure to repair replication-associated errors due to a defective MMR system allows persistence of mismatch mutations located throughout the genome, especially in regions of repetitive DNA known as microsatellites, giving rise to the phenomenon of microsatellite instability (MSI) (reviewed in (Velho et al. 2005)).
A high frequency of instability at microsatellites (MSI-H) is the hallmark of the most common form of hereditary susceptibility to CRC, known as Lynch syndrome (LS) (previously known as hereditary non-polyposis colorectal cancer syndrome, HNPCC), but is also observed in 15–20 % of sporadic colonic cancers (and rarely in rectal cancers). The mutational targets in MMR-deficient tumors are not random, in that mutations occur preferentially in repetitive sequences; nonetheless there is no a priori favoring of mutation site. However, the mutations in MMR-deficient CRC are positively selected, presumably on the basis that they confer a selective advantage for growth, survival and escape from immune surveillance. Interestingly, there is a strong association between PIK3CA mutations and MSI status (P = 0.0046) (Sjoblom et al. 2006), despite the fact that there is no repetitive nucleotide sequence within the PIK3CA coding region. On the other hand, the frequency of KRAS mutations is significantly lower in MSI-H colon cancers (Gupta et al. 2007), while PTEN is more frequently mutated in these tumors, particularly in exons 7 and 8 because of the poly-A tracts present in their sequence (Gupta et al. 2007). The co-mutation rate of PI3K/Akt pathway genes is, therefore, likely to be dramatically different between MSI-H and microsatellite stable (MSS) colon cancers and requires further investigation. Uncoupling the coexistence or mutual exclusivity of PI3K alterations will likely be critical in understanding the substantial evolutionary implications that drive the tumorigenic potential of CRC subtypes. A deeper understanding of this phenomenon will guide identification of novel, more effective individualized treatment options necessary to overcome the substantial burden of drug resistance and tumor recurrence.
6.4.2 Relevance of PI3K Pathway Mutations in Human Colorectal Cancer Therapy
The prognostic significance of PI3K-pathway genetic alterations is well established in CRC, yet it appears to be much more complex than initially appreciated. PIK3CA mutations have been associated with poorer survival of CRC patients (Amado et al. 2008), while PIK3CA gene amplification/gain was shown to be independent of PIK3CA point mutations, and was positively correlated with longer survival in patients who received adjuvant chemotherapy and/or radiotherapy (Engelman et al. 2008). However, both the clinical significance of PIK3CA gene amplification and the degree in which this genetic alteration is observed in CRC are unclear. According to recent high-throughput DNA copy number studies, the PIK3CA locus (3q26.32) is the target of frequent amplification in ovarian, breast, and brain tumors (Karapetis et al. 2008), but is very rarely, if ever, amplified in CRCs (Karapetis et al. 2008; Linardou et al. 2008).
The coexistence or mutual exclusivity of PI3K pathway mutations is also likely to have a great impact on the clinical utility of PIK3CA mutations. Interestingly, in a large study of 450 resectable colon cancer biopsies, PIK3CA mutation showed a clear association with higher colon cancer-specific mortality in both univariate and multivariate analyses (Nosho et al. 2008). However, the effect on patient’s survival differed significantly among patients with wild-type or mutant KRAS, with the latter group showing no association of PIK3CA mutation with poor prognosis. PTEN loss of expression has also been suggested to serve as a poor prognosis indicator in CRC. One study demonstrated that low PTEN-expressing tumors are significantly associated with shorter median progression-free survival (PFS) (Yuan and Cantley 2008). Likewise, another study showed that loss of PTEN expression was associated with larger tumor size and depth of invasion, lymphatic invasion, lymph node metastasis, higher Dukes’ staging, and reduced caspace-3 expression (Abubaker et al. 2009). Several studies have shown that there are multiple effector pathways downstream of PI3K/Akt activation, underlying the therapeutic challenge in treating PI3K-pathway-activated tumors. The degree in which the prognostic significance of PI3K-pathway mutations are influenced by inherent or acquired resistance to the conventional cancer therapeutic approaches is likely to be highly underrated. The standard first-line treatment options for metastatic CRC remain primarily based on traditional cytotoxic chemotherapies consisting of a 5-FU and folinic acid (FA, also known as leucovorin) backbone. In the initial studies, 5-FU/Leucovorin demonstrated a response rate of 20 % and a treatment-induced prolongation survival of 11 months compared with the median 5 month survival of best supportive care (BSC) (Chee and Sinicrope 2010). Subsequent trials demonstrated the survival benefit of adding the nonnephrotoxic platinum analogue oxaliplatin (FOLFOX) or the topoisomerase 1 inhibitor irinotecan (CPT-11)(FOLFIRI) to 5-FU/FA backbone, improving response rates to 35–53 %, PFS of 5–8 months and overall survival of 14–18 months (Laurent-Puig et al. 2009; Li et al. 2009; Loupakis et al. 2009). The first FDA-approved oral chemotherapeutic agent capecitabine is enzymatically converted to 5-FU in vivo, and can be substituted for 5-FU as it has been suggested to have milder side effects (Ogino et al. 2009).
Despite modest survival data, the overall response rate of palliative chemotherapy in CRC is low and often associated with severe side effects, raising the need for the development of targeted cancer therapies. Significant effort has been made in the development of inhibitors that target the VEGF and EGFR pathways in CRC. There are two main classes of EGFR inhibitors currently in clinical use: the anti-EGFR monoclonal antibodies and the small-molecule EGFR tyrosine kinase inhibitors. These inhibitors are not exclusive to the EGFR pathway and can block different RTKs, including VEGF (Chee and Sinicrope 2010). Two clinical trials have evaluated the efficacy of the anti-EGFR monoclonal antibodies panitumumab and cetuximab, and demonstrated that clinical benefit was restricted to wild-type KRAS tumors (De Roock et al. 2010; Li et al. 2010). Evaluation of the predictive and prognostic value of KRAS and PIK3CA mutations in relation to both 5-FU-based first-line chemotherapy treatment and second line cetuximab therapy indicated that KRAS mutations could predict for lack of response (P = 0.002) and shorter PFS (P = 0.09) (Souglakos et al. 2009), while PIK3CA mutations were associated with even lower PFS in response to cetuximab treatment (P = 0.01). Recently, a European consortium studied the effects of KRAS, BRAF, NRAS, and PIK3CA on the efficacy of cetuximab or panitumumab in a large cohort (n = 1,022) of patients with chemotherapy-refractory metastatic CRC (Poulogiannis et al. 2010). This study confirmed that patients with KRAS mutations do not benefit from cetuximab treatment. Importantly, in subset analysis this study showed that only PIK3CA exon 20 mutations are associated with a lack of response to cetuximab in KRAS-WT tumors, with a lower median PFS of 11.5 versus 24 weeks. While there is biologic reason to suspect that exon 20 PIK3CA mutations may predict cetuximab resistance in KRAS-WT CRC, this observation needs to be confirmed in a larger tumor series to exclude the possibility of any confounding factors reflective of the low number of tumors with exon-20 PIK3CA mutation included in the latter study (n = 9). In contrast, exon 9 PIK3CA mutations were associated with KRAS mutations and did not confer an independent adverse effect on cetuximab response rate. Other PI3K pathway perturbations, including loss of PTEN expression, have also been linked to lack of cetuximab response in metastatic CRC (Sullivan and Kozuch 2011; Naguib et al. 2011; Poulogiannis et al. 2012). However, tumor heterogeneity confounds the immunohistochemical (IHC) assessment of PTEN expression (19–36 %) and necessitates establishment of a more reliable and standardized protocol for PTEN IHC testing.
Despite considerable progress in selecting which CRC patients are suitable for anti-EGFR treatment, and basic understanding of the alternative mechanisms driving resistance to this therapy, most CRC patients who respond to these agents inevitably experience progressive disease after a few months of treatment. The relatively short response durations to second and third line CRC treatments highlight the need for both a better molecular characterization of individual patient tumors and the possibility of combination therapies such as Cetuximab with a pan-PI3K inhibitor which may delay the onset of resistance and translate to clinical benefits in both progression free and hopefully overall survival. Currently, there are ongoing early phase trials of pan-PI3K and PI3K-isoform-specific inhibitors, as well as Akt and mTOR inhibitors, alone or in combination in a wide range of solid tumors and clinical settings, as discussed in the following sections.
6.5 Inhibitors Targeting PI3K Pathways
Development of novel, targeted cancer therapies is rapidly replacing that of traditional, nonspecific cytotoxic drugs, and slowly new targeted therapies are making their way into clinical practice (Yun et al. 2009; Yun 2010). Prior successes in targeted therapy, such as imatinib for chronic myelogenous leukemia (CML), trastuzumab for breast cancer with amplification of HER2 (also known as ERBB2), and erlotinib and gefitinib for lung cancer that expresses mutant EGFR paved the way for future targeted therapy. While, conceptually, targeted therapy is ideal, many roadblocks exist to the development of new targeted therapies. First, the pathway of interest should be both central to maintenance of the malignancy and druggable. Second, the targeted therapy must not be too toxic to surrounding normal tissues. Third, the ideal targeted therapy would have a noninvasive monitoring method and mechanism to study resistance. As discussed previously, genetic and cell line/xenograft studies have shown that deregulated PI3K signaling is vital to the growth and survival of cancer cells, making the PI3K pathway one of the most attractive targets for anticancer therapy. Over the past decade, a number of PI3K pathway inhibitors have been developed and entered into the clinic. Table 6.1 shows a list of drugs in development to exploit the PI3K signaling pathways, and existing clinical trials can be found at http://www.clinicaltrials.gov/. In the following section, we will discuss four different classes of PI3K pathway inhibitors: PI3K inhibitors, AKT inhibitors, mTOR inhibitors, and dual PI3K/mTOR inhibitors.
6.5.1 PI3K Inhibitors
Like the majority of small molecule kinase inhibitors, all existing PI3K inhibitors belong to a class of ATP-competitive inhibitors. The PI3K inhibitors can be further divided into pan-PI3K inhibitors or isoform-specific PI3K inhibitors. The majority of PI3K inhibitors in clinical trials thus far are pan-PI3K inhibitors, inhibiting all of the catalytic subunit isoforms of class I PI3Ks: p110α, p110β, p110γ, and p110δ. Developing isoform-specific inhibitors is challenging because of the highly conserved nature of the ATP-binding pocket. Structural visualization techniques including X-ray crystallography are critical for imaging drug-PI3K complexes and informing rational isoform-specific inhibitor development. Indeed, the X-ray structures of the p110 subunit of PI3Kγ, and more recently of the human p110α/p85α complex, have been crucial in providing a detailed structural analysis of the ATP-binding cleft of class I PI3K, leading to the development of p110γ or p110α isoform-specific PI3K inhibitors (Walker et al. 1999, 2000; Huang et al. 2007).
While there are theoretical benefits to both pan-PI3K and isoform-specific PI3K inhibition, it remains unclear which type of inhibitor, if any, will be more effective clinically. The answer depends on several factors including toxicities resulting from complete inhibition of all PI3K isoforms with pan-PI3K inhibitors, identification of the tumor subtypes in which inhibition of only one or two of the PI3K isoforms will be sufficient, and the time to resistance development. One major potential flaw of PI3K inhibition are the multiple mechanisms by which malignant cells can activate AKT, the major downstream effector of the PI3K signaling cascade.
6.5.1.1 “First Generation” Pan-PI3K Inhibitors: Wortmannin and LY294002
Wortmannin and LY294002 are two well-known, first-generation PI3K inhibitors. Wortmannin was isolated from the fungus Penicillium wortmannin in1957 and is an irreversible inhibitor that forms a covalent bond in the ATP-binding pocket of the kinase (Yuan and Cantley 2008). It inhibits PI3K enzymatic activity in the nanomolar range; however, it is not a specific PI3K inhibitor, as it binds to other kinases such as DNA-PK, ATM, ATR, and mTOR. Furthermore, Wortmannin is extremely reactive, with a half-life of only a few minutes in serum, and causes liver dysfunction, lymphocytopenia, and hyperglycemia in animals (Ihle et al. 2004). In 1994, Eli Lilly (Indianapoli, IN) synthesized the reversible PI3K ATP-competitive inhibitor, LY294002. It was developed as a structural analogue of quercetin, a bioflavonoid produced by plants, which can inhibit several protein kinases including PI3K, similar to wortmannin. LY294002 is more stable, but less potent, than Wortmannin. Both Wortmannin and LY294002 substantially inhibit growth of most cancer cell lines when administered as single agents, particularly in cases of excess PI3K activity (Markman et al. 2010) and sensitize tumor cells to other targeted therapeutics such as chemotherapy and radiation (Hu et al. 2002). However, these compounds have not progressed to clinical trials because of unfavorable pharmacokinetic properties, poor selectivity, and high toxicity in animal models (Vlahos et al. 1994).
One approach to bring wortmannin to the clinic involved increasing its stability by PEGylation (Cleary and Shapiro 2010). PWT-458 (Pfizer) is a PEGylated derivative of wortmannin that has a higher therapeutic index in preclinical animal models compared to wortmannin (Yu et al. 2005). Upon intravenous administration, the polyethylene-glycol (PEG) moiety is cleaved, releasing 17-hydroxywortmannin. PWT-458 inhibited AKT kinase and its downstream effectors at nontoxic doses. Inhibition of AKT signaling was accompanied by a slowing of xenograft growth. Moreover, PTW-458 improved the anticancer effects of paclitaxel and PEGylated rapamycin in certain xenograft models (Zhu et al. 2006).
Similar efforts have been applied to improve the pharmacological properties of LY294002. SF1126 (Semafore Phamaceuticals) is a water-soluble prodrug of LY294002 (Garlich et al. 2008; Nutley et al. 2005). The RGD (Arg-Gly-Asp) targeting peptide attached to SF1126 enables the drug to target specific integrins within the tumor compartment. The integrin-targeting RGD peptide moiety causes the drug to preferentially accumulate in endothelial cells and tumor cells. SF1126 inhibits all class I PI3K isoforms and other closely related kinases such as DNA-PK and mTOR. It blocks the phosphorylation and activation of AKT in cell lines with IC50 values in the low micromolar range. In preclinical studies, SF1126 has shown antitumor activity in xenograft models of brain, neuroblastoma, non-small cell lung, prostate, myeloma, renal, and colon carcinoma. In addition to its direct activity on cancer cells, it has demonstrated anti-angiogenic activity in xenografted glioma cells by substantially reducing microvessel density. SF1126 is currently in phase I clinical trials. XL147 (under co-development by Exelixis and Sanofi-Aventis) selectively inhibits PI3K without inhibiting mTOR or DNA-PK (Yun et al. 2009). The compound inhibits PI3K signaling in cultured tumor cells and blocks VEGF-induced tubule formation in cell lines. Oral administration slows tumor growth or causes shrinkage of breast, lung, ovarian, prostate, and glioma tumors in xenografts. XL147 is currently in phase I clinical trials and its dose-limiting toxicity is rash, elevated liver function tests, and fatigue. In Phase I clinical trials, increased tumor growth inhibition was achieved by combining XL147 with cytotoxic (carboplatin and paclitaxel) or targeted anticancer agents (erlotinib) without significantly increased toxicity.
6.5.1.2 “Second Generation” Pan-PI3K Inhibitors
The new generation of PI3K compounds was designed to improve upon the pharmaceutical limitations of wortmannin and LY294002. PX-866 (Oncothyreon) is a structural analogue of wortmannin and functions as an irreversible inhibitor of PI3K by making a covalent bond with the PI3K molecule similar to wortmannin (Ihle et al. 2004, 2005; Howes et al. 2007). Compared to Wortmannin, PX-866 exhibits increased stability, reduced toxicity, and enhanced biological activity. In humans and preclinical models, PX-866 is metabolized to produce an active metabolite, 17-OH, that is a more potent PI3K inhibitor than the parent drug and retains the same irreversible mechanism of action. In biochemical assays, PX-866 and the 17-OH metabolite inhibit all four PI3K isoforms and have the greatest potency for PI3K α and β, the two family members that are most strongly associated with solid tumors such as breast, colon, ovarian, and prostate cancers. Preclinical studies demonstrate that PX-866 is efficacious in numerous mouse xenograft models of lung, ovarian, and CRC as a single agent and in combination with chemotherapy, radiation, and targeted cancer drugs, such as EGFR inhibitors. In these studies, PX-866 sustained inhibition of the PI3K pathway, a property that is attributable to its unique, irreversible mechanism of action. Preliminary results from Phase I clinical trials showed several patients with stable disease, with mild side effects including abdominal pain and mild diarrhea (Ihle et al. 2009a). Oncothyreon is currently evaluating PX-866 in Phase I/II and Phase II clinical studies in solid tumors.
BKM120 (Novartis) is an oral pyrimidine-derived pan-PI3K inhibitor. BKM120 inhibits all class I PI3K isoforms at nanomolar concentrations without inhibitory activity against members of the other classes of PI3K or mTOR. In vitro experiments showed a strong anti-proliferative effect of BKM120 on human cancer cell lines exhibiting aberrant PI3K pathway activity. In vivo, BKM120 demonstrated significant antitumor activity in human tumor xenograft models with good correlation between BKM120 treatment and inhibition of the PI3K pathway (Lee et al. 2006; Seki et al. 2004). BKM120 is in phase I clinical trials with colorectal, breast, ovarian, and endometrial cancers patients. Recent preliminary data from phase I trials with 35 patients with advanced solid tumors demonstrated the clinical safety and tolerability of BKM120, as well as its favorable pharmacokinetic profile. The reported side effects were mood alteration, hyperglycemia, and rash (Bendell et al. 2012).
6.5.1.3 Isoform-Specific PI3K Inhibitors
The four isoforms of PI3K, α, β, γ, and δ have distinct biological functions (reviewed in (Liu et al. 2009)). For example, PI3Kα is involved in tumorigenesis and insulin signaling, PIK3β plays a role in platelet aggregation, PI3Kγ is expressed in leukocytes and is a component of the inflammatory response, and PI3Kδ is implicated in allergic responses and hematological cancers. Therefore, a pan-PI3K inhibitor used as an anticancer agent may generate undesirable toxic side effects due to inhibition of all isoforms. p110α-specific inhibitors are of great interest for treating cancers that have PIK3CA mutations. Considering that more than 20 % of CRC have PIK3CA alterations, it will certainly be important to develop p110α-specific inhibitors and evaluate them in CRC lines and animal models with wild-type or mutant PIK3CA. Preclinical models suggest potential advantages of p110α-specific inhibition over inhibition of other PI3K isoforms in certain tumor types. For example, p110α has a critical role in angiogenesis among other Class I PI3K members (Graupera et al. 2008). Thus, specific inhibition of p110α represents a potential method of blocking angiogenesis, a known hallmark of cancer. Since p110α plays a major role in insulin signaling and glucose metabolism, several side effects of p110α can be expected, such as hyperglycemia or glucose intolerance. However, these side effects might be lightened by treatment with peroxisome proliferation-activated receptor gamma (PPARγ) agonists (Ihle et al. 2005). BYL719 (Novartis), a selective inhibitor for p110α, is currently in Phase I clinical trials in patients with advanced solid tumors harboring PIK3CA mutations. While potentially undesirable, a predictable physiological change such as hyperglycemia may offer a minimally invasive clinical surrogate of target inhibition.
The p110β-specific inhibitors are also of interest in treatment of some cancers. Several reports show that p110β is the dominant isoform carrying PI3K activity in PTEN-deficient tumors of brain, breast, prostate, and endometrium both in vitro and in vivo (Jia et al. 2008; Oda et al. 2008; Wee et al. 2008). Since p110β may play a lesser role in insulin response, it is possible that this class of compounds would show fewer side effects compared to p110α-specific inhibitors.
p110δ is mainly expressed in cells of the immune system, where it regulates B-cell maturation and function (Jou et al. 2002; Okkenhaug et al. 2002; Zhang et al. 2011). Therefore, selective inhibitors of p110δ are an attractive therapeutic option in patients with B-cell malignancies. The p110δ-specific inhibitor CAL-101 (Calistoga Phamaceuticals) is being tested in a phase I dose-escalation trial of patients with relapsed or refractory hematologic malignancies (Fruman and Rommel 2011).
Although major advances have been made in the identification of isoform-specific p110 inhibitors, it remains to be seen whether mutant-specific PIK3CA inhibition can be translated into clinical benefit. Ideally, mutant-specific PIK3CA inhibitors would interfere with the oncogenic versions of p110α and leave the important normal functions of wild-type p110α unaffected. Unfortunately, the design of mutant-specific ATP-competitive inhibitors is complicated by the fact that PIK3CA mutations commonly observed in cancers do not alter the ATP-binding site geometry in a manner that can be clearly exploited during drug design. In comparison to wild-type PIK3CA, the crystal structure of PIK3CA containing the hotspot mutation H1047R in the p110α kinase domain (the most commonly observed PIK3CA mutation) revealed no significant structural differences in the ATP-binding site (Mandelker et al. 2009). Structural and biochemical data suggest the H1047R mutation alters the way p110α interacts with lipid membranes, allowing it easier access to the PIP2 substrate, thereby increasing PI3K pathway activity. Knowledge of the proposed mechanism of action of the H1047R mutation has not yet been exploited to develop novel mutation-specific inhibitors. No crystal structure is currently available of PI3K containing the second most common hotspot mutation E545K in the p110α helical domain. Nevertheless, similarly to the H1047R mutation, the E545K mutation is not predicted to alter the ATP-binding cleft in a structurally significant manner. Thus, great difficulty exists in designing mutant-specific ATP-competitive PI3K inhibitors. The success of mutant-specific p110α inhibition may depend on alternative inhibitory approaches such as allosteric kinase inhibitors, or antagonists of protein–protein interactions, to provide the desirable activity as well as selectivity profile.
6.5.2 AKT Inhibitors
AKT is another attractive target in inhibiting the PI3K signaling pathway in cancer. AKT is the central node of the PI3K signaling pathway, and both ATP-competitive inhibitors and allosteric inhibitors targeting AKT kinases are under active clinical development. Most ATP-competitive inhibitors are nonselective and target all three isoforms of AKT. GSK690693 (GlaxoSmithKline) is an ATP-competitive AKT kinase inhibitor that targets all three AKT isoforms at low nanomolar range and is active against additional kinases from the cyclic AMP-dependent, cGMP-dependent, and protein kinase C (PKC) family. GSK690693 was recently terminated during clinical phase I trial because of high toxicity. An allosteric dual inhibitor of AKT1 and AKT2 developed by Merck has potent antitumor activity in tumor xenograft models and its analogue MK2206 (Merck) is in Phase I study in patients with locally advanced or metastatic solid tumors (Yap et al. 2011). The most clinically advanced allosteric AKT inhibitor is an alkylphospholipid, perifosine (KRX-0401, Keryx Biopharmaceuticals) (Hilgard et al. 1997; Kondapaka et al. 2003; Van Ummersen et al. 2004). It inhibits AKT activity by disrupting the binding of its PH domain to PIP3, thereby preventing its membrane translocation and activation by PDK1. In vitro, perifosine inhibits growth of melanoma, colon, lung, prostate, and breast cancer cells in association with inhibition of AKT activity (Kondapaka et al. 2003; Crul et al. 2002). Perifosine has also been found to sensitize cancer cells to apoptosis and cell-cycle arrest induced by radiation in vitro and in vivo (Caron et al. 2005; Vink et al. 2006).
In 2010, perifosine finished clinical phase II and is currently in phase III testing. In a phase II trial of metastatic CRC, perifosine in combination with capecitabine doubled time to progression for metastatic CRCs (Bendell et al. 2011), and this trial led to the FDA assigning Perifosine fast-track status. Another AKT inhibitor, VQD-002 (VioQuest Pharmaceuticals), is a water-soluble tricyclic nucleotide that demonstrated antitumor activity against a wide spectrum of cancers in preclinical and clinical studies. A recent study showed that VQD-002 could play a role in reversing drug resistance in cisplatin treated ovarian cancer (Yang et al. 2008). VQD-002 is currently being tested in phase I/II clinical trials in patients with both solid and hematological malignancies.
The distinct functions of AKT1 and AKT2 in cancers spurred the development of isoform-specific AKT inhibitors, with the promise of effective antitumor activity and fewer toxic side effects compared to compounds that inhibit all three AKT isoforms. In an AKT1 null mouse, glucose homeostasis is unperturbed, but the animals are smaller, consistent with a role for AKT1 in cell growth. In contrast, mice without AKT2 have mild growth defects and show a diabetic phenotype, consistent with data indicating that AKT2 plays an important role in insulin signaling (Cho et al. 2001b; George et al. 2004; Engelman 2009). In this case, it is plausible that AKT1-specific inhibition could shrink tumors with minimal impact on glucose homeostasis. Thus, recent drug-discovery efforts have focused on the development of isoform-specific AKT inhibitors.
Although AKT is the major PI3K downstream effector, PI3K can activate AKT-independent pathways, including the Bruton tyrosine kinase (BTK), the Tec families of non-RTKs, serum-and glucocorticoid-regulated kinase (SGKs), and regulators of small GTPase that are implicated in cell polarity and migration. For example, AKT was a less essential effector of cell survival than SGK3 in a subset of cancers with PIK3CA mutations (Morrow et al. 2005). Further, a recent comprehensive analysis of cancer cells carrying mutant PIK3CA showed that many of these cells exhibit minimal increased activation of AKT and downstream signaling (Vasudevan et al. 2009). Thus, AKT inhibitors alone may not provide adequate inhibition of non-AKT effectors of the PI3K pathway. Additionally, inhibition of AKT may actually increase AKT-independent PI3K signaling via loss of negative feedback loops. The prevalence and importance of AKT-independent effectors of PI3K must be more fully elucidated prior to further clinical testing.
6.5.3 mTOR Inhibitors
mTOR is an important downstream effector of PI3K that regulates protein synthesis, cell proliferation, and angiogenesis. Therefore, mTOR inhibition is another promising approach toward blocking aberrant PI3K signaling in cancer cells, and mTOR inhibitors have been in clinical use for several years. Rapamycin (sirolimus, Wyeth), the prototypical allosteric mTOR inhibitor, is a bacterially derived natural product originally used as antifungal agent. It was later found to have immunosuppressive properties, and was approved for clinical use as an immunosuppressive agent in 1999. Rapamycin binds to its intracellular receptor, FK506-binding protein 12 (FKBP12), which then binds directly to mTORC1, inhibiting mTOR-mediated phosphorylation of its downstream targets, S6K and 4EBP1. Later, derivatives of rapamycin, such as CCI-779 (termsirolimus/Torisel; Wyeth) and RAD001 (Everolimus/Afinitor; Novartis) were developed as anticancer drugs (Granville et al. 2006). These rapamycin analogues (referred to as rapalogues) inhibit mTOR through the same mechanism as rapamycin, but possess more favorable pharmacological properties (Liu et al. 2009). Results from clinical studies with CCI-779 and RAD001 used as single agents showed that these drugs improved survival in patients with advanced renal cell carcinoma (RCC), leading to FDA-approval of in 2007 (CCI-779) and 2009 (RAD001) respectively.
There are several possible mechanisms underlying the limited success of rapalogues outside of RCC and breast cancer. First, the negative feedback loop that is blocked upon mTORC1 inhibition may activate upstream receptor tyrosine signaling through IGF-1R or IRS1, resulting in increased PI3K–Akt signaling (Wan et al. 2007; Baselga 2011). Indeed, tissue samples taken from patients with colon or breast cancer after 4 weeks of treatment with RAD-001 showed higher levels of activated AKT compared to pretreatment samples (O’Reilly et al. 2006). In another study, tumor materials from patients treated with rapalogues also showed increased AKT activity (Tabernero et al. 2008). Second, rapalogues only partially inhibit mTORC1 target phosphorylation. For example, 4E-BP is rephosphorylated and is refractory to long-term rapamycin treatment while S6K phosphorylation remained permanently inhibited under those conditions. Persistent 4E-BP phosphorylation may allow cancer cells to continue proliferating and growing independent of AKT dependency. Last but not least, rapalogues cannot inhibit mTORC2 in acute treatment settings. These data imply that ATP-competitive mTOR inhibitors capable of targeting both mTORC1 and mTORC2 might show broader efficacy than rapalogues, sparkling enthusiasm for mTOR catalytic site inhibitors.
The first reported catalytic mTOR inhibitor was PP242, which potently inhibited both mTORC1 and mTORC2 (Apsel et al. 2008). In a preclinical study, PP242 sustained 4E-BP dephosphorylation and suppressed tumor growth in a mouse model of AKT-driven lymphangiogenesis, whereas rapamycin was ineffective (Hsieh et al. 2010). Interestingly, improved efficacy of PP242 may be the result of more effective mTORC1 inhibition, rather than its additional inhibition of mTORC2 (Feldman et al. 2009). INK128 (Intellikine), a derivative of PP242, is currently in Phase I trials (Hsieh and Ruggero 2010). Three additional mTOR catalytic site inhibitors—TORKi CC223 (Celgene), OS1027 (OSI Pharmaceuticals), and AZD8055 (AstraZeneca)—have been shown to inhibit proliferation of a variety of cancer cell lines and human xenograft models more effectively than rapamycin (Chresta et al. 2010) and each of recently entered Phase I trials. Despite the promising results in preclinical studies involving mTOR catalytic inhibitors, several general concerns exist related to mTOR inhibition. First, these compounds may not inhibit AKT T308 phosphorylation by PDK1. This is concerning because previous studies suggested that loss of AKT S473 phosphorylation was not able to block all downstream effectors of AKT signaling. Indeed, a report demonstrated that the mTOR catalytic site inhibitor PP242 had minimal effects on the phosphorylation state of several AKT substrates despite effectively inhibiting AKT S473 phosphorylation (Feldman et al. 2009). Second, inhibition of mTORC1 may activate AKT-independent PI3K signaling due to loss of feedback inhibition, suggesting a role for dual PI3K/mTOR inhibitors. Third, mTOR is not exclusively regulated by PI3K signaling and is involved in additional cellular functions including protein synthesis, cell growth, survival, and metabolism. These processes can be affected by inhibition of mTOR kinase activity, potentially reducing the therapeutic index. Finally, the genetic factors determining the differential sensitivity of cells to mTOR inhibitors are not clear. Despite mounting cell line and xenograft data the role for mTOR inhibitors in CRC remains to be determined, and response CRC patients enrolled in phase I studies will be important.
6.5.4 Dual PI3K/mTOR Inhibitors
The catalytic domains of the p110 subunits (α, β, δ, and γ) and mTOR are structurally similar, and many PI3K inhibitors under development exhibit concomitant mTOR inhibition (Garcia-Echeverria and Sellers 2008). When compared with other types of PI3K pathway inhibitors, dual PI3K/mTOR inhibitors have the potential advantage of inhibiting all class IA PI3K isoforms (p110α, β, and δ), mTORC1, and mTORC2. The broader spectrum of inhibition offered by dual PI3K/mTOR inhibitors has the added benefit of overcoming feedback inhibition normally observed when either mTOR or PI3K inhibitors are administered alone (O’Reilly et al. 2006; Fan et al. 2007). Numerous dual PI3K/mTOR inhibitors such as NVP-BEZ235 (Novartis), NVP-BGT226 (Novartis), and XL765 (Exelixis) are currently in Phase I/II clinical trials. NVP-BEZ235, an imidazoquinazoline derivative, was generated by structure-based design (Maira et al. 2008). Preclinical data showed that NVP-BEZ235 has effective anti-proliferative activity against tumor xenografts that have aberrant PI3K signaling, especially in the presence of PTEN loss or gain-of-function PIK3CA mutations (Serra et al. 2008). In addition dual PI3K/mTOR inhibitors have been shown to possess anti-angiogenic properties (Schnell et al. 2008). Further, unlike other inhibitors of the PI3K/AKT pathway, in vivo efficacy experiments in mice or rats treated with NVP-BEZ235 demonstrated no statistically significant changes in blood glucose levels. These preclinical data establish the feasibility of effectively blocking the PI3K pathway in vivo without serious effects on glucose regulation (Maira et al. 2008). After promising phase I trials with NVP-BEZ235 the drug is now in phase II study. Early reports on Phase I trials of XL756, another dual mTOR/PI3K inhibitor, demonstrated inhibition of AKT phosphorylation and reduced tumor growth. Five out of 19 patents showed clinical benefit with disease stabilization for at least 3 months, and for longer than 6 months in two cases (Molckovsky and Siu 2008). Due to the fact that the dual PI3K/mTOR inhibitors inhibit multiple kinases, one major concern with this class of inhibitors is off-target toxicities. However, recent clinical data at the 2010 American Society of Clinical Oncology (ASCO) annual meeting indicated there were no significant differences in terms of toxicity profiles among dual PI3K/mTOR, Pan-PI3K, and isoform-specific PI3K inhibitors. The most common side effects reported with dual PI3K/mTOR inhibitors were diarrhea, nausea, vomiting, and fatigue. Interestingly, insulin resistance-hyperinsulinemia or hyperglycemia, originally predicted to be one of the most likely toxicities resulting from on-target effects of PI3K inhibitors, not been widely observed in clinical trials to date.
The ideal combination, sequence, and tumor type for dual PI3K/mTOR, Pan-PI3K, AKT, mTOR and isoform-specific PI3K inhibitors in cancer remains to be seen. Underlying the development of PI3K-modulating drugs is the need for an ongoing paradigm shift in oncology with improved molecular tumor characterization at the individual patient level. The generation of large molecularly annotated tumor registries may increase the identification of patient subsets likely to benefit from PI3K inhibition and streamline pipeline drug development and clinical trial design.
6.5.4.1 Strategies for Targeting PI3K Pathways in CRC Therapy
6.5.5 Single-Agent Therapy
One of the hallmarks of cancer is the accumulation of genomic alterations, and most malignancies accumulate numerous genetic alterations during tumorigenesis and progression. Despite the fact that multiple mutations occur in each cancer during tumor progression, growth, and survival are sometimes highly dependent on one or a few oncogenes, and their growth and survival can often be compromised by the inactivation of a single oncogene. This phenomenon, dubbed as “oncogene addiction” has provided a rationale for targeted cancer therapy (Weinstein and Joe 2008). Recent clinical data suggested that oncogenes that are mutated or amplified represent attractive targets for therapy. This principle is exemplified by the successes of targeted therapies such as imatinib for CML; trastuzumab for breast cancer with amplification of HER2 (also known as ERBB2); and erlotinib and gefitinib for non-small cell lung cancers that express mutant EGFR.
The high frequency of mutations in the PI3K pathway in human cancers strongly supports the critical role of the PI3K pathway during tumorigenesis. Tumors with oncogenic PI3K mutations may be highly susceptible to single agents that target PI3K signaling pathway, and there is mounting preclinical evidence to support this hypothesis. NVP-BEZ 235 (Novartis), a dual PI3K/mTOR inhibitor, inhibited the growth of lung adenocarcinomas in transgenic mice that expressed p110α with a H1047R mutation (Engelman et al. 2008). NVP-BEZ235 or an allosteric AKT inhibitor (AKTi-1/2) suppressed growth of human breast tumor xenografts with PI3KCA mutations (Serra et al. 2008; She et al. 2008). Considering more than 20 % of CRCs have PIK3CA genetic alterations, single agents targeting PI3KCA may be an effective strategy in this subgroup of patients.
Preclinical in vitro and in vivo data have revealed the potential efficacy of single agent PI3K inhibition in CRC treatment. The reversible PI3K inhibitor LY294002 blocked PI3K signaling and specifically inhibited proliferation of CRC cell lines, HCT116 and DLD1with PIK3CA mutations, but not cells with wild-type PIK3CA (Samuels et al. 2005). The GSK3 inhibitors, lithium chloride and SB216763 selectively decreased the proliferation of HCT116 cells with oncogenic PIK3CA mutations (Yoong et al. 2011). Further, oral treatment with lithium preferentially inhibited the growth of xenografts of HCT116 with PIK3CA mutations as compared to isogenic HCT116 containing only wild-type PIK3CA (Yoong et al. 2011). The irreversible PI3K inhibitor Wortmannin reduced anchorage-independent growth of CRC cells in a soft agar assay (Khaleghpour et al. 2004), and small interfering RNA-mediated knockdown of PI3K p85α in CRC cells induced G1-phase arrest (Sun et al. 2009). Ongoing studies will be investigating the effects of p110α isoform-specific inhibitors and p110α mutant-specific inhibitors in CRC cells with PIK3CA mutations.
6.5.6 Combination Therapy
Despite the moderate successes of single agent targeted cancer therapies, preclinical and clinical data suggest a clear role for combination therapy. Combination therapies have the potential advantages of delaying the development of resistance, improving response rate, and improving harder clinical endpoints such as progression-free and overall survival. Most clinically effective targeted therapies are directed against RTKs, such as KIT, EGFR, VEGFR, and HER2, which modulate multiple downstream intracellular pathways. RTK inhibition therefore blocks multiple signaling pathways, not only the PI3K pathway. Despite preclinical evidence of single agent PI3K inhibition in CRC, it remains to be seen whether inhibition of the PI3K pathway alone will offer advantages over upstream RTK inhibition. Other concerns in targeting PI3K pathway components alone is the cross talk between many of the RTK signaling pathways such as the RAS/RAF/MEK/ERK and PI3K/AKT pathways, as well as complex signaling feedback loops.
One of the major pitfalls of targeted therapies is the emergence of resistance. When cancer cells are treated with drugs that block a single molecular target, they are often able to activate alternative pathways as escape mechanisms to overcome the blockade and therefore the effectiveness of these drugs. Recent studies have shown that inhibition of mTORC1 leads to activation of the ERK signaling pathway capable of driving proliferation and growth (Carracedo et al. 2008). Further, mutations in PIK3CA often coexist with other genetic lesions, such as KRAS or BRAF mutations in CRC. KRAS can directly activate the RAF/MEK/ERK and PI3K/AKT signaling pathways by directly binding to RAF proteins and the PI3K subunit p110. Therefore, simultaneous inhibition of MEK/ERK and PI3K/AKT may have potential therapeutic benefit in KRAS-mutant cancers. Engelman et al. showed that KRAS-driven mice lung tumors did not respond when a dual mTOR/PI3K inhibitor (NVP-BEZ235) was administered after tumors were established. However, concomitant inhibition of the PI3K and RAF pathways with NVP-BEZ235 and AZD6244, an inhibitor of MEK, inhibits tumor growth in these mice (Engelman et al. 2008). In human NSCLC cell lines and animal models combination therapy with the MEK inhibitor PD-0329501, and mTOR inhibitor rapamycin, or its derivative AP 23573 (ARIAD Pharmaceuticals/Merck), showed a synergistic effect in the inhibition of cell proliferation and protein translation, indicating that both pathways converge to regulate cell growth and ribosomal biogenesis (Legrier et al. 2007).
Several reports involving CRC cell lines support the advantage of combination therapy. KRAS-mutant CRC cell lines were resistant to MEK inhibition when PTEN deletions or activating PI3KCA mutations exist (Wee et al. 2009). The combination of RTK and MEK inhibitors led to dual inhibition of PI3K and MEK signaling, marked growth suppression and apoptosis of KRAS-mutant CRC in vitro and xenograft models in vivo. The combination of the mTOR inhibitor, rapamycin, and the MEK inhibitor, PD89059, caused cell-cycle arrest and induced apoptosis in KRAS-mutant CRC cell lines (Zhang et al. 2009). Taken together, these studies support the idea that rational combinations of targeted treatments, especially those blocking both the PI3K and RAS/RAF/MEK pathways, to circumvent, reverse, or even stop resistance are necessary for optimal use of molecular targeted therapies in CRC (Engelman 2009; She et al. 2005). Indeed, many pharmaceutical companies are actively looking for promising combination therapies with existing targeted drugs or nonspecific cytotoxic drugs. In a recent phase I trial, the combination of PI3K and MEK inhibitors from Genentech, GDC-0941 and GDC-0973 respectively, was effective against advanced solid tumors at tolerable doses for patients. Additionally, mTOR and MEK inhibitor combinations as well as AKT inhibitor (MK-2206) and MEK inhibitor (AZD6244) combinations are in clinical trials.
6.6 Targeting Angiogenesis via the PI3K Pathway Inhibitors
Angiogenesis, the physiological process involving the growth of new blood vessels from preexisting vessels, plays an essential role in tumor growth and metastasis (Folkman 2007). Tumor growth requires angiogenesis when the size of the tumor reaches 1–2 mm in diameter (Folkman 2007). Endothelial cells express numerous cell-surface RTK receptors such as VEGFR1-3, TIE-1/2, PDGFR-beta, and ERBB1–4 to integrate the VEGFs secreted by tumor and stromal cells (Yuan and Cantley 2008; Hofer and Schweighofer 2007). Molecularly targeted agents against VEGF have been developed, and in clinical trials were shown to augment the efficacy of cytotoxic chemotherapy in patients with advanced CRC (Chee and Sinicrope 2010). Based on improved survival, the anti-VEGF monoclonal antibody bevacizumab (Avastin) was approved by the FDA for the treatment of advanced CRC in 2004. The success of bavacizumab in CRC treatment underscores the idea that angiogenesis inhibition is an effective form of CRC therapy.
Numerous studies using conditional or germline knockout mice suggest the strong connection between the PI3K pathway and angiogenesis. PI3K/AKT signaling can induce expression of VEGF and suppress the expression of anti-angiogenic protein TSP-1 in cancer and endothelial cells (Niu et al. 2004; Wen et al. 2001). FOXO1 germline deletion results in defects in arterial development and early vessel remodeling (Furuyama et al. 2004). Moreover, somatic deletion of all three FOXO genes in mice showed that FOXO regulates endothelial cell homeostasis (Paik et al. 2007). Complete loss of PTEN in endothelial cells resulted in a phenotype indicative of impaired vascular remodeling, resulting in embryonic lethality, whereas heterozygous deletion of PTEN in the endothelium increased tumor growth by enhancing tumor angiogenesis (Hamada et al. 2005). Constitutive AKT1 activation in endothelial cells by transgenic expression of a myristoylated AKT1 caused abnormal vessel patterning and congestion (Sun et al. 2005). A conditional mouse model with homozygous deletion of the class IA PI3K regulatory subunits (p85α, p55α, p50α, and p85β) in the endothelium impaired vessel integrity during development and decreased the rate of tumor growth (Yuan et al. 2008). In another study, embryos with kinase-dead p110α developed gross vascular defects indicating p110α is essential for endothelial cell migration and angiogenesis (Graupera et al. 2008). Overall, the results from genetic mouse models strongly suggest that inhibiting the PI3K pathway may have anti-angiogenic effects in reducing tumor growth. In fact, the traditional PI3K inhibitor LY2904002 inhibited the expression of VEGF in both endothelial cells and ovarian cancer cells (Skinner et al. 2004; Jiang et al. 2000) and blocked EGF-induced expression of VEGF and leptin in the CRC cell line HT-29 (Cascio et al. 2009). Furthermore, LY294001 showed in vivo anti-angiogenic activity by decreasing the microvessel density in tumor tissue of a mouse U87 xenograft model (Su et al. 2003). Newer PI3K inhibitors including SF1126 (Garlich et al. 2008), ZSTK474 (Kong et al. 2009), and PI103 (Raynaud et al. 2007) have also demonstrated anti-angiogenic activity in xenograft models, as we discussed previously. A dual PI3K/mTOR inhibitor, NVP-BEZ235, blocked VEGF-induced angiogenesis in mice (Schnell et al. 2008), and inhibited the growth and proliferation of cancer cells with wild type and mutant p110α (Serra et al. 2008). The mTOR inhibitor rapamycin and its analogues are among the most extensively studied drugs in the clinic as anti-angiogenic agents. Rapamycin markedly reduced production of VEGF (Zhong et al. 2000), and abrogated the response of vascular endothelial cells to stimulation by VEGF in vivo (Guba et al. 2002). In RCC, loss of the von-Hippel-Lindau tumor suppressor (VHL) that normally inhibits HIF1A causes enhanced vascularization, and an mTOR analogue, CCI-779, sensitized the cancer cells to death (Thomas et al. 2006). The efficacy of CCI-779 in RCC likely results from rapamycin inhibition of mTORC1-dependent translation and action of HIF1A, thereby decreasing VEGF production (Hudson et al. 2002; Bernardi et al. 2006). Recently, the catalytic mTOR inhibitors, OSI-027 and OXA-01, have been shown to significantly reduce angiogenesis and tumor growth compared to rapamycin alone (Falcon et al. 2011). Taken together the mounting preclinical data suggest targeting the PI3K pathway in the clinic may have the dual effect of blocking both tumor growth and angiogenesis.
6.7 Future Directions
Since the discovery of PI3K more than 20 years ago, our knowledge of the PI3K pathway in various cellular processes and tumorigenesis has experienced unprecedented advances. The PI3K pathway is unquestionably important in cancers such as CRC, and has demonstrated clear potential as an anticancer target. A number of targeted agents that inhibit the key components of the PI3K pathway are already in clinical trials. The question remains as to how PI3K inhibitors can be strategically applied in patients to maximize patient benefit and minimize toxicity. To understand the optimal role of PI3K inhibitors further work is needed. Rather than testing compounds in mixed patient populations, which occur during the traditional cancer clinical trial, the number of genotype-directed and Bayesian hypothesis-testing trials must be increased. Multiple preclinical studies demonstrated that cancers with PIK3CA mutations might be most sensitive to PI3K inhibition, while cancers with KRAS mutations might be inherently more resistant to single agent PI3K inhibitors (Dan et al. 2010; Sos et al. 2009; Ihle et al. 2009b). Mutation-specific patient clinical trial entry will both address proof of principle and tease out parameters related to treatment failure, ultimately bringing personalized treatment in the clinic. The complexity of PI3K signaling due to numerous downstream targets, feedback loops, and cross talk with multiple pathways leading to drug resistance will likely emerge with single agent therapy. In order to figure out the most effective combination therapies to prevent development of drug resistance, preclinical and clinical studies will need to focus on potential resistance mechanisms of PI3K inhibitors. Furthermore, it is imperative to improve preclinical cancer models and assays capable of predicting drug response or potential drug resistance in clinical trials. For example, development of genetically engineered CRC mouse models with PIK3CA mutations alone, or in combination with other mutations such as APC and/or KRAS, would be an especially valuable tool for understanding drug resistance mechanisms and selection of the most effective method of PI3K pathway inhibition. Finally, although there are growing numbers of isoform-specific and allosteric PI3K pathway inhibitors, the majority of inhibitors in the pathway are ATP-competitive pan inhibitors which may not possess the potency and selectivity necessary for efficacy in patients. The importance of high throughput structural biology techniques, such as X-ray crystallography and kinome wide selectivity profiling will continue aid development of isoform-specific, mutant-specific, and allosteric inhibitors of key components of the PI3K pathway. Direct knowledge of the three dimensional structure of the target affords the best tool for rational inhibitor design and optimization of target selectivity. Crystal structures of p110α mutants, p110β, p110δ, and mTOR will be essential to the advancement of future strategies targeting the PI3K pathway. The success of PI3K pathway inhibitors in CRC and other cancers depends upon the interplay of basic molecular understanding, rational inhibitor design, biomarker advances, and ultimately well-designed clinical trials.
References
Abubaker J, Bavi P, Al-Haqawi W, Jehan Z, Munkarah A, Uddin S, Al-Kuraya KS (2009) PIK3CA alterations in Middle Eastern ovarian cancers. Mol Cancer 8:51
Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM (2011) INPP4B: the new kid on the PI3K block. Oncotarget 2:321–328
Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P (1996) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399:333–338
Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M (1997) 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 7:776–789
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26:1626–1634
Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA (2008) Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol 4:691–699
Bader AG, Kang S, Zhao L, Vogt PK (2005) Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 5:921–929
Baselga J (2011) Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist 16(suppl 1):12–19
Bendell JC, Nemunaitis J, Vukelja SJ, Hagenstad C, Campos LT, Hermann RC, Sportelli P, Gardner L, Richards DA (2011) Randomized placebo-controlled phase II trial of perifosine plus capecitabine as second- or third-line therapy in patients with metastatic colorectal cancer. J Clin Oncol 29:4394–4400
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J (2012) Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 30(3):282–290
Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP (2006) PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 442:779–785
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463:899–905
Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM (2002) The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem 277:33895–33900
Bhaskar PT, Hay N (2007) The two TORCs and Akt. Dev Cell 12:487–502
Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem 274:10963–10968
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Calera MR, Martinez C, Liu H, Jack AK, Birnbaum MJ, Pilch PF (1998) Insulin increases the association of Akt-2 with Glut4-containing vesicles. J Biol Chem 273:7201–7204
Caron RW, Yacoub A, Li M, Zhu X, Mitchell C, Hong Y, Hawkins W, Sasazuki T, Shirasawa S, Kozikowski AP, Dennis PA, Hagan MP, Grant S, Dent P (2005) Activated forms of H-RAS and K-RAS differentially regulate membrane association of PI3K, PDK-1, and AKT and the effect of therapeutic kinase inhibitors on cell survival. Mol Cancer Ther 4:257–270
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP (2008) Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 118:3065–3074
Cascio S, Ferla R, D’Andrea A, Gerbino A, Bazan V, Surmacz E, Russo A (2009) Expression of angiogenic regulators, VEGF and leptin, is regulated by the EGF/PI3K/STAT3 pathway in colorectal cancer cells. J Cell Physiol 221:189–194
Castro AF, Rebhun JF, Clark GJ, Quilliam LA (2003) Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem 278:32493–32496
Chee CE, Sinicrope FA (2010) Targeted therapeutic agents for colorectal cancer. Gastroenterol Clin North Am 39:601–613
Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV (2005) Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med 11:1188–1196
Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ (2001a) Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276:38349–38352
Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB III, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ (2001b) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292:1728–1731
Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, Hummersone M, Malagu K, Menear K, Jenkins R, Jacobsen M, Smith GC, Guichard S, Pass M (2010) AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 70:288–298
Cleary JM, Shapiro GI (2010) Development of phosphoinositide-3 kinase pathway inhibitors for advanced cancer. Curr Oncol Rep 12:87–94
Cook PJ, Doll R, Fellingham SA (1969) A mathematical model for the age distribution of cancer in man. Int J Cancer 4:93–112
Crul M, Rosing H, de Klerk GJ, Dubbelman R, Traiser M, Reichert S, Knebel NG, Schellens JH, Beijnen JH, ten Bokkel Huinink WW (2002) Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. Eur J Cancer 38:1615–1621
Cully M, You H, Levine AJ, Mak TW (2006) Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 6:184–192
Dan S, Okamura M, Seki M, Yamazaki K, Sugita H, Okui M, Mukai Y, Nishimura H, Asaka R, Nomura K, Ishikawa Y, Yamori T (2010) Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res 70:4982–4994
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241
Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME (2000) 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell 6:41–51
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11:753–762
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278:687–689
Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP (1998) Pten is essential for embryonic development and tumour suppression. Nat Genet 19:348–355
Diehl JA, Cheng M, Roussel MF, Sherr CJ (1998) Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12:3499–3511
Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ (2002) FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol 156:531–542
Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275:661–665
Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ (2005) Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol 25:1869–1878
Engelman JA (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 9:550–562
Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong KK (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14:1351–1356
Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM (2011) Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res 71:1573–1583
Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, Weiss WA (2007) A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res 67:7960–7965
Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM (2009) Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 7:e38
Folkman J (2007) Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 6:273–286
Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJ, Withers DJ, Vanhaesebroeck B (2006) Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441:366–370
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L (2007) PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 97:1139–1145
Fruman DA (2004) Phosphoinositide 3-kinase and its targets in B-cell and T-cell signaling. Curr Opin Immunol 16:314–320
Fruman DA, Rommel C (2011) PI3Kdelta inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discov 1:562–572
Fruman DA, Meyers RE, Cantley LC (1998) Phosphoinositide kinases. Annu Rev Biochem 67:481–507
Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Ikeda K, Motoyama N, Mori N (2004) Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem 279:34741–34749
Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11:1457–1466
Garcia-Echeverria C, Sellers WR (2008) Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 27:5511–5526
Garlich JR, De P, Dey N, Su JD, Peng X, Miller A, Murali R, Lu Y, Mills GB, Kundra V, Shu HK, Peng Q, Durden DL (2008) A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res 68:206–215
Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG (2003) Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest 112:197–208
Gasic GP (1994) Basic-helix-loop-helix transcription factor and sterol sensor in a single membrane-bound molecule. Cell 77:17–19
George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR, Williams RM, Acerini CL, Dunger DB, Barford D, Umpleby AM, Wareham NJ, Davies HA, Schafer AJ, Stoffel M, O’Rahilly S, Barroso I (2004) A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 304:1325–1328
Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 12:502–513
Gordan JD, Thompson CB, Simon MC (2007) HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12:108–113
Granville CA, Memmott RM, Gills JJ, Dennis PA (2006) Handicapping the race to develop inhibitors of the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin pathway. Clin Cancer Res 12:679–689
Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJ, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B (2008) Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453:662–666
Grothey A (2009) Medical treatment of advanced colorectal cancer in 2009. Ther Adv Med Oncol 1:55–68
Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M, Bruns CJ, Zuelke C, Farkas S, Anthuber M, Jauch KW, Geissler EK (2002) Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 8:128–135
Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J (2007) Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 129:957–968
Hamada K, Sasaki T, Koni PA, Natsui M, Kishimoto H, Sasaki J, Yajima N, Horie Y, Hasegawa G, Naito M, Miyazaki J, Suda T, Itoh H, Nakao K, Mak TW, Nakano T, Suzuki A (2005) The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev 19:2054–2065
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hilgard P, Klenner T, Stekar J, Nossner G, Kutscher B, Engel J (1997) D-21266, a new heterocyclic alkylphospholipid with antitumour activity. Eur J Cancer 33:442–446
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279:577–580
Hofer E, Schweighofer B (2007) Signal transduction induced in endothelial cells by growth factor receptors involved in angiogenesis. Thromb Haemost 97:355–363
Howes AL, Chiang GG, Lang ES, Ho CB, Powis G, Vuori K, Abraham RT (2007) The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Mol Cancer Ther 6:2505–2514
Hsieh AC, Ruggero D (2010) Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin Cancer Res 16:4914–4920
Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, Ruggero D (2010) Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 17:249–261
Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB (2002) Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 62:1087–1092
Huang XF, Chen JZ (2009) Obesity, the PI3K/Akt signal pathway and colon cancer. Obes Rev 10:610–616
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM (2007) The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 318:1744–1748
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 22:7004–7014
Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, Kirkpatrick L, Powis G (2004) Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 3:763–772
Ihle NT, Paine-Murrieta G, Berggren MI, Baker A, Tate WR, Wipf P, Abraham RT, Kirkpatrick DL, Powis G (2005) The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther 4:1349–1357
Ihle NT, Lemos R, Schwartz D, Oh J, Halter RJ, Wipf P, Kirkpatrick L, Powis G (2009a) Peroxisome proliferator-activated receptor gamma agonist pioglitazone prevents the hyperglycemia caused by phosphatidylinositol 3-kinase pathway inhibition by PX-866 without affecting antitumor activity. Mol Cancer Ther 8:94–100
Ihle NT, Lemos R Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G (2009b) Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res 69:143–150
Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M (2005) Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 65:4562–4567
Inoki K, Zhu T, Guan KL (2003a) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590
Inoki K, Li Y, Xu T, Guan KL (2003b) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834
Inukai K, Funaki M, Ogihara T, Katagiri H, Kanda A, Anai M, Fukushima Y, Hosaka T, Suzuki M, Shin BC, Takata K, Yazaki Y, Kikuchi M, Oka Y, Asano T (1997) p85alpha gene generates three isoforms of regulatory subunit for phosphatidylinositol 3-kinase (PI 3-Kinase), p50alpha, p55alpha, and p85alpha, with different PI 3-kinase activity elevating responses to insulin. J Biol Chem 272:7873–7882
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ (2008) Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 454:776–779
Jiang BH, Zheng JZ, Aoki M, Vogt PK (2000) Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci USA 97:1749–1753
Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Wang D, Ihle JN (2002) Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol 22:8580–8591
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359:1757–1765
Khaleghpour K, Li Y, Banville D, Yu Z, Shen SH (2004) Involvement of the PI 3-kinase signaling pathway in progression of colon adenocarcinoma. Carcinogenesis 25:241–248
Kishimoto H, Hamada K, Saunders M, Backman S, Sasaki T, Nakano T, Mak TW, Suzuki A (2003) Physiological functions of Pten in mouse tissues. Cell Struct Funct 28:11–21
Kohn AD, Summers SA, Birnbaum MJ, Roth RA (1996) Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem 271:31372–31378
Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK (2003) Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther 2:1093–1103
Kong D, Okamura M, Yoshimi H, Yamori T (2009) Antiangiogenic effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor. Eur J Cancer 45:857–865
Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y, Ui M, Hazeki O, Katada T (1997) Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J Biol Chem 272:24252–24256
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, Bouche O, Reid J, Stone S, Penault-Llorca F (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27:5924–5930
Lee YJ, Hong KH, Yun J, Oh SP (2006) Generation of activin receptor type IIB isoform-specific hypomorphic alleles. Genesis 44:487–494
Legrier ME, Yang CP, Yan HG, Lopez-Barcons L, Keller SM, Perez-Soler R, Horwitz SB, McDaid HM (2007) Targeting protein translation in human non small cell lung cancer via combined MEK and mammalian target of rapamycin suppression. Cancer Res 67:11300–11308
Lengauer C, Kinzler KW, Vogelstein B (1997) Genetic instability in colorectal cancers. Nature 386:623–627
Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396:643–649
Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R (1998) The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res 58:5667–5672
Li XH, Zheng HC, Takahashi H, Masuda S, Yang XH, Takano Y (2009) PTEN expression and mutation in colorectal carcinomas. Oncol Rep 22:757–764
Li FH, Shen L, Li ZH, Luo HY, Qiu MZ, Zhang HZ, Li YH, Xu RH (2010) Impact of KRAS mutation and PTEN expression on cetuximab-treated colorectal cancer. World J Gastroenterol 16:5881–5888
Liang J, Slingerland JM (2003) Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2:339–345
Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S (2008) Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol 9:962–972
Liu P, Cheng H, Roberts TM, Zhao JJ (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 8:627–644
Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, Canestrari E, Funel N, Schiavon G, Petrini I, Magnani M, Tonini G, Campani D, Floriani I, Cascinu S, Falcone A (2009) PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 27:2622–2629
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 7:1851–1863
Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, Amzel LM (2009) A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci USA 106:16996–17001
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261–1274
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10:151–162
Markman B, Atzori F, Perez-Garcia J, Tabernero J, Baselga J (2010) Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol 21:683–691
Mayo LD, Donner DB (2001) A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 98:11598–11603
Mizoguchi M, Nutt CL, Mohapatra G, Louis DN (2004) Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol 14:372–377
Molckovsky A, Siu LL (2008) First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American society of clinical oncology meeting. J Hematol Oncol 1:20
Morrow CJ, Gray A, Dive C (2005) Comparison of phosphatidylinositol-3-kinase signalling within a panel of human colorectal cancer cell lines with mutant or wild-type PIK3CA. FEBS Lett 579:5123–5128
Naguib A, Cooke JC, Happerfield L, Kerr L, Gay LJ, Luben RN, Ball RY, Mitrou PN, McTaggart A, Arends MJ (2011) Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study: associations with clinicopathological and dietary factors. BMC Cancer 11:123
Niu Q, Perruzzi C, Voskas D, Lawler J, Dumont DJ, Benjamin LE (2004) Inhibition of Tie-2 signaling induces endothelial cell apoptosis, decreases Akt signaling, and induces endothelial cell expression of the endogenous anti-angiogenic molecule, thrombospondin-1. Cancer Biol Ther 3:402–405
Nosho K, Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Zepf D, Yan L, Longtine JA, Fuchs CS, Ogino S (2008) PIK3CA mutation in colorectal cancer: relationship with genetic and epigenetic alterations. Neoplasia 10:534–541
Nutley BP, Raynaud FI, Wilson SC, Fischer PM, Hayes A, Goddard PM, McClue SJ, Jarman M, Lane DP, Workman P (2005) Metabolism and pharmacokinetics of the cyclin-dependent kinase inhibitor R-roscovitine in the mouse. Mol Cancer Ther 4:125–139
O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66:1500–1508
Oda K, Okada J, Timmerman L, Rodriguez-Viciana P, Stokoe D, Shoji K, Taketani Y, Kuramoto H, Knight ZA, Shokat KM, McCormick F (2008) PIK3CA cooperates with other phosphatidylinositol 3′-kinase pathway mutations to effect oncogenic transformation. Cancer Res 68:8127–8136
Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, Chan AT, Engelman JA, Kraft P, Cantley LC, Giovannucci EL, Fuchs CS (2009) PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol 27:1477–1484
Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJ, Vanhaesebroeck B (2002) Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297:1031–1034
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB (1999) NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401:82–85
Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA (2007) FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128:309–323
Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC Jr, Sonenberg N (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 371:762–767
Peterson RT, Schreiber SL (1998) Translation control: connecting mitogens and the ribosome. Curr Biol 8:R248–R250
Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA (2001) The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res 61:7426–7429
Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A (2005) PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24:6465–6481
Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8:224–236
Poulogiannis G, Ichimura K, Hamoudi RA, Luo F, Leung SY, Yuen ST, Harrison DJ, Wyllie AH, Arends MJ (2010) Prognostic relevance of DNA copy number changes in colorectal cancer. J Pathol 220:338–347
Poulogiannis G, Luo F, Arends MJ (2012) RAS signalling in the colorectum in health and disease. Cell Commun Adhes 19(1):1–9
Pullen N, Thomas G (1997) The modular phosphorylation and activation of p70s6k. FEBS Lett 410:78–82
Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G (1998) Phosphorylation and activation of p70s6k by PDK1. Science 279:707–710
Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, Henley A, Di-Stefano F, Ahmad Z, Guillard S, Bjerke LM, Kelland L, Valenti M, Patterson L, Gowan S, de Haven Brandon A, Hayakawa M, Kaizawa H, Koizumi T, Ohishi T, Patel S, Saghir N, Parker P, Waterfield M, Workman P (2007) Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res 67:5840–5850
Roche S, Downward J, Raynal P, Courtneidge SA (1998) A function for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in fibroblasts during mitogenesis: requirement for insulin- and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol 18:7119–7129
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304:554
Samuels Y, Diaz LA Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE (2005) Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 7:561–573
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22:159–168
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM (2000) Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287:1040–1046
Schnell CR, Stauffer F, Allegrini PR, O’Reilly T, McSheehy PM, Dartois C, Stumm M, Cozens R, Littlewood-Evans A, Garcia-Echeverria C, Maira SM (2008) Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res 68:6598–6607
Seki T, Hong KH, Yun J, Kim SJ, Oh SP (2004) Isolation of a regulatory region of activin receptor-like kinase 1 gene sufficient for arterial endothelium-specific expression. Circ Res 94:e72–e77
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J (2008) NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 68:8022–8030
She QB, Solit DB, Ye Q, O’Reilly KE, Lobo J, Rosen N (2005) The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 8:287–297
She QB, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, DeFeo-Jones D, Huber HE, Rosen N (2008) Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One 3:e3065
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314:268–274
Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH (2004) Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem 279:45643–45651
Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, Heynck S, Stuckrath I, Weiss J, Fischer F, Michel K, Goel A, Regales L, Politi KA, Perera S, Getlik M, Heukamp LC, Ansen S, Zander T, Beroukhim R, Kashkar H, Shokat KM, Sellers WR, Rauh D, Orr C, Hoeflich KP, Friedman L, Wong KK, Pao W, Thomas RK (2009) Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci USA 106:18351–18356
Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M, Silver J, Ogino S, Hooshmand S, Kwak E, Freed E, Meyerhardt JA, Saridaki Z, Georgoulias V, Finkelstein D, Fuchs CS, Kulke MH, Shivdasani RA (2009) Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 101:465–472
Stambolic V, Woodgett JR (2006) Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol 16:461–466
Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT (1998) Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 279:710–714
Stokoe D (2001) Pten. Curr Biol 11:R502
Su JD, Mayo LD, Donner DB, Durden DL (2003) PTEN and phosphatidylinositol 3′-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis: evidence for an effect on the tumor and endothelial compartment. Cancer Res 63:3585–3592
Suire S, Hawkins P, Stephens L (2002) Activation of phosphoinositide 3-kinase gamma by Ras. Curr Biol 12:1068–1075
Suire S, Coadwell J, Ferguson GJ, Davidson K, Hawkins P, Stephens L (2005) p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr Biol 15:566–570
Sullivan KM, Kozuch PS (2011) Impact of KRAS mutations on management of colorectal carcinoma. Patholog Res Int 2011:219309
Sun JF, Phung T, Shiojima I, Felske T, Upalakalin JN, Feng D, Kornaga T, Dor T, Dvorak AM, Walsh K, Benjamin LE (2005) Microvascular patterning is controlled by fine-tuning the Akt signal. Proc Natl Acad Sci USA 102:128–133
Sun Y, Zhao S, Tian H, Xie X, Xiao F, Li K, Song Y (2009) Depletion of PI3K p85alpha induces cell cycle arrest and apoptosis in colorectal cancer cells. Oncol Rep 22:1435–1441
Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J (2005) Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab 1:379–391
Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, Martinelli E, Ramon y Cajal S, Jones S, Vidal L, Shand N, Macarulla T, Ramos FJ, Dimitrijevic S, Zoellner U, Tang P, Stumm M, Lane HA, Lebwohl D, Baselga J (2008) Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 26:1603–1610
Taha C, Liu Z, Jin J, Al-Hasani H, Sonenberg N, Klip A (1999) Opposite translational control of GLUT1 and GLUT4 glucose transporter mRNAs in response to insulin. Role of mammalian target of rapamycin, protein kinase b, and phosphatidylinositol 3-kinase in GLUT1 mRNA translation. J Biol Chem 274:33085–33091
Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL (2006) Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med 12:122–127
Tran H, Brunet A, Griffith EC, Greenberg ME (2003) The many forks in FOXO’s road. Sci STKE 2003:RE5
Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA (2005) Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 132:2943–2954
Ueki K, Algenstaedt P, Mauvais-Jarvis F, Kahn CR (2000) Positive and negative regulation of phosphoinositide 3-kinase-dependent signaling pathways by three different gene products of the p85alpha regulatory subunit. Mol Cell Biol 20:8035–8046
Van Ummersen L, Binger K, Volkman J, Marnocha R, Tutsch K, Kolesar J, Arzoomanian R, Alberti D, Wilding G (2004) A phase I trial of perifosine (NSC 639966) on a loading dose/maintenance dose schedule in patients with advanced cancer. Clin Cancer Res 10:7450–7456
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, Boehm JS, Reiling JH, Silver S, Lu Y, Stemke-Hale K, Dutta B, Joy C, Sahin AA, Gonzalez-Angulo AM, Lluch A, Rameh LE, Jacks T, Root DE, Lander ES, Mills GB, Hahn WC, Sellers WR, Garraway LA (2009) AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16:21–32
Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S Jr, Duval A, Carneiro F, Machado JC, Hamelin R, Seruca R (2005) The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer 41:1649–1654
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302:1036–1038
Vink SR, Lagerwerf S, Mesman E, Schellens JH, Begg AC, van Blitterswijk WJ, Verheij M (2006) Radiosensitization of squamous cell carcinoma by the alkylphospholipid perifosine in cell culture and xenografts. Clin Cancer Res 12:1615–1622
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2:489–501
Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269:5241–5248
Voigt P, Dorner MB, Schaefer M (2006) Characterization of p87PIKAP, a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J Biol Chem 281:9977–9986
Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR (1998) Activation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alpha. Biochem J 331(Pt 1):299–308
Walker EH, Perisic O, Ried C, Stephens L, Williams RL (1999) Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402:313–320
Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL (2000) Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 6:909–919
Wan X, Harkavy B, Shen N, Grohar P, Helman LJ (2007) Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 26:1932–1940
Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H (2003) Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4:209–221
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C (2008) PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA 105:13057–13062
Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C, Stegmeier F (2009) PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res 69:4286–4293
Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr (2005) The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 8:25–33
Weinstein IB, Joe A (2008) Oncogene addiction. Cancer Res 68:3077–3080; discussion 3080
Welcker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM (2003) Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol Cell 12:381–392
Wen S, Stolarov J, Myers MP, Su JD, Wigler MH, Tonks NK, Durden DL (2001) PTEN controls tumor-induced angiogenesis. Proc Natl Acad Sci USA 98:4622–4627
West MJ, Stoneley M, Willis AE (1998) Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene 17:769–780
White MF (1998) The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem 182:3–11
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B (2007) The genomic landscapes of human breast and colorectal cancers. Science 318:1108–1113
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124:471–484
Yang H, Kong W, He L, Zhao JJ, O’Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, Cheng JQ (2008) MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res 68:425–433
Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW (2011) First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 29:4688–4695
Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, Manning BD (2011) Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 14:21–32
Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R (2004) A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 6:308–318
Yoong J, Michael M, Leong T (2011) Targeted therapies for gastric cancer: current status. Drugs 71:1367–1384
Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM (1998a) Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol 18:1379–1387
Yu J, Wjasow C, Backer JM (1998b) Regulation of the p85/p110alpha phosphatidylinositol 3′-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem 273:30199–30203
Yu K, Lucas J, Zhu T, Zask A, Gaydos C, Toral-Barza L, Gu J, Li F, Chaudhary I, Cai P, Lotvin J, Petersen R, Ruppen M, Fawzi M, Ayral-Kaloustian S, Skotnicki J, Mansour T, Frost P, Gibbons J (2005) PWT-458, a novel pegylated-17-hydroxywortmannin, inhibits phosphatidylinositol 3-kinase signaling and suppresses growth of solid tumors. Cancer Biol Ther 4:538–545
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27:5497–5510
Yuan TL, Choi HS, Matsui A, Benes C, Lifshits E, Luo J, Frangioni JV, Cantley LC (2008) Class 1A PI3K regulates vessel integrity during development and tumorigenesis. Proc Natl Acad Sci USA 105:9739–9744
Yun J (2010) Allosteric AKT inhibitors as a targeted cancer therapy. Cancer Biol Ther 9:504–506
Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N (2009) Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325:1555–1559
Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B (1998) Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J 17:5085–5094
Zhang YJ, Tian XQ, Sun DF, Zhao SL, Xiong H, Fang JY (2009) Combined inhibition of MEK and mTOR signaling inhibits initiation and progression of colorectal cancer. Cancer Invest 27:273–285
Zhang J, Roberts TM, Shivdasani RA (2011) Targeting PI3K signaling as a therapeutic approach for colorectal cancer. Gastroenterology 141:50–61
Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL (2000) Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60:1541–1545
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC (2001) HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 3:973–982
Zhou XP, Loukola A, Salovaara R, Nystrom-Lahti M, Peltomaki P, de la Chapelle A, Aaltonen LA, Eng C (2002) PTEN mutational spectra, expression levels, and subcellular localization in microsatellite stable and unstable colorectal cancers. Am J Pathol 161:439–447
Zhu T, Gu J, Yu K, Lucas J, Cai P, Tsao R, Gong Y, Li F, Chaudhary I, Desai P, Ruppen M, Fawzi M, Gibbons J, Ayral-Kaloustian S, Skotnicki J, Mansour T, Zask A (2006) Pegylated wortmannin and 17-hydroxywortmannin conjugates as phosphoinositide 3-kinase inhibitors active in human tumor xenograft models. J Med Chem 49:1373–1378
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Yun, J., Poulogiannis, G., Brower, E.T., Klempner, S., Cantley, L.L. (2013). The PI3K Pathway in Colorectal Cancers. In: Haigis, Ph.D., K. (eds) Molecular Pathogenesis of Colorectal Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8412-7_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8412-7_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8411-0
Online ISBN: 978-1-4614-8412-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)