
Abstract
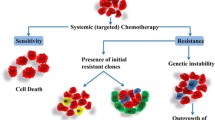
The high heterogeneity and genomic instability of malignant tumors explains why even responsive tumors contain cell clones that are resistant for many possible mechanisms involving intracellular drug inactivation, low uptake or high efflux of anticancer drugs from cancer cells, qualitative or quantitative changes in the drug target. Many tumors, however, are resistant because of insufficient exposure to anticancer drugs, due to pharmacokinetic reasons and inefficient and heterogeneous tumor drug distribution, related to a deficient vascularization and high interstitial pressure. Finally, resistance can be related to the activation of anti-apoptotic and cell survival pathways by cancer cells and often enhanced by tumor microenvironment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Although significant improvement of the therapy of several human malignancies have been achieved in the last two decades with the use of small molecules or antibodies designed to hit cancer-specific molecular targets, for the majority of solid human tumors at advanced stage pharmacological treatments have only a transient antitumor effect with small or no impact on patients’ survival. The major reason for this disappointing result is the phenomenon of drug resistance.
Although most recent research has been focused on molecular and cellular mechanism of resistance, it seems likely that in many cases the lack of antitumor activity is related to pharmacokinetic reasons, essentially associated with an insufficient drug concentration in the cancer cells.
The pharmacokinetic factors that can be involved in the failure of treatment can be dependent on the route of administration. For example, in the case of orally administered drugs the variable absorption as well as the first pass effect can be responsible for an insufficient bioavailability. For many anticancer drugs the variable metabolism can be a clinically relevant problem, also in consideration of potential interactions with other drugs interfering with the activity of cytochromes involved in drug biotransformation. These pharmacokinetic factors can be investigated by monitoring plasma drug levels, thus allowing, at least in theory, a dose adjustment or a change in the route of administration.
However, the drug plasma levels that for other classes of drugs predict the drug concentration at the pharmacological target, for anticancer drug are often meaningless. The circulating drug concentrations in fact do not necessarily mirror those present in the neoplastic tissue and its metastases because of tumor architecture and microenvironment (Fuso Nerini et al. 2014).
It is known that the vascularization of solid tumors is often inefficient, as the angiogenic process occurring in growing tumors leads to unorganized capillary network with fenestrated vessels and significant leakage of proteins responsible for an increase in the interstitial pressure. Probably further factors contributing to the high interstitial pressure are the inefficient lymphatic drainage as well as a significant infiltration of inflammatory and mesenchymal cells that represent common features of many solid tumors. This peculiar abnormal vascularization and tissue architecture is responsible for a low and heterogeneous tumor drug distribution (Minchinton and Tannock 2006). The evidence of this heterogeneous distribution of anticancer drugs was already available several years ago by examining different parts of tumors. It was demonstrated that in necrotic or hypovascularized and hypoxic parts of the tumor the drug concentrations were lower than those that were normally perfused by blood.
Recently new powerful technologies have been developed to investigate drug distribution in different parts of the tumor by simultaneous visualization and quantification of the drug levels. Among these new approaches, mass spectrometry imaging (MSI) is an interesting technique to obtain specific information on the spatial distribution of a drug linking qualitative molecular information of compounds to their spatial coordinates and distribution within the investigated tissue (Morosi et al. 2013).
MSI can be applied to visualize potentially every tissue component localizing the molecule of interest on the base of its molecular mass and fragmentation pattern, without the need to label the analyte, a great advantage over other imaging techniques such as fluorescence microscopy, positron emission tomography, magnetic resonance spectroscopy, and autoradiography (Sugiura and Setou 2010).
MSI was applied to localize drug distribution and penetration inside solid tumors, thanks to its superior spatial resolution (20–100 μm) and specificity allowing the detection of parent compound and metabolites simultaneously (Buck et al. 2015; Connell et al. 2015).
MSI can also provide the quantitative amounts of target compounds in well-defined region of interest of the examined tissue, ideally in a single pixel. This goal is extremely challenging depending on the technical possibilities and limitations of the MSI instrument hardware, but equally on the chosen calibration/standardization strategy (Giordano et al. 2016a; Rzagalinski and Volmer 2016).
MSI has also some limitation: it is only applicable to molecules that are ionizable, the selection of optimal matrix is arduous and driven by empirical results, moreover the interpretation of quantitative data is often complex especially because of ion suppression effect and the chemical noise from the matrix ions covering the drug ion signal and finally, the sensitivity is quite limited (Prideaux and Stoeckli 2012).
By applying this methodology it has become clear that drug distribution in tumors is very heterogeneous comparing different tumor models having different histopathological characteristics (Giordano et al. 2016b). Figure 1 shows the intratumor distribution of the anticancer drug paclitaxel by MSI. The drug distribution is homogeneus only in the ovarian cancer model while it is highly irregular in the other exemples of tumor types. As you can see in other models such as sarcoma or breast cancer, areas of the tumor where the drug is highly concentrated and areas where it is almost absent can be observed at the same time. Broad parts of the tumor where the drug is not present at all are present especially in mesothelioma xenograft. This aspect surely contributes to explain tumor resistance. Torok et al. recently correlate the antitumor activity of several antiangiogenic drugs with their intratumoral distribution data obtained by MSI (Torok et al. 2017). Moreover, Cesca et al. observed an enhancement in paclitaxel activity combined with bevacizumab associated with the improvement of its intratumor distribution (Cesca et al. 2016).

Paclitaxel distribution in different cancer models analyzed by MALDI mass spectrometry imaging. Light blue and green mark the drug presence on the basis of the color scale on the right
The possibility to superimpose MSI molecular images with histological and immune-histological evaluation could help understanding the factors that impair drug penetration. Finally, this technology could be used in future to evaluate the effect of therapeutic strategies aimed at modifying tumor microenvironment or altering drug properties to facilitate drug uptake and intratumor distribution.
2 Cellular Mechanism of Multidrug Resistance
The hallmarks of cellular anticancer drug resistance fall into distinct categories:
2.1 Drug Activation and Inactivation
Altered local drug metabolism and detoxification are key resistance mechanisms highly specific for each class of drugs. Lack of prodrug activation or epigenetic silencing – by promoter methylation – of genes involved in drug processing are examples of potential cause of resistance and are frequently tumor-specific. Classical examples could be the inactivation of platinum drugs by the thiol glutathione (Meijer et al. 1992) or metallothioneins (Amable 2016), or by epigenetic mechanisms, e.g., silencing – by methylation – of gene encoding for thymidine phosphorylase, an enzyme involved in the conversion of capecitabine (prodrug) to 5-fluorouracil (active drug) (Longley et al. 2003). For many DNA interacting agents, e.g., alkylating agents, anthracyclines, and platinum drugs, an important mechanism of inactivation is related to the overexpression of Glutathione–S–Transferases (GSTs) that metabolizes the drugs into inactive molecules (Kauvar et al. 1998).
2.2 Expression of Drug Efflux Pumps
Plasma membrane transport proteins are key players in the uptake of nutrients, such as sugars, amino acids, nucleosides, and inorganic ions and the efflux of xenobiotic toxins, including many anticancer drugs. Transport proteins can be classified into two major families, the solute carrier (SLC) and the ATP-binding cassette (ABC) transporters and can affect drug absorption, distribution, and excretion conferring sensitivity or resistance to anticancer drugs.
The importance of ABC transporters in cancer therapy has been well documented while the impact of the solute carriers (SLC) on cancer therapy has not been extensively characterized (Table 1).
So far, the most characterized transporters belong to ABC family and include ABCB1 (also known as MDR1 or P-glycoprotein), ABCC1 (MRP1), and ABCG2 (BCRP or MXR).
The ATP-binding cassette (ABC) transporters superfamily is a large group of membrane proteins that are responsible for the translocation of various cytotoxic molecules out of the cell reducing their intracellular concentration and thus their cytotoxicity. ABC transporters are codified by 48 genes (divided into 7 subfamilies) and characterized by a conserved quaternary structure containing 2 transmembrane domains (TMDs) and 2 nucleotide (ATP)-binding domains (NBDs). Their ATP-dependent activity is involved in the movement of a wide variety of xenobiotics – including drugs – lipids, peptides, and metabolic products across the plasma and intracellular membranes. The ABC transporters tissue distribution reflects the complex physiological network of these proteins and reveals their important role in absorption, excretion, and distribution of drugs.
P-glycoprotein (P-gp) is a 170 kDa glycoprotein coded by ABCB1 gene mapped to chromosome 7q21.1. Its presence in normal tissue (as liver, kidney, brain, intestine, etc.) reveals the ability of modulating bioavailability and transport of wide range of substrates, especially organic molecules containing aromatic groups, and suggests its role in determining multidrug resistance (MDR) phenotype. P-gp is overexpressed in many tumors (thus causing intrinsic drug resistance) and the expression of P-gp can also be induced by chemotherapy (thus resulting in the acquired development of MDR) (Thomas and Coley 2003). Expression of P-gp fluctuates with elevated expression level in untreated cancer into higher level upon relapse after chemotherapy and undetectable or low level in the expression in drug sensitive tumors. P-gp expression has been reported in 40% of breast cancer (Kao et al. 2001), 20% of ovarian cancer (Baekelandt et al. 2000), and in more than 50% of patients with acute myelogeneous leukemia experiencing relapse (Leith et al. 1999) and is now associated with treatment failure in kidney, liver, and colon cancer (Ambudkar et al. 2003). It confers the strongest resistance to the widest variety of compounds including vinca alkaloids, anthracyclines, and taxanes contributing to the failure of chemotherapy (Szakács et al. 2006). Recent reports have suggested that molecularly targeted therapies, such as some kinases inhibitors, are also substrates for drug efflux proteins (Shervington and Lu 2008).
MRP1 is a transporter involved in glutathione-linked organic compound transport and is normally expressed in the basolateral membrane of cells from testis, kidney, oropharyngeal mucosa, etc. Recently the overexpression of MRP1 has been correlated with chemoresistance in prostate, lung, and breast cancer (Holohan et al. 2013) and confers resistance to several hydrophobic compounds that are also P-gp substrates.
Screens carried out with the NCI60 cell panel indicate that there is a strong correlation between the expression of several transporter (including ABC superfamily) and decrease in chemosensitivity.
The goal of reversing MDR in the clinic through the pharmacological inhibition of ABC transporter has been pursued for years and different generation of inhibitors have been developed. Only P-gp inhibitors have been evaluated in clinical studies. However, the results of clinical trials using modulators of P-gp have been disappointing mainly because of the required marked reduction of the anticancer drug doses. In addition it is now very clear that resistance is not only due to the overexpression of P-gp, but also to many other mechanisms, thus making unrealistic to reverse tumor resistance by pump inhibition.
The resistance of tumors originating from tissues expressing high levels of P-gp (such as colon, kidney, or the adrenocortex) often extends to drugs that are not subject to P-gp-mediated transport, suggesting that “intrinsically resistant” cancer is also protected by non-Pgp-mediated mechanisms (Szakács et al. 2006).
Evidence linking P-gp expression with poor clinical outcome is more conclusive for breast cancer, sarcoma, and certain types of leukemia. P-gp expression in patients with AML has consistently been associated with reduced chemotherapy response rates and poor survival (Pallis and Russell 2004), in contrast with MRP1 whose expression is not a significant factor in drug resistance in AML (Leith et al. 1999).
The expression of these transporters as a mechanism of acquired resistance after a specific treatment is also responsible for the phenomenon of cross-resistance that must be taken into account especially for a clinical perspective.
2.3 DNA Damage Repair
The cellular response to DNA damage involves several mechanisms aimed at repairing the damage, thus restoring DNA integrity or alternatively activating cell death pathways.
Many chemotherapeutic drugs induce DNA damage either directly (for example, platinum-based drugs) or indirectly (for example, topoisomerase inhibitors). Without repair, this damage can result in genetic instability, acquisition of further mutations, and cytotoxic effects (Holohan et al. 2013). The different sensitivity to DNA damaging agents can be due to a different ability to repair drug-induced damage. For example, cancer cells deficient in Homologous Recombination (HR), e.g., because of mutations of BRCA1 or BRCA2 genes, are very sensitive to DNA damage caused by platinum drugs. HR deficient cancers are very sensitive to PARP inhibitors that cause an accumulation of DNA single strand breaks. In HR proficient cells, DNA single strand breaks generate DNA double strand breaks during DNA replication that are repaired before mitosis. Instead in HR deficient cells the inhibition of base excision repair by PARP inhibitors leads to unrepairable DNA double strand breaks with consequent cell death. This is the concept of synthetic lethality whose application has shown promising results in patients with breast and ovarian tumors (Farmer et al. 2005). Even in this case resistance can occur because of the acquisition of secondary mutations restoring HR normal function (Ashworth 2008).
Other pathways involved in DNA repair that have been reported to be relevant for drug resistance are nucleotide-excision repair (NER) and mismatch repair (MMR). The first one is required for the repair of DNA damage caused by many DNA-damaging drugs, such as platinum-based drugs. High expression of a key component, excision repair cross-complementing 1 (ERCC1), has been linked with poor responses to chemotherapy in non-small-cell lung carcinoma (NSCLC), gastric, and ovarian cancer (Kirschner and Melton 2010). Notably, testicular cancers, which are very sensitive to cisplatin treatment, have very low levels of ERCC1 (Usanova et al. 2010).
The mismatch repair (MMR) system, whose key players are MLH1 and MSH2 genes, is crucial for maintaining genomic integrity, and its deficiency has been linked to high mutation rate of a large number of genes including those involved in immunogenicity and drug sensitivity. This explains why hypermethylation of MLH1 causes resistance to cisplatin and carboplatin (Fink et al. 1998). More recently, a synthetic lethal interaction between MSH2-targeted short interfering RNA (siRNA) and methotrexate was identified in MMR-deficient cancer cells; methotrexate caused the accumulation of oxidative lesions such as 8-oxoguanine (8-oxoG) in MSH2-deficient cells, which resulted in a loss of viability through apoptosis. This has led to an on-going Phase II clinical trial with methotrexate in patients with MSH2-deficient metastatic colorectal cancer using measurement of 8-oxoG lesions as a biomarker (Holohan et al. 2013).
Another DNA protein involved in resistance is O-6-methylguanine-DNA-methyl transferase (MGMT), whose overexpression is responsible for the resistance to methylating agents such as temozolomide. The hypomethylation of the promoter of the MGMT gene has been associated with resistance of human glioblastoma to temozolomide (Thomas et al. 2017).
2.4 Deregulation of Apoptosis
The resistance to anticancer drugs can be due to the fact that in cancer cells, apoptotic pathways are frequently dysfunctional. Numerous intrinsic adaptive responses can be triggered and promote survival of cancer cells exposed to DNA-damaging agents. Anti-apoptotic BCL-2 family members, inhibitor of apoptosis proteins (IAPs), and the caspases are key proteins of this mechanism. Mutations, amplifications, chromosomal translocations, and overexpression of the genes encoding for these proteins have been associated with various malignancies and linked to resistance to chemotherapy and targeted therapies.
BCL-2 family proteins have a pivotal role in dictating cell fate following chemotherapy treatment. The balance between the anti-apoptotic BCL-2 family members (such as BCL-XL and MCL1) and pro-apoptotic family members (such as BAX, BAD, and BAK, as well as various BH3-only proteins) is critical in determining the activation or not of a common pathway generally stimulated from all cytotoxic drugs that causes the mitochondrial outer membrane permeabilization (MOMP) and finally cell death. Overexpression of one or more anti-apoptotic proteins or underexpression of one or more pro-apoptotic proteins or a combination of both dysregulate apoptosis. For example, the overexpression of Bcl-2 protected prostate cancer cells from apoptosis while led to inhibition of TRAIL-induced apoptosis in neuroblastoma, glioblastoma, and breast carcinoma cells. In colorectal cancers bax(G)8 frameshift mutations could contribute to resistance of cells to anticancer treatments. In the case of chronic lymphocytic leukemia (CLL), the malignant cells have an anti-apoptotic phenotype with high levels of anti-apoptotic Bcl-2 and low levels of pro-apoptotic proteins such as Bax (Wong 2011). The IAPs are a group of structurally and functionally similar proteins that regulate apoptosis by inhibiting caspase activity and promoting degradation of active caspases. Dysregulated IAP expression has been reported in many cancers. For example, drug resistance correlated with the expression of cIAP-2 in pancreatic cells while Apollon was found to be upregulated in gliomas and was responsible for cisplatin and camptothecin resistance (Chen et al. 1999).
The caspases are a large group of proteins that play a central role in apoptosis. As they work at two levels, initiator caspases (e.g., caspase-2, -8, -9, and -10) responsible for the initiation of the apoptotic pathway and effector caspases (caspase-3, -6, and -7) responsible in the actual cleavage of cellular components during apoptosis. Therefore low levels of these proteins or impairment function can decrease apoptosis thus contributing to resistance.
The p53 protein, also called tumor protein 53 (or TP 53), is one of the best known tumor suppressor proteins whose mutation acquired oncogenic property. Defects in the p53 tumor suppressor gene have been linked to more than 50% of human cancers (Bai and Zhu 2006) because it is not only involved in the induction of apoptosis but it is also a key player in cell cycle regulation, development, differentiation, gene amplification, DNA recombination, chromosomal segregation, and cellular senescence. A recent study reported that some target genes of p53 involved in apoptosis and cell cycle regulation are aberrantly expressed in melanoma cells and possibly related to resistance (Avery-Kiejda et al. 2011).
Several abnormalities in the death signaling pathways that can lead to evasion of the extrinsic pathway of apoptosis have been identified. Death receptors such as Fas, DR3, Trail-1, Trail-2, and ligands of the death receptors are key players in the regulation of extrinsic pathway of apoptosis. Downregulation or impairment of receptor function, abnormal expression of decoy receptors, as well as a reduced level in the death signals can contribute to impaired signaling and hence a reduction of apoptosis and acquisition of drug resistance.
2.5 Alteration of Drug Target
One of the most common drug resistance mechanisms involves genetic alterations of drug target such as mutations or changes in expression levels. These modifications could exist at low levels before drug treatment and undergo positive selection during exposure to chemotherapy. In oncology, genomic characterization of cancer has highlighted the importance of driver somatic mutations that give rise to an unusual reliance of cancer cells on a particular molecular pathways and their specific oncogenic kinases. These proteins are targets for many drugs and can be altered by different mechanisms.
Main categories of this type of resistance could be:
-
Downregulation of gene expression (e.g., effect of doxorubicin on topoisomerase IIα) (Di Nicolantonio et al. 2005)
-
Gene amplification (e.g., BCR-ABL amplification detected both in vitro and in imatinib-resistant CML specimens) (Gorre et al. 2001)
-
Mutations in gatekeeper residue (e.g., EGFR-T790M in NSCLC after treatment with tyrosine kinase inhibitor (TKI) gefitinib and erlotinib (Bell et al. 2005); BCR-ABL T315 in CML patient treated with imatinib) (Gorre et al. 2001)
-
Mutations that alter the conformation (e.g., mutations in ALK – F1174L, C1156Y, and L1152R – confer an increase in ATP affinity and clinical resistance to the ALK inhibitor crizotinib) (Choi et al. 2010)
-
Alternative spliced form (e.g., p61BRAFV600E detected in both vemurafenib in vitro-resistant cells and from resistant patient tumor biopsies produces enhanced dimerization with other RAF family members and resistance to vemurafenib but not MEK inhibitors) (Poulikakos et al. 2011).
A possible consequence of the inhibition of specific target is also the activation of a so-called bypass signaling mechanism. This results in the activation of a critical downstream signaling effector – normally activated by the kinase and extinguished by a kinase inhibitor – through a parallel mechanism that is indifferent to the kinase-directed therapy. An illustrative example of bypass-mediated resistance has been described in EGFR-mutant NSCLC. The bypass resistance mechanisms may also involve the modulation of positive or negative feedback loops and an example could be the augmentation of AKT signaling by MEK inhibitors (Garraway and Jänne 2012).
3 Conclusion
This review article shortly summarizes the main mechanisms of resistance to anticancer drugs. In order to define possible strategies to counteract resistance mechanisms, it can be useful to divide them in pre-target, target related, and post-target mechanisms.
As far as pre-target mechanisms – that can also be classified as pharmacokinetics mechanisms – we envisage the possibility to circumvent them by increasing drug doses and improving drug delivery testing combinations with modulators of drug transport and of tumor microenvironment.
On the other side, a deeper and deeper knowledge of target related mechanisms, e.g., target mutations, downregulation, amplification, is instrumental to identify alternative drugs, equally effective against cancer cells, to circumvent these types of resistance.
Finally as regards post-target mechanisms, the modulation of apoptosis appears to be a potentially feasible approach as recently demonstrated by Croce and Reed (2016).
References
Amable L (2016) Cisplatin resistance and opportunities for precision medicine. Pharmacol Res 106:27–36. doi:10.1016/j.phrs.2016.01.001
Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM (2003) P-glycoprotein: from genomics to mechanism. Oncogene 22:7468–7485. doi:10.1038/sj.onc.1206948
Ashworth A (2008) Drug resistance caused by reversion mutation. Cancer Res 68:10021–10023. doi:10.1158/0008-5472.CAN-08-2287
Avery-Kiejda KA, Bowden NA, Croft AJ et al (2011) P53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer 11:203. doi:10.1186/1471-2407-11-203
Baekelandt MM, Holm R, Nesland JM et al (2000) P-glycoprotein expression is a marker for chemotherapy resistance and prognosis in advanced ovarian cancer. Anticancer Res 20:1061–1067
Bai L, Zhu WG (2006) p53: structure, function and therapeutic applications. J Cancer Mol 2:141–153
Bell DW, Gore I, Okimoto RA et al (2005) Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 37:1315–1316. doi:10.1038/ng1671
Buck A, Halbritter S, Späth C et al (2015) Distribution and quantification of irinotecan and its active metabolite SN-38 in colon cancer murine model systems using MALDI MSI. Anal Bioanal Chem 407:2107–2116. doi:10.1007/s00216-014-8237-2
Cesca M, Morosi L, Berndt A et al (2016) Bevacizumab-induced inhibition of angiogenesis promotes a more homogeneous intratumoral distribution of paclitaxel, improving the antitumor response. Mol Cancer Ther 15:125–135. doi:10.1158/1535-7163.MCT-15-0063
Chen Z, Naito M, Hori S et al (1999) A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem Biophys Res Commun 264:847–854. doi:10.1006/bbrc.1999.1585
Choi YL, Soda M, Yamashita Y et al (2010) EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 363:1734–1739. doi:10.1056/NEJMoa1007478
Connell JJ, Sugihara Y, Török S et al (2015) Localization of sunitinib in in vivo animal and in vitro experimental models by MALDI mass spectrometry imaging. Anal Bioanal Chem 407:2245–2253. doi:10.1007/s00216-014-8350-2
Croce CM, Reed JC (2016) Finally, an apoptosis-targeting therapeutic for cancer. Cancer Res 76:5914–5920. doi:10.1158/0008-5472.CAN-16-1248
Di Nicolantonio F, Mercer SJ, Knight LA et al (2005) Cancer cell adaptation to chemotherapy. BMC Cancer 5:78. doi:10.1186/1471-2407-5-78
Farmer H, McCabe N, Lord CJ et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917–921. doi:10.1038/nature03445
Fink D, Aebi S, Howell SB (1998) The role of DNA mismatch repair in drug resistance. Clin Cancer Res 4:1–6
Fuso Nerini I, Morosi L, Zucchetti M et al (2014) Intratumor heterogeneity and its impact on drug distribution and sensitivity. Clin Pharmacol Ther 96:224–238. doi:10.1038/clpt.2014.105
Garraway LA, Jänne PA (2012) Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov 2:214–226. doi:10.1158/2159-8290.CD-12-0012
Giordano S, Morosi L, Veglianese P et al (2016a) 3D mass spectrometry imaging reveals a very heterogeneous drug distribution in tumors. Sci Rep 6:37027. doi:10.1038/srep37027
Giordano S, Zucchetti M, Decio A et al (2016b) Heterogeneity of paclitaxel distribution in different tumor models assessed by MALDI mass spectrometry imaging. Sci Rep 6:39284. doi:10.1038/srep39284
Gorre ME, Mohammed M, Ellwood K et al (2001) Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293:876–880. doi:10.1126/science.1062538
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13:714–726. doi:10.1038/nrc3599
Kao CH, Tsai SC, Liu TJ et al (2001) P-glycoprotein and multidrug resistance-related protein expressions in relation to technetium-99m methoxyisobutylisonitrile scintimammography findings. Cancer Res 61:1412–1414
Kauvar LM, Morgan AS, Sanderson PE, Henner WD (1998) Glutathione based approaches to improving cancer treatment. Chem Biol Interact 111–112:225–238
Kirschner K, Melton DW (2010) Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res 30:3223–3232
Leith CP, Kopecky KJ, Chen IM et al (1999) Frequency and clinical significance of the expression of the multidrug resistance proteins MDR1/P-glycoprotein, MRP1, and LRP in acute myeloid leukemia: a southwest oncology group study. Blood 94:1086–1099
Longley DB, Harkin DP, Johnston PG (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3:330–338. doi:10.1038/nrc1074
Meijer C, Mulder NH, Timmer-Bosscha H et al (1992) Relationship of cellular glutathione to the cytotoxicity and resistance of seven platinum compounds. Cancer Res 52:6885–6889
Minchinton AI, Tannock IF (2006) Drug penetration in solid tumours. Nat Rev Cancer 6:583–592. doi:10.1038/nrc1893
Morosi L, Zucchetti M, D’Incalci M, Davoli E (2013) Imaging mass spectrometry: challenges in visualization of drug distribution in solid tumors. Curr Opin Pharmacol 13:807–812. doi:10.1016/j.coph.2013.06.003
Pallis M, Russell N (2004) Strategies for overcoming p-glycoprotein-mediated drug resistance in acute myeloblastic leukaemia. Leukemia 18:1927–1930. doi:10.1038/sj.leu.2403511
Poulikakos PI, Persaud Y, Janakiraman M et al (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480:387–390. doi:10.1038/nature10662
Prideaux B, Stoeckli M (2012) Mass spectrometry imaging for drug distribution studies. J Proteome 75:4999–5013. doi:10.1016/j.jprot.2012.07.028
Rzagalinski I, Volmer DA (2016) Quantification of low molecular weight compounds by MALDI imaging mass spectrometry – a tutorial review. Biochim Biophys Acta. doi:10.1016/j.bbapap.2016.12.011. pii:S1570-9639(16)30276-X [Epub ahead of print]
Shervington A, Lu C (2008) Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Investig 26:535–542. doi:10.1080/07357900801904140
Sugiura Y, Setou M (2010) Imaging mass spectrometry for visualization of drug and endogenous metabolite distribution: toward in situ pharmacometabolomes. J Neuroimmune Pharmacol 5:31–43. doi:10.1007/s11481-009-9162-6
Szakács G, Paterson JK, Ludwig JA et al (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234. doi:10.1038/nrd1984
Thomas H, Coley HM (2003) Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control J Moffitt Cancer Center 10:159–165
Thomas A, Tanaka M, Trepel J et al (2017) Temozolomide in the era of precision medicine. Cancer Res 77:823–826. doi:10.1158/0008-5472.CAN-16-2983
Torok S, Rezeli M, Kelemen O et al (2017) Limited tumor tissue drug penetration contributes to primary resistance against angiogenesis. Inhibitors. Theranostics 7(2):400–412. doi:10.7150/thno.16767
Usanova S, Piée-Staffa A, Sied U et al (2010) Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol Cancer 9:248. doi:10.1186/1476-4598-9-248
Wong RSY (2011) Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res CR 30:87. doi:10.1186/1756-9966-30-87
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Colmegna, B., Morosi, L., D’Incalci, M. (2017). Molecular and Pharmacological Mechanisms of Drug Resistance:An Evolving Paradigm. In: Mandalà, M., Romano, E. (eds) Mechanisms of Drug Resistance in Cancer Therapy. Handbook of Experimental Pharmacology, vol 249. Springer, Cham. https://doi.org/10.1007/164_2017_20
Download citation
DOI: https://doi.org/10.1007/164_2017_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-10506-8
Online ISBN: 978-3-030-10507-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)