
Abstract
It is generally accepted that recent advances in anticancer agents have contributed significantly to the improvement of both the disease-free survival and quality of life in cancer patients. However, in many instances, a favorable initial response to treatment changes afterwards, thereby leading to cancer relapse and recurrence. This phenomenon of acquired resistance to therapy, it is a major problem for totally efficient anticancer therapy. The failure to obtain an initial response reflects a form of intrinsic resistance. Specific cell membrane transporter proteins are implicated in intrinsic drug resistance by altering drug transport and pumping drugs out of the tumor cells. Moreover, the gradual acquisition of specific genetic and epigenetic abnormalities in cancer cells could contribute greatly to acquired drug resistance. A critical issue in the clinical setting, is that the problem of drug resistance appears to have a negative effect on also the new molecularly-targeted anticancer drugs. Several ongoing efforts are being made by the medical community aimed to the identification of such resistance mechanisms and the development of novel drugs that could overcome them. In this review, the major drug resistance mechanisms and strategies to overcome them are critically discussed, and also possible future directions are suggested.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drug resistance can be defined as the decrease in the efficacy and potency of a drug to produce therapeutic merits and represents a major impediment to the disease treatment and overall patient survival. Of note, resistance to anticancer treatments can be manifested by local or loco-regional, as well as distant tumor metastases leading in the paradox of therapy-induced metastasis (TIM) [1,2,3]. In many cases, tumors such as renal cancer, hepatocellular carcinoma and malignant melanoma often exhibit intrinsic resistance to chemotherapy, without prior exposure to anticancer agents, so the initial response to treatment is poor [1]. In other settings, the initial optimism after good treatment response is often followed by poor results and a devastating outcome, as tumors initially sensitive to therapy, later become unresponsive due to development of acquired drug resistance. Currently, surgery and/or radiotherapy represent the optimal treatment modalities for the management of localized tumors. Systemic treatments are required for hematologic malignancies or metastatic tumors. Current forms of systemic treatment are chemotherapy, immunotherapy and anti-angiogenic agents [6].
The interaction between the drug and the tumor microenvironment is a complex phenomenon. Cancers have the ability to develop remarkable resistance to various treatments which target different molecular pathways [6]. Recent evidence suggests that radio- or chemotherapy for breast cancer result in a stem cell-like phenotype in non-stem tumor cells. Therefore, as it has also been suggested by a recent extensive meta-analysis, there is an urgent need to identify basic factors that determine drug resistance in cancer stem cells [7]. Of particular note, several lines of evidence have demonstrated that chemotherapy can potentially increase the levels of circulating endothelial progenitor cells (EPCs) that promote tumor growth and metastasis [3, 8].
In the “new era of targeted chemotherapy”, molecules and metabolic pathways implicated specifically in the growth and proliferation of cancer cells are blocked using molecularly-targeted drugs e.g., imatinib (Gleevec) which specifically targets BCR-ABL in chronic myeloid leukemia, aiming at achieving maximum treatment response and minimum toxicity compared to other types of cancer treatment. Of importance, the more targeted a drug is, the lower the probability to elicit drug resistance [9]. The largely quantitative difference between the conventional and the molecularly-targeted drugs, that provides some therapeutic margin, is that the targets of the former are mainly cellular (e.g., cell proliferation and DNA replication) or components (e.g., microtubules, topoisomerases) that are both in normal and cancer cells [10]. As a result, the molecularly-targeted drugs are less toxic than the conventional drugs, and they achieve effective treatment at remarkably lower doses than the maximum tolerated dose [11]. However, both types of drugs (i.e., conventional and molecularly-targeted) suffer from the problem of intrinsic and acquired drug resistance [12].
In the present review, the main factors that contribute to a compromized effectiveness of systemic anticancer drug regimens, as well as the potential mechanisms underlying drug resistance, are discussed.
Intrinsic and acquired drug resistance
Drug resistance may arise due to intrinsic and/or acquired factors. Intrinsic resistance can be attributed to: (a) drug breakdown, (b) altered expression and/or function of the drug target, (c) altered drug transport across the cellular membrane or (d) reduced interaction efficiency between the drug and its molecular target [4, 5]. Intrinsic cellular resistance can be mediated through ATP-dependent membrane transporters or nuclear receptors, e.g. sxr [14]. In addition, cellular metabolic processes, such as ceramide glycosylation decrease efficacy of chemotherapeutic agents [15] Also, cell cycle regulators and DNA damage repair factors enhance cross-drug resistance, by inhibiting drug accumulation, reducing influx, increasing efflux through cell membrane transporters, or inactivating drugs [9, 16]. Of interest, inactivation of tumor-associated genes including the tumor suppressor gene TP53 has been shown to result in resistance to chemotherapeutic drugs [17].
On the other hand, acquired drug resistance is influenced by genetic or environmental factors that facilitate the development of drug-resistant cancer cell clones or induce mutations of enzymes involved in relevant metabolic pathways [1, 5].
Genetic determinants of acquired drug resistance
Genetic instability in the form of aneuploidy, deletions, point mutations, chromosomal translocations and gene amplifications is a key factor in several aspects of cancer pathogenesis [18], including intratumor heterogeneity, which fosters primary cancers, distant metastatic lesions and cancer relapse after therapeutic failure [19, 20].
Mathematical and computational models indicate a positive correlation between chemotherapeutic resistance and the number of spontaneous genetic mutations [21, 22], which can be utilized in adjusting the administered dose or determining the type of administered treatment [23, 24]. Other models used paradigm prokaryotic organisms, such as Escherichia coli, in an effort to detect mutations in drug-resistant cancer cells and investigate their role in drug resistance [25,26,27,28,29,30,31]. These studies demonstrated that drug resistance usually results in a pattern of random mutations rather than a drug-specific effect [32,33,34,35].
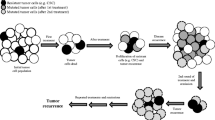
Overall, resistance to drugs is directly dependent on the stability of the genetic material of the tumor cells and the level of genomic instability, the mechanism(s) of action of a chemotherapeutic drug, the dose of the administered drug and the treatment intervals [21, 22, 24]. Alterations in the genetic material such as inactive mutations may occur either before or during treatment, in small subpopulations of cancer cells. It is also possible that cancers intrinsically sensitive to chemotherapy contain at least one drug-resistant cell clone, expansion of which leads to acquired resistance and possibly recurrence. Therefore, combinatorial drug therapy provides a powerful rationale for reducing the likelihood of the development of multiple resistant clones, especially for patients undergoing adjuvant anticancer therapy with micrometastases and low-tumor burdens [36, 37].
There is an increasing interest in studying cancer clonal cell subpopulations and the evolution of resistant variants [38,39,40]. These studies demonstrate an intratumor heterogeneity and changes in the distribution of clonal subpopulations following treatment administration [41]. Furthermore, gene amplification (i.e., increase in gene copy number) of specific drug resistance-relevant genes was found to be associated with enhanced resistance to many molecularly-targeted drugs [18, 42].
Epigenetic determinants of acquired drug resistance
Epigenetic factors, such as DNA methylation and chromatin-remodeling, contribute greatly to drug tolerance [43,44,45]. Zeller at al. [46] identified a series of genes that exhibited promoter hypermethylation in the cisplatin resistant ovarian cancer cells compared to their drug sensitive counterparts. Given that hypermethylation of gene promoters can be associated with transcriptional gene silencing [47], demethylation of several of these genes led to gene re-activation and restored chemosensitivity in cancer cells. In another study by Bhatla et al. [48], inhibition of histone deacetylation and DNA methylation resulted in the activation of genes preferentially methylated and repressed in relapsed pediatric acute lymphoblastic leukemia and drug sensitivity. Of importance, targeting of DNA methyltransferases effectors of DNA methylation and histone modification was found to reverse chemoresistance in heterogeneous multiple myeloma [49].
Intrinsic drug resistance-associated membrane proteins
Multidrug resistance (MDR) is largely dependent on the activity of membrane transporter proteins, referred to as “drug resistance-associated membrane proteins” or “DRAMPs” which act either directly by extruding drug molecules out of cells to reduce intracellular accumulation, or indirectly by affecting net accumulation of drugs through physico-chemical processes [4, 50].
Two major classes of DRAMPs have been identified: (i) the ATP-binding cassette (ABC) transporter superfamily, which pumps hydrophobic chemotherapeutic drugs out of tumor cells thereby reducing the net intracellular accumulation and thus the efficacy of the drugs into tumor cells, and (ii) the solute carrier transporters which increase chemoresistance by interfering with the cellular uptake of hydrophilic anticancer agents [4, 50]. Approximately, 50 ABC transporters have been identified in the human genome which catalyze the active transport of diverse chemical compounds including anticancer drugs in an ATP-dependent way by a pair of cytoplasmic nucleotide-binding domains (NBD) [51]. There are three broad groups of ABC transporters implicated in MDR, namely, P-glycoprotein, ABCG2, and the multidrug resistance-associated proteins (MRPs) [50, 52] discussed below.
P-glycoprotein
Overexpression of the protein P-glycoprotein (or ABCB1/MDR1), an ATP-dependent efflux pump, results to MDR in several types of cancer (e.g., multiple myeloma, leukemia) [53,54,55,56], through the active translocation of drug molecules out of the tumor cells [50, 57]. The final prognosis in epithelial and solid tumors, as well as in blood malignancies, was particularly unfavorable due to enhanced ABCB1 efflux potential [58,59,60]. ABCB1 overexpression has been demonstrated in cases of chemotherapeutic failure [59,60,61,62,63]. Moreover, a high level of ABCB1 gene amplification was observed in MDR murine melanoma cells [64]. P-glycoprotein exhibits a very broad substrate specificity, including anthracyclines, vinca alkaloids, or taxanes, epipodophyllotoxins, which is the biochemical basis for its “MDR” property [65].
MDR-associated protein
The MDR-associated proteins (MRPs) constitute a group of 13 members, including MRP1 (ABCC1) [50, 52]. MRP1 overexpression was shown to result to resistance to anticancer agents. The presence of reduced glutathione (GSH) is a prerequisite for the transport of unmodified chemotherapeutic agents via MRP1 [66]. A peptidomimetic glutathione-conjugate of ethacrynic acid (EA), GS-EA, was found to inhibit MRP1-mediated efflux of drugs in ovarian cancer cells which display overexpression of MRP1. In addition, resistance of these cells to methotrexate was reversed in part [67].
ABCG2
Another member of the broad ABC superfamily, ABCG2, was overexpressed in human-derived breast cancer cells resistant to adriamycin [68]. Furthermore, it has been reported that hypoxia can regulate ABCG2 expression. Stem cells or cancer cells in hypoxic environment exhibit resistance to drugs due to enhanced ABCG2 expression [69]. ABCG2 is responsible for cell resistance to many anticancer drugs, with camptothecins being the most prominent example [70, 71]. However, FL118, a camptothecin analogue, was able to overcome effectively ABCG2-induced resistance [72]. The substrates of ABCG2 include many molecularly targeted chemotherapy drugs, such as Gefitinib an inhibitor of epidermal growth factor receptor (EGFR), and Imatinib. However, its importance in clinical practice remains to be investigated.
Classic chemotherapeutic drugs
Methotrexate is an anticancer molecularly-targeted cytostatic drug, used either alone or in combination with other agents, to treat a variety of malignancies such as breast, lung, skin, and head and neck cancer. It is also used for the treatment of severe rheumatoid arthritis and psoriasis. Methotrexate exerts its anticancer effect through inhibiting the expression of its biochemical target dihydrofolate reductase (DHFR), a key enzyme in DNA synthesis, which facilitates cancer cell growth and proliferation. Molecular studies have demonstrated enhanced DHFR expression in cells that display resistance toward methotrexate. In methotrexate-resistant cancer cells, increased DHFR gene copies were identified [41].
Some anticancer agents, such as vinca alkaloids and taxanes prevent cell division through altering the dynamic instability of microtubules [73]. Taxanes have been successfully used as anticancer agents over the last 25 years by destabilizing microtubules; whereas, vinca alkaloids are implicated in the inhibition of microtubule function. The synergistic effect of these agents was tested experimentally and it was observed that their microtubule-specific activity was enhanced [74]. Resistance against the taxane paclitaxel was exerted by β III-tubulin isoforms [75]. In several types of epithelial tumors, βIII-tubulin expression was linked to poor response toward paclitaxel treatment and overall patient outcome [76].
Of particular note, an initial favorable clinical response was followed by resistance to taxane treatment. This could be explained by a gradual change in microtubule dynamics and functionality [77]. Particularly, the microtubule-associated protein (MAP)-Tau interferes with the binding of taxanes to microtubules. Down regulation of MAP-Tau was shown to lead to alteration of cancer cells’ chemosensitivity, rendering them more vulnerable to paclitaxel [78].
Anticancer drugs like camptothecin and epipodophyllotoxin target key enzymes involved in DNA replication and transcription such as topoisomerases. Camptothecin is a cytotoxic alkaloid administered to patients with leukemia [79, 80]. Experimental studies show that resistance to camptothecin and treatment failure are due to the activity of the enzyme topoisomerase type I [81]. Moreover, overexpression of topoisomerases type II was associated with altered efficacy of molecularly-targeted drugs [82, 83], such as Adriamycin, in chemoresistant leukemia cell lines [80].
Genetic alterations, such as mutations, in molecular drug targets like genes or proteins contribute greatly to acquired drug resistance, thereby leading to limited effectiveness or complete ineffectiveness of chemotherapy, mainly in advanced cancers [65]. For instance, Bcr-Abl kinase domain point mutations could impair or abolish imatinib binding in chemoresistant patients with chronic myeloid leukemia (CML) [84, 85].
DNA damage repair
Important determinants of response to many chemotherapy drugs and targeted therapies represent the DNA damage repair (DDR) pathways which include a complex of proteins, like Nucleotide Excision Repair (NER) machinery that processes and removes the so-called bulky lesions, such as those induced by UV light (thymine dimers and 6,4-photoproducts) and chemotherapeutic drugs such as cisplatin [86].
Two sub-pathways are involved in NER: the global genomic NER (GG-NER or GGR) which repairs DNA damage in transcriptionally silent loci and the transcription-coupled NER (TC-NER or TCR) which repairs lesions located in the transcriptionally active DNA regions. The NER pathway consists of some basic steps, including the identification of DNA damage, DNA unfolding, and other processes such as incision, polymerization, degradation, and ligation [87]. One of the most important genes related to NER is ERCC1, overexpression of which is usually associated with DDR induced by platinum and alkylating agent-based treatment and is correlated with negative outcomes in patients receiving cisplatin-based treatment in advanced non-small cell lung cancer (NSCLC) [88]. DNA damage caused by platinum-based drugs can also be recognized by specific proteins, such as mismatch repair (MMR) complexes, thereby resulting to transduction of DNA damage signals and various downstream effectors [89].
In addition, the DNA damage repair protein O6-methylguanyl DNA methyltransferase (MGMT) is associated with resistance to chemotherapy with DNA alkylating anticancer drugs, such as nitrosoureas, carmustine and temozolomide in central nervous system tumors [87, 90]. The crosstalk and the signals generated between the effector molecules involved in DDR lead to either cell death or cell survival. One very important effector is the well-known TP53 gene which triggers a major tumor abrogation mechanism via primarily the initiation of cell death, and plays a cardinal role in carcinogenesis when mutated [91]. If DNA damage is extensive and impossible to be repaired, then the apoptotic pathway becomes activated [17]. Moreover, DNA damage can result in apoptosis through the TP73 gene, a TP53-related gene [89]. There is a close relationship between oncogenesis and drug resistance/sensitivity modulated by a pathway dependent on TP53, mutations of which are detected in many human cancers [92, 93].
Cancer stem cells
Cancer stem cells have stem-like properties and they exhibit higher resistant to chemotherapy in contrast to the differentiated tumor cells. Factors that affect CSCs’ resistance to drugs include: (a) induction of pathways implicated in stem cell maintenance; (b) activation and elevated expression of ABC transporter proteins (e. g., ABCB1 and ABCG2); (c) overexpression of detoxification enzymes such as certain aldehyde dehydrogenase (ALDH) isoforms; (d) inhibition of apoptotic pathways, like the ones mediated by the pro-apoptotic TP53; (e) enhanced DNA damage repair capacity, thereby reducing the effectiveness of DNA-damaging chemotherapeutic agents; and (f) increased influence of the tumor microenvironmental niche [7, 94].
Cancer microenvironment
The cellular environment can affect greatly drug response, where the cell kinetic parameters and proliferation rate constitute important determinants of therapeutic effectiveness. Anticancer drugs and new biological agents that target these determinants have been shown to exert their antineoplastic effect by tranquilizing cancer cells [95]. Increased treatment effectiveness is usually achieved by targeting rapidly proliferating cancer cells [96]. According to a study by Hirst and Denekamp, the most effective chemotherapies are those that rapidly neutralize highly proliferating cells or selectively affect cell division [97]. Different treatments are required in the hypoxic regions of tumors or in regions with slow cell proliferation. Combination therapies with more than one anticancer agents do not necessarily guarantee treatment success because a drug can counteract or overlap the effect of another drug so that the combined effect is lower than predicted. There is a positive correlation between tumor cell proliferation and tumor vasculature, which ensures continuous blood supply to the growing tumor cells, as well as vascular permeability [97]. Reduced blood flow leads to deprivation of nutrients essential for the increased energy demands of the cell in the proliferation phase, thereby leading to delay or inhibition of proliferation. The slow cell proliferation and the poor blood supply are associated with potential resistance to molecularly-targeted drugs [98].
Novel chemosensitizing agents have been developed to counteract the phenomenon of resistance to many molecularly-targeted drugs [99]. Cell adhesion, cytokine activity, growth factors, cell proximity, oxygen and energy supply to the cells, as well as other factors, are suggested to be implicated in reduced anticancer drug response, decreased chemotherapeutic effectiveness and subsequent rapid tumor recurrence [100, 101].
Another factor which regulates the accumulation of oxygen to tumor cells is the hypoxia- inducible factor 1 (HIF1), a transcription factor [102]. HIF-1 promotes the activation of genes implicated in hypoxia signaling and inhibition of cell proliferation. HIF-1 is also implicated in the intracellular metabolism, pH regulation, inhibition of autophagy and cell death [102].
Oxygen deficiency was shown to be associated with the activation of genes encoding proteins that induce resistance to anticancer drugs, such as P-glycoprotein, especially in solid tumors [103]. Specific anticancer drugs can inhibit oxygen supply, allowing cancer cells to enter a dormant state [104]. Cells that survive drug treatment may potentially proliferate under hypoxic conditions, leading to tumor relapse in a short span of time. Additionally, the sufficient oxygenation of normal tissues is important in the mobility of anticancer drugs and favorable response to drug treatment [105]. Therefore, low oxygen concentration in tumor tissues is linked to low activity of anticancer drugs, poor tumor response to chemotherapy or rapid tumor relapse [106].
The cytotoxicity of anticancer drugs is largely affected by the pH of the tumor microenvironment. Molecules passively diffuse across the cell membrane, more effectively in the uncharged (non-ionized) form. Accordingly, alkalization of the extracellular environment increases the uptake and cytotoxicity of drugs such as Doxorubicin, with a pKa value of almost 9 [107, 108]. On the other hand, a microenvironment with acidic pH can also inhibit the active transport of certain drugs (e.g., methotrexate) [109].
Drug uptake and drug activation
Plasma membrane proteins play a very important role in intrinsic drug sensitivity or resistance, given that antineoplastic drug molecules may be expelled from cancer cells either through passive diffusion or facilitated diffusion mediated by membrane proteins [110]. The majority of antimetabolite drugs need metabolic activation to produce therapeutically effective nucleotides or nucleosides intracellularly through the activity of phosphoribosyl transferases and kinases [111].
Another component of the redistribution of anticancer drugs is the binding of drug molecules to specific intracellular proteins [112]. In blood malignancies, these proteins have been found to largely contribute to significant drug resistance and treatment failure. For example, overexpression of a major vault protein, LRP, was observed in acute myeloid leukemia (AML) patients [113]. Moreover, changes in intracellular chemical processes affect drug kinetics, leading to treatment failure [114].
Moses Judah Folkman proved first that angiogenesis and cancer are closely related [118]. Drug molecules are delivered to the target tumor cells through blood vessels. Abnormal vasculature and ultimately necrosis largely affect drug penetration and effectiveness [119, 120]. Currently, there is a number of chemotherapeutic drugs targeting the potent angiogenic factor, vascular epithelial growth factor (VEGF), such as Bevacizumab, Aflibercept, Pazopanib, Sunitinib and Sorafenib. The drugs normalize tumor vasculature, in order to increase the distribution of anticancer agents and achieve maximum therapeutic efficacy [121]. Moreover, the various physical barriers, such as the blood–brain barrier (BBB) and the blood-testis barriers, prevent the diffusion of chemotherapeutic drugs. Of note, overexpression of P-glycoprotein was observed in the endothelial cells of these barriers [122].
Strategies to overcome resistance mechanisms
Many factors and parameters must be taken into consideration for the effective cancer treatment using antineoplastic drugs. Clinicians are mainly concerned with the route of drug administration as well as the maximum tolerated dose able to destroy cancer cells while minimizing adverse effects [115]. The “maximum tolerated dose”, also called “maximum tolerable dose” or the “maximally tolerated dose” (MTD), can be defined as the highest single dose of an agent or treatment that does not cause significant or intolerable toxicity/adverse effects. For many drugs, the optimal dose does not necessarily coincide with the MTD; thus, determination of the optimal dose poses a great challenge [116, 117].
A novel, promising modality of drug administration has emerged, the so-called ‘metronomic chemotherapy’, that is, the repetitive administration of chemotherapeutic agents at low doses. It has been demonstrated that metronomic chemotherapy can be extremely beneficial in many cases; however, extended research is needed to confirm these results [124, 125]. One therapeutic strategy to overcome drug resistance is ‘treatment holiday’, where a patient’s chemotherapy treatment is stopped for some time in order to avoid selection for drug-resistant tumor cells that could lead to cancer recurrence and relapse [115].
Of importance, special emphasis should be given to the development and optimization of molecularly-targeted drugs designed to block key genes or gene products implicated in chemoresistance. For example, resistance to chemotherapy caused by P-glycoprotein can be potentially counteracted by adding to a chemotherapy regimen the molecularly-targeted drug verapamil, a candidate competitive inhibitor of P-glycoprotein [54, 56, 126]. Moreover, a positive correlation was observed between cytidine deaminase activity in blast cells obtained from acute leukemia patients and the development of resistance to the antimetabolite drug cytosine arabinoside (ara-C) [114]. Therefore, an effective way of blocking the deamination of ara-C would circumvent resistance to ara-C.
Other potential future directions
The apparent complexity of cancer drug resistance, as discussed throughout the manuscript, leads to the suggestion that there is a pressing need for the design of novel therapeutic regimens. Unraveling the mechanisms underlying patients’ response to anticancer drugs and identification of their genetic profile would enable the development of new, personalized drugs to prolong patients’ overall survival, as well as quality of life.
Conclusions
Cancer patients’ chemotherapeutic response and outcome depends on multiple redundant and diverse biological processes and molecular mechanisms that affect the sensitivity of cancer cells to chemotherapy drugs. Multiple molecular determinants of intrinsic and acquired resistance, including genetic/epigenetic factors, as well as membrane transporter proteins that act at the genomic or cellular level respectively, have been identified. The key genes/gene products found in this review study to be involved in chemoresistance were used to construct an interaction network (Fig. 1). These molecules are, in most cases, highly interconnected and some of them (TP53, TP73, VEGFA, HIF1A, ABCB1, ABCG2 and TOP1) act as ‘hubs’. This leads to the suggestion that these genes/proteins may play a central role in drug sensitivity/resistance by interacting with each other, either functionally or physically. Novel targeted therapies for cancer must be developed that would be directed toward cellular drug resistance, and specifically to the ‘hub’ genes. The goal of these therapies must be to achieve maximum chemotherapeutic effect by eliminating cancer cells, with reduced normal tissue toxicity.

An interaction network of the genes/gene products related to clinical chemoresistance with the usage of bioinformatics. The associations among them were investigated and visualized using STRING v10.5 [127]. HIF-1: HIF1A, sxr: NR1I2, VEGF: VEGFA, topoisomerase type I: TOP1, topoisomerase type II: TOP2A
Abbreviations
- MDR:
-
Multidrug resistance
- ABC:
-
Adenosine triphosphate-binding cassette
- DHFR:
-
Dihydrofolate reductase
- NER:
-
Nucleotide excision repair
- MTD:
-
Maximum tolerated dose
References
Gottesman MM (2002) Mechanisms of cancer drug resistance. Annu Rev Med 53:615–627. https://doi.org/10.1146/annurev.med.53.082901.103929
Clynes M (1998) Multiple drug resistance in cancer 2: molecular, cellular and clinical aspects. Kluwer Academic Publishers, Dodrecht
Ebos JM (2015) Prodding the beast: assessing the Impact of treatment-induced metastasis. Cancer Res 75(17):3427–3435. https://doi.org/10.1158/0008-5472.CAN-15-0308
Sherlach KS, Roepe PD (2014) Drug resistance associated membrane proteins. Front Physiol 5:108. https://doi.org/10.3389/fphys.2014.00108
Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B (2017) The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull 7(3):339–348. https://doi.org/10.15171/apb.2017.041
Gottesman MM, Ludwig J, Xia D, Szakacs G (2006) Defeating drug resistance in cancer. Discov Med 6(31):18–23
Pavlopoulou A, Oktay Y, Vougas K, Louka M, Vorgias CE, Georgakilas AG (2016) Determinants of resistance to chemotherapy and ionizing radiation in breast cancer stem cells. Cancer Lett 380(2):485–493. https://doi.org/10.1016/j.canlet.2016.07.018
Shaked Y, Henke E, Roodhart JM, Mancuso P, Langenberg MH, Colleoni M, Daenen LG, Man S, Xu P, Emmenegger U, Tang T, Zhu Z, Witte L, Strieter RM, Bertolini F, Voest EE, Benezra R, Kerbel RS (2008) Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell 14(3):263–273. https://doi.org/10.1016/j.ccr.2008.08.001
Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S (2014) Drug resistance in cancer: an overview. Cancers 6(3):1769–1792. https://doi.org/10.3390/cancers6031769
Joo WD, Visintin I, Mor G (2013) Targeted cancer therapy—are the days of systemic chemotherapy numbered? Maturitas 76(4):308–314. https://doi.org/10.1016/j.maturitas.2013.09.008
Kummar S, Gutierrez M, Doroshow JH, Murgo AJ (2006) Drug development in oncology: classical cytotoxics and molecularly targeted agents. Br J Clin Pharmacol 62(1):15–26. https://doi.org/10.1111/j.1365-2125.2006.02713.x
Groenendijk FH, Bernards R (2014) Drug resistance to targeted therapies: deja vu all over again. Mol Oncol 8(6):1067–1083. https://doi.org/10.1016/j.molonc.2014.05.004
Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5(3):219–234. https://doi.org/10.1038/nrd1984
Synold TW, Dussault I, Forman BM (2001) The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med 7(5):584–590. https://doi.org/10.1038/87912
Liu YY, Han TY, Giuliano AE, Cabot MC (2001) Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J 15(3):719–730. https://doi.org/10.1096/fj.00-0223com
Torgovnick A, Schumacher B (2015) DNA repair mechanisms in cancer development and therapy. Front Genet 6:157. https://doi.org/10.3389/fgene.2015.00157
Lowe SW, Ruley HE, Jacks T, Housman DE (1993) p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74(6):957–967
Fojo T (2007) Multiple paths to a drug resistance phenotype: mutations, translocations, deletions and amplification of coding genes or promoter regions, epigenetic changes and microRNAs. Drug Resist Updates 10 (1–2):59–67. https://doi.org/10.1016/j.drup.2007.02.002
Greaves M, Maley CC (2012) Clonal evolution in cancer. Nature 481(7381):306–313. https://doi.org/10.1038/nature10762
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194(4260):23–28
Goldie JH, Coldman AJ (1979) A mathematic model for relating the drug sensitivity of tumors to their spontaneous mutation rate. Cancer Treat Rep 63(11–12):1727–1733
Goldie JH, Coldman AJ (1985) Genetic instability in the development of drug resistance. Semin Oncol 12(3):222–230
Coldman AJ, Goldie JH (1986) A stochastic model for the origin and treatment of tumors containing drug-resistant cells. Bull Math Biol 48(3–4):279–292
Woodhouse JR, Ferry DR (1995) The genetic basis of resistance to cancer chemotherapy. Ann Med 27(2):157–167
Angerer WP (2001) An explicit representation of the Luria-Delbruck distribution. J Math Biol 42(2):145–174
Dewanji A, Luebeck EG, Moolgavkar SH (2005) A generalized Luria-Delbruck model. Math Biosci 197(2):140–152. https://doi.org/10.1016/j.mbs.2005.07.003
Frank SA (2003) Somatic mosaicism and cancer: inference based on a conditional Luria-Delbruck distribution. J Theor Biol 223(4):405–412
Haeno H, Iwasa Y, Michor F (2007) The evolution of two mutations during clonal expansion. Genetics 177(4):2209–2221. https://doi.org/10.1534/genetics.107.078915
Iwasa Y, Nowak MA, Michor F (2006) Evolution of resistance during clonal expansion. Genetics 172(4):2557–2566. https://doi.org/10.1534/genetics.105.049791
Komarova NL, Mironov V (2005) On the role of endothelial progenitor cells in tumor neovascularization. J Theor Biol 235(3):338–349. https://doi.org/10.1016/j.jtbi.2005.01.014
Komarova NL, Wodarz D (2005) Drug resistance in cancer: principles of emergence and prevention. Proc Natl Acad Sci USA 102(27):9714–9719. https://doi.org/10.1073/pnas.0501870102
Beketic-Oreskovic L, Duran GE, Chen G, Dumontet C, Sikic BI (1995) Decreased mutation rate for cellular resistance to doxorubicin and suppression of mdr1 gene activation by the cyclosporin PSC 833. J Natl Cancer Inst 87(21):1593–1602
Chen G, Jaffrezou JP, Fleming WH, Duran GE, Sikic BI (1994) Prevalence of multidrug resistance related to activation of the mdr1 gene in human sarcoma mutants derived by single-step doxorubicin selection. Cancer Res 54(18):4980–4987
Dumontet C, Duran GE, Steger KA, Beketic-Oreskovic L, Sikic BI (1996) Resistance mechanisms in human sarcoma mutants derived by single-step exposure to paclitaxel (Taxol). Cancer Res 56(5):1091–1097
Jaffrezou JP, Chen G, Duran GE, Kuhl JS, Sikic BI (1994) Mutation rates and mechanisms of resistance to etoposide determined from fluctuation analysis. J Natl Cancer Inst 86(15):1152–1158
Chen KG, Wang YC, Schaner ME, Francisco B, Duran GE, Juric D, Huff LM, Padilla-Nash H, Ried T, Fojo T, Sikic BI (2005) Genetic and epigenetic modeling of the origins of multidrug-resistant cells in a human sarcoma cell line. Cancer Res 65(20):9388–9397. https://doi.org/10.1158/0008-5472.CAN-04-4133
Matsumoto Y, Takano H, Fojo T (1997) Cellular adaptation to drug exposure: evolution of the drug-resistant phenotype. Cancer Res 57(22):5086–5092
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New Engl J Med 366(10):883–892. https://doi.org/10.1056/NEJMoa1113205
Lee AJ, Swanton C (2012) Tumour heterogeneity and drug resistance: personalising cancer medicine through functional genomics. Biochem Pharmacol 83(8):1013–1020. https://doi.org/10.1016/j.bcp.2011.12.008
Swanton C (2012) Intratumor heterogeneity: evolution through space and time. Cancer Res 72(19):4875–4882. https://doi.org/10.1158/0008-5472.CAN-12-2217
Alt FW, Kellems RE, Bertino JR, Schimke RT (1992) Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. 1978. Biotechnology 24:397–410
Wang YC, Juric D, Francisco B, Yu RX, Duran GE, Chen GK, Chen X, Sikic BI (2006) Regional activation of chromosomal arm 7q with and without gene amplification in taxane-selected human ovarian cancer cell lines. Genes Chromosomes Cancer 45(4):365–374. https://doi.org/10.1002/gcc.20300
Matei D, Fang F, Shen C, Schilder J, Arnold A, Zeng Y, Berry WA, Huang T, Nephew KP (2012) Epigenetic resensitization to platinum in ovarian cancer. Cancer Res 72(9):2197–2205. https://doi.org/10.1158/0008-5472.CAN-11-3909
Balch C, Nephew KP (2013) Epigenetic targeting therapies to overcome chemotherapy resistance. Adv Exp Med Biol 754:285–311. https://doi.org/10.1007/978-1-4419-9967-2_14
Wilting RH, Dannenberg JH (2012) Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updates 15 (1–2):21–38. https://doi.org/10.1016/j.drup.2012.01.008
Zeller C, Dai W, Steele NL, Siddiq A, Walley AJ, Wilhelm-Benartzi CS, Rizzo S, van der Zee A, Plumb JA, Brown R (2012) Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 31(42):4567–4576. https://doi.org/10.1038/onc.2011.611
Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25(10):1010–1022. https://doi.org/10.1101/gad.2037511
Bhatla T, Wang J, Morrison DJ, Raetz EA, Burke MJ, Brown P, Carroll WL (2012) Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood 119(22):5201–5210. https://doi.org/10.1182/blood-2012-01-401687
Issa ME, Takhsha FS, Chirumamilla CS, Perez-Novo C, Vanden Berghe W, Cuendet M (2017) Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin Epigenetics 9:17. https://doi.org/10.1186/s13148-017-0319-5
Huang Y (2007) Pharmacogenetics/genomics of membrane transporters in cancer chemotherapy. Cancer Metastasis Rev 26(1):183–201. https://doi.org/10.1007/s10555-007-9050-6
Gottesman MM, Ambudkar SV (2001) Overview: ABC transporters and human disease. J Bioenerg Biomembr 33(6):453–458
Glavinas H, Krajcsi P, Cserepes J, Sarkadi B (2004) The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv 1(1):27–42
Campos L, Guyotat D, Archimbaud E, Calmard-Oriol P, Tsuruo T, Troncy J, Treille D, Fiere D (1992) Clinical significance of multidrug resistance P-glycoprotein expression on acute nonlymphoblastic leukemia cells at diagnosis. Blood 79(2):473–476
Dalton WS, Grogan TM, Meltzer PS, Scheper RJ, Durie BG, Taylor CW, Miller TP, Salmon SE (1989) Drug-resistance in multiple myeloma and non-Hodgkin’s lymphoma: detection of P-glycoprotein and potential circumvention by addition of verapamil to chemotherapy. J Clin Oncol 7(4):415–424. https://doi.org/10.1200/JCO.1989.7.4.415
Marie JP, Zittoun R, Sikic BI (1991) Multidrug resistance (mdr1) gene expression in adult acute leukemias: correlations with treatment outcome and in vitro drug sensitivity. Blood 78(3):586–592
Miller TP, Grogan TM, Dalton WS, Spier CM, Scheper RJ, Salmon SE (1991) P-glycoprotein expression in malignant lymphoma and reversal of clinical drug resistance with chemotherapy plus high-dose verapamil. J Clin Oncol 9(1):17–24. https://doi.org/10.1200/JCO.1991.9.1.17
Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM (2003) P-glycoprotein: from genomics to mechanism. Oncogene 22(47):7468–7485. https://doi.org/10.1038/sj.onc.1206948
Bradshaw DM, Arceci RJ (1998) Clinical relevance of transmembrane drug efflux as a mechanism of multidrug resistance. J Clin Oncol 16(11):3674–3690. https://doi.org/10.1200/JCO.1998.16.11.3674
Clarke R, Leonessa F, Trock B (2005) Multidrug resistance/P-glycoprotein and breast cancer: review and meta-analysis. Semin Oncol 32(6 Suppl 7):S9–S15. https://doi.org/10.1053/j.seminoncol.2005.09.009
Mahadevan D, List AF (2004) Targeting the multidrug resistance-1 transporter in AML: molecular regulation and therapeutic strategies. Blood 104(7):1940–1951. https://doi.org/10.1182/blood-2003-07-2490
Fisher GA, Sikic BI (1995) Clinical studies with modulators of multidrug resistance. Hematol/Oncol Clin N Am 9(2):363–382
Sikic BI (1993) Modulation of multidrug resistance: at the threshold. J Clin Oncol 11(9):1629–1635. https://doi.org/10.1200/JCO.1993.11.9.1629
Sikic BI (1997) Pharmacologic approaches to reversing multidrug resistance. Semin Hematol 34(4 Suppl 5):40–47
Capranico G, De Isabella P, Castelli C, Supino R, Parmiani G, Zunino F (1989) P-glycoprotein gene amplification and expression in multidrug-resistant murine P388 and B16 cell lines. Br J Cancer 59(5):682–685
Shukla S, Chen ZS, Ambudkar SV (2012) Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist Updates 15 (1–2):70–80. https://doi.org/10.1016/j.drup.2012.01.005
Chang XB (2007) A molecular understanding of ATP-dependent solute transport by multidrug resistance-associated protein MRP1. Cancer Metastasis Rev 26(1):15–37. https://doi.org/10.1007/s10555-007-9041-7
Burg D, Wielinga P, Zelcer N, Saeki T, Mulder GJ, Borst P (2002) Inhibition of the multidrug resistance protein 1 (MRP1) by peptidomimetic glutathione-conjugate analogs. Mol Pharmacol 62(5):1160–1166
Chen YN, Mickley LA, Schwartz AM, Acton EM, Hwang JL, Fojo AT (1990) Characterization of adriamycin-resistant human breast cancer cells which display overexpression of a novel resistance-related membrane protein. J Biol Chem 265(17):10073–10080
Robey RW, Polgar O, Deeken J, To KW, Bates SE (2007) ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev 26(1):39–57. https://doi.org/10.1007/s10555-007-9042-6
Bates SE, Robey R, Miyake K, Rao K, Ross DD, Litman T (2001) The role of half-transporters in multidrug resistance. J Bioenerg Biomembr 33(6):503–511
Ross DD, Yang W, Abruzzo LV, Dalton WS, Schneider E, Lage H, Dietel M, Greenberger L, Cole SP, Doyle LA (1999) Atypical multidrug resistance: breast cancer resistance protein messenger RNA expression in mitoxantrone-selected cell lines. J Natl Cancer Inst 91(5):429–433
Westover D, Ling X, Lam H, Welch J, Jin C, Gongora C, Del Rio M, Wani M, Li F (2015) FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol Cancer 14:92. https://doi.org/10.1186/s12943-015-0362-9
Dumontet C, Sikic BI (1999) Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J Clin Oncol 17(3):1061–1070. https://doi.org/10.1200/JCO.1999.17.3.1061
Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB (2003) Mechanisms of Taxol resistance related to microtubules. Oncogene 22(47):7280–7295. https://doi.org/10.1038/sj.onc.1206934
Seve P, Mackey J, Isaac S, Tredan O, Souquet PJ, Perol M, Lai R, Voloch A, Dumontet C (2005) Class III beta-tubulin expression in tumor cells predicts response and outcome in patients with non-small cell lung cancer receiving paclitaxel. Mol Cancer Ther 4(12):2001–2007. https://doi.org/10.1158/1535-7163.MCT-05-0244
Yusuf RZ, Duan Z, Lamendola DE, Penson RT, Seiden MV (2003) Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr Cancer Drug Targets 3(1):1–19
Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, Stec J, Ayers M, Ross JS, Zhang P, Buchholz TA, Kuerer H, Green M, Arun B, Hortobagyi GN, Symmans WF, Pusztai L (2005) Microtubule-associated protein tau: a marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci USA 102(23):8315–8320. https://doi.org/10.1073/pnas.0408974102
Wagner P, Wang B, Clark E, Lee H, Rouzier R, Pusztai L (2005) Microtubule Associated Protein (MAP)-Tau: a novel mediator of paclitaxel sensitivity in vitro and in vivo. Cell Cycle 4(9):1149–1152. https://doi.org/10.4161/cc.4.9.2038
Andoh T, Ishii K, Suzuki Y, Ikegami Y, Kusunoki Y, Takemoto Y, Okada K (1987) Characterization of a mammalian mutant with a camptothecin-resistant DNA topoisomerase I. Proc Natl Acad Sci USA 84(16):5565–5569
Deffie AM, Batra JK, Goldenberg GJ (1989) Direct correlation between DNA topoisomerase II activity and cytotoxicity in adriamycin-sensitive and -resistant P388 leukemia cell lines. Cancer Res 49(1):58–62
Tanizawa A, Pommier Y (1992) Topoisomerase I alteration in a camptothecin-resistant cell line derived from Chinese hamster DC3F cells in culture. Cancer Res 52(7):1848–1854
Beck WT, Morgan SE, Mo YY, Bhat UG (1999) Tumor cell resistance to DNA topoisomerase II inhibitors: new developments. Drug Resist Updates 2(6):382–389. https://doi.org/10.1054/drup.1999.0110
Xu Y, Villalona-Calero MA (2002) Irinotecan: mechanisms of tumor resistance and novel strategies for modulating its activity. Ann Oncol 13(12):1841–1851
Lackner MR, Wilson TR, Settleman J (2012) Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol 8(8):999–1014. https://doi.org/10.2217/fon.12.86
O’Hare T, Eide CA, Deininger MW (2007) Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 110(7):2242–2249. https://doi.org/10.1182/blood-2007-03-066936
Wang D, Lippard SJ (2005) Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4(4):307–320. https://doi.org/10.1038/nrd1691
Kaina B, Christmann M (2002) DNA repair in resistance to alkylating anticancer drugs. Int J Clin Pharmacol Ther 40(8):354–367
Ceppi P, Volante M, Novello S, Rapa I, Danenberg KD, Danenberg PV, Cambieri A, Selvaggi G, Saviozzi S, Calogero R, Papotti M, Scagliotti GV (2006) ERCC1 and RRM1 gene expressions but not EGFR are predictive of shorter survival in advanced non-small-cell lung cancer treated with cisplatin and gemcitabine. Ann Oncol 17(12):1818–1825. https://doi.org/10.1093/annonc/mdl300
Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22(47):7265–7279. https://doi.org/10.1038/sj.onc.1206933
Gerson SL (2004) MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer 4(4):296–307. https://doi.org/10.1038/nrc1319
Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362(6423):847–849. https://doi.org/10.1038/362847a0
Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362(6423):849–852. https://doi.org/10.1038/362849a0
Fan S, el-Deiry WS, Bae I, Freeman J, Jondle D, Bhatia K, Fornace AJ Jr, Magrath I, Kohn KW, O’Connor PM (1994) p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res 54(22):5824–5830
Carnero A, Garcia-Mayea Y, Mir C, Lorente J, Rubio IT, LLeonart ME (2016) The cancer stem-cell signaling network and resistance to therapy. Cancer Treat Rev 49:25–36. https://doi.org/10.1016/j.ctrv.2016.07.001
Tannock I (1978) Cell kinetics and chemotherapy: a critical review. Cancer Treat Rep 62(8):1117–1133
Tannock IF (1968) The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer 22(2):258–273
Hirst DG, Denekamp J (1979) Tumour cell proliferation in relation to the vasculature. Cell Tissue Kinetics 12(1):31–42
Ljungkvist AS, Bussink J, Rijken PF, Kaanders JH, van der Kogel AJ, Denekamp J (2002) Vascular architecture, hypoxia, and proliferation in first-generation xenografts of human head-and-neck squamous cell carcinomas. Int J Radiat Oncol Biol Phys 54(1):215–228
Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS (2000) Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 19(38):4319–4327. https://doi.org/10.1038/sj.onc.1203782
Shain KH, Dalton WS (2001) Cell adhesion is a key determinant in de novo multidrug resistance (MDR): new targets for the prevention of acquired MDR. Mol Cancer Ther 1(1):69–78
Wang GL, Semenza GL (1995) Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 270(3):1230–1237
Pouyssegur J, Dayan F, Mazure NM (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441(7092):437–443. https://doi.org/10.1038/nature04871
Rice GC, Hoy C, Schimke RT (1986) Transient hypoxia enhances the frequency of dihydrofolate reductase gene amplification in Chinese hamster ovary cells. Proc Natl Acad Sci USA 83(16):5978–5982
Rice GC, Ling V, Schimke RT (1987) Frequencies of independent and simultaneous selection of Chinese hamster cells for methotrexate and doxorubicin (adriamycin) resistance. Proc Natl Acad Sci USA 84(24):9261–9264
Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP (2002) Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 62(12):3387–3394
Kennedy KA (1987) Hypoxic cells as specific drug targets for chemotherapy. Anti-Cancer Drug Des 2(2):181–194
Raghunand N, He X, van Sluis R, Mahoney B, Baggett B, Taylor CW, Paine-Murrieta G, Roe D, Bhujwalla ZM, Gillies RJ (1999) Enhancement of chemotherapy by manipulation of tumour pH. Br J Cancer 80(7):1005–1011. https://doi.org/10.1038/sj.bjc.6690455
Raghunand N, Mahoney BP, Gillies RJ (2003) Tumor acidity, ion trapping and chemotherapeutics. II. pH-dependent partition coefficients predict importance of ion trapping on pharmacokinetics of weakly basic chemotherapeutic agents. Biochem Pharmacol 66(7):1219–1229
Cowan DS, Tannock IF (2001) Factors that influence the penetration of methotrexate through solid tissue. Intl J Cancer 91(1):120–125
Cooper GM (2000) The cell: a molecular approach, 2nd edn. Sinauer Associates, Boston University, Sunderland (MA)
Spears CP (1995) Clinical resistance to antimetabolites. Hematol/Oncol Clin N Am 9(2):397–413
Kickhoefer VA, Rajavel KS, Scheffer GL, Dalton WS, Scheper RJ, Rome LH (1998) Vaults are up-regulated in multidrug-resistant cancer cell lines. J Biol Chem 273(15):8971–8974
List AF, Spier CS, Grogan TM, Johnson C, Roe DJ, Greer JP, Wolff SN, Broxterman HJ, Scheffer GL, Scheper RJ, Dalton WS (1996) Overexpression of the major vault transporter protein lung-resistance protein predicts treatment outcome in acute myeloid leukemia. Blood 87(6):2464–2469
Steuart CD, Burke PJ (1971) Cytidine deaminase and the development of resistance to arabinosyl cytosine. Nature 233(38):109–110
Carlson RW, Sikic BI (1983) Continuous infusion or bolus injection in cancer chemotherapy. Ann Internal Med 99(6):823–833
Cassidy J (1994) Chemotherapy administration: doses, infusions and choice of schedule. Ann Oncol 5(Suppl 4):25–29 (discussion 29–30)
Marangolo M, Bengala C, Conte PF, Danova M, Pronzato P, Rosti G, Sagrada P (2006) Dose and outcome: the hurdle of neutropenia (Review). Oncol Rep 16(2):233–248
Ribatti D (2008) Judah Folkman, a pioneer in the study of angiogenesis. Angiogenesis 11(1):3–10. https://doi.org/10.1007/s10456-008-9092-6
Kyle AH, Huxham LA, Yeoman DM, Minchinton AI (2007) Limited tissue penetration of taxanes: a mechanism for resistance in solid tumors. Clin Cancer Res 13(9):2804–2810. https://doi.org/10.1158/1078-0432.CCR-06-1941
Minchinton AI, Tannock IF (2006) Drug penetration in solid tumours. Nat Rev Cancer 6(8):583–592. https://doi.org/10.1038/nrc1893
Matsumoto S, Batra S, Saito K, Yasui H, Choudhuri R, Gadisetti C, Subramanian S, Devasahayam N, Munasinghe JP, Mitchell JB, Krishna MC (2011) Antiangiogenic agent sunitinib transiently increases tumor oxygenation and suppresses cycling hypoxia. Cancer Res 71(20):6350–6359. https://doi.org/10.1158/0008-5472.CAN-11-2025
Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR (1989) Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci USA 86(2):695–698
Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA, van der Valk MA, Robanus-Maandag EC, te Riele HP et al (1994) Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77(4):491–502
Benzekry S, Pasquier E, Barbolosi D, Lacarelle B, Barlesi F, Andre N, Ciccolini J (2015) Metronomic reloaded: theoretical models bringing chemotherapy into the era of precision medicine. Semin Cancer Biol 35:53–61. https://doi.org/10.1016/j.semcancer.2015.09.002
Pasquier E, Kavallaris M, Andre N (2010) Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 7(8):455–465. https://doi.org/10.1038/nrclinonc.2010.82
Callaghan R, Luk F, Bebawy M (2014) Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy? Drug Metabol Dispos 42(4):623–631. https://doi.org/10.1124/dmd.113.056176
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45(D1):D362–D368. https://doi.org/10.1093/nar/gkw937
Acknowledgements
Dr. A.G. Georgakilas acknowledges funding from DAAD Grant “DNA Damage and Repair and Their Relevance to Carcinogenesis” (No. 57339330).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Nikolaou, M., Pavlopoulou, A., Georgakilas, A.G. et al. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis 35, 309–318 (2018). https://doi.org/10.1007/s10585-018-9903-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-018-9903-0