
Abstract
The ability to identify population structure, estimate rates of hybridization, and genetic sources of gene flow is critical when attempting to conserve wild populations. Recently diverged species with few pre- or post-zygotic isolating mechanisms are prone to exchange genetic material during secondary contact events, potentially causing the breakup of important coadapted genes and resulting in maladapted populations. Such events are especially exacerbated when domestic versions come into contact with their wild congeners and exchange genetic variation that had been under artificial selection. Being able to genetically identify individuals to populations or species, and thus potential hybrids, is essential when attempting to assess impacts from hybridization. Until recently, molecular methods often resulted in insufficient marker coverage to confidently assign individuals to populations for organisms comprised of closely related taxa. Advances in partial genome sequencing methods (e.g., ddRAD-seq, sequence capture) and decreasing sequencing costs have made it possible to readily access thousands of genetic markers across hundreds of samples, providing a population genomics across the landscapes of wild systems. Here, I review what landscape-level sampling coupled with thousands of nuclear markers has uncovered for a group of recently radiated ducks of the Mallard Complex (genus Anas). Deploying the latest population genomics approaches, researchers have been able to reconstruct complex evolutionary histories, assign individuals to species with confidence, as well as identify genetic hybrids. These population genomics studies have produced findings that are in contrast to what was thought to be known for many of these species. Among results, studies consistently found that the problem of hybridization for many of these species was due to feral mallard populations. In fact, the result of these anthropogenic hybridization events is the formation of hybrid swarms on Hawaii, North America, Eurasia, and New Zealand. Wildlife biologists are now incorporating these population genomics-based results into their management planning, demonstrating the need and importance of population genomics in wildlife conservation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Anas
- Conservation genetics
- ddRAD-seq
- Hybridization
- Partial genome sequencing
- Population genomics
- Wildlife management
1 Introduction
1.1 Background
For conservation to work, it is essential to understand the biological unit for which efforts are being undertaken; however, this is often easier said than done. Specifically, management of wild populations requires an understanding of how many potentially evolutionarily independent groups exist as to devise specific conservation plans for each group (Oyler-McCance et al. 2016; Peters et al. 2016; Allendorf 2017; Ralls et al. 2018). Among issues faced for many organisms as habitats and environments continue to change is the increasing events of secondary contact with other closely related sister species or even their domestic conspecifics (Randi 2008; Kidd et al. 2009; Tufto 2017; Wang et al. 2017; Heikkinen et al. 2019). Thus, determining the source of hybridization and potential outcomes of admixture are important aspects in the field of conservation genetics (Crispo et al. 2011; Nadeau and Kawakami 2019).
The exchange of genetic material between two populations, or gene flow, is an important and sometimes necessary process in the speciation process (Dobzhansky 1940; Hoskin et al. 2005; Rundle and Nosil 2005; Schluter 2009) and can even result in positive outcomes by increasing the adaptive potential of a population or species (i.e., adaptive introgression; Hedrick 2013; Hamilton and Miller 2016; Nadeau and Kawakami 2019; vonHoldt et al. 2018; Nadachowska-Brzyska et al. 2019; Qiao et al. 2019; Owens and Samuk 2020) or even increasing overall biodiversity (i.e., hybrid speciation; Mallet 2007; Jacobsen and Omland 2011a; Schumer et al. 2014; Lavretsky et al. 2015b; Lamichhaney et al. 2018). In addition, conservation efforts for highly fragmented populations often require the direct movement of individuals to artificially reinvigorate gene flow (Ralls et al. 2018). Typically, however, gene flow is considered as a negative player in conservation as the most foreseen consequence(s) are often the creation of perpetual hybrid zone(s) (Barton and Hewitt 1989) and inhibition of the speciation process (Mallet 2005, 2007), including a reversal of speciation (Seehausen 2006; Webb et al. 2011; Kearns et al. 2018) and outright extinction through introgressive hybridization (Rhymer 2006) (also see Fig. 1 in Crispo et al. 2011). Though gene flow is becoming more relevant and clearly an important player in the evolution of many organisms (Mallet 2007; Nadeau and Kawakami 2019; vonHoldt et al. 2018), it is artificially induced secondary contact events (a.k.a. anthropogenic gene flow; McFarlane and Pemberton 2019) that are now of conservation concern for many organisms (Lande 1998; Puigcerver et al. 2014; Lavretsky et al. 2015b; Skoglund et al. 2015; Wayne and Shaffer 2016; Söderquist et al. 2017; Leitwein et al. 2018; McFarlane and Pemberton 2019).

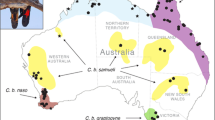
Picture representations of all mallard-like ducks and their respective distributions; note wild mallards have a Holarctic distribution. Hawaiian Islands are within the inset
Identifying genetically admixed individuals and their true parental populations with confidence is essential when attempting to conserve populations facing high frequencies of hybridization. Moreover, understanding the true biological outcome of hybridization (see Crispo et al. 2011), including whether such events have negative (i.e., extinction by introgressive hybridization; Rhymer 2006) or potentially positive (i.e., adaptive introgression; vonHoldt et al. 2018; Qiao et al. 2019; Owens and Samuk 2020) outcome(s), is paramount. However, determining shared genetic variability due to admixture is often complicated for recent radiations, including between wild and domestic congeners as much of the genome may be shared simply due to ancestry (Orr et al. 2004; Seehausen 2004; Wu and Ting 2004; Nosil et al. 2009; Via 2009; Nosil and Schluter 2011). Being able to assign samples to their true genetic population cluster(s) can help distinguish what genetic variability is due to shared ancestry versus gene flow (Lavretsky et al. 2019b). In general, population genomics has the potential to advance our understanding of even the most complex interactions (DaCosta and Sorenson 2014; Andrews et al. 2016; McKinney et al. 2017; Nadeau and Kawakami 2019; McFarlane and Pemberton 2019).
In this chapter, I recount how the integration of genetic information from population genomics studies has transformed our understanding of the evolutionary histories and contemporary population structure of worldwide populations of closely related ducks of the Mallard Complex. The Mallard Complex successfully radiated around the world and is comprised of 14 species of mallard-like ducks (Fig. 1). Across the continents and Islands that they successfully adapted to, these mallard-like ducks are ecologically, culturally, and economically important. Their importance has made it a priority to understand the genetic health and adaptive potential of respective populations and species by local conservation private and public organizations.
Among the species, the mallard (Anas platyrhynchos) is one of the most ubiquitous and well-known ducks in the world, and wildlife, in general. Its success is best explained by its adaptive nature and the fact that it was domesticated 4,000 years ago, providing a long, intertwined history with humans. Being domesticated, many forms of the mallard have been introduced around the world. Population genomics has revealed that the expansion of these domestic mallards resulted in secondary contact with their wild congeners and ultimately resulted in high levels of anthropogenic gene flow and the formation of feral x wild hybrid swarms in many instances. Finally, I discuss the potential that historical museum samples and ancient DNA techniques provide in understanding how populations have changed, including how hybridization has shaped the genetic diversity of contemporary populations. In general, population genomics studies coupling landscape-level sampling have been able to provide important insight into the evolution and population structure and identify previously unknown levels of hybridization for a sweep of wild populations of ducks. The Mallard Complex provides a unique system to study how radiations occur and the importance of gene flow on these processes.
1.2 History of the Mallard Complex
Of the many avian orders, waterfowl (order Anseriformes) experience the highest rates of hybridization (Johnsgard 1960; Livezey 1986; Lijtmaer et al. 2003), with 30–40% of species being capable of hybridizing (Grant and Grant 1992) and about 20% producing viable hybrids (Scherer and Hilsberg 1982). For example, the Mallard Complex is comprised of 13–14 closely related species of mallard-like ducks (Fig. 1) believed to have radiated out of Africa in the last million years (Palmer 1976; Johnson and Sorenson 1999). With some hybridization events producing 100% viable offspring (Avise et al. 1990), concerns over the possibility of introgressive extinction for many of the endemic mallard-like ducks have been raised over the years. Therefore, there has been a growing need to assess and delimitate individuals to species when assessing conservation risks for many of these species.
First proposed by Palmer (1976), the “out of Africa hypothesis” suggests an African origin for the mallard clade, followed by a northward and eastward radiation through Eurasia, with a stepwise progression through the South Pacific, and perhaps a single colonization event into North America. Several phylogenies have been reconstructed from either plumage characteristics (Livezey 1991), allozymes (Browne et al. 1993) or mitochondrial molecular markers (Johnson and Sorenson 1999; McCracken et al. 2001; Kulikova et al. 2004, 2005). However, these have been largely inconclusive, and it was not until Lavretsky et al. (2014b) who coupled multiple nuclear introns, multiple samples per species, and the coalescence method as implemented in the program ∗BEAST (Drummond and Rambaut 2007; Drummond et al. 2012; Bouckaert et al. 2014) that provided more robust phylogenetic relationships that largely confirmed the out of Africa evolutionary history for the Mallard Complex.
The onset for speciation of the Mallard Complex is estimated to be approximately 1 million years ago, with the most recent divergences estimated at 150,000 years ago (Lavretsky et al. 2014a, b). Within this single radiation, you can find species divergence proceeding in allopatry, parapatry, undergoing secondary contact, and potentially evolving via hybrid speciation (Lavretsky et al. 2014a, b, 2015a, b; Peters et al. 2016). With the exception of the dichromatic mallard, the remaining species are all monochromatic, where males and females show a similar phenotype (Fig. 1). With mallards being the only species with obvious sex-based plumage differences, phylogenetic comparisons have led to several speculations into the evolution of dimorphism in the mallard clade including that it was gained once (Johnson and Sorenson 1999; McCracken et al. 2001; Kulikova et al. 2004, 2005) or lost several times (Omland 1997). As a result of their relatively recent divergence, widespread incomplete lineage sorting has resulted in the retention of much of the genome among taxa, and likely contributing to the fact that viable hybrid offspring are produced in sympatry (Avise et al. 1990; Rhymer 2006; Lavretsky et al. 2014b).
Currently, with the exception of the Holarctic mallard, the remaining species are endemic to a single continent or island group (Haddon 1984; Rhymer et al. 1994; Kulikova et al. 2004) (Fig. 1). Unfortunately, environmental degradation and both human-facilitated (i.e., release programs) and natural expansion of the mallard’s range have caused formerly allopatric species to come into secondary contact leading to hybridization. For example, mallard introductions in the Hawaiian Islands and New Zealand have resulted in nearly complete introgression of mallard alleles into the endemic species (i.e., Hawaiian duck or koloa (A. wyvilliana; Fowler et al. 2009; Wells et al. 2019) and New Zealand grey duck (A. superciliosa superciliosa; Rhymer et al. 1994)), respectively. Furthermore, the mallard has expanded its range westward in both Eurasia and North America, and accumulating evidence demonstrates that mallards are likely to outcompete and to hybridize with native species, including spot-billed ducks (A. zonorhyncha; Kulikova et al. 2004) and American black ducks (A. rubripes; Rhymer and Simberloff 1996), respectively. With a variety of evolutionary histories and ability to successfully interbreed, the Mallard Complex is an ideal example of an adaptive or rapid radiation in which secondary contact events can have real implications into the adaptive potential of the invaded species.
Advancing our understanding of the evolutionary history and consequences of interspecific gene flow was not possible with confidence until advancement in partial genome sequencing attained sufficient marker coverage to be able to taxonomically assign samples with confidence. Specifically, coupling landscape-level sampling with double-digest restriction site-associated DNA sequencing (ddRAD-seq) methods (Peterson et al. 2012; Catchen et al. 2017; McKinney et al. 2017) has been truly transformative for gaining insight into the genetic histories of species within the Mallard Complex (Fig. 2). Finally, with ducks being well represented in museum collections, using ancient DNA methods on historical specimens have recently permitted researchers to understand genetic change through time, helping to understand timing, cause, and extent in changes of population structure and contemporary gene flow (Lavretsky et al. 2020).

Population structure of the New World mallard clade that includes mallards (MALL), American black duck (ABDU), Mexican duck (MEDU), West Gulf Coast mottled duck (MODUWGC), and Florida mottled duck (MODUFL) and as determined using (a) 17 nuclear introns (adapted from Lavretsky et al. 2014a) or (b) 15,687 biallelic ddRAD-seq nuclear SNPs (adapted from Lavretsky et al. 2019a). Note the clear increased resolution of population structure using thousands of ddRAD-seq loci. For American black ducks and mallards, genetic diagnosability was achieved once a sufficient sample size of pure parental was sampled (see Fig. 4; also see Lavretsky et al. (2019b)). These results demonstrate the importance of attaining sufficient statistical diagnosability across genetic markers when dealing with systems as closely related as these mallard-liked ducks
1.3 Mitochondrial DNA and the Onset of Fear of Hybrid Swarms
Avian researchers have generally focused on mtDNA. Maternally inherited and having no recombination (Giles et al. 1980; Watanabe et al. 1985), mtDNA has a more rapid sorting rate and shorter coalescent intervals relative to biparentally inherited, recombining nuclear DNA. This makes it particularly useful for recently diverged populations (Moore 1995; Zink and Barrowclough 2008). However, being maternally inherited and potentially under strong selection, its appropriateness for phylogenetics and phylogeography has been questioned (Hurst and Jiggins 2005; Bazin et al. 2006; Edwards and Bensch 2009; Jacobsen and Omland 2011b).
Mitochondrial DNA has played a significant role in shaping our understanding of the evolution of the Mallard Complex. In particular, mtDNA was important in gaining insight into the relationship between New World (NW) and Old World (OW) mallard populations (Johnson and Sorenson 1999; Kulikova et al. 2005). Whereas Eurasian mallard populations are all characterized as possessing OW A haplotypes, those in North America are paraphyletic with a substantial proportion carrying OW A and NW B mtDNA haplotypes. Hypotheses to explain the co-occurrence of OW A and NW B mtDNA haplotypes in North America were (a) secondary contact and widespread bi-directional gene flow between invading Eurasian mallards (Johnson and Sorenson 1999; McCracken et al. 2001; Kulikova et al. 2004, 2005) and/or (b) incomplete lineage sorting from a dichromatic ancestor that invaded the New World and diverged into several monochromatic species (Omland 1997). Regardless, consensus from early work was that bi-directional gene flow with endemic NW mallard-like ducks (Fig. 1) was a big reason for why OW A and NW B mtDNA haplotypes were present in North America. In fact, there was a growing concern for the possibility of genetic extinction via widespread introgressive hybridization across monochromatic species from these results. Concern that endemic North American mallard-like ducks were likely hybrid swarms continued until recent advances in various genomics methods permitted for the access of the genome (i.e., thousands of nuclear loci), and making it possible to assign individuals to populations with confidence. Coupling population assignments with nuclear markers and mtDNA confirmed OW A mtDNA haplotypes in wild populations of mallard-like ducks were in fact the result of gene flow with domestic mallards. Note that all domestic mallards are known to carry OW A mtDNA haplotypes as the origins of domestication for mallards are in Eurasia (Kiple 2001; Huang et al. 2013). For example, comparing mitochondrial sequences to local domestic mallard populations, OW A mtDNA haplotypes recovered in Hawaii’s Hawaiian duck populations (Fowler et al. 2009; Lavretsky et al. 2019a, b) and Florida’s mottled ducks (A. fulvigula) (Bielefeld et al. 2016; Peters et al. 2016) was the result of gene flow with local populations of park mallards. Similarly, OW A mtDNA haplotypes found in North American wild mallards, American black ducks, and West Gulf Coast mottled ducks were the result of gene flow with domestic game-farm mallards, which have been released for sport hunting in North America since the 1920s (Lavretsky et al. 2019a, b, 2020). Thus, the true story was only revealed by applying landscape- and genomic-level sampling. While gene flow is the culprit for why OW A mtDNA haplotypes are now widespread in North American mallard-like ducks, it was not due to gene flow with wild mallards coming from Eurasia, but rather domestic ones that were released in the respective areas.
1.4 Hybridization Versus Gene Flow
Avian lineages are especially prone to hybridization, even between species with relatively deep divergences (Grant and Grant 1997; Rheindt and Edwards 2011; Ottenburghs et al. 2015; Ottenburghs 2019). The high rates of hybridization in birds are attributable to their dispersal ability (Greenwood 1980), chromosomal stasis (Ellegren 2010), and relatively low levels of reinforcement (Grant and Grant 1997). The rapid evolutionary history, as well as the extent of gene flow among species within the Mallard Complex, makes this system ideal to study the interplay among various evolutionary mechanisms at the earliest stages of species divergence. Moreover, whereas many taxa within the Mallard Complex evolved in allopatry, the mallard has responded to anthropogenic influences (e.g., releases from game-farms and altered landscapes) and can now be found in sympatry with most of the other species. This secondary contact has resulted in widespread hybridization with the American black duck (Avise et al. 1990), Mexican duck (A. diazi; Hubbard 1977; Lavretsky et al. 2015a), mottled duck (McCracken et al. 2001; Williams et al. 2005a), Chinese spot-billed duck (A. zonorhyncha; Kulikova et al. 2004; Wang et al. 2019), New Zealand (NZ) grey duck (A. superciliosa superciliosa; Rhymer et al. 1994), Hawaiian duck (Griffin and Browne 1990; Lavretsky et al. 2015b), and yellow-billed duck (A. undulata; Stephens et al. 2019). Because all hybridization events involve mallards, this group provides natural replicates to understand how true gene flow impacts the genomes of endemic species and overall consequences on their adaptive and evolutionary trajectories.
From a conservation standpoint, determining the extent that hybridization events translate into true gene flow is critical (Fig. 3). I define hybridization as an event in which pure parental taxa interbreed and make a potentially viable F1 hybrid. If that hybrid does not breed into either of the parental populations, then the biological outcome of said mating event is simply lost breeding potential for both parental taxa. Conversely, gene flow requires the F1 hybrid to breed back into one or both parental populations, as to effectively move genes between the parental taxa (Fig. 3). While both events may be of concern for conservation biologists, the inability for the hybrid to breed and effectively move genes between parental taxa would be evidence of hybrid breakdown, assortative mating, or other potential post-zygotic isolating processes. Thus, determining the number and hybrid types (F1, F2-backcross, F3-backcross, etc.) found on the landscape is critical when attempting to determine the extent and potential genetic polluting from intraspecific hybridization events. For example, if hybrids are relegated to the F1 generation with none or few backcrosses, then one can conclude the ultimate consequence of hybridization is lost breeding potential without gene flow. Conversely, if a variety of backcrosses are identified in a population, then the ultimate outcome of hybridization is gene flow. If this is the case, then determining the number of generational backcrossed hybrids, the geographical locations of these hybrids, as well as whether these backcrosses tend to breed with one of the parental populations are important next steps to best inform proper conservation action(s).

Schematic and biologically relevant potential outcomes of hybridization: (a) hybridization results in an F1 hybrid that does not breed into parental gene pools and, thus, is simply lost breeding potential. (b) Hybridization results in an F1 hybrid that does breed into parental gene pools (denoted by arrows), thus effectively moving genes between species
Often, breeding experiments are necessary to establish expected genetic assignments of various generational classes of hybrids; however, such experiments are often not possible with wild populations (Lavretsky et al. 2016). I note that breeding experiments are ideal to carefully understand the genetic admixture effect(s) on morphology and ecology of a species (Grabenstein and Taylor 2018). There are a variety of methods that allow to assign individuals to hybrid status (Nielsen et al. 2006; VÄHÄ and Primmer 2006; Corbett-Detig and Nielsen 2017; Wringe et al. 2017; Janzen et al. 2018). Recently, a method that permits the use of empirical molecular data collected from wild individuals was designed to simulate breeding experiments and based on the available dataset was created (Lavretsky et al. 2016). In short, a parental gene pool is established with samples that are genetically vetted as pure parental. A single random allele or SNP is chosen from each parental gene pool and across all available markers to simulate F1 genotypes. Next, subsets of F1 hybrids are then backcrossed into each parental gene pool for a determined number of times, and thus establishing gene pools for F2 through FX backcrossed generations. Assignment probabilities are estimated in programs like STRUCTURE (Pritchard et al. 2000) or ADMIXTURE (Alexander et al. 2009; Alexander and Lange 2011) for a combined dataset of the simulated and empirical genotypes (Fig. 4). This can be done multiple times to establish expected average and range of assignment probabilities for each hybrid generation. For example, simulation outcomes for American black ducks and mallards resulted in six identifiable indices that included individuals with (a) ≥95% black duck assignment as pure black duck, (b) ≥98% mallard assignment as pure mallard, (c) 27–72% interspecific assignment as F1 hybrids, (d) 10–27% as F2-black duck backcrosses, (e) 2–27% black duck assignment as F2-mallard backcrosses, and (f) 5–10% mallard assignment as F3-black duck backcrosses (Fig. 4a). In the end, such analyses allow researchers to gain a more robust estimate of assignment probabilities of what a pure parental or hybrid is expected given the available molecular data.

(a) The average and range of assignment probabilities from ADMIXTURE results at K = 2 and 3 across 25 simulated replications of hybridization (F1) and 9 generations of backcrossing (F2-F10) using genetically vetted American black ducks (ABDU) and mallards (MALL) – each K is based on 250 independent ADMIXTURE analyses. (b) Empirical data for western (WEST), Mississippi flyway (MISS), and Atlantic flyway (ATL) samples originally identified by USFWS as Mallards, American black ducks, or putative hybrids. Within geographical region, samples in all three phenotypic classes are aligned by interspecific assignment probability from high to low. Based on expected assignment probabilities as determined from simulations, I recategorized samples by assignment probabilities and found that ~80% of all phenotypically identified mallards and black ducks, as well as only ~60% of phenotypically identified hybrids, are correct. Figure adapted from Lavretsky et al. (2019b)
Simulated “breeding” experiments help determine the number of hybrid classes and subsequently allow researchers to assign individuals to those classes. The proportion of individuals falling into each hybrid class provides an estimate of the relative rate of hybridization across the sampled landscape of the specie(s). For example, using simulations to genetically vet North American mallards and American black ducks demonstrated the limitations of US Fish and Wildlife Service’s phenotypic key being used to assign pure and hybrid status to samples (Lavretsky et al. 2019b). Specifically, of those individuals phenotypically assigned as pure American black duck or pure mallard, 20% should have been identified as hybrids in each set. Similarly, only ~60% of all samples assigned as phenotypic American black duck x mallard hybrids were correct, with the remaining samples actually being either pure American black duck (~28% of samples) or pure mallard (12% of samples) (Fig. 4). Thus, these results clearly demonstrated the ineffectiveness in correctly identifying individuals and particularly hybrids with the current sweep of phenotypic traits for American black ducks and mallards. Finally, overlaying empirical and simulated data to determine the true number of each generational class provided the means to determine the rate of hybridization across North America. As expected, American black duck and mallard hybridization was highest in eastern North America and with evidence for a variety of hybrid classes present on the landscape. However, despite a century and a half of secondary contact between American black ducks and mallards resulting in some of the highest rates of hybridization (i.e., ~25%), Lavretsky et al. (2019b) concluded that American black ducks are not the hybrid swarm once hypothesized and that gene flow into American black duck was somehow limited (e.g., assortative mating). Once again, the ability to genetically identify samples, and even between taxa that are very closely related like American black ducks and mallards, can illuminate inconsistencies in current methods and datasets, and even previous notions (e.g., American black ducks are a hybrid swarm) that are used to guide and make important management decisions. Moreover, these results demonstrate how advances in genomic methods provide the capacity to genetically establish phenotypic traits that are truly informative (e.g., see the genetically vetted key created for Florida mottled ducks; Bielefeld et al. 2016). These and similar methods offer a powerful approach for examining concerns of hybridization in conservation efforts and without the requirement of captive breeding.
The depletion of native populations makes it susceptible to genetic swamping from even small numbers of introduced species (Childs et al. 1996; Rhymer 2006; Russo et al. 2018). Although hybridization is prevalent in birds, and ducks especially (Cade 1983; Rhymer 2006), species extinction due to complete genetic swamping, while concerning (Rhymer and Simberloff 1996; Buerkle et al. 2003), has been identified in few systems (Rhymer and Simberloff 1996; Salzburger et al. 2002; Wells et al. 2019; Lavretsky et al. 2020). Examples of true hybrid swarms may be rare because in general, the backcrossing of hybrids back into large parental populations may prevent the persistence of large numbers of admixed individuals (e.g., Lavretsky et al. 2016). For example, between American black ducks and mallards where Lavretsky et al. (2019b) found that clear outlier regions between the parental species decreased in genetic differentiation when comparing the genomic landscape of several generations of backcrossed individuals (Fig. 5). Thus, having a parental gene pool to which hybrids can continuously backcross into is not only important for conservation but may be an important mechanism that decreases the potential negative effects of gene flow, in general. As habitat continues to be depleted, domestic forms increasing on the landscape, as well as overall global change is bring many closely related taxa together, and thus understanding the geographical extent of hybridization and whether these events result in true gene flow is essential. Genomics methods that readily provide sufficient marker coverage have made even the most difficult or complex relationships possible to tease apart (Wayne and Shaffer 2016; Catchen et al. 2017; McKinney et al. 2017).

(a) Hypothesized mating between mallards (MALL) and American black ducks (ABDU) and subsequent backcrosses into American black ducks with expected genomic contributions of offspring. (b) Empirical ΦST estimates across markers along the Z-chromosome and chromosome 1 between genetically vetted samples that are consistent with expected offspring from each respective hypothesized mating event. Genomes depicted to be largely ancestrally shared (denoted as genus Anas), with species specificity identified at a few highly selected regions. Note the clear decrease in outlier regions with each subsequent backcross that correspond with proportional replacement of mallard alleles with American Black ducks until F3 (or second generation backcrosses), which are genetically identical to a pure American black ducks. Figure adapted from Lavretsky et al. (2019b)
2 How Population Genomics Has Increased Our Understanding of the Mallard Complex and Its Implications for Conservation
2.1 The History of the New World Mallard Complex: How the Phenotype Lies and What Genetics Has Revealed
Seven mallard-like ducks make North America home. Of these, the mallard, American black duck, Mexican duck, and two subspecies of mottled ducks are found on mainland North America, while the Laysan and Hawaiian ducks are found on islands making up the Hawaiian Archipelago (Fig. 1). All but the mallard are endemic to either the Hawaiian Islands or mainland North America (Baldassarre 2014). Concern over genetic swamping, the resulting hybrid swarm, and eventual genetic extinction for all the endemic mallard-like ducks has spurred over four decades of research into understanding rates of hybridization between mallards and each of the other North American taxa (Rhymer and Simberloff 1996; Rhymer 2006). Cause for concern was due to the high prevalence of what appeared to be individuals carrying phenotypic traits of the mallard (e.g., green iridescence in head, top and/or bottom secondary white wing bars, curl feathers in the rump) and of the respective monochromatic taxa in populations of Mexican ducks (Hubbard 1977), American black ducks (Brodsky and Weatherhead 1984; Ankney et al. 1987; Avise et al. 1990; Kirby et al. 2000), mottled ducks (Bielefeld et al. 2010, 2016), and Hawaiian ducks (Griffin and Browne 1990; Livezey 1993; Engilis et al. 2002a). The presentation of these mallard traits in significant proportions heightened concern over the possible genetic extinction of these endemic monochromatic ducks. Any advances made in molecular methods since the 1980s have been applied towards attempting to determine rates of hybridization and gene flow. However, early attempts with allozymes (Browne et al. 1993), microsatellites (Williams et al. 2002, 2005a, b; Mank et al. 2004; Fowler et al. 2009), and Sanger sequencing of single and multiple loci (Avise et al. 1990; Johnson and Sorenson 1999; McCracken et al. 2001; Kulikova et al. 2004, 2005; Lavretsky et al. 2014a) resulted in largely inconclusive findings (see Fig. 3a as example). In each case, the authors determined that too much of the genetic variation was shared among the taxa to be able to confidently identify hybrids, let alone assign samples to their respective taxon. Paraphyly and intermixed mtDNA haplotypes further suggested to researchers that these species likely were represented by highly admixed individuals (Avise et al. 1990; McCracken et al. 2001; Lavretsky et al. 2014a, b; Peters et al. 2014). In general, additive effects from ancestry (i.e., incomplete lineage sorting) and extensive hybridization were used to explain the extent of shared molecular diversity in this group of birds.
The lack of more definitive molecular work had important implication onto taxonomic revisions and conservation efforts for many of the taxa within the Mallard Complex. Until recently, many taxonomic and conservation decisions remained informed through largely phenotypic work and the basic premise regarding the general lack of non-paraphyletic genetic markers as putatively the result of widespread admixture. For example, the taxonomy of Mexican duck has been defined by phenotypic work done in the 1970s, which presumed that the clinal-like presence of mallard-like traits in Mexican ducks was the result of extensive introgressive hybridization (Hubbard 1977). As a result of these phenotypic-based conclusions and a general lack of definitive molecular results, the Mexican duck has been relegated to subspecies status. Similarly, conservation efforts for the endangered Hawaiian duck largely surrounded the need to remove mallard-like traits from then captive Hawaiian duck population prior to release. However, no matter the effort to breed Hawaiian duck looking individuals together, a proportion of juvenile males always displayed mallard-like phenotypic characters (Engilis and Pratt 1993; Engilis et al. 2002a). Similarly, breeding experiments attempting to “breed out” mallard characters by specifically mating individuals that were especially American black duck-looking continuously resulted in broods with a proportion of males still displaying mallard-like characters (Kirby et al. 2004). Once again, understanding whether mallard-like characters displayed in many of these monochromatic species was due to the fact that these represented hybrid swarms or simply a case of ancestry remained unknown until advances in genomic methods permitted researchers to genetically identify between pure and hybrid individuals.
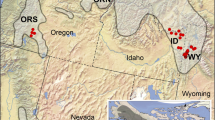
The population genomics approaches required to properly answer questions about hybridization in the Mallard Complex were optimized in the mid-2010s, and specifically, it was advancements made in the reduced genomic representation methods (e.g., RADs, ddRAD-seq, SeqCap) that opened the door to accessing sufficient sized genomic datasets. First in 2015, Lavretsky et al. (2015a) applied a ddRAD-seq method to sample 3,695 polymorphic loci – 3,523 loci (316,175 base pairs (bp) assigned to autosomes and 172 loci (15,869 bp) assigned to the Z-sex chromosome) – across 105 Mexican ducks from six Mexican states (N = 92 individuals) and two US states (N = 13 individuals), as well as 17 mallards sampled across North America. The authors demonstrated that Mexican ducks and mallards were genetically distinguishable and identified no samples in Mexico and only a handful of samples from the USA as genetic hybrids. The latter finding was in stark contrast to the notion that Mexican ducks were largely a Mexican duck x mallard hybrid swarm as suggested with phenotypic data (Hubbard 1977). In fact, the landscape perspective that Lavretsky et al. (2015a) achieved showed that Mexican ducks biogeography naturally followed an isolation-by-distance pattern and was the result of sequential founder events from north to south (Fig. 6b-3). Moreover, Lavretsky et al. (2015a) identified specific genetic regions on several autosomal and Z-sex chromosome that were under divergent selection in mallards or Mexican ducks. More recent work further demonstrated that Mexican ducks retained mallard diversity due to ancestry (i.e., ILS) and not extensive gene flow, as well as provided more definitive demarcation of several genetic markers on autosomal chromosomes 2 and 14 that were best explained by divergent selection in Mexican ducks specifically (Figs. 7 and 8a; Lavretsky et al. 2019a). Finally, ongoing work linking phenotype with genetics has surprisingly revealed that Mexican ducks display mallard-like traits and shared mallard genetic diversity that follow a cline from north to south but that this is explained by retained ancestry and not gene flow from mallards (Brown et al., unpublished data). Specifically, having genetically vetted samples, the researchers determine that juvenile or hatch-year genetically pure male Mexican ducks displayed mallard-like characters and that the proportion of hatch-year males that displayed such characters decreased southward. Thus, while Hubbard (1977) correctly characterized the clinal variation in regard to mallard-like plumage displayed by Mexican ducks across their range, these patterns were not indicative of a hybrid swarm but rather due to retained ancestry. Specifically, the evolution of the Mexican duck was likely the result of a mallard population that isolated and adapted to the Chihuahuan Desert 200,000–500,000 years before present and expanded southward through sequential founder events losing dichromatism in the process (Fig. 6b-3). In the end, coupling genomics methods with landscape-level sampling proved to resolve the evolutionary history of the Mexican duck, including establishing that Mexican ducks showed some of the lowest rates of hybridization within the Mallard Complex. Moreover, Mexican ducks harbor genomic regions under divergent selection and which are at species-level differences that would suggest that taxonomic revisions for this duck may be warranted.

The evolutionary and contemporary histories of mallard-like ducks from North America as determined from population genomics

Pairwise estimates of relative differentiation (ΦST) across New World mallard-like ducks for 3,017 and 177 ddRAD-seq loci that assigned to autosomes or the Z-sex chromosome, respectively. Note no ddRAD-seq loci were recovered on chromosome 17. Coupling species pair analyses and partial genome sequencing provides a mean to explore regions under divergent selection in specific species. Here, a ~21 Mbp region (positions 1.7E7–3.8E7 bp) on the Z-sex chromosome and a ~11 Mbp region (1.0E8–1.2E8 bp) on chromosome 1 were identified to be under divergent selection in mallards. An outlier locus on chromosome 14 (position ~1.6E7) was detected in all four comparisons involving Mexican ducks, suggesting directional selection at this or a linked locus in Mexican ducks only. Additional outliers were detected in pairwise comparisons involving other species. Finally, genomic patterns as observed between the two mottled ducks are consistent with instance of strict allopatric speciation. Figure adapted from Lavretsky et al. (2019a)

Chromosomally aligned ΦST estimates of (a) 3,037 autosomal and 163 Z-linked ddRAD-seq loci for pairwise comparisons between genetically vetted wild mallards, wild American Black Ducks, and feral x wild mallard hybrids (i.e., hybrid swarm). Markers identified in BayeScan analyses as putatively under divergent selection are denoted in red, arrows denoting loci potentially under artificial selection. Adapted from Lavretsky et al. (2019b). (b) Sequence captured 2,103 autosomal and 99 Z-linked loci for pairwise comparisons between historical and contemporary mallards and American black ducks. Brackets denote the same loci demarcated as outliers between American black ducks and wild mallards, and red circles + arrows denote loci found as under artificial selection in (a), which was adapted from Lavretsky et al. (2020). Note no markers were recovered for chromosome 17
Applying similar methods to study the evolution and population structure of mottled ducks (Peters et al. 2016; Ford et al. 2017) and American black ducks (Lavretsky et al. 2019b, 2020) proved to be once again fruitful, with researchers being able to establish that none of these monochromatic taxa are already or on their way to becoming a hybrid swarm. Instead, data determined that these ducks are all very closely related, with much of the genome shared due to ancestry from and not gene flow with the mallard (Fig. 6b; Lavretsky et al. 2019a). More specific analyses also revealed that while mottled ducks from Florida and the West Gulf Coast diverged in allopatry from each other and the mallard (Peters et al. 2016), the American black duck and mallard likely diverged under punctuated events of secondary contact (Lavretsky et al. 2020) (Fig. 6b). Importantly, attaining sufficient marker coverage and across hundreds of samples was required to finally determine rates of hybridization and potential effects of gene flow. In short, the rates of hybridization with mallards were highest for black ducks (i.e., ~25%; Lavretsky et al. 2019b), followed by mottled ducks (i.e., 5–8%; Peters et al. 2016; Ford et al. 2017), and Mexican ducks (i.e., 2–5%; Lavretsky et al. 2015a); however, gene flow into each of the monochromatic species remained low (Lavretsky et al. 2019a), suggesting that post-zygotic isolating mechanisms likely evolved within this recent radiation. In fact, pairwise species comparisons across ~3,194 ddRAD-seq loci demarcated several outlier regions on the Z-sex and other autosomal chromosomes harboring genes under divergent selection in one or more of the taxa (Fig. 7; Lavretsky et al. 2019a). Among these sites, a ~21 Mbp region on the Z-sex chromosome was recovered to harbor genes under divergent selection in mallards that may be playing an important role in the evolution of dichromatism in this group (Figs. 7 and 8a). These results are consistent with a growing body of evidence suggesting that sex chromosomes are often involved in early stages of species divergence, including harboring genes linked to phenotypic variation in other taxa (Minvielle et al. 2000; Sæther et al. 2007; Phadnis and Orr 2009; Pryke 2010; Ellegren et al. 2012; Martin et al. 2013; Sutter et al. 2013; Ruegg et al. 2014; Lavretsky et al. 2015a).
2.2 The Curious Case of the Hawaiian Duck: Conservation Implications When a Hybrid Species Meets Its Feral Parent
With Laysan ducks now relegated to the Laysan and Midway Atoll Islands, the endangered Hawaiian duck is the only remaining endemic duck on the main Hawaiian Islands (Engilis et al. 2004; Pyle and Pyle 2017). Recent molecular work provided strong evidence that Hawaiian ducks represent a homoploid hybrid species (Fig. 6a). Specifically, Lavretsky et al. (2015b) determined that the Hawaiian duck’s evolutionary history was the result of an ancestral hybridization event between vagrant mallards and once prevalent Laysan ducks and dated the admixture event to the Pleistocene-Holocene boundary; the authors analyzed molecular variation across 19 nuclear introns and used coalescent analyses to estimate the ancestral gene flow event to ~3,000 years before present (95% HPD = 0–207,000 years before present). More recent work by Wells et al. (2019) using thousands of ddRAD-seq loci also found evidence that pure Hawaiian ducks shared near 50:50 coancestry with Laysan ducks and mallards and the only one to show such patterns of all mallard-like ducks (Lavretsky et al. 2014b, 2015b). Together, Hawaiian ducks are genetically (Lavretsky et al. 2014b, 2015b), phenotypically, and ecologically (Engilis et al. 2002b; Uyehara et al. 2008) distinct from all other mallard-like ducks and thus satisfy all primary criteria used in avian taxonomy for species designations (Gill 2014; Sangster 2014) and as a result likely represent a young hybrid species. In fact, it was likely the combination of mallard and Laysan duck molecular variation, including predator aversion that would only be innate in the mallard that permitted the Hawaiian duck to endure past Polynesian settlement where other Island life (e.g., Laysan ducks) could not.
The Hawaiian duck remained largely in allopatry from both of its wild parental taxa, until domestic mallards were first imported to the Hawaiian Islands for food and hunting beginning in the 1800s (Engilis et al. 2004; Pyle and Pyle 2017). Later, mallards were commercially farmed on O’ahu during the 1930s and 1940s, and multiple feral populations became established on Kaua’i, O’ahu, Maui, and Hawai’i (Engilis and Pratt 1993). As a result of these actions, genetic extinction through ongoing hybridization with feral mallards has been primary concern for their conservation (USFWS 2012). Historically, Hawaiian ducks occurred on the main Hawaiian Islands of Kaua’i, Ni’ihau, O’ahu, Maui, Moloka’i, and Hawai’i but were extirpated from all islands except Kaua‘i and Ni’ihau by the 1960s (Engilis et al. 2002a). Hawaiian ducks were captive-reared and reintroduced onto O’ahu and Hawai’i until the late 1980s and onto Maui in 1989. However, feral populations of domestic mallards were not dealt with prior to attempted reintroductions and may have been the reason that biannual waterbird surveys suggested an increasing number of Hawaiian duck-mallard hybrids through time (USFWS 2012). In fact, earlier molecular work confirmed that hybridization between Hawaiian ducks and local mallards was occurring on O’ahu (Browne et al. 1993; Fowler et al. 2009). However, despite early molecular efforts to understand hybridization, much of the hybrid identification done by USFWS and state agency personnel was based on phenotype. As with the case for other New World mallard-like ducks (see above), we now know that Hawaiian ducks, and primarily first year males, naturally display mallard-like characters as by-product of their recent mallard ancestry (Lavretsky et al. 2015b). Thus, whether the increasing number of hybrids across Hawaiian Islands was real or simply due to overestimation resulting from nondiagnostic phenotypic traits remained unknown until recently.
Wells et al. (2019) set to determine the extent of true hybridization rates between Hawaiian ducks and mallards across Hawaiian Islands, potential mallard source (i.e., domestic vs. wild), whether hybridization rates have increased through time, and whether the presence/absence of mallard-like traits in the phenotype of an individual can be confidently applied to identify hybrids. Sampling included assaying 3,114 autosomal and 194 Z-linked ddRAD-seq loci across 425 Hawaiian ducks obtained across 5 Hawaiian Islands, as well as 30 samples of each Laysan ducks and wild North American mallards. Sampling effort of Hawaiian ducks was nearly a decade apart, permitting the researchers to test for changing hybridization rates across time. First, the authors confirmed that pure Hawaiian ducks persist on Kauaʻi in large numbers and with relatively little evidence of mallard-Hawaiian duck hybridization during the past decade (Fig. 9). This finding was incredibly informative for conservation biologists attempting to determine whether pure Hawaiian ducks even existed (USFWS 2012). Unfortunately, Wells et al. (2019) reported that all reintroduced populations on Hawai’i, Maui, and O’ahu constituted hybrid swarms – i.e., not a single sample among reintroduced populations was identified as a pure Hawaiian duck (i.e., ≥95% Kaua’i Hawaiian duck ancestry; Fig. 9). The authors were able to determine that the extensive hybridization was primarily with non-wild, local feral mallards. Furthermore, the authors were able to genotype ducks collected from 1998 to 2015. First, a decreasing trend in the number of hybrids on Kauaʻi where removal efforts of mallards and potential hybrids were underway was found. Conversely, all sampled reintroduced population showed no change in overall admixture proportions across samples or in the overall proportion of admixed individuals. Thus, the authors could conclude that all sampled sites across Hawaiian Islands in which reintroductions were attempted eventually failed due to extensive hybridization with these feral mallards and untimely became true hybrid swarms as early as 1998. Finally, further molecular assessment of Hawaiian ducks that were culled due to the presence of mallard-like traits on Kauaʻi revealed only a handful of these to be true hybrids and the remaining as hatch-year males. Once again, mallard-like traits displayed by Hawaiian ducks were found to be due to shared mallard ancestry and not contemporary hybridization as with the other monochromatic mallard-like taxa on mainland North America (see above).

Location for 425 Hawaiian ducks and putative hybrids sampled across Hawaiian Islands. Individual ancestry proportions as estimated in the program STRUCTURE using 3,114 ddRAD-seq autosomal loci assuming both K = 3 and K = 4 populations. Included in analyses are Laysan ducks and North American mallards. Pie charts represent the proportion of samples with the putative New World “B” mtDNA lineage (yellow) and the putative Old World “A” lineage (blue) for a subset of ducks on each island. Note that all reintroduced populations are now genetically identified as hybrids (i.e., hybrid swarm; Fig. 6a). Additionally, most reintroduced populations are largely identified carrying Old World A mtDNA haplotypes that are associated with mallards from Eurasia. Figure adapted from Wells et al. (2019)
Island populations and those that have recently declined are more susceptible to genetic swamping by an introduced species (Childs et al. 1996; Rhymer 2006; Grabenstein and Taylor 2018). The absence of large native populations of Hawaiian duck on O’ahu, Maui, Moloka’i, and Hawai’i likely precipitated the formation of hybrid swarms on these islands. Hawaiian duck reintroductions involved relatively few individuals, and captive-reared Hawaiian ducks were introduced on islands with established populations of feral mallards (Engilis et al. 2002a). Together, the evidence presented by Wells et al. (2019) clearly demonstrated that all reintroduction efforts were set up to fail by not dealing with the feral mallard problem prior to attempting Hawaiian duck reintroductions. Given the lack of large, pure populations of Hawaiian ducks into which hybrids can backcross, hybrid swarms will likely persist on these islands. Thus, in the absence of additional genetic contributions from Kaua’i Hawaiian ducks, time alone is unlikely to decrease hybrid individuals comprising these hybrid swarms. Thus, these molecular results establish that future conservation efforts to reestablish pure Hawaiian ducks outside Kauaʻi will need to remove the identified hybrid swarms, as well as feral mallards. Hawaiian duck reintroduction efforts represent an example of what happens to a small, isolated founder population confronted with a large population of a nonreproductively isolated congener. These results demonstrate the potential amalgamating effects of gene flow during secondary contact, spatial variation in the extent and consequences of hybridization, and the importance of considering such effects during conservation planning.
2.3 The Genetic and Conservation Consequences of Feral Mallards
Along with changing habitat, the direct release of domesticated individuals into the wild is a practice used worldwide to augment various wildlife populations (Laikre and Ryman 1996; Lichatowich and Lichatowich 2001; Waples and Drake 2004; Champagnon et al. 2012). Where these releases are common and intensive, concern for wild populations has increased because breeding with released or feral conspecifics can cause a loss of genetic variation leading to a loss of adaptation and overall fitness within wild population (Frankham 2005; Araki et al. 2009; Crispo et al. 2011; Grabenstein and Taylor 2018). In general, movement of these artificially selected, maladaptive traits into wild populations can reduce the adaptability and capacity of that population to survive in the wild (Araki et al. 2007; Evans and Evans 2007; Haccou et al. 2013; Corbi et al. 2018). In fact, modeling effects of gene flow between domestic and wild congeners, Tufto (2017) showed a slow, additive effect of increasing maladaptation in wild populations through continued interaction with their respective domestic counterpart and with negative outcomes taking time to be observable. As anthropogenic gene flow impacts more species, it is increasingly critical to monitor geographic regions where wild and domestic [feral] populations interact to assess for any possible genetic effects. Doing so ensures the adaptive qualities, and subsequent continued conservation of wild populations occurs.
Humans and mallards have been closely linked since their domestication in central China shortly after 500 BC (Kiple 2001; Huang et al. 2013). Around the world, domestic mallard stocking has been extensively practiced throughout history and most intense at the turn of the twentieth century (Heusmann 1991; Champagnon et al. 2012, 2013). While naturally found across the Holarctic, the intentional or accidental release of mallards has increased their range to include the entire world outside the Poles (Baldassarre 2014). Feral mallards now pose a genetic threat to global populations of wild mallard and mallard-like taxa. In all cases of mallard introductions, >25,000 domestic mallards were intentionally introduced, with the most extreme cases as within North America and Eurasia; these releases are now in the tens-of-millions (Heusmann 1974; Braithwaite and Miller 1975; Brooke and Siegfried 1991; Heusmann 1991; Tamisier 1992; Engilis and Pratt 1993; Dean 2000; Engilis et al. 2004; Guay and Tracey 2009; Bielefeld et al. 2010; Dyer and Williams 2010; Čížková et al. 2012; Pyle and Pyle 2017). For the Mallard Complex, secondary contact has always been considered to be occurring between wild populations and specifically with wild mallards. However, applying thousands of molecular markers and landscape-level sampling efforts has been transformative in correcting this early dogma, and more definitively determining that introgressive hybridization has not been with wild but rather domestic strains of the mallard. First, we learned that Kaua‘i is home to the last remaining pure population of the endangered Hawaiian duck (Wells et al. 2019) and that extensive introgressive hybridization with local, feral mallards has resulted in the formation of hybrid swarms across the Islands of Hawai’i, Maui, Moloka’i, and O’ahu (Figs. 6a and 9). Similarly alarming are the rates of widespread introgression between domestic and wild mallards in Eurasia and North America, where stocking practices still annually augment wild populations with nearly six million (Rueness et al. 2017) and two-hundred thousand (USFWS 2013) game-farm mallards, respectively, for the last 100 years. The substantial annual influx of these domesticated forms has significantly changed the genetic composition of Eurasian (Champagnon et al. 2010; Söderquist et al. 2014; Söderquist et al. 2017) and North American (Lavretsky et al. 2019b, 2020) mallards. Similarly, there is strong evidence of widespread introgression between domestic mallards and Pacific black ducks (Anas superciliosa) in Australia (Guay and Tracey 2009) and New Zealand (Hitchmough et al. 1990; Rhymer et al. 1994; Williams 2017). In fact, stocking practices of game-farm mallards in New Zealand have resulted in a current population of five million feral mallard birds (Williams 1981; Guay et al. 2015). A couple of recent studies assaying several molecular markers across range-wide sampled yellow-billed ducks determined that while wild mallards do not pose a genetic threat (Brown et al. 2019), yellow-billed ducks are now genetically threatened by domestic mallards that have been released and with feral populations recently establishing across Africa (Stephens et al. 2019). In general, domestic mallards differ in fertility, overall morphology, and biology from their wild counterparts (Desforges and Wood-Gush 1975a, b; Miller 1977; Paulke and Haase 1978; Cheng et al. 1979, 1982; Söderquist et al. 2013), with traits optimized for domestic settings. Understanding how the movement of their genetics and associated maladaptive traits may be decreasing the adaptability of wild populations will continue to grow in importance and particularly when devising conservation plans.
2.4 Attaining a Historical Perspective to Reconstruct Evolutionary Histories
Significant advances in ancient DNA (aDNA) extraction and sequence capture techniques have made it possible to isolate thousands of genetic markers to full genomes from historical and ancient samples, opening the possibility to attain a genetic perspective over large time scales (Grover et al. 2012; Mitchell et al. 2014; Orlando et al. 2015; Leonardi et al. 2017). These methods have been instrumental in understanding evolutionary histories of variety of organisms, including humans (Callaway 2016; Kuhlwilm et al. 2016), but are also now being used to understand the genetic turnover of populations and species (Cooper et al. 1996; Loreille et al. 2001; Leonard et al. 2002; Willerslev and Cooper 2005; Grealy et al. 2017; Leonardi et al. 2017; Rawlence et al. 2017; Lindqvist and Rajora 2019; Pont et al. 2019; Fenderson et al. 2020). In fact, the ability to not only determine the genetic diversity that was lost in a species but attaining full genomes for extinct species may make the idea of de-extinction possible (Shapiro 2017). For conservation efforts, rather than attempting to breed diversity back into an endangered species, we may be able to one day simply add the lost genetic diversity back into the species. Regardless, increasingly efficient aDNA methods have made museum specimens even more important not only to understand evolutionary histories but also understand how populations have genetically changed through time. For example, by understanding how populations responded to changing habitat in the past can undoubtedly help refine conservation plans for those populations in the future (Fenderson et al. 2020).
The first use of aDNA methods in the Mallard Complex was by Cooper et al. (1996), who were able to isolate a small piece of mitochondrial DNA from some Anas subfossils on Hawai’i. Doing so, Cooper and colleagues determined that these subfossils were indeed Laysan ducks, providing the first support that Laysan ducks were once widespread across the Hawaiian Islands. Next, Mank et al. (2004) assayed three microsatellite markers in historical mallards and American blacks and reported an 18-fold reduction in differentiation (Gst) between the two sampled in 1998 (0.008) versus 1940 (0.146). The authors concluded that a century of hybridization must have led to a loss of genetic distinctiveness. The numbers of usable samples were low with these early methods as they required the creation of primers as to amplify targeted DNA using PCR. Often, however, ancient and historical samples are highly degraded, posing limitations for PCR-based methods (Keyser-Tracqui and Ludes 2005). More recently, coupling sequence capture methods with high-throughput sequencing has made the isolation and sequencing of aDNA more accessible and reliable across any specimens with endogenous DNA (Briggs and Heyn 2012; Knapp et al. 2012; Schubert et al. 2012; Lindqvist and Rajora 2019). For example, Lavretsky et al. (2020) used these recently developed aDNA methods on American black ducks and mallards from 1860 to 1915 to revisit the hypothesis that American black ducks are closely related to mallards due to widespread hybridization as suggested by Mank et al. (2004). To do so, a bait capture array was first designed from 3,446 nuclear loci initially isolated from contemporary samples using ddRAD-seq methods (Lavretsky et al. 2019b). Across the 69 historical samples and another 39 contemporary samples, a total of 2,202 markers (140,477 base pairs (bp) across the Z-sex (99 markers; 6,122 bp) and 28 autosomal (2,103 markers; 134,355 bp) chromosomes) were isolated (Fig. 8b), resulting in a recovery rate of 64%; similar recovery rates using RAD-based bait arrays were reported in early work (Souza et al. 2017). Additionally, the authors were able to off target sequence 641 base pairs of the mtDNA control region across samples. Mitochondrial DNA is often obtained as bycatch in sequence capture datasets (Griffin et al. 2014; Gasc et al. 2016) due to its stability and abundance in samples (Picardi and Pesole 2012). In contrast to the results presented by Mank et al. (2004), Lavretsky et al. (2020) reported an overall increase in divergence, including the maintenance of all known outlier positions across the genomes of these two ducks (Fig. 8b). Moreover, there was no significant change in the sampled genome of historical and contemporary American black ducks (Fig. 8b), providing additional evidence that genetically pure American black ducks today are the same as those from 150 years ago (Fig. 6b-5). These results are clearly contradicting the notion that today’s American black ducks are simply a hybrid swarm. The authors suggest that earlier work by Mank et al. (2004) suffered from the total number of markers analyzed (i.e., three microsatellites). Given the evident genomic heterogeneity across their captured markers (Fig. 8b), using a few markers as done by Mank et al. (2004) is unlikely to provide a complete picture and demonstrates the importance in maximizing genetic data. Finally, in addition to important information for the conservation of the American black ducks, the sequencing of historical mallards and comparing to contemporary populations provided further evidence that today’s eastern mallards are indeed genetically different from both historical and contemporary western mallards. In the context of conservation, these results demonstrated opportunities that aDNA methods coupled with museum specimens towards understanding particular evolutionary histories, as well as determining lost genetic variation of specific populations or species.
Finally, ddRAD and related methods (e.g., RAD, GBS, etc.; Andrews et al. 2016) are inherently biased by the possibility of allelic dropout due to mutations in enzymatic cut-sites (Graham et al. 2015; Lowry et al. 2017; Catchen et al. 2017). These biases are not present in sequence capture datasets as they do not require enzymatic cut-sites to be present or intact to work. Lavretsky et al. (2020) were able to provide highly similar results between ddRAD and sequence capture datasets across a variety of estimates and analyses (e.g., Fig. 8), demonstrating that known biases with restriction enzyme-based techniques (e.g., allelic dropout; Graham et al. 2015; Lowry et al. 2017; Catchen et al. 2017) may have little or no effect on population-level statistics for species with very shallow divergence, such as between mallards and black ducks.
3 Integrating Population Genomics Results into Wildlife Management
Advances in molecular methods and high-throughput sequencing technology will continue to advance the field of population genomics, making the use of these methods possible for any organism. Though there is no doubt that lowering costs associated with full genomes will one day make it possible to be applied towards population genomics, partial genome sequencing methods like ddRAD-seq and related methods (e.g., RAD, GBS, etc.; Andrews et al. 2016) provide a perfect balance between data and cost. Today, a ddRAD-seq library can be attained for as little as $25–40 per sample and an Illumina HiSeq X capable of sequencing up to ~200 samples on a single lane. Importantly, methods like ddRAD-seq require no previous genetic information on the organism and are more forgiving when dealing with degraded DNA as compared to other genomic methods (Graham et al. 2015). Thus, the lowering cost and universal applicability of these partial genome sequencing methods make them ideal to study the population genetics of any wild population (Oyler-McCance et al. 2016). Moreover, although still proportionally a small amount of the genome (i.e., ddRAD datasets often represent <0.03% of the genome; Lavretsky et al. 2015a, 2019a), the thousands of markers remain a powerful means to screen for loci putatively under selection (e.g., Figs. 7 and 8; Andrews et al. 2016; Catchen et al. 2017).
Proper wildlife conservation requires an understanding of the population in question (Oyler-McCance et al. 2016; Peters et al. 2016; Allendorf 2017; Ralls et al. 2018). The fields of population and conservation genetics have illuminated the potential pitfalls when management decisions are made without truly knowing the genetic constitution of the population or species being managed. Understanding fine-scale population structure and hybridization rates requires a population genomics approach in which datasets are represented by samples spanning the taxon’s geographic range, and a maximum number of loci. Informative and decisive research into the evolution and population structure of the Mallard Complex, which harbors some of the most complex scenarios as often the case for recent and/or rapid radiations, was not possible until a landscape- and genomic-level sampling scheme was achieved (Lavretsky et al. 2019a). For example, it was not until a genomic perspective that provided sufficient marker coverage to genetically identify individuals to species with confidence that the issue of feral mallards, and not wild mallards, to the conservation of many of these mallard-like ducks was realized. Wildlife biologists now incorporated this problematic feral population into decision-making regarding future management efforts. Similarly, it was not until pure parental and genetic hybrids could be genetically determined that understanding whether the expression of particular mallard-like traits were due to ancestry or contemporary gene flow. A recent study that genetically vetted phenotypic traits between mallards, Florida mottled ducks, and their hybrids reported that a key character used to identify hybrids was in fact found in 10% of genetically “pure” mottled ducks (i.e., whitewing bar over and under secondaries; Bielefeld et al. 2016). By determining which samples were genetic hybrids, Bielefeld et al. (2016) were able to identify those phenotypic traits that were indeed diagnostic of hybrids. Doing so, the authors were able to construct a genetically vetted phenotypic field key that increased the ability of wildlife biologists to correctly identify hybrids from 60% to >90%. In general, applying a landscape- and genomic-scale approach, research into Hawaiian ducks (Wells et al. 2019), Mexican ducks (Lavretsky et al. 2015a), American black ducks (Lavretsky et al. 2019b), and yellow-billed ducks (de Souza et al. 2019) provided the same discrepancies in hybrid identification using nongenetically vetted phenotypic traits versus the traits expressed by true genetic hybrids. These studies demonstrate that many phenotypic traits once considered to be indicative of hybrids are simply due to stochastic processes independently acting on ancestral mallard variation in each of these species. Estimating rates of hybridization plays important roles in taxonomic evaluation and conservation efforts. Thus, the capacity to determine true genetic hybrids is critical and can either validate current practices or identify which species cohort requires reevaluation in regard to hybrid identification.
With lowering costs and increasing efficiency in wet lab and sequencing methods, as well as developing user-friendly bioinformatics pipelines, the future for the field of conservation genetics is bright. Integrating knowledge gained from molecular work continues to be a powerful tool for conservation (Andrews et al. 2016; Oyler-McCance et al. 2016; McKinney et al. 2017). Given that population genomics of non-model, wild systems is achievable for any organism, attaining a molecular understanding regarding the evolutionary and population genetics of even the most complex systems can now be realized.
4 Future Perspectives
Landscape-level and genome-wide population genomic datasets continue to expand our understanding regarding the dynamics of wild populations, including potential issues of hybridization. Among the opportunities afforded by the growing field of population genomics is being able to assign genetic purity to samples with confidence, which has been instrumental in advancing our understanding of evolutionary and contemporary population dynamics of specific taxa. Here, I also demonstrate how identifying hybrids and establishing hybridization rates can be key in establishing whether the expression of shared non-molecular traits (phenotypic and/or biological) across species is truly due to introgressive hybridization or simply ancestry (i.e., ILS).
Although much of this chapter focuses on how advances in population genomics has opened the possibilities for conservation, these same datasets can be applied to advance our understanding of the speciation process in wild systems (Lavretsky et al. 2015a, 2019a; Nadeau and Kawakami 2019), how the domestication process impacts genomic variation (Cornejo et al. 2018; Wu et al. 2018), as well as how species may adapt to today’s ever-changing climates (Fenderson et al. 2020). Among these efforts, aDNA methods are especially promising as they open historical and ancient samples for analysis, providing a means to understand how species have responded to changing climates in the past. Similarly, genomic data from landscape-level sampling of contemporary samples coupled with Gradient Forest analyses (Ellis et al. 2012) can now be used to assign the genetic niche space of a species given its standing genetic variability. Importantly, such models now make it possible to model forward expected responses to specific climatic shifts given available genetic variation of a population (Fitzpatrick and Keller 2015; Bay et al. 2018). In short, populations with lacking variation will show contracting ranges as survival in a changing landscape will require substantial increases in standing molecular variation to adapt. These models will allow researchers to build specific genetic niche maps for their favorite organism and be able to determine where habitat may be most critical under different climatic models. Such analyses are sure to be promising when attempting to predict species range responses to climate change and using this information to better inform where habitat work may be most warranted.
As full genome [re-]sequencing becomes increasingly approachable, partial genome sequencing can still offer important information to guide sampling efforts for genomic analyses. In general, if starting out with little knowledge regarding some species of interest, researchers can use partial genome sequencing on many samples to understand true population structure, hybridization rates, and hybrid identification. Having genetically vetted samples will then help inform sampling efforts for full genome sequencing by ensuring that representative pure parental and various hybrid classes are in fact used, thus decreasing the chance of mistakenly sequencing samples with incorrectly presumed origins (Lavretsky et al. 2019b; Leitwein et al. 2019). Among the opportunities afforded with full genomes is the possibility to better understand potential interactions between selection and gene flow and, in particular, the consequence of domestic variant introgression into wild populations. Having full genome sequences for pure parental and various classes of hybrids, researchers can identify haplotype block organization, number of recombination events, and types of parental variation in hybrids (Tang et al. 2006; Corbett-Detig and Nielsen 2017; Schaefer et al. 2017; Janzen et al. 2018; Leitwein et al. 2019). First, one would expect any genetic variation linked to putatively maladaptive traits to decrease in size due to recombination and lost over some number of generational backcrosses (Leitwein et al. 2019). Conversely, introgressed neutral variation is expected to simply show increasing fragmentation due to recombination events that arise with each generation of backcrossing (Janzen et al. 2018). Thus, these methods show promise to identify maladaptive versus neutral molecular variants that are moved between species during gene flow events, as well as establish the true number of generations since the initial hybridization event. Such information is invaluable when attempting to understand how hybridization may actually be impacting the adaptive potential, including survival and fecundity of a species.
5 Conclusions
Population genomics has opened possibilities to better refine the conservation of many organisms that was once impossible. Continued advances in wet lab and statistical analyses will undoubtedly further unlock the potential of genomics for conservation. In addition to groundbreaking and important research, translating these findings to not only the biologists themselves (Garner et al. 2016; Funk et al. 2019) but also the general public is almost as critically important as the data itself (Holderegger et al. 2019). Thus, efforts to transfer findings from genetic data to those directly implementing on the ground conservation work, as well as making the general public understand the benefits of wild lands and wildlife, are just as important endeavors.
References
Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics. 2011;12:246.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–64.
Allendorf FW. Genetics and the conservation of natural populations: allozymes to genomes. Mol Ecol. 2017;26:420–30.
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet. 2016;17:81–92.
Ankney DC, Dennis DG, Bailey RC. Increasing mallards, decreasing American black ducks: coincidence or cause and effect? J Wildl Manag. 1987;51:523–9.
Araki H, Ardren WR, Olsen E, Cooper B, Blouin MS. Reproductive success of captive-bred steelhead trout in the wild: evaluation of three hatchery programs in the Hood River. Conserv Biol. 2007;21:181–90.
Araki H, Cooper B, Blouin MS. Carry-over effect of captive breeding reduces reproductive fitness of wild-born descendants in the wild. Biol Lett. 2009;5:621–4.
Avise JC, Ankney DC, Nelson WS. Mitochondrial gene trees and the evolutionary relationship of mallard and black ducks. Evolution. 1990;44:1109–19.
Baldassarre G. Ducks, geese, and swans of North America. Baltimore: Johns Hopkins University Press; 2014.
Barton NH, Hewitt GM. Adaption, speciation and hybrid zones. Nature. 1989;341:497–503.
Bay RA, Harrigan RJ, Le Underwood V, Gibbs HL, Smith TB, Ruegg K. Genomic signals of selection predict climate-driven population declines in a migratory bird. Science. 2018;359:83–6.
Bazin E, Glémin S, Galtier N. Population size does not influence mitochondrial genetic diversity in animals. Science. 2006;312:570–2.
Bielefeld RR, Brasher MG, Moorman TE, Gray PN. Mottled duck (Anas fulvigula). In: Poole A, editor. The birds of North America online. Ithaca: Cornell Lab of Ornithology; 2010.
Bielefeld RR, Engilis A, Feddersen JC, Eadie JM, Tringali MD, Benedict RJ. Is it a mottled duck? The key is in the feathers. Wildl Soc Bull. 2016;40:446–55.
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537.
Braithwaite L, Miller B. The mallard, Anas platyrhynchos, and mallard-black duck, Anas superciliosa rogersi, hybridization. Wildl Res. 1975;2:47–61.
Briggs AW, Heyn P. Preparation of next-generation sequencing libraries from damaged DNA. In: Ancient DNA. Berlin: Springer; 2012. p. 143–54.
Brodsky LM, Weatherhead PJ. Behavioral and ecological factors contributing to American black duck-mallard hybridization. J Wildl Manag. 1984;48:846–52.
Brooke RK, Siegfried WR. Birds introduced to the fynbos biome of South Africa. In: Groves RH, di Castri F, editors. Biogeography of mediterranean invasions. Cambridge: Cambridge University Press; 1991. p. 359–64.
Brown JI, Lavretsky P, Cumming GS, Peters JL. Strong population structure and limited gene flow between yellow-billed ducks and mallards in southern Africa. The Condor. 2019.
Browne RA, Griffin CR, Chang PR, Hubley M, Martin AE. Genetic divergence among populations of the Hawaiian duck, Laysan duck, and mallard. Auk. 1993;110:49–56.
Buerkle C, Wolf D, Rieseberg L. The origin and extinction of species through hybridization. In: Population viability in plants. Berlin: Springer; 2003. p. 117–41.
Cade TJ. Hybridization and gene exchange among birds in relation to conservation. In: Schonewald-Cox CM, Chambers SM, MacBryde B, Thomas WL, editors. Genetics and conservation: a reference for managing wild animals and plant populations. Menlo Park, CA: Benjamin/Cummings; 1983. p. 288–309.
Callaway E. Ancient DNA pinpoints dawn of Neanderthals: sequencing of 430,000-year-old DNA pushes back species’ divergence from humans. Nature. 2016;531:286–7.
Catchen JM, Hohenlohe PA, Bernatchez L, Funk WC, Andrews KR, Allendorf FW. Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol Ecol Resour. 2017;17:362–5.
Champagnon J, Guillemain M, Elmberg J, Folkesson K, Gauthier-Clerc M. Changes in Mallard Anas platyrhynchos bill morphology after 30 years of supplemental stocking. Bird Study. 2010;57:344–51.
Champagnon J, Elmberg J, Guillemain M, Gauthier-Clerc M, Lebreton J-D. Conspecifics can be aliens too: a review of effects of restocking practices in vertebrates. J Nat Conserv. 2012;20:231–41.
Champagnon J, Crochet PA, Kreisinger J, Čížková D, Gauthier-Clerc M, Massez G, et al. Assessing the genetic impact of massive restocking on wild mallard. Anim Conserv. 2013;16:295–305.
Cheng KM, Shoffner RN, Phillips RE, Lee FB. Mate preference in wild and domesticated (game-farm) mallards: II. Pairing success. Anim Behav. 1979;27:417–25.
Cheng KM, Burns JT, McKinney F. Forced copulation in captive mallards (Anas platyrhynchos): II. Temporal factors. Anim Behav. 1982;30:695–9.
Childs MR, Echelle AA, Dowling TE. Development of the hybrid swarm between Pecos pupfish (Cyprinodontidae: Cyprinodon pecosensis) and sheepshead minnow (Cyprinodon variegatus): a perspective from allozymes and mtDNA. Evolution. 1996;50:2014–22.
Čížková D, Javůrková V, Champagnon J, Kreisinger J. Duck’s not dead: does restocking with captive bred individuals affect the genetic integrity of wild mallard (Anas platyrhynchos) population? Biol Conserv. 2012;152:231–40.
Cooper A, Rhymer J, James HF, Olson SL, Mclntosh CE, Sorenson MD, et al. Ancient DNA and island endemics. Nature. 1996;381:484.
Corbett-Detig R, Nielsen R. A hidden Markov model approach for simultaneously estimating local ancestry and admixture time using next generation sequence data in samples of arbitrary ploidy. PLoS Genet. 2017;13:e1006529.
Corbi J, Baack EJ, Dechaine JM, Seiler G, Burke JM. Genome-wide analysis of allele frequency change in sunflower crop–wild hybrid populations evolving under natural conditions. Mol Ecol. 2018;27:233–47.
Cornejo OE, Yee M-C, Dominguez V, Andrews M, Sockell A, Strandberg E, et al. Population genomic analyses of the chocolate tree, Theobroma cacao L., provide insights into its domestication process. Commun Biol. 2018;1:167.
Crispo E, Moore JS, Lee-Yaw JA, Gray SM, Haller BC. Broken barriers: human-induced changes to gene flow and introgression in animals. Bioessays. 2011;33:508–18.
DaCosta JM, Sorenson MD. Amplification biases and consistent recovery of loci in a double-digest RAD-seq protocol. PLoS One. 2014;9:e106713.
de Souza SG, Symes CT, Smit-Robinson H, Mollett JM. Minimal evidence of interspecific hybridisation between the yellow-billed duck and introduced mallard in central and northwestern South Africa. Ostrich. 2019;90:285–301.
Dean W. Alien birds in southern Africa: what factors determine success? S Afr J Sci. 2000;96:9–14.
Desforges M, Wood-Gush D. A behavioural comparison of domestic and mallard ducks. Habituation and flight reactions. Anim Behav. 1975a;23:692–7.
Desforges M, Wood-Gush D. A behavioural comparison of domestic and mallard ducks. Spatial relationships in small flocks. Anim Behav. 1975b;23:698–705.
Dobzhansky T. Speciation as a stage in evolutionary divergence. Am Nat. 1940;74:312–21.
Drummond A, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Dyer J, Williams M. An introduction most determined: mallard (Anas platyrhynchos) to New Zealand. Notornis. 2010;57:178–95.
Edwards S, Bensch S. Looking forwards or looking backwards in avian phylogeography? A comment on Zink and Barrowclough 2008. Mol Ecol. 2009;18:2930–3.
Ellegren H. Evolutionary stasis: the stable chromosomes of birds. Trends Ecol Evol. 2010;25:283–91.
Ellegren H, Smeds L, Burri R, Olason PI, Backström N, Kawakami T, et al. The genomic landscape of species divergence in Ficedula flycatchers. Nature. 2012;491:756–60.
Ellis N, Smith SJ, Pitcher CR. Gradient forests: calculating importance gradients on physical predictors. Ecology. 2012;93:156–68.
Engilis A Jr, Pratt TK. Status and population trends of Hawaii’s native waterbirds, 1977-1987. Wilson Bull. 1993;105:142–58.
Engilis A Jr, Pyle R, David R. Status and occurrence of migratory birds in the Hawaiian Islands: Part 1 – Anseriformes: Anatidae (Waterfowl). Bishop Museum Occasional Paper 81. 2004.
Engilis A, Uyehara KJ, Giffin JG. Hawaiian duck: Anas wyvilliana birds of North America, Incorporated. 2002a.
Engilis AJ, Uyehara K, Giffin J. Hawaiian duck (Anas wyvilliana). In: Poole A, Gill F, editors. The Birds of North America. Philadelphia: The Birds of North America, Inc.; 2002b.
Evans CS, Evans L. Representational signalling in birds. Biol Lett. 2007;3:8–11.
Fenderson LE, Kovach AI, Llamas B. Spatiotemporal landscape genetics: investigating ecology and evolution through space and time. Mol Ecol. 2020;29:218–46.
Fitzpatrick MC, Keller SR. Ecological genomics meets community-level modelling of biodiversity: mapping the genomic landscape of current and future environmental adaptation. Ecol Lett. 2015;18:1–16.
Ford RJ, Selman W, Taylor SS. Hybridization between Mottled Ducks (Anas fulvigula maculosa) and Mallards (A. platyrhynchos) in the western Gulf Coast region. Condor. 2017;119:683–96.
Fowler A, Eadie J, Engilis A. Identification of endangered Hawaiian ducks (Anas wyvilliana), introduced North American mallards (A. platyrhynchos) and their hybrids using multilocus genotypes. Conserv Genet. 2009;10:1747–58.
Frankham R. Genetics and extinction. Biol Conserv. 2005;126:131–40.
Funk W, Forester BR, Converse SJ, Darst C, Morey S. Improving conservation policy with genomics: a guide to integrating adaptive potential into US Endangered Species Act decisions for conservation practitioners and geneticists. Conserv Genet. 2019;20:115–34.
Garner BA, Hand BK, Amish SJ, Bernatchez L, Foster JT, Miller KM, et al. Genomics in conservation: case studies and bridging the gap between data and application. Trends Ecol Evol. 2016;31:81–3.
Gasc C, Peyretaillade E, Peyret P. Sequence capture by hybridization to explore modern and ancient genomic diversity in model and nonmodel organisms. Nucleic Acids Res. 2016;44:4504–18.
Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A. 1980;77:6715–9.
Gill FB. Species taxonomy of birds: which null hypothesis? Auk. 2014;131:150–61.
Grabenstein KC, Taylor SA. Breaking barriers: causes, consequences, and experimental utility of human-mediated hybridization. Trends Ecol Evol. 2018;33(3):198–212.
Graham CF, Glenn TC, McArthur AG, Boreham DR, Kieran T, Lance S, et al. Impacts of degraded DNA on restriction enzyme associated DNA sequencing (RADSeq). Mol Ecol Resour. 2015;15:1304–15.
Grant PR, Grant BR. Hybridization of bird species. Science. 1992;256:193–7.
Grant PR, Grant BR. Hybridization, sexual imprinting, and mate choice. Am Nat. 1997;149:1–28.
Grealy A, Rawlence NJ, Bunce M. Time to spread your wings: a review of the avian ancient DNA field. Gene. 2017;8:184.
Greenwood PJ. Mating systems, philopatry and dispersal in birds and mammals. Anim Behav. 1980;28:1140–62.
Griffin CR, Browne R. Genetic variation and hybridization in Hawaiian Duck and Mallards in Hawaii. U.S. Fish and Wildlife Service, Hawaiian and Pacific Islands National Wildlife Refuge Complex. 1990.
Griffin HR, Pyle A, Blakely EL, Alston CL, Duff J, Hudson G, et al. Accurate mitochondrial DNA sequencing using off-target reads provides a single test to identify pathogenic point mutations. Genet Med. 2014;16:962–71.
Grover CE, Salmon A, Wendel JF. Targeted sequence capture as a powerful tool for evolutionary analysis. Am J Bot. 2012;99:312–9.
Guay PJ, Tracey JP. Feral Mallards: a risk for hybridisation with wild Pacific black ducks in Australia? Victorian Nat. 2009;126:87–91.
Guay P-J, Williams M, Robinson RW. Lingering genetic evidence of North American mallards (Anas platyrhynchos) introduced to New Zealand. N Z J Ecol. 2015;39:103–9.
Haccou P, Lu B, Snow A, Stewart C, Strasburg J, van Tienderen P, et al. Introgression of crop alleles into wild and weedy populations. Annu Rev Ecol Evol Syst. 2013;44:325–45.
Haddon M. A re-analysis of hybridization between mallards and grey ducks in New Zealand. Auk. 1984;101:190–1.
Hamilton JA, Miller JM. Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv Biol. 2016;30:33–41.
Hedrick PW. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol. 2013;22:4606–18.
Heikkinen ME, Ruokonen M, White TA, Alexander MM, Gündüz İ, Dobney KM, et al. Long-term reciprocal gene flow in wild and domestic geese reveals complex domestication history. bioRxiv. 2019:706226.
Heusmann HW. Mallard-black duck relationships in the Northeast. Wildl Soc Bull. 1974;2:171–7.
Heusmann HW. The history and status of the mallard in the Atlantic Flyway. Wildl Soc Bull. 1991;19:14–22.
Hitchmough RA, Williams M, Daugherty CH. A genetic analysis of mallards, grey ducks, and their hybrids in New Zealand. New Zeal J Zool. 1990:467–72.
Holderegger R, Balkenhol N, Bolliger J, Engler JO, Gugerli F, Hochkirch A, et al. Conservation genetics: linking science with practice. Mol Ecol. 2019;28:3848–56.
Hoskin CJ, Higgie M, McDonald KR, Moritz C. Reinforcement drives rapid allopatric speciation. Nature. 2005;437:1353–6.
Huang Y, Li Y, Burt DW, Chen H, Zhang Y, Qian W, et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet. 2013;45:776–83.
Hubbard JP. The biological and taxonomic status of the Mexican Duck. Santa Fe, NM: New Mexico Department of Game and Fish Bulletin; 1977. p. 1–56.
Hurst GDD, Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc R Soc B Biol Sci. 2005;272:1525–34.
Jacobsen F, Omland KE. Increasing evidence of the role of gene flow in animal evolution: hybrid speciation in the yellow-rumped warbler complex. Mol Ecol. 2011a;20:2236–9.
Jacobsen F, Omland KE. Species tree inference in a recent radiation of orioles (Genus Icterus): multiple markers and methods reveal cytonuclear discordance in the northern oriole group. Mol Phylogenet Evol. 2011b;61:460–9.
Janzen T, Nolte AW, Traulsen A. The breakdown of genomic ancestry blocks in hybrid lineages given a finite number of recombination sites. Evolution. 2018;72:735–50.
Johnsgard PA. Hybridization in the Anatidae and its taxonomic implications. Condor. 1960;62:25–33.
Johnson KP, Sorenson MD. Phylogeny and biogeography of dabbling ducks (genus: Anas): a comparison of molecular and morphological evidence. Auk. 1999;116:792–805.
Kearns AM, Restani M, Szabo I, Schrøder-Nielsen A, Kim JA, Richardson HM, et al. Genomic evidence of speciation reversal in ravens. Nat Commun. 2018;9:1–13.
Keyser-Tracqui C, Ludes B. Methods for the study of ancient DNA. In: Goodwin W, editor. Forensic DNA typing protocols. Berlin: Springer; 2005. p. 253–64.
Kidd A, Bowman J, Lesbarreres D, Schulte-Hostedde A. Hybridization between escaped domestic and wild American mink (Neovison vison). Mol Ecol. 2009;18:1175–86.
Kiple KF. The Cambridge world history of food, vol. 2. Cambridge: Cambridge University Press; 2001.
Kirby RE, Reed A, Dupuis P Obrecht HH III, Quist WJ. Description and identification of American black duck, mallard, and hybrid wing plumage. In: Biological science report. U.S. Geological Survey, Biological Resources Division. 2000. p. 26.
Kirby RE, Sargeant GA, Shutler D. Haldane’s rule and American black duck × mallard hybridization. Can J Zool. 2004;82:1827–31.
Knapp M, Clarke AC, Horsburgh KA, Matisoo-Smith EA. Setting the stage–building and working in an ancient DNA laboratory. Ann Anat. 2012;194:3–6.
Kuhlwilm M, Gronau I, Hubisz MJ, de Filippo C, Prado-Martinez J, Kircher M, et al. Ancient gene flow from early modern humans into Eastern Neanderthals. Nature. 2016;530:429–33.
Kulikova IV, Zhuravlev YN, McCracken KG. Asymmetric hybridization and sex-biased gene flow between eastern spot-billed ducks (Anas zonorhyncha) and mallards (A. platyrhynchos) in the Russian Far East. Auk. 2004;121:930–49.
Kulikova IV, Drovetski SV, Gibson DD, Harrigan RJ, Rohwer S, Sorenson MD, et al. Phylogeography of the mallard (Anas platyrhynchos): hybridization, dispersal, and lineage sorting contribute to complex geographic structure. The Auk. 2005;122:949–65.
Laikre L, Ryman N. Effects on intraspecific biodiversity from harvesting and enhancing natural populations. Ambio. 1996;25:504–9.
Lamichhaney S, Han F, Webster MT, Andersson L, Grant BR, Grant PR. Rapid hybrid speciation in Darwin’s finches. Science. 2018;359:224–8.
Lande R. Anthropogenic, ecological and genetic factors in extinction and conservation. Res Popul Ecol. 1998;40:259–69.
Lavretsky P, Hernández Baños BE, Peters JL. Rapid radiation and hybridization contribute to weak differentiation and hinder phylogenetic inferences in the New World mallard complex (Anas spp.). Auk. 2014a;131:524–38.
Lavretsky P, McCracken KG, Peters JL. Phylogenetics of a recent radiation in the mallards and allies (Aves: Anas): inferences from a genomic transect and the multispecies coalescent. Mol Phylogenet Evol. 2014b;70:402–11.
Lavretsky P, Dacosta JM, Hernández-Baños BE, Andrew Engilis J, Sorenson MD, Peters JL. Speciation genomics and a role for the Z chromosome in the early stages of divergence between Mexican ducks and mallards. Mol Ecol. 2015a;24:5364–78.
Lavretsky P, Engilis A, Eadie JM, Peters JL. Genetic admixture supports an ancient hybrid origin of the endangered Hawaiian duck. J Evol Biol. 2015b;28:1005–15.
Lavretsky P, Peters JL, Winker K, Bahn V, Kulikova I, Zhuravlev YN, et al. Becoming pure: identifying generational classes of admixed individuals within lesser and greater scaup populations. Mol Ecol. 2016;25:661–74.
Lavretsky P, DaCosta JM, Sorenson MD, McCracken KG, Peters JL. ddRAD-seq data reveal significant genome-wide population structure and divergent genomic regions that distinguish the mallard and close relatives in North America. Mol Ecol. 2019a;28:2594–609.
Lavretsky P, Janzen T, McCracken KG. Identifying hybrids & the genomics of hybridization: Mallards & American black ducks of eastern North America. Ecol Evol. 2019b;9:3470–90.
Lavretsky P, McInerney NR, Mohl J, Brown JI, James H, McCracken KG, et al. Assessing changes in genomic divergence following a century of human mediated secondary contact among wild and captive-bred ducks. Mol Ecol. 2020;29:578–95.
Leitwein M, Gagnaire PA, Desmarais E, Berrebi P, Guinand B. Genomic consequences of a recent three-way admixture in supplemented wild brown trout populations revealed by local ancestry tracts. Mol Ecol. 2018;27:3466–83.
Leitwein M, Cayuela H, Ferchaud AL, Normandeau É, Gagnaire PA, Bernatchez L. The role of recombination on genome-wide patterns of local ancestry exemplified by supplemented Brook Charr populations. Mol Ecol. 2019;28:4755–69.
Leonard JA, Wayne RK, Wheeler J, Valadez R, Guillén S, Vila C. Ancient DNA evidence for Old World origin of New World dogs. Science. 2002;298:1613–6.
Leonardi M, Librado P, Der Sarkissian C, Schubert M, Alfarhan AH, Alquraishi SA, et al. Evolutionary patterns and processes: lessons from ancient DNA. Syst Biol. 2017;66:e1–e29.
Lichatowich J, Lichatowich JA. Salmon without rivers: a history of the Pacific salmon crisis. Washington, DC: Island Press; 2001.
Lijtmaer DA, Mahler B, Tubaro PL, Dunn P. Hybridization and postzygotic isolation patterns in pigeons and doves. Evolution. 2003;57:1411–8.
Lindqvist C, Rajora OP. Paleogenomics: genome-scale analysis of ancient DNA. Cham: Springer; 2019.
Livezey BC. A phylogenetic analysis of recent anseriform genera using morphological characters. Auk. 1986;103:737–54.
Livezey BC. A phylogenetic analysis and classification of recent dabbling ducks (Tribe Anatini) based on comparative morphology. Auk. 1991;108:471–507.
Livezey BC. Comparative morphometrics of Anas ducks, with particular reference to the Hawaiian duck Anas wyvilliana, Laysan duck A. laysanensis, and Eaton’s Pintail A. eatoni. Wild. 1993;44:75–100.
Loreille O, Orlando L, Patou-Mathis M, Philippe M, Taberlet P, Hänni C. Ancient DNA analysis reveals divergence of the cave bear, Ursus spelaeus, and brown bear, Ursus arctos, lineages. Curr Biol. 2001;11:200–3.
Lowry DB, Hoban S, Kelley JL, Lotterhos KE, Reed LK, Antolin MF, et al. Breaking RAD: an evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol Ecol Resour. 2017;17:142–52.
Mallet J. Hybridization as an invasion of the genome. Trends Ecol Evol. 2005;20:229–37.
Mallet J. Hybrid speciation. Nature. 2007;446:279–83.
Mank JE, Carlson JE, Brittingham MC. A century of hybridization: decreasing genetic distance between American black ducks and mallards. Conserv Genet. 2004;5:395–403.
Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, et al. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 2013;23:1817–28.
McCracken KG, Johnson WP, Sheldon FH. Molecular population genetics, phylogeography, and conservation biology of the mottled duck (Anas fulvigula). Conserv Genet. 2001;2:87–102.
McFarlane SE, Pemberton JM. Detecting the true extent of introgression during anthropogenic hybridization. Trends Ecol Evol. 2019;34(4):315–26.
McKinney GJ, Larson WA, Seeb LW, Seeb JE. RADseq provides unprecedented insights into molecular ecology and evolutionary genetics: comment on Breaking RAD by Lowry et al.(2016). Mol Ecol Resour. 2017;17:356–61.
Miller DB. Social displays of mallard ducks (Anas platyrhynchos): effects of domestication. J Comp Physiol Psychol. 1977;91:221.
Minvielle F, Ito S, Inoue-Murayama M, Mizutani M, Wakasugi N. Brief communication. Genetic analyses of plumage color mutations on the Z chromosome of Japanese quail. J Hered. 2000;91:499–501.
Mitchell KJ, Wood JR, Scofield RP, Llamas B, Cooper A. Ancient mitochondrial genome reveals unsuspected taxonomic affinity of the extinct Chatham duck (Pachyanas chathamica) and resolves divergence times for New Zealand and sub-Antarctic brown teals. Mol Phylogenet Evol. 2014;70:420–8.
Moore WS. Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution. 1995;49:718–26.
Nadachowska-Brzyska K, Burri R, Ellegren H. Footprints of adaptive evolution revealed by whole Z chromosomes haplotypes in flycatchers. Mol Ecol. 2019;28(9):2290–304.
Nadeau NJ, Kawakami T. Population genomics of speciation and admixture. In: Rajora OP, editor. Population genomics: concepts, approaches and applications. Cham: Springer Nature Switzerland AG; 2019. p. 613–53.
Nielsen EE, Bach LA, Kotlicki P. HYBRIDLAB (version 1.0): a program for generating simulated hybrids from population samples. Mol Ecol Notes. 2006;6:971–3.
Nosil P, Schluter D. The genes underlying the process of speciation. Trends Ecol Evol. 2011;26:160–7.
Nosil P, Harmon LJ, Seehausen O. Ecological explanations for (incomplete) speciation. Trends Ecol Evol. 2009;24:145–56.
Omland KE. Examining two standard assumptions of ancestral reconstructions: repeated loss of dichromatism in dabbling ducks (Anatini). Evolution. 1997;51:1636–46.
Orlando L, Gilbert MTP, Willerslev E. Reconstructing ancient genomes and epigenomes. Nat Rev Genet. 2015;16:395.
Orr HA, Masly JP, Presgraves DC. Speciation genes. Curr Opin Genet Dev. 2004;14:675–9.
Ottenburghs J. Multispecies hybridization in birds. Avian Res. 2019;10:20.
Ottenburghs J, Ydenberg RC, Van Hooft P, Van Wieren SE, Prins HH. The Avian Hybrids Project: gathering the scientific literature on avian hybridization. Ibis. 2015;157:892–4.
Owens GL, Samuk K. Adaptive introgression during environmental change can weaken reproductive isolation. Nat Clim Chang. 2020;10:58–62.
Oyler-McCance SJ, Oh KP, Langin KM, Aldridge CL. A field ornithologist’s guide to genomics: practical considerations for ecology and conservation. Auk. 2016;133:626–48.
Palmer RS. Handbook of North American birds. New Haven, CT: Yale University Press; 1976.
Paulke E, Haase E. A comparison of seasonal changes in the concentrations of androgens in the peripheral blood of wild and domestic ducks. Gen Comp Endocrinol. 1978;34:381–90.
Peters JL, Winker K, Millam KC, Lavretsky P, Kulikova I, Wilson RE, et al. Mito-nuclear discord in six congeneric lineages of Holarctic ducks (genus Anas). Mol Ecol. 2014;23:2961–74.
Peters JL, Lavretsky P, DaCosta JM, Bielefeld RR, Feddersen JC, Sorenson MD. Population genomic data delineate conservation units in mottled ducks (Anas fulvigula). Biol Conserv. 2016;203:272–81.
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135.
Phadnis N, Orr HA. A single gene causes both male sterility and segregation distortion in Drosophila hybrids. Science. 2009;323:376–9.
Picardi E, Pesole G. Mitochondrial genomes gleaned from human whole-exome sequencing. Nat Methods. 2012;9:523.
Pont C, Wagner S, Kremer A, Orlando L, Plomion C, Salse J. Paleogenomics: reconstruction of plant evolutionary trajectories from modern and ancient DNA. Genome Biol. 2019;20:29.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Pryke SR. Sex chromosome linkage of mate preference and color signal maintains assortative mating between interbreeding finch morphs. Evolution. 2010;64:1301–10.
Puigcerver M, Sanchez-Donoso I, Vilá C, Sardá-Palomera F, García-Galea E, Rodríguez-Teijeiro JD. Decreased fitness of restocked hybrid quails prevents fast admixture with wild European quails. Biol Conserv. 2014;171:74–81.
Pyle R, Pyle P. The birds of the Hawaiian Islands: occurrence, history, distribution, and status. BP Bishop Museum, Honolulu, HI, USA. Version 2 (January 1, 2017). 2017.
Qiao H, Liu W, Zhang Y, Zhang YY, Li QQ. Genetic admixture accelerates invasion via provisioning rapid adaptive evolution. Mol Ecol. 2019.
Ralls K, Ballou JD, Dudash MR, Eldridge MD, Fenster CB, Lacy RC, et al. Call for a paradigm shift in the genetic management of fragmented populations. Conserv Lett. 2018;11:e12412.
Randi E. Detecting hybridization between wild species and their domesticated relatives. Mol Ecol. 2008;17:285–93.
Rawlence NJ, Kardamaki A, Easton LJ, Tennyson AJ, Scofield RP, Waters JM. Ancient DNA and morphometric analysis reveal extinction and replacement of New Zealand’s unique black swans. Proc R Soc B Biol Sci. 2017;284:20170876.
Rheindt FE, Edwards SV. Genetic introgression: an integral but neglected component of speciation in birds. Auk. 2011;128:620–32.
Rhymer JM. Extinction by hybridization and introgression in anatine ducks. Acta Zoolog Sin. 2006;52(Suppl):583–6.
Rhymer JM, Simberloff D. Extinction by hybridization and introgression. Annu Rev Ecol Syst. 1996;27:83–109.
Rhymer JM, Williams MJ, Braun MJ. Mitochondrial analysis of gene flow between New Zealand mallards (Anas platyrhynchos) and grey ducks (A. superciliosa). Auk. 1994;111:970–8.
Ruegg K, Anderson EC, Boone J, Pouls J, Smith TB. A role for migration-linked genes and genomic islands in divergence of a songbird. Mol Ecol. 2014;23:4757–69.
Rueness EK, Cadahia-Lorenzo L, Hoel K, Søderquist P, Velle G, Asmyhr MG, et al. Assessment of the risks associated with the import and release of hand-reared mallards for hunting purposes. Opinion of the Panel on Alien Organisms and Trade in Endangered Species (CITES) of the Norwegian Scientific Committee for Food Safety, VKM Report 2017:23.
Rundle HD, Nosil P. Ecological speciation. Ecol Lett. 2005;8:336–52.
Russo I-RM, Hoban S, Bloomer P, Kotzé A, Segelbacher G, Rushworth I, et al. ‘Intentional Genetic Manipulation’ as a conservation threat. Conserv Genet Resour. 2018;11:237–47.
Sæther SA, Sætre G-P, Borge T, Wiley C, Svedin N, Andersson G, et al. Sex chromosome-linked species recognition and evolution of reproductive isolation in flycatchers. Science. 2007;318:95–7.
Salzburger W, Baric S, Sturmbauer C. Speciation via introgressive hybridization in East African cichlids? Mol Ecol. 2002;11:619–25.
Sangster G. The application of species criteria in avian taxonomy and its implications for the debate over species concepts. Biol Rev. 2014;89:199–214.
Schaefer NK, Shapiro B, Green RE. AD-LIBS: inferring ancestry across hybrid genomes using low-coverage sequence data. BMC Bioinformatics. 2017;18:203.
Scherer VS, Hilsberg T. Hybridisierung und verwandtschaftsgrade innerhalb der Anatidae – eine systematische un evolutionstheoretische betrachtung. J Ornitol. 1982;123:357–80.
Schluter D. Evidence for ecological speciation and its alternative. Science. 2009;323:737–41.
Schubert M, Ginolhac A, Lindgreen S, Thompson JF, Al-Rasheid KA, Willerslev E, et al. Improving ancient DNA read mapping against modern reference genomes. BMC Genomics. 2012;13:178.
Schumer M, Rosenthal GG, Andolfatto P. How common is homoploid hybrid speciation? Evolution. 2014;68:1553–60.
Seehausen O. Hybridization and adaptive radiation. Trends Ecol Evol. 2004;19:198–207.
Seehausen O. Conservation: losing biodiversity by reverse speciation. Curr Biol. 2006;16:R334–7.
Shapiro B. Pathways to de-extinction: how close can we get to resurrection of an extinct species? Funct Ecol. 2017;31:996–1002.
Skoglund P, Ersmark E, Palkopoulou E, Dalén L. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr Biol. 2015;25:1515–9.
Söderquist P, Gunnarsson G, Elmberg J. Longevity and migration distance differ between wild and hand-reared mallards Anas platyrhynchos in Northern Europe. Eur J Wildl Res. 2013;59:159–66.
Söderquist P, Norrström J, Elmberg J, Guillemain M, Gunnarsson G. Wild mallards have more “Goose-Like” bills than their ancestors: a case of anthropogenic influence? PLoS One. 2014;9:e115143.
Söderquist P, Elmberg J, Gunnarsson G, Thulin C-G, Champagnon J, Guillemain M, et al. Admixture between released and wild game birds: a changing genetic landscape in European mallards (Anas platyrhynchos). Eur J Wildl Res. 2017;63:98.
Souza CA, Murphy N, Villacorta-Rath C, Woodings LN, Ilyushkina I, Hernandez CE, et al. Efficiency of ddRAD target enriched sequencing across spiny rock lobster species (Palinuridae: Jasus). Sci Rep. 2017;7:6781.
Stephens K, Measey J, Reynolds C, Le Roux JJ. Occurrence and extent of hybridisation between the invasive mallard duck and native yellow-billed duck in South Africa. Biol Invasions. 2019:1–15.
Sutter A, Beysard M, Heckel G. Sex-specific clines support incipient speciation in a common European mammal. Heredity. 2013;110:398–404.
Tamisier A. The Camargue: a model of environmental decline. In: Finlayson CM, Hollis GE, Davis TJ, editors. Managing Mediterranean wetlands and their birds. Proceedings of the Symposium of Grado, Italy. Slimbridge: IWRB; 1992. p. 106–8.
Tang H, Coram M, Wang P, Zhu X, Risch N. Reconstructing genetic ancestry blocks in admixed individuals. Am J Hum Genet. 2006;79:1–12.
Tufto J. Domestication and fitness in the wild: a multivariate view. Evolution 2017;71(9):2262–70.
U.S. Fish & Wildlife Service. Recovery plan for Hawaiian waterbirds. Portland, OR: U.S. Fish & Wildlife Service; 2012.
USFWS. Review of captive-reared mallard regulations on shooting preserves – final report. Washington: Division of Migratory Bird Management, US Fish and Wildlife Service; 2013.
Uyehara KJ, Engilis A, Dugger BD. Wetland features that influence occupancy by the endangered Hawaiian duck. Wilson J Ornithol. 2008;120:311–9.
Vähä J-P, Primmer CR. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol Ecol. 2006;15:63–72.
Via S. Natural selection in action during speciation. Proc Natl Acad Sci U S A. 2009;106:9939–46.
vonHoldt BM, Brzeski KE, Wilcove DS, Rutledge LY. Redefining the role of admixture and genomics in species conservation. Conserv Lett. 2018;11:e12371.
Wang H, Vieira FG, Crawford JE, Chu C, Nielsen R. Asian wild rice is a hybrid swarm with extensive gene flow and feralization from domesticated rice. Genome Res. 2017;27:1029–38.
Wang W, Wang Y, Lei F, Liu Y, Wang H, Chen J. Incomplete lineage sorting and introgression in the diversification of Chinese spot-billed ducks and mallards. Curr Zool. 2019;65:589–97.
Waples RS, Drake J. Risk/benefit considerations for marine stock enhancement: a pacific salmon perspective. Stock enhancement and sea ranching: developments, pitfalls and opportunities, 2nd ed. 2004. p. 260–306.
Watanabe T, Mizutani M, Wakana S, Tomita T. Demonstration of the maternal inheritance of avian mitochondrial DNA in chicken-quail hybrids. J Exp Zool. 1985;236:245–7.
Wayne RK, Shaffer HB. Hybridization and endangered species protection in the molecular era. Mol Ecol. 2016;25:2680–9.
Webb WC, Marzluff JM, Omland KE. Random interbreeding between cryptic lineages of the Common Raven: evidence for speciation in reverse. Mol Ecol. 2011;20:2390–402.
Wells CP, Lavretsky P, Sorenson MD, Peters JL, DaCosta JM, Turnbull S, et al. Persistence of an endangered island endemic, an introduced congener, and multiple hybrid swarms across the main Hawaiian Islands. Mol Ecol. 2019;28:5203–16.
Willerslev E, Cooper A. Review paper. Ancient DNA. Proc R Soc B Biol Sci. 2005;272:3–16.
Williams M. The duckshooter’s bag: an understanding of New Zealand’s wetland gamebirds. Wellington: Wetland Press; 1981.
Williams M. The changing relative abundance of grey duck (Anas superciliosa) and mallard (A. platyrhynchos) in New Zealand. Notornis. 2017;64:211–28.
Williams CL, Brust RC, Rhodes OE Jr. Microsatellite polymorphism and genetic structure of Florida mottled duck populations. Condor. 2002;104:424–31.
Williams CL, Brust RC, Fendley TT, Tiller GR, Rhodes OE. A comparison of hybridization between mottled ducks (Anas fulvigula) and mallards (A. platyrhynchos) in Florida and South Carolina using microsatellite DNA analysis. Conserv Genet. 2005a;6:445–53.
Williams CL, Fedynich AM, Pence DB, Olin E, Rhodes J. Evaluation of allozyme and microsatellite variation in Texas and Florida mottled ducks. Condor. 2005b;107:155–61.
Wringe BF, Stanley RR, Jeffery NW, Anderson EC, Bradbury IR. parallel new hybrid: an R package for the parallelization of hybrid detection using new hybrids. Mol Ecol Resour. 2017;17:91–5.
Wu C-I, Ting C-T. Genes and speciation. Nat Rev Genet. 2004;5:114–22.
Wu D, Ding X, Wang S, Wójcik J, Zhang Y, Tokarska M, et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat Ecol Evol. 2018;2:1139–45.
Zink RM, Barrowclough GF. Mitochondrial DNA under siege in avian phylogeography. Mol Ecol. 2008;17:2107–21.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lavretsky, P. (2020). Population Genomics Provides Key Insights into Admixture, Speciation, and Evolution of Closely Related Ducks of the Mallard Complex. In: Hohenlohe, P.A., Rajora, O.P. (eds) Population Genomics: Wildlife. Population Genomics. Springer, Cham. https://doi.org/10.1007/13836_2020_76
Download citation
DOI: https://doi.org/10.1007/13836_2020_76
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63488-9
Online ISBN: 978-3-030-63489-6
eBook Packages: Computer ScienceComputer Science (R0)