
Abstract
The resolution of conservation genetic analyses has been limited until recently due to technological and computational challenges associated with genotyping multiple loci at once. In this review, we focus on how the development of high-throughput genotyping methods have enabled conservation genomics studies of wolves in North America. The gray wolf (Canis lupus) historically had a Holarctic distribution across widely varying environments, yet during the early twentieth century many populations declined due to direct persecution and other anthropogenic disturbances. First, we discuss genetic substructure and adaptive uniqueness among genetically and environmentally defined wolf ecotypes. Second, we focus on the new conservation implications revealed by studies having increased genomic resolution of the dynamics of reintroduced and re-established wolves, specifically Mexican and Pacific Northwest wolves. Mexican wolves, a distinct subspecies of North American wolf that inhabit a small area within the southwestern U.S. and Mexico, remain endangered despite decades since a reintroduction program began. How biologists and management agencies use scientific data to define the historical range of Mexican wolves will be critical to future reintroduction efforts. In the Pacific Northwest, admixture occurs between the distinct and declining coastal wolf ecotype and the more abundant reintroduced interior wolves. If coastal wolves obtain protection, then the Pacific Northwest wolves may also warrant protection. Therefore, more precise policies are needed for the management of admixed populations when one source is protected. We recommend that future conservation efforts should provide full protection for distinct ecotypes, support scientifically rigorous definitions of historical range to inform restoration, and enhance the legal status of admixed populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Until recently, large-scale genome-wide population-level explorations of demography, natural selection, and gene flow have been limited in non-model organisms in part because of the effort and cost of collecting genome-wide single nucleotide polymorphism (SNP) genotyping data. For some non-model systems, the genome of a phylogenetically close species can provide resources such as DNA sequence and gene annotations for addressing questions of evolutionary and conservation importance. For example, the genome sequence of the domestic dog (Canis lupus domesticus), the fifth mammal species with a complete genome (Lindblad-Toh et al. 2005), enabled the early development of a canine SNP microarray containing ~ 127,000 SNPs (Affymetrix v2 Canine GeneChip). This array has been used to examine the genetic diversity and infer origins of domestic dogs (Boyko et al. 2010; vonHoldt et al. 2011), and more recently to study population structure and admixture in canids of North America (NA) and Europe (vonHoldt et al. 2011; Pilot et al. 2014; Schweizer et al. 2016b). The canine SNP genotyping array is based on pre-screening of marker variability in a small panel of canines, which may result in bias when ascertainment is not properly accounted for in studies that compare variability in dogs to wild canids (see vonHoldt et al. 2010). However, such comparisons are not the intent of studies featured in this review. One benefit of genotyping arrays is that they generally have a higher fraction of polymorphic loci than other genomic methods such as RADsEq. Additionally, methods such as sequence capture and whole genome sequencing have been used to identify adaptive variation and patterns of demographic history in NA canids (e.g. Schweizer et al. 2016a; Fan et al. 2016; vonHoldt et al. 2016).
Genome-wide analyses can be applied to new as well as long-standing dilemmas in conservation management. For example, large numbers of loci can improve the power and accuracy of inference concerning demography, local adaptation, inbreeding depression, hybridization, and disease susceptibility (Allendorf et al. 2010; Steiner et al. 2013). Surveys of genome-wide neutral markers enable a more robust reconstruction of the demographic histories of species and populations. Accurate inference of demography can then facilitate more precise predictions of how anthropogenic influences might lead to changes in detrimental and adaptive genetic variation (Bay et al. 2017). Genome-wide data also provide higher resolution for genome scans and quantitative trait loci (QTL) mapping, approaches that identify candidate genes under selection or underlying phenotypic variation, respectively. Furthermore, population genomic approaches can assess detrimental genetic variants that may potentially diminish fitness, long-term viability, or reduce the adaptive potential of populations (e.g. Robinson et al. 2016). Together, genome-wide neutral and non-neutral variation can be used to identify populations and species of conservation concern (for further discussion see Ouborg et al. 2010; Allendorf et al. 2010; Funk et al. 2012; Steiner et al. 2013).
As mentioned above, the development of large-scale genome-level data collection technologies has been instrumental to increased resolution in conservation genomics. Here, we briefly introduce some of the most relevant technologies for conservation genomics and/or those that have been instrumental in the recent study of NA gray wolves (Canis lupus). We refer the interested reader to more detailed reviews elsewhere (Allendorf et al. 2010; McMahon et al. 2014; Shafer et al. 2015; Bernatchez et al. 2017). High-density SNP arrays (e.g., Kranis et al. 2013) are custom DNA microarrays that are capable of genotyping thousands of SNPs from a large sample of individuals within a single experiment. These SNP arrays are relatively inexpensive, and can be used for closely related non-genome-enabled species (e.g., Schweizer et al. 2016b). Some qualifications for SNP arrays use is that they are most efficient in phylogenetically close species, the setup costs in a new organism may be prohibitively expensive, and allelic variation can be biased as a result of ascertainment of SNPs using a small panel of individuals (Nielsen and Signorovitch 2003; Clark et al. 2005; Rosenblum and Novembre 2007; Lachance and Tishkoff 2013; Malomane et al. 2018). Reduced-representation approaches, which use various techniques to reduce genome complexity prior to sequencing, are also appropriate methods for obtaining thousands of SNPs in non-model organisms (Good 2012; Andrews et al. 2016; Lowry et al. 2016; Catchen et al. 2017; McKinney et al. 2017). Some of these methods use restriction enzymes to cut genomic DNA molecules into fragments (restriction-associated DNA sequencing (RAD-Seq) and related protocols (see Andrews et al. 2016)). Another approach to subsample a fraction of positions within the genome is based on sequencing of the transcriptome. Messenger RNA (mRNA) transcripts is isolated from tissues, complementary DNA (cDNA) is generated and subsequently sequenced on a high-throughput platform (RNA-Seq; see Wang et al. 2009; Wilhelm and Landry 2009; Marguerat and Bähler 2009). Another genome reduction method encompasses selective enrichment of the genomic library for particular loci of interest (targeted sequence capture; see Cosart et al. 2011; Jones and Good 2016). A high-quality reference genome for the study species, or a closely related taxon, aids in the custom design of capture probe sets, yet designing targeted capture experiments in non-model species without a reference genome are also possible (see review Jones and Good 2016). Finally, whole-genome sequencing and de novo genome assembly in non-model organisms, which aims to sequence nearly every position within the nuclear and mitochondrial genomes, is now possible due to the recent advances in high-throughput sequencing and bioinformatics (see reviews Ellegren 2014; Fan et al. 2016; Goodwin et al. 2016; vonHoldt et al. 2016). Selecting the most appropriate method from the array of genomic approaches is dependent upon the questions being asked and nontrivial commitments of time and resources available (see review Oyler-McCance et al. 2016).
In this review, we synthesize results from studies on canids produced in the last decade, a period in which the rapid development of genome-wide genotyping approaches has most significantly affected the study of non-model organisms of conservation concern. We focus on two issues related to conservation genomics of NA gray wolves: (1) genetic substructure and adaptive uniqueness; and (2) the genetics of reintroduction and re-establishment of wolves. Within the latter, we focus on two adaptively distinct forms, the Mexican wolf and wolves of the Pacific Northwest. For each issue, we provide a summary of results from relevant studies, then discuss in detail the specific conservation relevance and how genomics has enabled improved resolution of population processes such as gene flow and selection. Our discussion of these genomic studies reveals that current conservation schemes do not provide adequate protection for diverse ecotypes, nor do they provide for full restoration of ecotypes to their historical range. Additionally, we discuss how current conservation policy for and management of admixed and hybrid populations, as identified by genomic analyses, is insufficient and needs a conceptual framework and restoration policy that is consistent with the evolutionary function of hybrids.
Genetic substructure and adaptive uniqueness
Artic wolves: local adaptation
Background
The gray wolf is historically the dominant predator in NA (Mech 1970) and can disperse over long distances averaging 50–100 km and up to several hundred kilometers before establishing territories (Mech 1970; Fritts 1983; Merrill and Mech 2000; Jimenez et al. 2017). Despite these characteristics, populations show striking morphologic and genetic differentiation at a local scale (Carmichael et al. 2007; Musiani et al. 2007; vonHoldt et al. 2011; Pilot et al. 2014; Schweizer et al. 2016a, b). The gray wolf geographic range, which spans from Mexico to the High Arctic in NA, is characterized by strong environmental gradients involving dramatic changes in temperature, precipitation and vegetation (Geffen et al. 2004; Muñoz Fuentes et al. 2009; Schweizer et al. 2016b). The varied environmental gradient may act as drivers for divergent natural selection in wolf populations resulting in patterns of local adaptation. For example, variation in cranial forms have been found to correspond to differences in prey size (Slater et al. 2009). Additionally, coat color varies across NA wolf populations and paler pelage is more common in northern regions (Musiani et al. 2007; Anderson et al. 2009), suggesting a response of coat color in some populations to differences in temperature and thermoregulatory differences among populations.
Initial genetic studies based on a small number of microsatellite loci showed weak patterns of differentiation with distance (Roy et al. 1994). However, ecological variables were not included in assessments of population structure until Geffen et al. (2004) and this analysis showed a substantial effect of climate and habitat on genetic variation. Further studies suggested a correlation between various habitat types, such as tundra and coastal forest, and wolf genetic partitions. The methods used by these studies increased in complexity with technological advances, from microsatellite and SNP genotyping arrays (Carmichael et al. 2007; Musiani et al. 2007; vonHoldt et al. 2011), to fully quantifying environmentally and genetically determined wolf ecotypes using canid SNP and custom capture arrays (Schweizer et al. 2016a, b). The resulting genetic divisions among wolf populations may reflect observed morphologic features related to diet (e.g., dentition, skull robustness and shape), vision (e.g., for open or closed terrain), metabolism, thermal regulation in response to ambient temperature, and locomotion (e.g., for migratory or territorial behavior) suggesting these genetic partitions may define ecological units (“ecotypes”). The most distinct ecotypes are the Mexican, rainforest, and Artic wolves. Inhabiting the arid lands of Southwest U.S. and Mexico, the Mexican wolf is an ecotype that is smaller in size and feeds on prey such as elk and native ungulates (Reed et al. 2006; Newsome et al. 2016). The rainforest wolf inhabits the temperate rainforest regions of British Columbia (Canada) and southeastern Alaska (U.S.) coasts, feeds on salmon and deer (Darimont et al. 2003), and is smaller in body size than other NA wolves, such as the Forest wolf ecotype (Fig. 1). Finally, the caribou feeding Arctic wolf is the largest of the NA wolves (Fig. 1).

Examples of the varying habitat and ecology of wolves in North America. Coastal wolves live in temperate rainforests of southeast Alaska, USA, and British Columbia, Canada, and have diets composed of salmon and black-tailed deer. Forest wolves live in a subarctic climate and prey on elk, moose, and deer. Arctic wolves live in the high tundra of Canada and prey on caribou. Photo credits: coastal wolf, Steve Williamson; coastal habitat, Pixabay creative commons; forest wolves, Daniel Stahler/National Park Service; caribou, Pixabay creative commons; Arctic wolf, Marco Musiani. Orange regions on map indicate current range of the gray wolf. Map based on data from the International Union for Conservation of Nature Red List, http://www.iucnredlist.org
Given evidence showing NA wolves are morphologically and genetically differentiated on a local scale, Schweizer et al. (2016b) used a SNP genotyping array to detect genetic subdivision, then used multiple selection methods to identify outlier SNPs and their nearby genes as candidates involved in local adaptation (Fig. 2, Schweizer et al. 2016b). Using the results of this initial genome scan, Schweizer et al. (2016a) designed a targeted capture array to sequence 1040 candidate genes under selection and associated promotor regions of wolves from the six ecotypes. Also, as a demographic control, 5000 1-kb nongenic neutral regions (see Freedman et al. 2014) were sequenced. The six wolf ecotypes correspond to specific habitats that were environmentally and genetically defined within the study: West Forest, Boreal Forest, Arctic, High Arctic, British Columbia and Atlantic Forest. The genetically defined ecotypes were largely concordant with previous studies (Carmichael et al. 2007; Muñoz Fuentes et al. 2009; vonHoldt et al. 2011). NA wolves (n = 107) were resequenced at these genic and nongenic regions, and patterns of genetic variability within and among ecotypes were used to detect selective sweeps using Sweed (Pavlidis et al. 2013) and diversifying selection using BayeScan v2.1 (Foll and Gaggiotti 2008). Additionally, data for multiple environmental variables summarizing precipitation, temperature, and vegetation were extracted for each individual wolf’s location and used with genetic data to test for environmentally correlated selection using Bayenv (Coop et al. 2010). Genes within a 6-kb buffer on either side of outlier SNPs were identified using the Ensembl annotation gene set (CanFam3.1), and Ensembl’s Variant Effect Predictor (VEP) pipeline v77 (McLaren et al. 2010) was used to identify and annotate functional variants within genic regions. Lists of outlier genes were tested for enrichment of Gene Ontology (GO) categories using gProfileR (Reimand et al. 2007, 2011).

Environmentally driven functional variation among North American wolf populations. a The primary source of differentiation in North American wolves is related to habitat rather than distance or topographic boundaries, as determined by varied population structure and habitat classification methods. Sampling locations of wolves are shown on maps of annual precipitation and mean diurnal temperature range; these environmental variables were ranked highly important in Random Forest analysis (see Schweizer et al. 2016b). b Wolf ecotypes show evidence of enivornmentally correlated selection on non-synonymous SNPs. Each plot shows the mean reference allele frequency in each ecotype versus the mean value of the environmental variable the SNP was an outlier for in Bayenv analysis. Arctic and High Arctic populations are often at one end of the distribution of allele and environmental variable values. See key for ecotype designations. Figure reproduced from Schweizer et al. (2016a, b)
Schweizer et al. (2016b) found patterns of selection among ecotypes for genes related to morphology, vision, metabolism, and thermoregulation. Using Sweed, the authors identified regions with the genetic signatures of selective sweeps in each wolf ecotype. Furthermore, the authors found several genes containing putatively functional variants (either non-synonymous variants or variants in predicted transcription factor binding sites) that varied significantly with environmental variables quantifying precipitation, temperature, and vegetation (Fig. 2B). These candidate genes are thought to relate to olfactory (e.g. OR5B17), vision and hearing (e.g. PCDH15), pigmentation and immunity (e.g. CBD103), or metabolism-related (e.g. LIPG) functions in wolves (Fig. 2B; see Schweizer et al. 2016a for further discussion of gene function). Bayescan identified the fewest genes under selection, a result that may reflect the conservative model by which Bayescan identifies outliers. Nevertheless, there was relatively high overlap between significant genes with a P-value ≤ 0.05 for all selection tests (Sweed, Bayescan and Bayenv). Arctic and High Arctic wolves had the highest numbers of total candidate genes, microRNA categories (implicated in post-transcriptional regulation), and significantly enriched gene ontology (GO) categories, and the highest number of unique candidate genes (those not seen in any other ecotype). This result was not likely a reflection of differences in demographic history, since genetic variation in neutral regions was used to control for differences in population history for each of the selection tests (Schweizer et al. 2016a). Importantly, Schweizer et al. (2016a) could only confirm about 35% of genes identified by the SNP genotype array studies, suggesting a high rate of false positives in SNP tagging studies, or that other categories of DNA changes that were not assayed are experiencing selection. The high false positive rate implies that simple “tagging” SNP surveys where selection is inferred on nearby genes should be interpreted with caution unless followed by resequencing studies. Finally, this study found evidence of selection in promotor regions, implicating them in local adaptation as well, and suggesting again the importance of resequencing of both exons and flanking regions implicated in SNP tagging studies.
An important consideration in assessing adaptive potential in non-model organisms is which databases are appropriate for assigning putative function to genes or mutations. For example, Schweizer et al. (2016a) used existing gene annotation databases developed for the domestic dog to infer function in wolves. Given that the wolf and the dog are closely related (0.1% sequence divergence; Freedman et al. 2014), it is reasonable to assume similar gene function. Additionally, many databases, such as gProfiler for gene ontology enrichment (Reimand et al. 2007, 2011) and Ensembl’s Variant Effect Predictor for annotating functional effects of mutations (McLaren et al. 2010) develop predictions based on coding sequence similarity, which is very high between dog and wolf. The list of species supported by these databases is continually being updated, therefore increasing the likelihood that a non-model study species will have databases for a closely related species available.
Conservation implications
Several lines of evidence indicate that High Arctic and Arctic wolves may have evolved highly specific adaptations and regulatory responses to survive in their environment. First, the High Arctic and Arctic ecotypes exhibited a large number of significant GO-related categories (i.e. GO, KEGG pathway, Human Phenotype) and the greatest number of unique outlier genes in selective sweep regions (Schweizer et al. 2016a). Second, these two ecotypes were found to have the large number of significantly enriched microRNA categories. MicroRNAs are involved in post-transcriptional regulation, and have been implicated in adipocyte differentiation and extreme environment adaptation in several species (Griffiths-Jones 2004; Zaragosi et al. 2011; Hilton et al. 2013; Wu et al. 2013; Storey 2015). Third, positive selection was detected in Arctic and High Arctic wolves on genes influencing vision, immunity, pigmentation and metabolism. However, more thorough testing should be completed to verify and further develop these hypotheses; yet, these results suggest that Arctic and High Arctic wolves have adapted uniquely to the extreme environment in which they live.
The Arctic and High Arctic wolf ecotypes are threatened by the progressive loss of their main habitat, the tundra, which may disappear by the end of this century (Mech 2004; Gilg et al. 2012; Mahlstein and Knutti 2012). Three conceivable responses to this threat are: (1) extinction of the ecotype; (2) adaptation via standing variation; and (3) adaptive admixture of genes from wolves immigrating from boreal or more temperate forests. Wolves may begin to den at higher latitudes as the tree lines shift northward with changing climate (Grace et al. 2002; Heard and Williams 2011). Conservation measures to combat population decrease might allow standing genetic variation to persist and increase the likelihood that a rapid adaptive response would allow this ecotype to survive despite their changing habitat. However, a wait-and-see approach might be best for the short term, with the hope of an adaptive response from within Arctic populations. If the Arctic population begins a decline due to stress associated with a disappearing Tundra habitat and the projected associated shifts in the distribution of vegetation and associated prey species (Brotton and Wall 1997; Mech 2005), wolves from the south might migrate naturally following the northern advance of boreal habitat. These wolves may rescue the Arctic population, but further intermingling of wolf types may result in an eventual loss of regional differentiation. Unique adaptations that now exist in Arctic wolf populations might be lost if admixture and selection favor an array of variants from southern populations. Identification of the factors with greatest influence on the contemporary genetics of Arctic wolves may be particularly useful to inform their conservation in a changing environment.
Managers can use information on the extent and nature of local adaptation to inform conservation actions to preserve the evolutionary potential and adaptive capacity of populations. For example, the use of the relative number of genes and number of significantly enriched top-level GO categories summarizing those genes under selection could potentially add to metrics for ranking conservation priorities (Bonin et al. 2007; Gebremedhin et al. 2009). Similar to species diversity indices, the number of genes under selection provides a numerical ranking of adaptive diversity of each population. Therefore, the populations with the greatest number of unique genes, and possibly GO categories, could be argued to exhibit the greatest adaptive diversity and, therefore, deserve the greatest priority for conservation. These simple indices represent genome-wide measures of adaptive divergence that can readily be incorporated into conservation schemes to preserve species as they encounter diverse stressors imposed by changing environments. Of course, GO categories are related and hierarchical, and the number of genes under selection may be influenced by factors other than adaptive potential (such as genetic linkage or demographic history), so further testing of the robustness of this approach would be necessary before extensive use for conservation management. Note that environmental factors and other means to formulate genetic indices of adaptation have been discussed (Razgour et al. 2017).
The genetics of reintroduction and re-establishment of wolves
Mexican wolves: historical range delimitation
Background
The Mexican wolf (Canis lupus baileyi) was once spread throughout much of Mexico and southwestern U.S., but was extirpated in the wild by the 1980s (Shaw 1983). Both morphologic (Bogan and Melhop 1983) and genetic evidence support the Mexican wolf as a subspecies of gray wolf (Wayne et al. 1992; Vilà et al. 1999; vonHoldt et al. 2011, 2016; Fan et al. 2016). This subspecies is the most genetically divergent wolf in NA (Wayne et al. 1992; Vilà et al. 1999; vonHoldt et al. 2011, 2016; Fan et al. 2016) with the lineage likely resulting from one of the earliest migrations of Canis lupus into the New World (Leonard et al. 2005; vonHoldt et al. 2011; Fan et al. 2016). A reintroduction program, which re-established the Mexican wolf populations from captive individuals (Hedrick et al. 1997), was initiated by the U.S. Fish and Wildlife Service (USFWS) in 1998. However, this program and the recovery of the Mexican wolf have been plagued by several recent controversies.
The designation of the Mexican wolf as a separate subspecies has been questioned for several reasons (Cronin et al. 2014, 2015; Fredrickson et al. 2015). First, the extant population was founded by 3 captive lineages (Hedrick et al. 1997) and, although admittedly improbable based on previously published genetic data (Moreno et al. 1996; Hedrick et al. 1997), Cronin et al. (2014) suggested that the Mexican wolf founders may have included dog or coyote ancestry due to previous admixture events. However, genetic analysis of the 3 captive lineages using microsatellite and mtDNA analysis (Hedrick et al. 1997) found an absence of dog admixture (Moreno et al. 1996; Hedrick et al. 1997). This conclusion was subsequent confirmed with genomic data (Fitak 2014, Fan et al. 2016). Second, Cronin et al. (2014) argued that subspecies designation is of questionable validity because Mexican wolves share haplotypes with wolves in other areas and with coyotes (Leonard et al. 2005; Hailer and Leonard 2008). Mexican wolves were historically and are currently part of a monophyletic clade consisting of the mitochondrial haplotype of extant Mexican wolves and closely-related haplotypes found in museum specimens (referred to as the “southern clade”) that extended further north into the southern Rockies and Greater Plains (Leonard et al. 2005). The wide distribution of the southern clade implies that gene flow was naturally extensive across the recognized limit of the subspecies and that Mexican wolves may have admixed with other wolf populations to the north. Generally with highly mobile species, large zones of intergradation may characterize subspecies boundaries (Schweizer et al. 2016b) and admixed individuals within this zone might enhance the adaptive potential of reintroduced stocks (Hedrick 2013). Contrary to Cronin et al. (2014), although Mexican wolves are geographically and genetically discrete now, it might be more biologically appropriate to encourage a wider geographical range in the reintroduction program (see discussion in Hendricks et al. 2017). Finally, despite these arguments against subspecies statue, the Mexican wolf, which was previously listed under the umbrella of gray wolf at the species level, was recently reclassified by the USFWS with a subspecies designation allowing it to have an independent endangered status from other populations of gray wolves (U.S. Fish and Wildlife Service).
A second controversy concerning the Mexican wolf recovery program involves the legal framework for defining a historical range and its use to inform reintroduction. As of early 2018, the reintroduced population hovered at ~ 110 individuals despite the continued release of captive wolves (http://www.fws.gov/southwest/es/mexicanwolf/). This number stands in stark contrast to the 1700 individuals of other C. lupus subspecies in Wyoming (U.S.), Idaho (U.S.), and Montana (U.S.; http://www.fws.gov/mountain-prairie/es/grayWolf.php), which is in part a result of the successful reintroduction of gray wolves to Yellowstone National Park (U.S.) and central Idaho (U.S.). Supplementary recovery locations for the Mexican wolf may be crucial for successful re-establishment of this predator to arid lands of the U.S. (Smith et al. 2003). However, additional locations in the southern portion of their range (i.e., Mexico) are limited due to anthropogenic disturbance of habitat once occupied by Mexicans wolves (Araiza et al. 2012) and the underestimation of the defined historical geographic range limits (Hendricks et al. 2016). Although the validity of both issues has been questioned (Heffelfinger et al. 2017a, b), they nonetheless may be factors limiting the success of this recovery program.
The USFWS-defined historical range for the Mexican wolf may be underestimated as evidenced by several factors. The delineation of the current historical range is based on the previously accepted range plus an arbitrary 200-mile northward extension (dashed line in Fig. 3; Parsons 1996). This historical range was determined by species delimitations based on traditional morphological analysis of a relatively small number of historical specimens that post-date the period of time when the subspecies was already in decline (Young and Goldman 1944; Shaw 1983; Bogan and Melhop 1983; Nowak 1995). More modern and precise methods of determining species delimitation using genetics have shown that the genetic structure of NA gray wolves is strongly influenced by their habitat distribution (see Section “Ecological units in North American wolves”; Geffen et al. 2004; Pilot et al. 2006, 2010; Carmichael et al. 2007; Musiani et al. 2007; Koblmüller et al. 2009; Muñoz Fuentes et al. 2009; vonHoldt et al. 2011; Stronen et al. 2014; Schweizer et al. 2016a, b). The Mexican wolf represents a physically smaller form inhabiting more arid ecosystems (Nowak 1995) and has DNA haplotypes belonging to the “southern clade” (Leonard et al. 2005). This haplotype has been found well outside of the topologically-defined range delineation (Leonard et al. 2005), consistent with a larger historical geographic range. Furthermore, wolves often exhibit natal habitat homing, whereby they disperse over large distances until they encounter habitats with a similar prey base and context to their natal habitat (Geffen et al. 2004). Ecologically suitable habitat exists outside of the USFWS-defined historical range (Carroll et al. 2014). However, Mexican wolves have not been allowed to use any lands outside of their reintroduction sites due to the limits of the previously defined historical range and unsuitable of previously habitable lands.

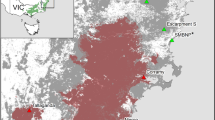
a Comparison of species distribution model and previously defined historical range of the Mexican wolves (Canis lupus baileyi). Maxent modeling identified areas with suitable abiotic conditions only (shades of red). Areas unsuitable due to modern human habitat alterations are shown in blue. Differences between the distribution of suitable habitat and the previously defined historical range (dashed lines) may represent inaccuracies in the previously defined historical range, which were verified through historical location records (gray circles) and new genetic data (green circle). b Genealogical species distribution models of Mexican wolves (gray circles) and closely related (“southern clade” lineage) wolves (yellow circles). Areas unsuitable due to modern human habitat alterations are shown in blue. Figure reproduced from Hendricks et al. (2016)
Hendricks et al. (2016) used a three-tiered approach that incorporated morphological, ecological, and molecular traits to more fully quantify and estimate the historical range of the Mexican wolf. The authors created a species distribution model in MaxEnt (Phillips et al. 2006) using presence-only location data to predict a suitable habitat. Previously published data revealed that one historical specimen from southern California, which was classified as another subspecies of wolf (Southern Rocky Mountain or C. l. youngi; Grinnell et al. 1937), was maternally of Mexican wolf ancestry and was captured within an area similar to Mexican wolf habitat. To determine the nuclear ancestry of this southern California specimen and, therefore, its subspecies assignment, Hendricks and colleagues used previously published data from the Affymetrix Canine SNP mapping array (vonHoldt et al. 2011) to discover SNPs that are highly differentiated between Northern Rocky Mountain wolves and Mexican wolves. These SNPs were genotyped in the museum specimen, which was found to have a diagnostic Mexican wolf mtDNA haplotype and SNP markers suggesting a high proportion of Mexican wolf ancestry (Hendricks et al. 2016). This specimen, plus other specimens that were previously classified as belonging to the “southern clade” that existed outside of the historical range (Leonard et al. 2005), and all verified Mexican wolf individuals within the historical range were used to produce a genealogically-based distribution model. Both topologically and genealogically-based models also identified geographic areas that should be considered high priority for continued reintroduction efforts since human density and associated disturbances are likely to have minimal impact on reintroduced wolves in those areas (Fig. 3a, b).
Based on the multi-trait data set and topological distribution model, Hendricks et al. (2016) showed that the historical range of Mexican wolves likely extended beyond the boundary currently recognized by the USFWS (Parsons 1996). First, there was historically a wide distribution of the “southern clade” in the American West (Leonard et al. 2005), revealing that individuals with Mexican wolf ancestry coexisted with Northern Rocky Mountain wolves (C. l. irremotus) outside of the defined Mexican wolf historical range and, therefore, these areas may represent appropriate habitat for both wolf ecotypes. Second, although the southern California specimen may have been a migrant rather than resident wolf, the ecological models identify this specimen’s locality as suitable habitat under current climate conditions (green circle on Fig. 3a). Additionally, since the southern California specimen was collected prior to extirpation in 1922 (Grinnell et al. 1937), yet shows Mexican wolf ancestry, the habitat of the sampling locality was likely historically suitable for Mexican wolves. Third, the ecological models also reveal that large portions of the historical range in Mexico are currently unsuitable due to human activity (blue areas in Fig. 3a, b).
Conservation implications
Despite a recent ruling that extends the Mexican Wolf Experimental Population Area (US Fish and Wildlife Service 2015), the USFWS prohibits natural reintroduction and expansion of Mexican wolves to areas in northern Arizona, New Mexico, Southern California, and Western Texas. This prohibition limits the movement of a subspecies that had naturally occurred across much of the southwestern U.S. and inhibits admixture for the foreseeable future.
The establishment of populations at or beyond the northern, rather than the southern, limit of the historical range may be an appropriate plan to increase recovery success and metapopulation resilience (Carroll et al. 2014). First, previous studies have suggested that the probability of anthropogenic wolf mortality is high within Mexico (Araiza et al. 2012). This study found only a few possible reintroductions sites due to restricted overlap between suitable habitat for wolves and area with limited interaction with humans. Second, increasing aridity in the southwestern U.S. due to climate change is projected (Notaro et al. 2012), and therefore, more northern habitat may be able to sustain wolf populations and their prey base, and increase metapopulation resilience into the future (Carroll et al. 2014). Third, connectively of U.S. and Mexican populations may be limited by physical barriers at international borders (Peters et al. 2018), which would likely decrease gene flow and genetic connectivity between recovery areas if wolves are reintroduced into Mexico as proposed. Anthropogenic factors leading to reduced dispersal dynamics should therefore be mitigated due to the potential loss of genetic variation in small effective populations (see review Frankham 2005). Additionally, more appropriate reintroduction site may exist that are not currently being considered. For example, the Grand Canyon Ecoregion has suitable habitat, low anthropogenic activity, connectivity with other suitable areas, and protected habitat within a U.S. National Park (Sneed 2001; Carroll et al. 2006, 2014).
Given the close proximity of Mexican wolf habitats to a southern-expanding population of Northern Rocky Mountain wolves now in the U.S., admixture zones may develop between these subspecies. Such admixture occurred historically as shown by genetic analysis (Leonard et al. 2005). Importantly, admixture may lead to enhanced opportunities for selection to craft appropriate phenotypes resilient to future environmental challenges, such as new diseases and climate change (Hamilton and Miller 2016). Although mutation plays an important role in evolutionary change, adaptive variation may also derive from crosses with related subspecies or species, called adaptive introgression. Evidence for adaptive introgression is found in the Great Lakes hybrid zone between gray wolves and coyotes (vonHoldt et al. 2016). Further, genomic analysis of data from the canine SNP microarray as well as complete genome sequences, the Mexican wolves was found to have the lowest genetic variation of any wolves indicating that there is limited standing variation for future adaptation (vonHoldt et al. 2011; Fan et al. 2016). Allowing for northern migration of Mexican wolves, may restore natural connectivity that historically existed, which may lead to increase in genetic diversity and adaptive potential (see discussion in Wayne and Shaffer 2016).
Defining the historical range of a taxon is critical for estimating a wide diversity of biological factors that may help inform conservation efforts, such as extinction probabilities, ecological requirements, and species interactions. An underestimation of historical range could, therefore, lead to prolonging species endangerment and increase the expense of recovery efforts. The geographic distribution of specimens assigned by modern morphologic techniques, combined with those assigned by phylogenetic analysis of historical specimens, defines a range of environments inhabited historically by the subspecies. This approach provides direct insight into the distribution of lineages defining the historical legacy of the Mexican wolf and captures the likely distribution it occupied prior to dramatic decline over the last century.
Pacific Northwest wolves: admixture between ecotypes
Background
Coastal wolves are a phenotypically distinct wolf ecotype that is found in the coastal habitats of British Columbia (BC) and the Alexander Archipelago in southeast Alaska (AK). Mitochondrial DNA sequencing, microsatellite loci, and SNP have shown that these coastal wolves are genetically differentiated from wolves interior to the Pacific coastal mountain ranges of NA (Weckworth et al. 2005; Muñoz Fuentes et al. 2009; vonHoldt et al. 2011; Stronen et al. 2014; Schweizer et al. 2016b). Despite this genetic evidence, the subspecies designation of the Alexander Archipelago wolves (C. l. ligoni) has been debated (Cronin et al. 2014, 2015; Weckworth et al. 2015). In 2015, the Alexander Archipelago wolves were considered for protection under the U.S. Endangered Species Act (ESA) as a result of a 60% decline in the population over one year due to human mediated habitat alteration (Jewell et al. 2015). Although ultimately not listed, this wolf population still deserves consideration for protection as a unique ecotype not found outside this area (Muñoz Fuentes et al. 2009; Schweizer et al. 2016a, b).
By the mid-1930s, wolves were extirpated in the U.S. portion of the Pacific Northwest (PNW) region of NA (Bailey 1936; Verts and Carraway 1998). Wolves have recently naturally re-colonizing the PNW, including the U.S states of Oregon (OR) and Washington (WA). Given the long distance dispersal capabilities of wolves (Mech 1970; Fritts 1983; Merrill and Mech 2000; Jimenez et al. 2017), these re-established wolves in OR and WA are likely to be migrants from adjacent wolf populations rather than from released privately-held captive wolves in each state. These adjacent populations consist of two ecotypes, the coastal ecotype and the Northern Rocky Mountain forest ecotype. If Alexander Archipelago wolves attain protected status under ESA, and if coastal ancestry is found within WA and/or OR, the management of wolves of the PNW is not straightforward. If admixture is a result of natural patterns of wolf dispersal, historical genetic connectivity is preserved, and adaptive potential is maintained, then protection status should be considered for the admixed population (Wayne and Shaffer 2016).
To assess the genetic source of the re-established population and their suitability to areas of reintroduction, Hendricks et al. (2018) used three complementary approaches. First, the authors sequenced a portion of the mtDNA control region in individuals from Washington, Oregon, and surrounding populations to establish maternal lineages. Second, the authors obtained single nucleotide polymorphisms (SNPs) through targeted DNA capture (Schweizer et al. 2016a) to estimate local population structure, ancestry, and relatedness among individuals. Third, the authors used ecological niche models based on climate predictors to assess habitat preference of re-established wolf packs in the PNW region. The ecological niche models identified appropriate habitat for the NRM and coastal wolf ecotypes. Finally, the authors mapped centroid locations of existing WA and OR packs as of 2015 to assess potential genetic barriers associated with environmental differences.
Hendricks et al. (2018) report the first cases of admixture between coastal and NRM wolves in the contiguous U.S. Analyses with both mitochondrial and nuclear DNA markers revealed that the Oregon population shares ancestry with NRM forest wolves only (Fig. 4a, b). However, the WA individuals have a more complex ancestry with some individuals of MT ancestry only and several other individuals with admixed NRM and coastal ancestry (Fig. 4a, b). For example, sample WAWedge8 with coastal mtDNA ancestry showed admixed nuclear ancestry of 53% Alberta, 35% coastal and 11% Montana (Hendricks et al. 2018). Ecological niche modelling of NRM and coastal wolf distributions revealed that the states of WA and OR contain environments suitable for both ecotypes (Fig. 4c). Although wolf packs have established in both environmental types, only one pack exists in the more western, coastal habitat. Furthermore, one wolf pack, containing an admixed individual, created a territory in an area deemed less suitable environment by the models for both the coastal and NRM populations, implying that admixed individuals might be well-suited to establish in these areas.

Population genomics of wolves in the Pacific Northwest. a Distribution of mtDNA control region haplotypes, with size of pie charts indicating relative sampling size and colored proportional to abundance of six haplotypes (see key). b Population assignment at K = 2 to K = 5 for 75 unrelated individuals, as determined by running Admixture on a set of 18,508 non-genic LD-pruned SNPs. The lowest cross-validation error rate occurred at K = 3, which shows the naturally re-established Montana population, the reintroduced Yellowstone National Park and Idaho population, and the coastal population (Alaska and British Columbia). Higher values of K are also biologically meaningful and therefore shown. BC: British Columbia. c Ecological niche model for coastal and interior wolves generated from MaxEnt. Warmer and cooler colors indicate greater habitat suitability for interior and coastal wolves, respectively. Figure reproduced from Hendricks et al. (2018)
Conservation implications
Wolf packs in WA that have a dominant coastal ancestry should be a priority for conservation given their unique evolutionary heritage and adaptations. Furthermore, continued migration from coastal rainforest and NRM forest source populations into WA and OR may benefit the Pacific Northwest population for several reasons. Wolves that migrate into the PNW may continue to add to the existing genetic diversity in the region. The addition of unrelated genetically diverse migrants who subsequently mate would help avoid inbreeding. If inbreeding does occur, it can lead to the expression of deleterious recessive alleles and cause inbreeding depression as shown in Scandinavian and Isle Royale wolves (Fredrickson et al. 2007; Räikkönen et al. 2009). Continued migration from adjacent areas into the PNW may also decrease the likelihood of wolf hybridization with coyotes or dogs. In the PNW, a combination of multiple factors, such as individual dispersing wolves, low wolf density populations, and the presence of coyotes, may lead to an increased likelihood of coyote-wolf hybridization (see vonHoldt et al. 2011). However, maintaining high wolf density and intact pack structure may decrease the likelihood of wolf-coyote and/or wolf-dog hybridization. If humans encourage continued wolf migration and allow higher wolf density, particularly in western WA, wolves may provide ecosystem and human services such as regulating prey abundance, providing carrion for use by other species in the community, and increasing ecotourism that benefits local economies (Smith et al. 2003). Finally, migration from the coastal population may aid in the preservation of adaptations for the coastal environment and restore historical connectively of the PNW population to its surrounding areas.
Given that the PNW population has admixed ancestry, with coastal influences apparent in Washington wolves, the admixed individuals/populations qualify for protection according to the decision tree criteria presented by Wayne and Shafer (2016). First, this admixture event is not due to recent anthropogenic influences, but has resulted from natural patterns of wolf dispersal between two native populations. Second, although not explicitly tested, these admixed individuals are likely ecological surrogates for the declining coastal wolves and likely do not function differently than native populations. Third, the healthy coastal habitats along western Washington (see Fig. 4c) may select for alleles unique to coastal wolves while simultaneously decreasing the genomic contribution from the NRM (non-endangered) wolf. Furthermore, admixed wolves in Washington may be a southern genetic refugium for coastal wolf ecotype if the populations in British Columbia were to decline or be genetically swamped by inland ancestry. Therefore, the natural expansion and protection of the coastal wolves in the contiguous U.S. should be an emphasis of wolf management in the PNW in order to restore ecological processes, and enable the evolutionary process for adaptation to coastal environments.
Summary and conclusions
The age of genomics is enabling scientists and management agencies to reformulate conservation goals in light of evolutionary and ecological principles. Here, we explore current issues in conservation by presenting three case examples of genomic studies of NA canids. First, distinct ecotypes, defined by environmental and genomic data, and exhibiting evidence of adaptive potential should not be excluded for consideration of full protection under regulatory legislation such as the ESA. In the case of NA canids, Arctic and High Arctic wolves have been identified as harboring unique candidate genes under selection and significantly enriched GO and microRNA categories suggesting adaptive diversity to their extreme environment. In general, managers can inform conservation actions to maintain the evolutionary potential and adaptive capacity of populations by using information on the extent and nature of local adaptation.
Second, much like the characterization of subspecies, taxonomic units, or ecotypes, historical distributions are most accurate when defined using the power of multiple data types. Further, approaches that use strict definitions of ranges, especially ranges based on descriptions of a few specimens, likely underestimate or misrepresent the fluidity of species boundaries. Additionally, historical range may be less important when considering reintroduction sites given future climate change predictions are shifting habitat suitability for many species. As discussed above, the Mexican wolf recovery program exemplifies some of these issues. Confounding and conflicting interpretations of scientific evidence, with regards to defining reintroduction sites within and outside of the USFWS-defined historical range, have hindered recovery. In this case, there is evidence for possible reintroduction sites north of the currently defined historical range that should be fully characterized with regard to prey abundance. These additional reintroduction sites would also allow for the restoration of demographic processes, such as admixture and potentially enhance the evolutionary potential of ecotypes (Hendricks et al. 2016). The combination of phylogenetic, morphometric, and ecological methods should be used to rigorously define historical range and inform the restoration of highly endangered populations.
Third, admixed populations require case-by-case evaluation (Allendorf et al. 2001) using evolutionary principles and guidelines as those suggested by recent reviews and commentaries (Arnold 2016; Wayne and Shaffer 2016; vonHoldt et al. 2017). For example, wolves of the PNW have recently been shown to be an admixed population consisting of coastal and NRM wolf ecotypes (Hendricks et al. 2018). Currently, these admixed populations are not protected under the ESA. If coastal wolves receive protection under the ESA, then the naturally reestablished wolves of PNW may warrant protection as well. Given that the PNW wolf populations have coastal ancestry and their probable adaptations to the coastal habitats, they may likely restore a missing role to the ecosystem of the U.S. portions of the PNW. This provides a case example of where more precise policies and legal verbiage are needed for the management of admixed populations, particularly when one source population is protected.
Here, we discuss several examples of how genomics has illuminated adaptive uniqueness and, therefore, identify new scientific challenges to conservation issues of NA canids. Specifically, we highlight issues concerning ecotype definition and preservation of adaptive capacity, historical range delimitations, and the legal handling of admixed populations. We hope that these case studies provide evidence to further promote an integrated research-to-application framework with the goal of bridging the gap between conservation, genomics, and legal implementation of the best available science.
References
Allendorf FW, Leary RF, Spruell P, Wenburg JK (2001) The problems with hybrids: setting conservation guidelines. Trends Ecol Evol 16:613–622. https://doi.org/10.1016/S0169-5347(01)02290-X
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709. https://doi.org/10.1038/nrg2844
Anderson TM, vonHoldt BM, Candille SI et al (2009) Molecular and evolutionary history of melanism in North American Gray Wolves. Science 323:1339–1343. https://doi.org/10.1126/science.1165448
Andrews KR, Good JM, Miller MR et al (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17:81–92. https://doi.org/10.1038/nrg.2015.28
Araiza M, Carrillo L, List R et al (2012) Consensus on criteria for potential areas for wolf reintroduction in Mexico. Conserv Biol 26:630–637. https://doi.org/10.1111/j.1523-1739.2012.01888.x
Arnold ML (2016) Divergence with genetic exchange. Oxford University Press, Oxford
Bay RA, Rose N, Barrett R et al (2017) predicting responses to contemporary environmental change using evolutionary response architectures. Am Nat 189:463–473. https://doi.org/10.1086/691233
Bailey V (1936) The Mammals and Life Zones of Oregon. North Am Fauna. https://doi.org/10.3996/nafa.55.0001
Bernatchez L, Wellenreuther M, Araneda C et al (2017) Harnessing the power of genomics to secure the future of seafood. Trends Ecol Evol 32:665–680. https://doi.org/10.1016/j.tree.2017.06.010
Bogan MA, Melhop P (1983) Systematic relationships of gray wolves (Canis lupus) in southwestern North America. Occas Pap Mus Southwest Biol 1:21
Bonin A, Nicole F, Prompanon F et al (2007) Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol 21:697–708. https://doi.org/10.1111/j.1523-1739.2007.00685.x
Boyko AR, Quignon P, Li L et al (2010) A simple genetic architecture underlies morphological variation in dogs. Plos Biol 8:e1000451. https://doi.org/10.1371/journal.pbio.1000451
Brotton J, Wall G (1997) Climate change and the Bathurst caribou herd in the northwest territories. Canada Climatic Change 35:35–52. https://doi.org/10.1023/A:1005313315265
Carmichael LE, Krizan J, Nagy JA et al (2007) Historical and ecological determinants of genetic structure in arctic canids. Mol Ecol 16:3466–3483. https://doi.org/10.1111/j.1365-294X.2007.03381.x
Carroll C, Fredrickson RJ, Lacy RC (2014) Developing metapopulation connectivity criteria from genetic and habitat data to recover the endangered Mexican wolf. Conserv Biol 28:76–86. https://doi.org/10.1111/cobi.12156
Carroll C, Phillips MK, Lopez-Gonzalez CA, Schumaker NH (2006) Defining recovery goals and strategies for endangered species: the wolf as a case study. BioSci 56:25–37. https://doi.org/10.1641/0006-3568(2006)056[0025:DRGASF]2.0.CO;2
Catchen JM, Hohenlohe PA, Bernatchez L et al (2017) Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol Ecol Resour 17:362–365. https://doi.org/10.1111/1755-0998.12669
Clark AG, Hubisz MJ, Bustamante CD et al (2005) Ascertainment bias in studies of human genome-wide polymorphism. Genome Res 15:1496–1502. https://doi.org/10.1101/gr.4107905
Coop G, Witonsky D, Di Rienzo A, Pritchard JK (2010) Using environmental correlations to identify loci underlying local adaptation. Genetics 185:1411–1423. https://doi.org/10.1534/genetics.110.114819
Cosart T, Beja-Pereira A, Chen S et al (2011) Exome-wide DNA capture and next generation sequencing in domestic and wild species. BMC Genom 12:347. https://doi.org/10.1186/1471-2164-12-347
Cronin MA, Canovas A, Bannasch DL et al (2014) Single nucleotide polymorphism (SNP) variation of wolves (Canis lupus) in Southeast Alaska and comparison with wolves, dogs, and coyotes in North America. J Hered 106:26–36. https://doi.org/10.1093/jhered/esu075
Cronin MA, Cánovas A, Bannasch DL , Fredrickson et al (2015) Wolf subspecies: reply to Weckworth et al. J Hered 106:417–419. https://doi.org/10.1093/jhered/esv029
Darimont CT, Reimchen TE, Paquet PC (2003) Foraging behaviour by gray wolves on salmon streams in coastal British Columbia. Can J Zool 81:349–353. https://doi.org/10.1139/z02-246
Ellegren H (2014) Genome sequencing and population genomics in non-model organisms. Trends Ecol Evol 29:51–63. https://doi.org/10.1016/j.tree.2013.09.008
Fan Z, Silva P, Gronau I et al (2016) Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res 26:163–173. https://doi.org/10.1101/gr.197517.115
Fitak RR (2014) Conservation genomics of the endangered Mexican wolf and de novo SNP marker development in pumas using next-generation sequencing
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993. https://doi.org/10.1534/genetics.108.092221
Frankham R (2005) Genetics and extinction. Biol Cons 126:131–140. https://doi.org/10.1016/j.biocon.2005.05.002
Fredrickson RJ, Siminski P, Woolf M, Hedrick PW (2007) Genetic rescue and inbreeding depression in Mexican wolves. Proc R Soc B 274:2365–2371. https://doi.org/10.1098/rspb.2007.0785
Fredrickson RJ, Hedrick PW, Wayne RK et al (2015) mexican wolves are a valid subspecies and an appropriate conservation target. J Hered 106:415–416. https://doi.org/10.1093/jhered/esv028
Freedman AH, Gronau I, Schweizer RM et al (2014) Genome sequencing highlights the dynamic early history of dogs. PLoS Genet 10:e1004016. https://doi.org/10.1371/journal.pgen.1004016
Fritts SH (1983) Record dispersal by a wolf from Minnesota. J Mammal 64:166–167
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496. https://doi.org/10.1016/j.tree.2012.05.012
Gebremedhin B, Ficetola GF, Naderi S et al (2009) Frontiers in identifying conservation units: from neutral markers to adaptive genetic variation. Anim Conserv 12:107–109. https://doi.org/10.1111/j.1469-1795.2009.00255.x
Geffen E, Anderson MJ, Wayne RK (2004) Climate and habitat barriers to dispersal in the highly mobile grey wolf. Mol Ecol 13:2481–2490. https://doi.org/10.1111/j.1365-294X.2004.02244.x
Gilg O, Kovacs KM, Aars J et al (2012) Climate change and the ecology and evolution of Arctic vertebrates. Ann NY Acad Sci 1249:166–190. https://doi.org/10.1111/j.1749-6632.2011.06412.x
Good JM (2012) Reduced representation methods for subgenomic enrichment and next-generation sequencing. In: Molecular methods for evolutionary genetics. Humana Press, Totowa, pp 85–103
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17:333–351. https://doi.org/10.1038/nrg.2016.49
Grace J, Berninger F, Nagy L (2002) Impacts of climate change on the tree line. Ann Bot 90:537–544. https://doi.org/10.1093/aob/mcf222
Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32:D109–D111. https://doi.org/10.1093/nar/gkh023
Grinnell J, Dixon JS, Linsdale JM (1937) Fur-bearing mammals of California: Their natural history, systematic status, and relations to man. University of California press
Hailer F, Leonard JA (2008) Hybridization among three native North American Canis species in a region of natural sympatry. PLoS ONE 3:e3333. https://doi.org/10.1371/journal.pone.0003333
Hamilton JA, Miller JM (2016) Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv Biol 30:33–41. https://doi.org/10.1111/cobi.12574
Heard DC, Williams TM (2011) Distribution of wolf dens on migratory caribou ranges in the Northwest Territories, Canada. Can J Zool 70:1504–1510. https://doi.org/10.1139/z92-207
Hedrick PW (2013) Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol Ecol 22:4606–4618. https://doi.org/10.1111/mec.12415
Hedrick PW, Miller PS, Geffen E, Wayne RK (1997) Genetic evaluation of the three captive mexican wolf lineages. Zoo Biol 16:47–69. https://doi.org/10.1002/(SICI)1098-2361(1997)16:1%3C47::AID-ZOO7%3E3.0.CO;2-B
Heffelfinger JR, Nowak RM, Paetkau D (2017a) Clarifying historical range to aid recovery of the Mexican wolf. Jour Wild Mgmt 43:255. https://doi.org/10.1002/jwmg.21252
Heffelfinger JR, Nowak RM, Paetkau D (2017b) Revisiting revising Mexican wolf historical range: a reply to Hendricks et al. J Wild Manag 81:1334–1337. https://doi.org/10.1002/jwmg.21371
Hendricks SA, Clee PRS, Harrigan RJ et al (2016) Re-defining historical geographic range in species with sparse records: implications for the Mexican wolf reintroduction program. Biol Cons 194:48–57. https://doi.org/10.1016/j.biocon.2015.11.027
Hendricks SA, Koblmüller S, Harrigan RJ et al (2017) Defense of an expanded historical range for the Mexican wolf: A comment on Heffelfinger et al. J Wild Mgmt 81:1331–1333. https://doi.org/10.1002/jwmg.21336
Hendricks SA, Schweizer RM, Harrigan RJ et al (2018) Natural re-colonization and admixture of wolves (Canis lupus) in the US Pacific Northwest: challenges for the protection and management of endangered taxa. Heredity https://doi.org/10.1038/s41437-018-0094-x
Hilton C, Neville MJ, Karpe F (2013) MicroRNAs in adipose tissue: their role in adipogenesis and obesity. Int J Obes (Lond) 37:325–332. https://doi.org/10.1038/ijo.2012.59
Jewell DM, Ashe M, Haskett M (2015) Re: Petition to List on an Emergency Basis the Alexander Archipelago Wolf (Canis Lupus Ligoni) as Threatened or Endangered Under the Endangered Species Act
Jimenez MD, Bangs EE, Boyd DK et al (2017) Wolf dispersal in the Rocky Mountains, Western United States: 1993–2008. J Wild Manag 81:581–592. https://doi.org/10.1002/jwmg.21238
Jones MR, Good JM (2016) Targeted capture in evolutionary and ecological genomics. Mol Ecol 25:185–202. https://doi.org/10.1111/mec.13304
Kranis A, Gheyas AA, Boschiero C et al (2013) Development of a high density 600K SNP genotyping array for chicken. BMC Genomics 14:59. https://doi.org/10.1186/1471-2164-14-59
Lachance J, Tishkoff SA (2013) SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it. BioEssays 35:780–786. https://doi.org/10.1002/bies.201300014
Leonard JA, Vilà C, Wayne RK (2005) Legacy lost: genetic variability and population size of extirpated US grey wolves (Canis lupus). Mol Ecol 14:9–17. https://doi.org/10.1111/j.1365-294X.2004.02389.x
Lindblad-Toh K, Wade CM, Mikkelsen TS et al (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature Publishing Group 438:803–819. https://doi.org/10.1038/nature04338
Lowry DB, Hoban S, Kelley JL et al (2016) Breaking RAD: An evaluation of the utility of restriction site associated DNA sequencing for genome scans of adaptation. Mol Ecol Resour. https://doi.org/10.1111/1755-0998.12596
Mahlstein I, Knutti R (2012) September Arctic sea ice predicted to disappear near 2 °C global warming above present. J Geophys Res. https://doi.org/10.1029/2011JD016709
Malomane DK, Reimer C, Weigend S et al (2018) Efficiency of different strategies to mitigate ascertainment bias when using SNP panels in diversity studies. BMC Genomics 19:22. https://doi.org/10.1186/s12864-017-4416-9
Marguerat S, Bähler J (2009) RNA-seq: from technology to biology. Cell Mol Life Sci 67:569–579. https://doi.org/10.1007/s00018-009-0180-6
McKinney GJ, Larson WA, Seeb LW, Seeb JE (2017) RADseq provides unprecedented insights into molecular ecology and evolutionary genetics: comment on Breaking RAD by Lowry et al. (2016). Mol Ecol Resour 17:356–361. https://doi.org/10.1111/1755-0998.12649
McLaren W, Pritchard B, Rios D et al (2010) Deriving the consequences of genomic variants with the ensembl API and SNP effect predictor. Bioinformatics 26:2069–2070. https://doi.org/10.1093/bioinformatics/btq330
McMahon BJ, Teeling EC, Höglund J (2014) How and why should we implement genomics into conservation? Evol Appl 7:999–1007. https://doi.org/10.1111/eva.12193
Mech LD (1970) The Wolf. American Museum of Natural History Natural History Press, Stroud
Mech LD (2004) Is climate change affecting wolf populations in the high arctic? Clim Change 67:87–93. https://doi.org/10.1007/s10584-004-7093-z
Mech L (2005) Decline and recovery of a High Arctic wolf-prey system. 58(3):305–307
Merrill SB, Mech LD (2000) Details of Extensive Movements by Minnesota Wolves (Canis lupus)
Moreno JG, Matocq MD, Roy MS et al (1996) Relationships and genetic purity of the endangered Mexican wolf based on analysis of microsatellite loci. Conserv Biol 10:376–389. https://doi.org/10.1046/j.1523-1739.1996.10020376.x
Muñoz Fuentes V, Darimont CT, Wayne RK et al (2009) Ecological factors drive differentiation in wolves from British Columbia. J Biogeogr 36:1516–1531. https://doi.org/10.1111/j.1365-2699.2008.02067.x
Musiani M, Leonard JA, Cluff D et al (2007) Differentiation of tundra/taiga and boreal coniferous forest wolves: genetics, coat colour and association with migratory caribou. Mol Ecol 16:4149–4170. https://doi.org/10.1111/j.1365-294X.2007.03458.x
Newsome TM, Boitani L, Chapron G et al (2016) Food habits of the world’s grey wolves. Mamm Rev 46:255–269. https://doi.org/10.1111/mam.12067
Nielsen R, Signorovitch J (2003) Correcting for ascertainment biases when analyzing SNP data: applications to the estimation of linkage disequilibrium. Theor Popul Biol 63:245–255. https://doi.org/10.1016/S0040-5809(03)00005-4
Notaro M, Mauss A, Williams JW (2012) Projected vegetation changes for the American Southwest: combined dynamic modeling and bioclimatic-envelope approach. Ecol Appl 22:1365–1388. https://doi.org/10.1890/11-1269.1
Nowak RM (1995) Another look at wolf taxonomy. In: Carbyn LN, Fritts SH, Seip DR (eds) Ecology and conservation of wolves in a changing. Occasional Publication No. 35, Edmonton, Alberta, pp 375–397
Ouborg NJ, Pertoldi C, Loeschcke V et al (2010) Conservation genetics in transition to conservation genomics. Trends Genet 26:177–187. https://doi.org/10.1016/j.tig.2010.01.001
Oyler-McCance SJ, Oh KP, Langin KM, Aldridge CL (2016) A field ornithologist’s guide to genomics: practical considerations for ecology and conservation. The Auk 133:626–648. https://doi.org/10.1642/AUK-16-49.1
Parsons D (1996) Case study: the Mexican wolf. N. M. J Science
Pavlidis P, Živkovic D, Stamatakis A, Alachiotis N (2013) SweeD: likelihood-based detection of selective sweeps in thousands of genomes. Mol Biol Evol 30:2224–2234. https://doi.org/10.1093/molbev/mst112
Peters R, Ripple WJ, Wolf C et al (2018) Nature divided, scientists united: US–Mexico border wall threatens biodiversity and binational conservation. BioSci 24:171–176. https://doi.org/10.1093/biosci/biy063
Pilot M, Jedrzejewski W, Branicki W et al (2006) Ecological factors influence population genetic structure of European grey wolves. Mol Ecol 15:4533–4553. https://doi.org/10.1111/j.1365-294X.2006.03110.x
Pilot M, Branicki W, Jędrzejewski W et al (2010) Phylogeographic history of grey wolves in Europe. BMC Evol Biol 10:104. https://doi.org/10.1186/1471-2148-10-104
Pilot M, Greco C, vonHoldt BM et al (2014) Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity 112:428–442. https://doi.org/10.1038/hdy.2013.122
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecolo Modell 190:231–259
Räikkönen J, Vucetich JA, Peterson RO, Nelson MP (2009) Congenital bone deformities and the inbred wolves (Canis lupus) of Isle Royale. Biol Cons 142:1025–1031. https://doi.org/10.1016/j.biocon.2009.01.014
Razgour O, Taggart JB, Manel S et al (2017) An integrated framework to identify wildlife populations under threat from climate change. Mol Ecol Resour 91:2437. https://doi.org/10.1111/1755-0998.12694
Reed JE, Ballard WB, Gipson PS et al (2006) Diets of Free-Ranging Mexican Gray Wolves in Arizona and New Mexico. Wildl Soc Bull 34:1127–1133. https://doi.org/10.2193/0091-7648(2006)34%5B1127:DOFMGW%5D2.0.CO;2
Reimand J, Kull M, Peterson H et al (2007) g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 35:W193–W200. https://doi.org/10.1093/nar/gkm226
Reimand J, Arak T, Vilo J (2011) g:Profiler—a web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res 39:W307–W315. https://doi.org/10.1093/nar/gkr378
Robinson JA, Vecchyo DO-D, Fan Z et al (2016) genomic flatlining in the endangered island fox. Curr Biol 1–25. https://doi.org/10.1016/j.cub.2016.02.062
Rosenblum EB, Novembre J (2007) Ascertainment bias in spatially structured populations: a case study in the eastern fence lizard. J Hered 98:331–336. https://doi.org/10.1093/jhered/esm031
Roy MS, Geffen E, Smith D et al (1994) Patterns of differentiation and hybridization in North American wolflike canids, revealed by analysis of microsatellite loci. Mol Biol Evol 11:553–570. https://doi.org/10.1093/oxfordjournals.molbev.a040137
Schweizer RM, Robinson JA, Harrigan RJ et al (2016a) Targeted capture and resequencing of 1040 genes reveal environmentally driven functional variation in grey wolves. Mol Ecol 25:357–379. https://doi.org/10.1111/mec.13467
Schweizer RM, vonHoldt BM, Harrigan RJ et al (2016b) Genetic subdivision and candidate genes under selection in North American grey wolves. Mol Ecol 25:380–402. https://doi.org/10.1111/mec.13364
Shafer ABA, Wolf JBW, Alves PC et al (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30:78–87. https://doi.org/10.1016/j.tree.2014.11.009
Shaw H (1983) The Wolf in the Southwest: the making of an endangered species. University of Arizona Press
Slater GJ, Dumont ER, Van Valkenburgh B (2009) Implications of predatory specialization for cranial form and function in canids. J Zool 278:181–188. https://doi.org/10.1111/j.1469-7998.2009.00567.x
Smith DW, Peterson RO, Houston D (2003) Yellowstone after Wolves. Bioscience 53:330–340
Sneed PG (2001) The feasibility of gray wolf reintroduction to the Grand Canyon ecoregion
Steiner CC, Putnam AS, Hoeck PEA, Ryder OA (2013) Conservation genomics of threatened animal Species. Ann Rev Anim Biosci 1:261–281. https://doi.org/10.1146/annurev-animal-031412-103636
Storey KB (2015) Regulation of hypometabolism: insights into epigenetic controls. J Exp Biol 218:150–159. https://doi.org/10.1242/jeb.106369
Stronen AV, Navid EL, Quinn MS et al (2014) Population genetic structure of gray wolves (Canis lupus) in a marine archipelago suggests island-mainland differentiation consistent with dietary niche. BMC Ecol 14:11. https://doi.org/10.1186/1472-6785-14-11
U.S. Fish and Wildlife Service Endangered and threatened wildlife and plants; removing the gray wolf (Canis lupus) from the list of endangered and threatened wild- life and maintaining protections for the Mexican wolf (Canis lupus baileyi) by listing it as endangered
Vilà C, Amorim IR, Leonard JA et al (1999) Mitochondrial DNA phylogeography and population history of the grey wolf (Canis lupus). Mol Ecol 8:2089–2103. https://doi.org/10.1046/j.1365-294x.1999.00825.x
Verts BJ, Carraway LN (1998) Land Mammals of Oregon. Univ of California Press
vonHoldt BM, Stahler DR, Bangs EE et al (2010) A novel assessment of population structure and gene flow in grey wolf populations of the Northern Rocky Mountains of the United States. Mol Ecol 19:4412–4427. https://doi.org/10.1111/j.1365-294X.2010.04769.x
vonHoldt BM, Pollinger JP, Earl DA et al (2011) A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res 21:1294–1305. https://doi.org/10.1101/gr.116301.110
vonHoldt BM, Cahill JA, Fan Z et al (2016) Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Science Advances 2:e1501714–e1501714. https://doi.org/10.1126/sciadv.1501714
vonHoldt BM, Brzeski KE, Wilcove DS, Rutledge LY (2017) Redefining the role of admixture and genomics in species conservation. Conserv Lett 16:613. https://doi.org/10.1111/conl.12371
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. https://doi.org/10.1038/nrg2484
Wayne RK, Shaffer HB (2016) Hybridization and endangered species protection in the molecular era. Mol Ecol 25:2680–2689. https://doi.org/10.1111/mec.13642
Wayne RK, Lehman N, Allard MW, Honeycutt RL (1992) Mitochondrial DNA variability of the gray wolf: genetic consequences of population decline and habitat fragmentation. Conserv Biol 6:559–569. https://doi.org/10.1046/j.1523-1739.1992.06040559.x
Weckworth BV, Talbot S, Sage GK et al (2005) A signal for independent coastal and continental histories among North American wolves. Mol Ecol 14:917–931. https://doi.org/10.1111/j.1365-294X.2005.02461.x
Weckworth BV, Dawson NG, Talbot SL, Cook JA (2015) Genetic distinctiveness of Alexander Archipelago Wolves (Canis lupus ligoni). J Hered 106:412–414. https://doi.org/10.1093/jhered/esv026
Wilhelm BT, Landry J-R (2009) RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods 48:249–257. https://doi.org/10.1016/j.ymeth.2009.03.016
Wu C-W, Biggar KK, Storey KB (2013) Dehydration mediated microRNA response in the African clawed frog Xenopus laevis. Gene 529:269–275. https://doi.org/10.1016/j.gene.2013.07.064
Young S, Goldman EA (1944) The Wolves of North America. 2 vols. American Wildlife Institute, Washington
Zaragosi L-E, Wdziekonski B, Brigand KL et al (2011) Small RNA sequencing reveals miR-642a-3p as a novel adipocyte-specific microRNA and miR-30 as a key regulator of human adipogenesis. Genome Biol 12:R64. https://doi.org/10.1186/gb-2011-12-7-r64
Acknowledgements
Support was provided to SAH by the National Institute of Health (P30GM103324); NSF (DEB-1316549); and the Bioinformatics and Computational Biology program at the University of Idaho, and to RMS by the NSF (DGE-1144087, DGE-0707424, 1612859).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hendricks, S.A., Schweizer, R.M. & Wayne, R.K. Conservation genomics illuminates the adaptive uniqueness of North American gray wolves. Conserv Genet 20, 29–43 (2019). https://doi.org/10.1007/s10592-018-1118-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-018-1118-z