
Abstract
Rapid environmental change makes adaptive potential—the capacity of populations to evolve genetically based changes in response to selection—more important than ever for long-term persistence of at-risk species. At the same time, advances in genomics provide unprecedented power to test for and quantify adaptive potential, enabling consideration of adaptive potential in estimates of extinction risk and laws protecting endangered species. The U.S. Endangered Species Act (ESA) is one of the most powerful environmental laws in the world, but so far, the full potential of genomics in ESA listing and recovery decisions has not been realized by the federal agencies responsible for implementing the ESA or by conservation geneticists. The goal of our paper is to chart a path forward for integrating genomics into ESA decision making to facilitate full consideration of adaptive potential in evaluating long-term risk of extinction. For policy makers, managers, and other conservation practitioners, we outline why adaptive potential is important for population persistence and what genomic tools are available for quantifying it. For conservation geneticists, we discuss how federal agencies can integrate information on the effect of adaptive potential on extinction risk—and the related uncertainty—into decisions, and suggest next steps for advancing understanding of the effect of adaptive potential on extinction risk. The mechanisms and consequences of adaptation are incredibly complex, and we may never have a complete understanding of adaptive potential for any organism. Nevertheless, we argue that the best available evidence regarding adaptive potential can now be incorporated by federal agencies into modeling and decision making processes, while at the same time conserving genome-wide variation and striving for a deeper understanding of adaptive potential and its effects on population persistence to improve decision making into the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The unprecedented rate of global environmental change means that many species and populations will have to adapt (see Box 1 for glossary of genetic terms in bold) to this change, or go extinct (Bell and Collins 2008; Hoffmann and Sgro 2011). The dramatic increase in human population size and associated rapid environmental change has been termed the “Great Acceleration” by the International Geosphere–Biosphere Programme (Steffen et al. 2015). Human-caused climate change, in particular, has already impacted biodiversity at all levels of biological organization and on every continent, including extinction of many species, with many more extinctions projected (Penuelas et al. 2013; Scheffers et al. 2016). Three primary mechanisms allow persistence in the face of this rapid environmental change: dispersal, phenotypic plasticity, and genetically-based adaptation to changing conditions (Dawson et al. 2011; O’Connor et al. 2012; Nicotra et al. 2015). In addition, epigenetic variation may play a role in buffering populations from environmental change (Bernatchez 2016; Verhoeven et al. 2016). Thus, species with greater adaptive potential—the capacity to evolve genetically-based changes in traits in response to changing environmental conditions—will be more resilient to climate and other environmental change.
Despite recognition of the critical importance of adaptive potential for persistence in the face of environmental change, it has been difficult or impossible to quantify for the vast majority of species. Adaptive potential is ultimately determined by the amount of additive genetic variation for adaptive traits within and among populations (see section below on “What determines adaptive potential of a species?”). Thus, genetic differences among individuals within populations, as well as genetic differences among populations, contribute to the overall adaptive potential of a species. The traditional approach for quantifying additive genetic variation within populations is to estimate the proportion of variance in a trait that is heritable using controlled breeding (Falconer and MacKay 1996). The gold standard for testing for adaptive differences among populations is a reciprocal transplant experiment. In these experiments, individuals from two different populations are transplanted to the environment of the other population to test whether individuals have greater fitness in their native environment compared to the foreign environment, demonstrating local adaptation (Clausen et al. 1948). Controlled breeding designs and reciprocal transplant experiments, however, are not feasible for most species of conservation concern, especially mobile or large endangered animals with small population sizes.
Fortuitously, the genomics revolution provides more power than ever to test for and quantify adaptation and adaptive potential to improve implementation of conservation policy (Black et al. 2001; Luikart et al. 2003; Beaumont and Balding 2004; Allendorf et al. 2010). For the first time, population genomics provides a means of testing for adaptation in species for which controlled breeding and reciprocal transplant experiments are impractical or impossible. Population genomics is the use of genome-wide data (e.g., single-nucleotide polymorphisms [SNPs]) at thousands to millions of loci across the genome of a sample of organisms to make inferences about microevolutionary processes (gene flow, genetic drift, selection, and mutation; Black et al. 2001; Luikart et al. 2003). The field has been enabled by rapid advances in next-generation sequencing (NGS) technology and computational power (Glenn 2011; Catchen et al. 2013; Hohenlohe et al. 2013). Due to the huge number of loci included in population genomic studies (typically thousands to hundreds of thousands), various statistical approaches can be used to identify putatively adaptive loci (Beaumont and Balding 2004; Joost et al. 2007; Coop et al. 2010; Frichot et al. 2013; Forester et al. 2018). By contrast, traditional population genetic approaches, which use a much smaller number of loci (e.g., 10–20), have much less power to identify adaptive loci because they evaluate insufficient numbers of molecular markers.
In particular, genomics has tremendous potential to improve our ability to incorporate information on adaptive potential into laws protecting endangered species. Several countries have enacted such laws, including the Endangered Species Act in the United States (ESA; passed in 1973), the Biodiversity Law of Costa Rica (passed in 1992), the Endangered Species Protection Act of Australia (passed in 2002), Canada’s Species at Risk Act (passed in 2002), and the South African National Environmental Management Biodiversity Act (passed in 2004; Waples et al. 2013). Here, we focus on application of genomics in listing and recovery decisions under the ESA as a case study of how information on adaptive potential inferred from genomics and other approaches can be integrated into risk assessments to improve implementation of endangered species laws. The ESA is one of the most powerful environmental laws in the world, providing the statutory basis for listing and legal protection of species and subspecific units determined to be threatened or endangered (Carroll et al. 1996; Waples et al. 2013). Since the law was signed in 1973, 2318 species—with ranges inside and outside the U.S.—have been listed as threatened or endangered, and 53 of these species have recovered to the point where they could be delisted (https://ecos.fws.gov/ecp0/reports/delisting-report), indicating it can be an effective law for improving the conservation status of at-risk species.
Many factors are considered in ESA listing and recovery decisions, including information on the capacity of species to adapt to cope with new environmental stressors (Carroll et al. 1996; Shaffer and Stein 2000; Vucetich et al. 2006; see Box 2). However, given the past difficulty of quantifying adaptive potential, little information has typically been available for most species that are candidates for listing. Because genomics greatly increases the feasibility of characterizing adaptive potential in non-model species, it can improve ESA listing and recovery decisions by allowing estimation and integration of adaptive potential into models of extinction risk. Due to the nascency of genomics, it has had limited application to the ESA so far. Compared to evolution and ecology where the use of genomics is widespread, its application to conservation questions has lagged behind due to several previously discussed obstacles (Shafer et al. 2015). However, this is changing rapidly as genomic tools are increasingly applied to conservation problems (Garner et al. 2016). Now is a critical time to determine how best to use genomics to directly inform conservation policy, including ESA decisions.
The goal of this paper is to provide guidance on how genomics can be integrated into ESA decision making to facilitate full consideration of adaptive potential in evaluating long-term extinction risk. This paper is geared towards both conservation practitioners (e.g., policy makers and managers) as well as conservation geneticists. For conservation practitioners, our objectives are to explain: (1) what determines adaptive potential; (2) why adaptive potential is important to conservation; and (3) what genomic tools are available for quantifying adaptive potential. For conservation geneticists, our objectives are to: (1) explain how federal agencies make ESA listing and recovery decisions, and how information on adaptive potential can be incorporated into these decisions; (2) explain how these decisions can be made in the face of uncertainty about the effects of adaptive potential on extinction risk; and (3) suggest next steps for advancing understanding of the effect of adaptive potential on extinction risk. Although we focus on the ESA here, our discussion of ways to apply genomics to ESA decision making should be applicable to similar laws in other countries.

Adaptation and conservation
What determines adaptive potential of a species?
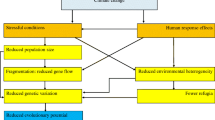
Adaptation is genetically-based change in traits that increases fitness. Adaptation results from selection on heritable phenotypic variation. The rate of adaptation in a given trait is proportional to the amount of additive genetic variation underlying that trait (Falconer and Mackay 1996). The adaptive potential of a species is determined by the amount of additive genetic variation within and among populations in fitness-related traits (Fig. 2). The ultimate source of all genetic variation is mutation (Dobzhansky and Wright 1941). Genetic drift, gene flow, and selection then act on variation generated by mutation to structure variation within and among populations. Genetic drift—random changes in allele frequencies due to the finite number of alleles passed on to the next generation—causes a reduction in genetic variation within populations and divergence in allele frequencies among populations. The effective size of a population (Ne), which is typically smaller than the observed number of individuals in a population, determines the rate of genetic drift (Wright 1938; Kimura and Crow 1963). Isolated populations with smaller Ne have more genetic drift and less adaptive potential. Gene flow is the movement of alleles among populations caused by dispersal and reproduction (Wright 1943; Slatkin 1987). Gene flow is predicted to cause an increase in genetic variation within populations, but a reduction in genetic divergence among populations. Finally, selection is caused by differences in fitness among genotypes (Fisher 1930; Haldane 1930; Wright 1931). Depending on the form of selection, it can decrease or increase genetic variation within and among populations. Divergent selection occurs when different alleles are favored in different environments at a given locus, and results in a reduction in within-population genetic variation and an increase in genetic differences among populations at the loci under selection. Ultimately, to conserve adaptive potential, it is necessary to conserve multiple large populations with minimal genetic drift, and allow gene flow among these populations so that they can exchange adaptive variants (Garant et al. 2007; Allendorf et al. 2013). In species or conservation units that exist as single populations, management should be focused on reducing threats and maximizing population size to minimize the probability of extinction.

Potential distribution of adaptive potential within and among populations, and genomic approaches for characterizing adaptive potential under these different scenarios. Although heat tolerance is most likely a polygenic trait, here, we show a single locus contributing to heat tolerance for the sake of simplicity. Each two-letter genotype represents a single diploid individual. If, for example, hot (denoted by h) and cold (denoted by c) tolerant alleles are segregating at a given locus within a population, then adaptive potential for heat tolerance allows the evolution of this trait within this population (a). In this case, genomic approaches could be used to estimate the heritability of heat tolerance or to identify loci underlying heat tolerance using a genome-wide association study (GWAS). Alternatively, if two different populations are fixed for alternative alleles at this locus due to strong divergent selection in hot (red) vs. cold (blue) environments, then there is no adaptive potential within populations, but there is adaptive potential in the entire metapopulation or species as a whole (b). In this case, heat tolerance could evolve in the population fixed for the c allele as long as immigration (natural or mediated by humans) into this population from the population fixed for the h allele occurs. Here, genomics could be used to test for loci with signatures of divergent selection and adaptive divergence using FST outlier tests. Finally, if a continuously distributed species is locally adapted to a climatic gradient, then an allele frequency cline should evolve at loci underlying thermal tolerance (c). In this case, genotype-by-environment associations (GEA) can be used to identify loci that are putatively involved in adaptation along this climatic gradient. (Color figure online)
Why is adaptation relevant to conservation?
Population genetic theory predicts that maintenance of additive genetic variation, the source of adaptive potential, is important for long term persistence (Burger and Lynch 1995). One well-known guideline for avoiding inbreeding depression and maintaining additive genetic variation is the “50/500 rule” (Franklin 1980). It states that a minimum Ne of 50 is needed to avoid significant inbreeding depression over the short-term, and a minimum Ne of 500 is needed to maintain adaptive potential over the long-term. This rule of thumb has been the subject of much debate (Jamieson and Allendorf 2012, 2013; Frankham et al. 2013), but the general idea that large Ne is needed to maintain adaptive potential, especially with rapid environmental change, is well accepted (Bell and Collins 2008; Hoffmann et al. 2017).
Some of the best evidence for the importance of adaptive potential in conservation comes from controlled laboratory experiments in model species. For example, Frankham et al. (1999, 2002) have demonstrated that small laboratory populations of Drosophila melanogaster are unable to evolve and persist in response to stressful environmental conditions. Populations that went through extreme bottlenecks of two individuals for one to three generations went extinct in high salt environments more frequently than outbred control populations (Frankham et al. 1999). Similarly, populations with low genetic variation maintained for 50 generations went extinct more often than populations with higher genetic variation (Frankham et al. 2002). Controlled laboratory experiments in flower beetles (Tribolium castaneum) yield similar results. Agashe et al. (2011) found that genetically diverse wheat-adapted flour beetle populations exposed to a novel suboptimal corn resource were less likely to go extinct than genetically depauperate populations. Moreover, genetically diverse populations recovered more quickly. Thus, laboratory studies support theoretical expectations that populations with more genetic variation are more likely to adapt and persist in novel stressful environments.
We also know that some populations have evolved in response to rapid environmental change (Hoffmann and Sgro 2011; Hansen et al. 2012; Penuelas et al. 2013; Scheffers et al. 2016; Cattau et al. 2017). Kovach et al. (2012) found evidence for the evolution of earlier run timing in a population of pink salmon (Oncorhynchus gorbuscha) in Auke Creek, Alaska. Late-migration phenotypes decreased from 30% to less than 10% in this population over a 40-year period during which temperatures in Auke Creek increased significantly. Moreover, a genetic marker for late migration timing decreased threefold during the same timeframe. In another example, Franks et al. (2016) found rapidly evolved changes in multiple phenotypic traits, including flowering time, in Brassica rapa in response to a multiyear drought in California. By comparing genome-wide differences between individuals sampled before vs. after the drought, they uncovered shifts in allele frequencies in many genes, some of which are related to drought stress and flowering time. These and many other examples demonstrate that rapid environmental change has already resulted in evolved adaptations (Hoffmann and Sgro 2011; Savage and Zamudio 2011; Hansen et al. 2012; Bataille et al. 2015; Scheffers et al. 2016; Cattau et al. 2017).
In contrast, other populations lack adaptive potential or have failed to evolve in response to environmental change. An example of the lack of adaptive potential comes from the intertidal copepod, Tigriopus californicus (Kelly et al. 2012). This species is highly adapted to local temperatures across a latitudinal gradient of 17° along the Pacific coast of North America. Less than one percent of quantitative variance in thermal tolerance is partitioned within populations; most variance in this trait is due to differences among populations. Moreover, heat tolerant phenotypes observed in low latitude populations did not evolve in high latitude populations after 10 generations of strong selection, indicating a lack of adaptive potential within populations. In chinook salmon (Oncorhynchus tshawytscha), Muñoz et al. (2015) found a lack of additive genetic variation for arrhythmic temperature of the heart, which constrains the thermal limit to a maximum of 24.5 °C. Based on projected increases in river temperatures, the authors estimated a 17% chance of catastrophic population decline by 2100, assuming behavioral and phenological changes do not buffer the population from increasing temperature. These examples suggest that many populations will likely lack sufficient additive genetic variation to adapt quickly enough to the pace of climate change, at least without aggressive between-population translocation efforts. Moreover, we know based on theory and lab studies, such as those described above, that reduction in population sizes and isolation caused by habitat loss, fragmentation, and other anthropogenic stressors will reduce the capacity of populations to adapt in response to climate or other environmental change. Thus, we need to be particularly cognizant of the erosion of adaptive potential for at-risk species with small population sizes. To determine the adaptive potential of a species, we first need to quantify heritable variation within populations and adaptive differences among them (Fig. 2).
Quantifying adaptive potential
Traditional approaches for quantifying adaptive potential
Within a population, adaptive potential for a given trait is determined by the heritability of that trait. Heritability can be thought of as the proportion of variance in a trait that is caused by genetic factors. The greater the heritability of a trait, the more it will evolve across generations in response to selection. In contrast, if heritability of a trait is zero, then that trait cannot evolve in response to selection. Traditionally, heritability is estimated using methods that rely on controlled breeding experiments, so that the identities of parents and offspring are known. For example, heritability can be estimated as the regression coefficient in a regression of family means against midparent values (the average trait value of both parents) for a given trait (Falconer and Mackay 1996). In addition to controlled breeding, these experiments often used controlled laboratory conditions (often termed a “common garden,” as early studies of heritability were typically done on plants) to control for the influence of the environment on phenotypic variation, making them difficult or impossible to implement in many species (Table 1).
Adaptive differences among populations can also increase the adaptive potential of a species. This is why several authors have emphasized the importance of delineating and maintaining adaptively divergent populations for long-term persistence of species (Ryder 1986; Waples 1991; Crandall et al. 2000; Fraser and Bernatchez 2001; Funk et al. 2012). Even when a trait is not heritable within populations, the trait can evolve if genetic differences among populations in that trait are moved among populations via gene flow (Fig. 2). Thus, it is important to quantify adaptive differences among populations in addition to heritable variation within populations. Three main lines of evidence are traditionally used to demonstrate adaptive differentiation among populations (Table 1). First, environmental differentiation that is accompanied by phenotypic divergence across sites or populations can provide support for local adaptation (Hanson et al. 2017). For example, tortoise populations in the Galapagos Archipelago can be found in two habitat types: mesic and xeric. Tortoises in mesic habitats have a dome-shaped shell, while tortoises in xeric habitats have a saddleback-shaped shell, indicating that tortoises may be locally adapted (Fritts 1984). This hypothesis is supported by data indicating that competition for resources is more intense and agonistic behaviors more common in xeric habitats, where the saddleback phenotype is at a competitive advantage (Fritts 1984). Saddleback tortoises have longer necks and forelimbs, and a carapace shape that allows for higher vertical reach, characteristics that increase both vertical feeding range and dominance during agonistic encounters.
The second traditional line of evidence for adaptive differences among populations comes from common garden experiments (Claussen et al. 1948; McKay et al. 2001; Thorpe et al. 2005). While environmental and phenotypic differences among populations can be useful for hypothesis generation, inferences based on these patterns alone can be confounded by phenotypic plasticity, which is the capacity of a single genotype to produce multiple phenotypes in response to different environmental conditions. A common garden experiment can be used to test whether phenotypic variation is genetically determined or due to plasticity. If phenotypic differences between individuals from different environments are maintained when they are reared in a common environment, it indicates the phenotype is genetically based, rather than plastic. Most variation in traits is due to a combination of genetic variation and plasticity.
The third traditional line of evidence for adaptive differences among populations comes from reciprocal transplant experiments (Berven 1982; Nagy and Rice 1997; Sork 2018). In these experiments, individuals are swapped between two different environments to test whether individuals are adapted to their local environment. If individuals do better in their native environment compared to the foreign environment, this suggests they are locally adapted to their native environment. However, in addition to being difficult or impossible to implement for many at-risk species, the use of controlled breeding and reciprocal transplant experiments to gauge overall adaptive potential within and between populations can be problematic (Hendry et al. 2011; Hoffmann et al. 2017). In particular, results of these experiments are only relevant to the trait, environment, and population that is studied, and can overestimate or underestimate adaptive potential (Harrisson et al. 2014).
Research on the threatened Italian agile frog (Rana latastei) provides an example of a combined, non-genomic (traditional) approach to testing for local adaptation and quantifying adaptive potential. This species shows differences in larval growth over short geographic distances as a function of temperature: tadpoles in colder environments take approximately 1 month longer for metamorphosis compared to tadpoles in warmer environments, which led Ficetola and De Bernardi (2005) to hypothesize that these frogs are adapted to their local temperature regime. Interestingly, a common garden experiment confirmed a genetically based difference in developmental rates among these populations, but in the opposite direction to that observed in the wild. Cold-adapted tadpoles developed faster than warm-adapted tadpoles held at the same temperature. This indicates selection for faster development in cold climates, despite slower growth in the field (Ficetola and De Bernardi 2005). These results were used to provide support for in situ conservation of cold-adapted populations, rather than transferring individuals between cold and warm populations, which could potentially disrupt local adaptation (Ficetola and De Bernardi 2005).
Genomic approaches for quantifying adaptive potential
While traditional methods for investigating adaptive potential can be useful in certain species, the necessity of controlled breeding and experiments means they will not be applicable for most at-risk populations and species. Fortunately, technological advances in next-generation sequencing provide novel opportunities to estimate adaptive potential in wild populations, providing stronger evidence than simple correlative approaches and avoiding the need for manipulative experiments (Table 1). Cost-effective genomic sequencing methods can be used in any species, even in cases where no prior genomic information is available (Andrews et al. 2016). These sequencing approaches produce large genomic datasets, for example, genotypes at thousands of SNPs for hundreds of individuals (Lowry et al. 2016; McKinney et al. 2017; Catchen et al. 2017). These genomic data can then be used in downstream analyses to estimate adaptive potential within and among populations. While many of these methods do not require a reference genome (i.e., an assembled genome), a high-quality genome assembly and subsequent gene annotation can improve both data quality (e.g., genotyping accuracy) and downstream inferences, such as the identification of candidate genes and calculation of linkage disequilibrium as a function of physical distance (Davey et al. 2011; Manel et al. 2016).
Within populations, genomic data can facilitate the estimation of adaptive potential by allowing for pedigree-free estimation of heritability, eliminating the need for controlled breeding or long-term field-based data collection. Instead, genomic data can be used directly to estimate the relatedness among all pairs of individuals in a focal population (Gienapp et al. 2017). Heritability is then estimated by testing the relationship between trait similarity and relatedness among individuals (Visscher et al. 2008; Sillanpää 2011). Sampling requirements can be high for these studies (e.g., 150–200 individuals and ~ 25,000 SNPs; Stanton-Geddes et al. 2013), and can be difficult to generalize since the number of individuals and markers needed can vary across species and populations due to differences in effective population sizes and linkage disequilibrium. However, since many species of conservation concern have small effective sizes, they can be good candidates for genomic-based heritability studies since the estimation error for heritability is proportional to the effective size (Visscher and Goddard 2015), and increased linkage disequilibrium among loci will reduce the number of SNPs required for robust estimates. For example, a recent study estimated heritability for four quantitative (continuously varying) traits in Corsican blue tits (Cyanistes caeruleus ogliastrae) using both genomic and pedigree-based approaches (Perrier et al. 2018). The authors found that 15,000 SNPs genotyped across 494 individuals was sufficient to match or surpass the accuracy of heritability estimates provided by a 7-year pedigree-based study of > 1600 individuals.
Genomic data can also be used within populations to identify the specific loci that underlie variation in fitness-related traits using genome-wide association studies (GWAS; Korte and Farlow 2013). Similar to heritability, GWAS generally requires large sample sizes (e.g. Hong and Park 2012), including the measurement of fitness-relevant traits in many individuals and dense genomic sampling (many genetic markers [e.g., SNP loci] across the genome so that most genes are physically linked to at least one marker). Again, GWAS in populations with small effective sizes will have the advantage of increased linkage disequilibrium, which will reduce the number of markers required to sample the genome effectively (McKinney et al. 2017). For example, Hess et al. (2016) used univariate and multivariate GWAS analyses to identify the genetic basis of the adult migration-timing phenotype in threatened Klickitat River steelhead (Oncorhynchus mykiss), part of the Middle Columbia River steelhead distinct population segment. Using a genomic data set of 15,239 SNPs genotyped in 237 individuals, this study identified 18 SNPs that explained ~ 60% of the variation in the adult migration-timing phenotype, information that can be used to inform conservation at pre-adult life stages and better assign adults to summer or winter-run phenotypes. In a similar study, Prince et al. (2017) used association mapping to determine the genetic basis of premature migration. They found that premature migration is associated with the same locus in multiple populations of steelhead and Chinook salmon (O. tshawytscha).
Estimating heritability and identifying loci that underlie trait variation using GWAS both require choosing the trait or traits to analyze. Given that adaptation to changing environmental conditions will likely involve many traits, a well-grounded understanding of the biology and natural history of the study species is essential to predict which traits will have the most important effects on fitness and population persistence in the face of environmental change and that should therefore be the focus of efforts to quantify adaptive potential. Researchers must also acknowledge uncertainty regarding which traits will be most important, and should include this uncertainty in models of extinction risk (see “Incorporating adaptive potential into Endangered Species Act decisions in the face of uncertainty” below).
Genomic data can also inform estimates of among-population adaptive potential. For these analyses, presumably adaptive loci with genetic signatures of divergent selection are identified, and then these loci are used to quantify adaptive potential (e.g., Bonin et al. 2007). Major approaches for identifying candidate adaptive loci include differentiation-based methods, genotype-environment associations, and transcriptomics (Table 1). Differentiation-based methods detect adaptive markers by distinguishing locus-specific patterns (caused by selection) from genome-wide patterns (caused by neutral processes such as genetic drift and gene flow; Luikart et al. 2003). These methods only require genomic data (they do not use environmental or phenotypic data) and are useful for detecting strong selection between populations, but do not uncover the potential environmental drivers of adaptation and are generally less useful for detecting contemporary selection (de Villemereuil et al. 2014). Additionally, these methods are typically dependent on theoretical population genetic models, which are violated in many empirical systems (Bierne et al. 2013).
Genotype-environment association (GEA) methods provide an alternative that does not require population genetic models and can be used with individual or population-based sampling designs. GEAs identify adaptive variation using associations between allele distributions and environmental variables hypothesized to drive selection, identified as a pattern of selected alleles at higher frequency in certain environments. The inclusion of environmental predictors improves power over differentiation-based methods, allows better detection of signals of contemporary selection, and identifies the environmental factor(s) underlying adaptation (Rellstab et al. 2015; Forester et al. 2018). When phenotypic data are available across multiple populations in different home environments, GEA can be combined with GWAS to link loci under selection to phenotypes, improve power to detect adaptive loci, and further strengthen inference of local adaptation (Berg and Coop 2014; Lasky et al. 2015). Although differentiation-based methods, GEA, and GWAS can identify different adaptive loci, the results of these different statistical tests can be combined in a common framework to increase power to infer patterns of local adaptation (François et al. 2016).
Finally, transcriptomics is another method of genomic data acquisition that quantifies gene expression in response to the environment and its effect on phenotypes (Alvarez et al. 2015). While this can be a more efficient means of surveying adaptive variation, since all RNA transcripts are by definition functional, transcriptomics is not easily applied in wild populations or at-risk species. Tissue requirements are more stringent than for genomic sequencing methods, and often require destructive sampling, for example to access internal organs. Additionally, robust transcriptomic studies require controlled, experimental treatments to reduce environmental effects and high-quality gene annotations so the function of transcripts is known. This limits the utility of field-based transcriptomic studies, which require careful design and execution since gene expression is highly sensitive to environmental conditions (Todd et al. 2016). Currently, field-based transcriptomic studies are most valuable for generating hypotheses for future research, limiting their utility for estimating adaptive potential in wild populations of at-risk species. Despite these limitations, field-based transcriptomic studies can be used effectively in species with adequate genomic resources to address conservation-relevant questions. For example, Thomas and Palumbi (2017) used temporal transcriptomic sampling of a reef-building coral (Acropora hyacinthus, which has extensive genomic resources available) subjected to a bleaching event to document long-term (> 12 month) disruption of the coral transcriptome despite the apparent recovery of the coral symbiont population. These lasting effects on species long after the return of normal environmental conditions has implications for ecosystem resiliency in the face of increasing extreme environmental events associated with climate change. In summary, despite the limitations of individual genomic methods, when combined, these methods finally allow the possibility of quantifying adaptive potential in species of conservation concern, providing the exciting opportunity of integrating this information into ESA decision making.
Endangered Species Act decisions and adaptive potential
How are Endangered Species Act listing and recovery decisions made?
Decision makers at the U.S. Fish and Wildlife Service (USFWS) and the National Marine Fisheries Service (NMFS), the two agencies responsible for administering the U.S. Endangered Species Act (ESA 1973, as amended), make decisions using the most up-to-date scientific information to evaluate extinction risk to species and consider that evaluation within the context of society’s willingness to tolerate risks (Doremus 1997; Waples et al. 2013). They face the difficult task of balancing species protection against the burden of regulations. For these decision makers, addressing the values of diverse stakeholders can be an uphill battle (Rohlf 1991; Ruhl 2004). In this section, we outline the important listing and recovery decisions faced by the conservation practitioners responsible for administering the ESA. Figure 3 details the steps involved in both listing and recovery decisions.

Flow chart showing steps involved in U.S. Endangered Species Act (ESA) listing decisions (yellow boxes) and recovery decisions (orange boxes). Blue boxes show possible outcomes of listing decisions. Green boxes show examples of ways in which information on adaptive potential (based on genomics and other data) and other inferences from genomics can inform different steps of listing and recovery decision workflows. In this paper, we focus on how information on adaptive potential can be incorporated into models of extinction risk to improve ESA listing and recovery decisions (shown in bold red font). See text for details on how genomics and other approaches can be used to infer adaptive potential and how uncertainty in these inferences can be included in decision making. (Color figure online)
A species can be listed as either an endangered or threatened species under the ESA depending on the degree of threat it faces (ESA 1973, Sect. 3, 4a). An endangered species is one that is in danger of extinction throughout all or a significant portion of its range. A threatened species is one that is likely to become endangered in the foreseeable future throughout all or a significant portion of its range. Thus, ultimately, the decision to list a species is based on its extinction risk. The ESA defines species broadly to include species, subspecies, varieties, and, for vertebrates, distinct population segments (DPS), defined as a population or group of populations that is discrete and significant in relation to the entire species. The USFWS and NMFS rely increasingly on genetics in defining species (USFWS and NMFS 1996). Accordingly, scientists’ conclusions about whether populations are genetically distinct have become extremely important in decision making (Brosi and Biber 2009). However, debate continues about how best to use increasingly detailed genomic information to identify species and how to determine if the entity meets the definition of threatened or endangered (e.g., Haig and D’Elia 2010; Regan et al. 2013; Keith et al. 2015; Boyd et al. 2017).
The ESA’s ultimate goal is to recover species so they no longer need protection under the ESA. Recovery plans describe the biological state at which protection is no longer needed, called recovery criteria, and the recommended steps to get there (Taylor et al. 2005; Neel et al. 2012). The ESA stipulates that recovery criteria be measurable and objective and that listing and delisting decisions be based on the best science. Both requirements inject a primary role for science, although how recovery criteria are set is not defined in the ESA (Doak et al. 2015). Defining recovery units, management sub-units of the listed entity, is optional, but, where used, sub-units should collectively encompass the entire listed entity and should each have recovery criteria (NMFS and USFWS 2010). Every recovery unit must be recovered before the species can be delisted.
Recovery is not likely to be a fast process; it takes time to address threats that were years in the making. The first milestone in recovery is halting the decline of the species. Next is stabilizing the species, followed by increasing numbers and distribution—finally to the point that it is secure in the wild and the intent of the recovery criteria is met. If the threats have been sufficiently reduced, delisting the species may be considered. The analysis to determine if a species no longer meets the definition of threatened or endangered is analogous to the status assessment the USFWS and NMFS undertake when first determining whether a species should be added to the endangered species list. The ESA requires monitoring of delisted species for at least 5 years to assess their ability to sustain themselves without the protective measures of the ESA. If threats to the species change or unforeseen events change the stability of the population, USFWS or NMFS may extend the monitoring period or re-list the species.
ESA biologists and decision makers, scientists, and other stakeholders often express concern about insufficient information for a particular ESA decision. Indeed, perfect information might lead to different, even better decisions, but the ESA has a strict policy on the luxury of perfect information. The ESA makes it clear that listing decisions are to be based on the “…best available scientific and commercial information…” (italics ours) (ESA 1973, Sect. 4b). Several statutory deadlines assure that ESA decisions are not postponed in favor of additional research. In particular, the 12-month finding, which is the bulk of the status assessment, dictates that USFWS and NMFS have 1 year to make their listing determinations. Thus, ESA decision makers almost always find themselves in the position of making tough decisions under high uncertainty. Given these rigid legal constraints, conservation geneticists need to understand how information on adaptive potential can improve ESA decision making so that they know what information is most important to provide to USFWS and NMFS decision makers.
How can information on adaptive potential improve Endangered Species Act decisions?
Incorporating information on adaptive potential into models of extinction risk can improve ESA listing and recovery decisions by increasing the accuracy of these models. On the one hand, if models do not allow for the possibility of adaptation in response to novel environmental stressors, then extinction estimates might be biased high, which could result in ESA listing of a species that actually has the capacity to evolve and persist in the face of environmental change. On the other hand, if the possibility that threats (e.g., habitat loss, invasive species, overexploitation, etc.) have decreased or are decreasing adaptive potential is not considered, then extinction estimates may be biased low, resulting in not listing a species for which listing is warranted. In the context of recovery decisions, an understanding of how to exploit available adaptive potential, or be conservative in the face of a lack of adaptive potential, could be beneficial in choosing optimal actions. Since ESA decisions are ultimately based on extinction risk, the quantification of adaptive potential with genomics—so that this information can be incorporated into models that predict extinction risk—is arguably the most important application of genomics in ESA decision making. Genomics has numerous other important applications in the ESA listing and recovery workflow, including delineating conservation units (Funk et al. 2012), inferring evolutionary history (Lemmon et al. 2012), quantifying hybridization (Payseur and Rieseberg 2016), and estimating Ne and gene flow (Waples et al. 2016; Fig. 3). However, as these applications of genomics to conservation policy have already been discussed in detail elsewhere, here we focus on discussing how incorporating information on adaptive potential, based on genomics and other approaches, can improve estimates of extinction risk and, therefore, ESA listing and recovery decision making.
In the context of ESA listing decisions, models can be developed to investigate how adaptive responses in specific traits may allow species to avoid extinction under rapid environmental change. A recent example in a population of sockeye salmon (Oncorhynchus nerka) from the Fraser River, Canada, used empirical data to parameterize an individual-based model to determine how evolution of migration timing impacted species persistence under a range of climate change projections (Reed et al. 2011). They found that, with evolution of earlier migration timing, the risk of extinction by 2100 was predicted to be only 17% of that faced by the population with no adaptive potential. This scenario simulated a heritability of 0.5 for migration timing and a 2 °C increase during this time frame (resulting in a 9% and 53% probability of extinction with vs. without adaptive potential, respectively). The authors concluded that the rates of evolution included in their models are plausible given estimated heritabilities and rates of microevolution in migration timing in salmon. A similar modeling approach could be used to assess the effect of adaptive potential on extinction risk in species status assessments of ESA candidate species.
Models have also been used to predict how gene flow of adaptive alleles among populations could improve adaptive potential and mitigate extinction in entire metapopulations or species. Creech et al. (2017) used simulations to investigate the spread of adaptive genotypes in desert bighorn sheep (Ovis canadensis nelsoni), a habitat specialist threatened by habitat loss and fragmentation due to climate change and other anthropogenic effects. They found that adaptation from standing genetic variation already present within populations had a much higher chance of spread and likelihood of persistence than adaptive variation arising from a new mutation, especially when landscapes were more highly connected. These results highlighted the importance of retaining high levels of genetic variation within populations, while maintaining the metapopulation structure that is characteristic of the subspecies across its range. Metapopulation models such as this and others (Converse et al. 2017) provide a means of assessing how spatial variation in adaptive alleles can influence extinction risk in an entire species, subspecies, or DPS.
If the listing process identifies a species as threatened or endangered, genomic data related to adaptive potential can inform specific recovery actions that mitigate extinction risk through the directional movement of “pre-adapted” individuals between populations to facilitate adaptation to changing conditions (Aitken and Whitlock 2013). This action, called assisted gene flow, has been advocated for long-lived, sessile species such as trees (e.g., Steane et al. 2014), and species that have a limited ability to track climate conditions to which they are adapted (Sgro et al. 2011). While such interventions include risks, for many populations and species that either lack the capacity for long-distance movement or have no available suitable habitats to disperse into, introduction of adaptive genetic variation may be the only possible path to persistence. In these cases, consideration of potentially far-reaching benefits and careful evaluation to minimize the risks of assisted gene flow can provide an important option for the management of vulnerable populations (Weeks et al. 2011; Aitken and Whitlock 2013). Thus, despite uncertainty, information on adaptive potential based on genomics and other sources can help inform ESA listing and recovery decisions.
Incorporating adaptive potential into Endangered Species Act decisions in the face of uncertainty
Decisions about endangered species management always will be made in the face of uncertainty, making recognition and quantification of uncertainty associated with information used in decision making as important as the information itself (Runge et al. 2011). However, uncertainty need not be paralyzing. Although the best available information may not be perfect, it can only lead to poor decision making if uncertainty associated with it is not recognized. Decision analysis is the application of decision science to render decisions that are more likely to achieve management objectives, are more robust to uncertainty, and are more transparent to those outside the decision-making process (Keeney 1992; Gregory et al. 2012; Converse et al. 2013; Garrard et al. 2017). All decisions are composed of a consistent set of components including: the decision to be made, the management objectives of the decision maker, the alternative management actions under consideration, models designed to predict the consequences of each alternative on the management objectives, and some approach to solving the decision (frequently known as optimization). In decision analysis, we break the decision into these components to identify and tackle impediments to the decision.
Decision making under uncertainty is the impetus for a large set of methods in decision analysis. General approaches to dealing with uncertainty in decision making include: (1) characterizing uncertainty and deciding in the face of that uncertainty; (2) characterizing uncertainty and choosing to delay a decision while further information is gathered; or (3) characterizing uncertainty and choosing to decide while simultaneously learning. The last of these can only occur for iterated decisions, and is known as adaptive management (Walters 1986; Williams et al. 2007; Runge 2011).
In the context of this paper, we are interested in decisions—either listing or recovery decisions—to maximize the long-term viability of some taxon. To predict viability, we may need to predict how adaptive potential affects extinction risk. Including information on adaptive potential will require recognizing the substantial uncertainty around it, although ignoring it has the potential to introduce bias and under-represent uncertainty.
With listing decisions, we are interested in whether the adaptive potential of the species could change extinction risk. Based on observed survival and birth rates, we can predict probability of persistence as well as uncertainty around that prediction, and a manager can decide based on that information. However, if we consider that survival or birth rates might improve due to adaptation, our predicted probability of persistence will increase, while our uncertainty will now reflect uncertainty about the degree to which adaptation might increase these rates. As discussed above (see section on “How can information on adaptive potential improve Endangered Species Act decisions?”), integrating adaptive potential can move decisions away or towards listing.
In recovery decisions, we are interested in considering which management actions might improve the status of a listed species. In these cases, the role of adaptive potential is likely to be more nuanced. For example, perhaps a translocation (e.g., assisted gene flow) is contemplated because of changing climate in the species’ range, and uncertainty about whether the species has the capacity to adapt. But a translocation will reduce the viability of the species in its existing range because some individuals will be removed, and establishment in the new location is uncertain. Should a manager do the translocation or not? Here, it could result in greater danger to the species to ignore adaptive potential.
For these reasons, it is critical to contemplate how uncertainty, including uncertainty about the effect of adaptive potential, can be integrated into decisions. For one-time decisions, a manager can decide immediately or can delay the decision to learn. Two issues must be considered here: first, is it legally or politically feasible to delay, and second, is it worthwhile to delay? Answering the first question will require analysis of the social aspects of the decision. Answering the second question will require analysis of the value of information (Runge et al. 2011; Williams et al. 2011; Johnson et al. 2014; Canessa et al. 2015). Value of information is a set of methods for evaluating the expected increase in management performance associated with learning. We anticipate how much management outcomes might improve if we had additional information. Calculating the value of information often will require elicitation of expert judgment, because we are anticipating the value of something that we do not yet know, and so the analysis does not lend itself to empirical approaches. Runge et al. (2011) provide an overview, an example, and a comprehensive review of value of information.
Whether we do delay decisions to learn, or plan to learn as we manage, uncertainty will remain. Therefore, we will ultimately need to make decisions in the face of uncertainty about how management actions will affect extinction risk. When we consider that the species we manage may be undergoing adaptation, uncertainty is likely to be substantial. When making decisions under uncertainty, we are primarily concerned with characterizing that uncertainty and understanding how the risk attitude of the decision maker should be accounted for in the analysis. Characterizing the uncertainty involves estimating the probability of various outcomes, given a management alternative, via some predictive model. Integrating the risk attitude of the decision maker involves recognizing that a manager may have a non-linear utility function, whereby, for example, an action resulting in a relatively high predicted probability of persistence but relatively high uncertainty may be less preferred than an action resulting in a lower predicted probability of persistence but with relatively low uncertainty, such that the risk of particularly poor outcomes is overall lower under the preferred action. This is akin to preferring an investment portfolio that is lower return but also lower risk. A thorough analysis of the uncertainty around probability of persistence is critical in allowing us to integrate uncertainty, and risk tolerance, into our decision making. And accounting for as many factors as possible that influence risk, including adaptive potential, will allow us to produce the most thorough analysis of the state of our population under the actions considered.
Advancing understanding of the effect of adaptive potential on extinction risk
One of the main challenges to improving models of extinction risk that incorporate adaptive potential is estimating adaptive potential in traits important for fitness in the face of environmental change (e.g., thermal tolerance, disease resistance, susceptibility to environmental contaminants, resistance to or tolerance of invasive species, etc.). To parameterize extinction risk models that allow evolution, at a minimum, modelers need to know, or at least hypothesize: (1) how traits affect survival and birth rates (which is both a measure of selection on these traits and necessary to parameterize demographic models); and (2) the heritability of these traits. Mark-recapture analysis can be used to test how traits affect survival and birth rates (White and Burnham 1999). Genomic and other approaches are necessary to quantify the heritability of these traits. As described above, genomics can be used to infer relatedness among individuals, and thereby allow estimation of heritability of traits within a population. However, more research is needed to figure out how to integrate inferences from multiple genomic analyses to inform models of extinction risk, since alone, most of these analyses do not provide all necessary information for parameterizing these models. For example, genome-wide association studies (GWAS) identify loci related to variation in a trait of interest, but they do not test whether the trait is related to fitness or is adaptive. Genotype-environment association (GEA) approaches, in contrast, identify loci that are related to specific environmental features and presumably adaptive, but they do not determine which phenotypic traits mediate the fitness effects of these loci. It will clearly be necessary to integrate these different types of genomic analyses to identify traits that increase fitness in response to specific environmental stressors and that are heritable, so that this information can be incorporated into models of extinction risk. This is an important frontier in conservation genomics to make genomics more useful for informing extinction risk.
Controlled experiments, while impractical for most species of conservation concern, will remain important for testing under what conditions adaptation can rescue populations from extinction. Although these experiments do not necessarily directly inform extinction risk for specific species of conservation concern, they are nonetheless often the only means of rigorously testing evolutionary theory on the potential of adaptation to reduce extinction probabilities. For example, research on the adaptive potential of two rainforest-restricted fruit fly species demonstrated very low additive genetic variation (the substrate for adaptation) for desiccation resistance, even though other traits maintained high levels of genetic variation (Hoffmann et al. 2003; Kellermann et al. 2006). This result calls into question the generalization that most traits will maintain sufficient additive genetic variation to ensure adaptive potential (Blows and Hoffmann 2005), and also illustrates that trait-specific measures of genetic variance are not necessarily indicative of overall adaptive capacity. In addition, experimental studies of model species allow testing management strategies as a proof of concept in the lab. For example, experimental evolution in yeast populations has provided evidence not only for the efficacy of evolutionary rescue (an increase in population growth and avoidance of extinction through adaptation from standing genetic variation, mutation, or gene flow), but also for the environmental, demographic, and selective conditions under which it is most likely to occur (Bell and Gonzalez 2009, 2011). Additional experimental studies such as these are needed to better characterize thresholds related to adaptive potential, including levels of additive genetic variance required for adaptive responses to different rates and magnitudes of environmental change, and to provide guidelines for management actions such as assisted gene flow.
Finally, ongoing studies of wild populations that leverage the power of genomics to inform adaptive potential are needed to better characterize evolutionary responses to climate and other environmental change. As genomic studies become more common, comparative genomics will be one avenue for investigating the mechanisms underlying loss of adaptive potential in threatened species and taxonomic groups and the resulting implications for extinction risk. For example, a comparative study of the genomes of 43 bird species, including eight species recovering from endangered or vulnerable status, showed loss of adaptive variation related to agrochemical pollution (Li et al. 2014). Meta-analyses will also be essential for developing a more general understanding of the genomic and environmental landscape of adaptive potential, including under what circumstances populations may adapt, or fail to adapt, to changing conditions (Merilä and Hendry 2014). The increasing use of genomic approaches to characterize adaptive potential in wild populations will facilitate these efforts.
Conclusions
Genomics has the potential to improve ESA listing and recovery decisions—and similar decisions in other countries—by providing information on adaptive potential for wild populations for which it is difficult or impossible to characterize adaptation using traditional approaches like controlled breeding or reciprocal transplant experiments. Incorporating this information into population models will lead to more accurate estimates of extinction risk, improving decision making and allocation of scarce conservation resources. In this paper, we provide specific guidelines on where in the listing and recovery decision making workflows this information is most pertinent. Although genomics, like any scientific tool, is imperfect, we cannot afford to be paralyzed by uncertainty in using this information to make decisions. A rich decision theoretic framework has already been developed for making management decisions in the face of uncertainty, which can readily be applied to decisions involving inference about adaptive potential. At the same time, conservation geneticists should continue striving to improve our understanding of the effects of adaptive potential on extinction risk using modeling, controlled experimental studies of model species, and case studies of wild populations. This will help reduce uncertainty to improve future management decisions. Finally, we urge conservation geneticists to develop partnerships with conservation practitioners charged with making tough decisions regarding the conservation management of small, at-risk populations to facilitate integration of the best science on the effects of adaptive potential on extinction risk into these decisions.
References
Agashe D, Falk JJ, Bolnick DI (2011) Effects of founding genetic variation on adaptation to a novel resource. Evolution 65:2481–2491
Aitken SN, Whitlock MC (2013) Assisted gene flow to facilitate local adaptation to climate change. Annu Rev Ecol Evol Syst 44:367–388. https://doi.org/10.1146/annurev-ecolsys-110512-135747
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709
Allendorf FW, Luikart G, Aitken SN (2013) Conservation and the genetics of populations, 2nd edn. Wiley, Oxford
Alvarez M, Schrey AW, Richards CL (2015) Ten years of transcriptomics in wild populations: what have we learned about their ecology and evolution? Mol Ecol 24:710–725
Andrews KR, Good JM, Miller MR et al (2016) Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet 17:81–92. https://doi.org/10.1038/nrg.2015.28
Bataille A, Cashins SD, Grogan L, Skerratt LF, Hunter D, McFadden M, Scheele B, Brannelly LA, Macris A, Harlow PS, Bell S, Berger L, Waldman B (2015) Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc R Soc B 282:20143127
Beaumont MA, Balding DJ (2004) Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol 13:969–980
Bell G, Collins S (2008) Adaptation, extinction and global change. Evol Appl 1:3–16
Bell G, Gonzalez A (2009) Evolutionary rescue can prevent extinction following environmental change. Ecol Lett 12:942–948. https://doi.org/10.1111/j.1461-0248.2009.01350.x
Bell G, Gonzalez A (2011) Adaptation and evolutionary rescue in metapopulations experiencing environmental deterioration. Science 332:1327–1330. https://doi.org/10.1126/science.1203105
Berg JJ, Coop G (2014) A population genetic signal of polygenic adaptation. PLOS Genet 10:e1004412. https://doi.org/10.1371/journal.pgen.1004412
Bernatchez L (2016) On the maintenance of genetic variation and adaptation to environmental change: considerations from population genomics in fishes. J Fish Biol 89:2519–2556
Berven KA (1982) The genetic basis of altitudinal variation in the wood frog Rana sylvatica. I. An experimental analysis of life history traits. Evolution 36:962–983
Bierne N, Roze D, Welch JJ (2013) Pervasive selection or is it… why are FST outliers sometimes so frequent? Mol Ecol 22:2061–2064. https://doi.org/10.1111/mec.12241
Black WCIV, Baer CF, Antolin MF, DuTeau NM (2001) Population genomics: genome-wide sampling of insect populations. Annu Rev Entomol 46:441–469
Blows MW, Hoffmann AA (2005) A reassessment of genetic limits to evolutionary change. Ecology 86:1371–1384. https://doi.org/10.1890/04-1209
Bonin A, Nicole F, Pompanon F et al (2007) Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol 21:697–708. https://doi.org/10.1111/j.1523-1739.2007.00685.x
Boyd C, DeMaster DP, Waples RS, Ward EJ, Taylor BL (2017) Consistent extinction risk assessment under the U.S. Endangered Species Act. Conserv Lett 10:328–336. https://doi.org/10.1111/conl.12269
Brosi BJ, Biber EG (2009) Statistical inference, Type II error, and decision making under the US Endangered Species Act. Front Ecol Environ 7:487–494
Burger R, Lynch M (1995) Evolution and extinction in a changing environment—a quantitative-genetic analysis. Evolution 49:151–163
Canessa S, Guillera-Arroita G, Lahoz-Monfort JJ, Southwell DM, Armstrong DP, Chadès I, Lacy RC, Converse SJ (2015) When do we need more data? A primer on calculating the value of information for applied ecologists. Methods Ecol Evol 6:1219–1228
Carroll R, Augspurger C, Dobson A, Franklin J, Orians G, Reid W, Tracy R, Wilcove D, Wilson J (1996) Strengthening the use of science in achieving the goals of the endangered species act: an assessment by the Ecological Society of America. Ecol Appl 6:1–11
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Catchen JM, Hohenlohe PA, Bernatchez L et al (2017) Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol Ecol Resour 17:362–365. https://doi.org/10.1111/1755-0998.12669
Cattau CE, Fletcher RJ Jr, Kimball RT, Miller CW, Kitchens WM (2017) Rapid morphological change of a top predator with the invasion of a novel prey. Nature Ecol Evol 2(1):108
Claussen J, Keck DD, Hiesey WM (1948) Experimental studies on the nature of species. III. Environmental responses of climatic races of Achillea. Carnegie Institution of Washington Publication, Washington
Converse SJ, Moore CT, Armstrong DP (2013) Demographics of reintroduced populations: estimation, modeling, and decision analysis. J Wildl Manag 77:1081–1093
Converse SJ, Bailey LL, Mosher BA, Funk WC, Gerber BD, Muths E (2017) A model to inform management actions as a response to chytridiomycosis-associated decline. Ecohealth 14:S144–S155
Coop G, Witonsky D, Di Rienzo A, Pritchard JK (2010) Using environmental correlations to identify loci underlying local adaptation. Genetics 185:1411–1423
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. Trends Ecol Evol 15:290–295
Creech TG, Epps CW, Landguth EL et al (2017) Simulating the spread of selection-driven genotypes using landscape resistance models for desert bighorn sheep. PLoS ONE 12:e0176960. https://doi.org/10.1371/journal.pone.0176960
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
Dawson TP, Jackson ST, House JI et al (2011) Beyond predictions: biodiversity conservation in a changing climate. Science 332:53–58. https://doi.org/10.1126/science.1200303
de Villemereuil P, Frichot É, Bazin É et al (2014) Genome scan methods against more complex models: when and how much should we trust them? Mol Ecol 23:2006–2019. https://doi.org/10.1111/mec.12705
Doak DF, Himes Boor GK, Bakker VJ, Morris WF, Louthan A, Morrison SA, Stanley S, Crowder LB (2015) Recommendations for improving recovery criteria under the US Endangered Species Act. Bioscience 65:189–199
Dobzhansky T, Wright S (1941) Genetics of natural populations. V. Relations between mutation rate and accumulation of lethals in populations of Drosophila pseudoobscura. Genetics 26:23–51
Doremus H (1997) Listing decisions under the Endangered Species Act: why better science isn’t always better policy. Wash Univ Law Q 75:1029–1153
[ESA] US Endangered Species Act of 1973, as amended, Pub. L. No. 93-205, 87 Stat. 884 (28 Dec 1973). http://www.fws.gov/endangered/esa-library/pdf/ESAall.pdf
Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics. Longman, Harlow
Ficetola GF, De Bernardi F (2005) Supplementation or in situ conservation? Evidence of local adaptation in the Italian agile frog Rana latastei and consequences for the management of populations. Anim Conserv 8:33–40. https://doi.org/10.1017/S1367943004001805
Fisher RA (1930) The genetical theory of natural selection. Clarendon Press, Oxford
Forester BR, Lasky JR, Wagner HH, Urban DL (2018) Comparing methods for detecting multilocus adaptation with multivariate genotype-environment associations. Mol Ecol 27:2215–2233
François O, Martins H, Caye K, Schoville SD (2016) Controlling false discoveries in genome scans for selection. Mol Ecol 25:454–469
Frankham R, Lees K, Montgomery ME, England PR, Lowe EH, Briscoe DA (1999) Do population size bottlenecks reduce evolutionary potential? Anim Conserv 2:255–260
Frankham R, Ballou JD, Briscoe AD (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Frankham R, Brook BW, Bradshaw CJA, Traill LW, Spielman D (2013) 50/500 rule and minimum viable populations: response to Jamieson and Allendorf. Trends Ecol Evol 28:187–188
Franklin IR (1980) Evolutionary change in small populations. In: Soulé ME, Wilcox BA (eds) Conservation biology: an evolutionary-ecological perspective. Sinauer Associates, Sunderland
Franks SJ, Kane NC, O’Hara NB, Tittes S, Rest JS (2016) Rapid genome-wide evolution in Brassica rapa populations following drought revealed by sequencing of ancestral and descendant gene pools. Mol Ecol 25:3622–3631
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10:2741–2752
Frichot E, Schoville SD, Bouchard G, Francois O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30:1687–1699
Fritts TH (1984) Evolutionary divergence of giant tortoises in Galapagos. Biol J Linn Soc 21:165–176. https://doi.org/10.1111/j.1095-8312.1984.tb02059.x
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496. https://doi.org/10.1016/j.tree.2012.05.012
Garant D, Forde SE, Hendry AP (2007) The multifarious effects of dispersal and gene flow on contemporary adaptation. Funct Ecol 21:434–443
Garner BA, Hand BK, Amish SJ, Bernatchez L, Foster JT, Miller KM, Morin PA, Narum SR, O’Brien SJ, Roffler G, Templin WD, Sunnucks P, Strait J, Warheit KI, Seamons TR, Wenburg J, Olsen J, Luikart G (2016) Genomics in conservation: case studies and bridging the gap between data and application. Trends Ecol Evol 31:81–83
Garrard GE, Rumpff L, Runge MC, Converse SJ (2017) Rapid prototyping for decision structuring: an efficient approach to conservation decision analysis. In: Bunnefeld N, Nicholson E, Milner-Gulland E (eds) Decision-making in conservation and natural resource management: models for interdisciplinary approaches. Cambridge University Press, Cambridge
Gienapp P, Fior S, Guillaume F, Lasky JR, Sork VL, Csillery K (2017) Genomic quantitative genetics to study evolution in the wild. Trends Ecol Evol 32:897–908
Glenn TC (2011) Field guide to next-generation DNA sequencers. Mol Ecol Resour 11:759–769
Gregory R, Failing L, Harstone M, Long G, McDaniels T, Ohlson D (2012) Structured decision making: a practical guide to environmental management choices. Wiley, Oxford
Haig SM, D’Elia J (2010) Avian subspecies and the U.S. Endangered Species Act. Ornithol Monogr 67:24–34
Haldane JBS (1930) A mathematical theory of natural and artificial selection (VI Isolation). Proc Cambridge Philos Soc 26:220–230
Hansen MM, Olivieri I, Waller DM, Nielsen EE, Grp GW (2012) Monitoring adaptive genetic responses to environmental change. Mol Ecol 21:1311–1329
Hanson JO, Rhodes JR, Riginos C, Fuller RA (2017) Environmental and geographic variables are effective surrogates for genetic variation in conservation planning. PNAS 114:12755–12760
Harrisson KA, Pavlova A, Telonis-Scott M, Sunnucks P (2014) Using genomics to characterize evolutionary potential for conservation of wild populations. Evol Appl 7:1008–1025. https://doi.org/10.1111/eva.12149
Hendry AP, Kinnison MT, Heino M et al (2011) Evolutionary principles and their practical application. Evol Appl 4:159–183. https://doi.org/10.1111/j.1752-4571.2010.00165.x
Hess JE, Zendt JS, Matala AR, Narum SR (2016) Genetic basis of adult migration timing in anadromous steelhead discovered through multivariate association testing. Proc R Soc B 283. https://doi.org/10.1098/rspb.2015.3064
Hoffmann AA, Sgro CM (2011) Climate change and evolutionary adaptation. Nature 470:479–485
Hoffmann AA, Hallas RJ, Dean JA, Schiffer M (2003) Low potential for climatic stress adaptation in a rainforest Drosophila species. Science 301:100–102. https://doi.org/10.1126/science.1084296
Hoffmann AA, Sgro CM, Kristensen TN (2017) Revisting adaptive potential, population size, and conservation. Trends Ecol Evol 32:506–517. https://doi.org/10.1016/j.tree.2017.03.012
Hohenlohe PA, Day MD, Amish SJ, Miller MR, Kamps-Hughes N, Boyer MC, Muhlfeld CC, Allendorf FW, Johnson EA, Luikart G (2013) Genomic patterns of introgression in rainbow and westslope cutthroat trout illuminated by overlapping paired-end RAD sequencing. Mol Ecol 22:3002–3013
Hong EP, Park JW (2012) Sample size and statistical power calculation in genetic association studies. Genomics Inform 10:117–122. https://doi.org/10.5808/GI.2012.10.2.117
Jamieson IG, Allendorf FW (2012) How does the 50/500 rule apply to MVPs? Trends Ecol Evol 27:578–584
Jamieson IG, Allendorf FW (2013) A school of red herring: reply to Frankham et al. Trends Ecol Evol 28:188–189
Johnson FA, Hagan G, Palmer WE, Kemmerer M (2014) Uncertainty, robustness, and the value of information in managing a population of northern bobwhites. J Wildl Manag 78:531–539
Joost S, Bonin A, Bruford MW, Despres L, Conord C, Erhardt G, Taberlet P (2007) A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol 16:3955–3969
Keeney RL (1992) Value-focused thinking: a path to creative decision making. Harvard University Press, Cambridge
Keith D, Akçakaya HR, Butchart SHM, Collen B, Dulvy NK, Homes EE, Hutchings JA, Keinath D, Schwartz MK, Shelton AO, Wapes RS (2015) Temporal correlations in population trends: conservation implications from time-series analysis of diverse animal taxa. Biol Conserv 192:247–257
Kellermann VM, Heerwaarden B van, Hoffmann AA, Sgrò CM (2006) Very low additive genetic variance and evolutionary potential in multiple populations of two rainforest Drosophila species. Evolution 60:1104–1108. https://doi.org/10.2307/4095411
Kelly MW, Sanford E, Grosberg RK (2012) Limited potential for adaptation to climate change in a broadly distributed marine crustacean. Proc R Soc B 279:349–356
Kimura M, Crow JF (1963) The measurement of the effective population number. Evolution 17:279–288
Korte A, Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9:29
Kovach RP, Gharrett AJ, Tallmon DA (2012) Genetic change for earlier migration timing in a pink salmon population. Proc R Soc B 279:3870–3878
Lasky JR, Upadhyaya HD, Ramu P et al (2015) Genome-environment associations in sorghum landraces predict adaptive traits. Sci Adv 1:e1400218. https://doi.org/10.1126/sciadv.1400218
Lemmon AR, Emme SA, Lemmon EM (2012) Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst Biol 61:727–744
Li S, Li B, Cheng C et al (2014) Genomic signatures of near-extinction and rebirth of the crested ibis and other endangered bird species. Genome Biol 15:557. https://doi.org/10.1186/s13059-014-0557-1
Lowry DB, Hoban S, Kelley JL et al (2016) Breaking RAD: an evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol Ecol Resour 17:142–152. https://doi.org/10.1111/1755-0998.12635
Luikart G, England PR, Tallmon D, Jordan S, Taberlet P (2003) The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet 4:981–994
Manel S, Perrier C, Pratlong M, Abi-Rached L, Paganini J, Pontarotti P, Aurelle D (2016) Genomic resources and their influence on the detection of the signal of positive selection in genome scans. Mol Ecol 25:170–184
McKay JK, Bishop JG, Lin JZ, Richards JH, Sala A, Mitchell-Olds T (2001) Local adaptation across a climatic gradient despite small effective population size in the rare sapphire rockcress. Proc R Soc B 268:1715–1721
McKinney GJ, Larson WA, Seeb LW, Seeb JE (2017) RADseq provides unprecedented insights into molecular ecology and evolutionary genetics: comment on Breaking RAD by Lowry et al. (2016). Mol Ecol Resour 17:356–361. https://doi.org/10.1111/1755-0998.12649
Merilä J, Hendry AP (2014) Climate change, adaptation, and phenotypic plasticity: the problem and the evidence. Evol Appl 7:1–14. https://doi.org/10.1111/eva.12137
Muñoz NJ, Farrell AP, Heath JW, Neff BD (2015) Adaptive potential of a Pacific salmon challenged by climate change. Nat Clim Change 5:163–166
Nagy ES, Rice KJ (1997) Local adaptation in two subspecies of an annual plant: implications for migration and gene flow. Evolution 51:1079–1089
[NMFS and USFWS] National Marine Fisheries Serivice and U.S. Fish and Wildlife Service (2010) Interim endangered and threatened species recover planning guidance, version 1.3. http://www.nmfs.noaa.gov/pr/recovery/
Neel MC, Leidner AK, Haines A, Goble DD, Scott JM (2012) By the numbers: how is recovery defined by the US Endangered. Species Act? BioScience 62:646–657
Nicotra AB, Beever EA, Robertson AL et al (2015) Assessing the components of adaptive capacity to improve conservation and management efforts under global change. Conserv Biol 29:1268–1278. https://doi.org/10.1111/cobi.12522
O’Connor MI, Selig ER, Pinsky ML, Altermatt F (2012) Toward a conceptual synthesis for climate change responses. Glob Ecol Biogeogr 21:693–703
Payseur BA, Rieseberg LH (2016) A genomic perspective on hybridization and speciation. Mol Ecol 25:2337–2360
Penuelas J, Sardans J, Estiarte M, Ogaya R, Carnicer J, Coll M, Barbeta A, Rivas-Ubach A, Llusia J, Garbulsky M, Filella I, Jump AS (2013) Evidence of current impact of climate change on life: a walk from genes to the biosphere. Glob Chang Biol 19:2303–2338
Perrier C, Delahaie B, Charmantier A (2018) Heritability estimates from genomewide relatedness matrices in wild populations: application to a passerine, using a small sample size. Mol Ecol Resour (in press)
Prince DJ, O’Rourke SM, Thompson TQ, Ali OA, Lyman HS, Saglam IK, Hotaling TJ, Spidle AP, Miller MR (2017) The evolutionary basis of premature migration in Pacific salmon highlights the utility of genomics for informing conservation. Sci Adv 3:e1603198
Reed TE, Schindler DE, Hague MJ, Patterson DA, Meir E, Waples RS, Hinch SG (2011) Time to evolve? Potential evolutionary responses of Fraser River sockeye salmon to climate change and effects on persistence. PLoS ONE 6:e20380
Regan TJ, Taylor BL, Thompson GG, Cochrane JF, Ralls K, Runge MC, Merrick R (2013) Testing decision rules for categorizing species’ extinction risk to help develop quantitative listing criteria for the U.S. Endangered Species Act. Conserv Biol 27:821–831
Rellstab C, Gugerli F, Eckert AJ et al (2015) A practical guide to environmental association analysis in landscape genomics. Mol Ecol 24:4348–4370. https://doi.org/10.1111/mec.13322
Rohlf DJ (1991) Six biological reasons the Endangered Species Act doesn’t work—and what to do about it. Conserv Biol 5:273–282
Ruegg K, Bay RA, Anderson EC, Saracco JF, Harrigan RJ, Whitfield M, Paxton EH, Smith TB (2018) Ecological genomics predicts climate vulnerability in an endangered southwestern songbird. Ecol Lett (in press)
Ruhl JB (2004) The battle over the Endangered Species Act methodology. FSU College of Law, Public Law Research Paper No. 99
Runge MC (2011) An introduction to adaptive management for threatened and endangered species. J Fish Wildl Manag 2:220–233
Runge MC, Converse SJ, Lyons JE (2011) Which uncertainty? Using expert elicitation and expected value of information to design an adaptive program. Biol Conserv 144:1214–1223
Ryder OA (1986) Species conservation and systematics: the dilemma of subspecies. Trends Ecol Evol 1:9–10
Savage AE, Zamudio KR (2011) MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci USA 108:16705–16710
Scheffers BR, De Meester L, Bridge TCL, Hoffmann AA, Pandolfi JM, Corlett RT, Butchart SHM, Pearce-Kelly P, Kovacs KM, Dudgeon D, Pacifici M, Rondinini C, Foden WB, Martin TG, Mora C, Bickford D, Watson JEM (2016) The broad footprint of climate change from genes to biomes to people. Science 354:aaf7671
Sgro C, Lowe A, Hoffmann A (2011) Building evolutionary resilience for conserving biodiversity under climate change. Evol Appl 4:326–337. https://doi.org/10.1111/j.1752-4571.2010.00157.x
Shafer ABA, Wolf JBW, Alves PC, Bergstrom L, Bruford MW, Brannstrom I, Colling G, Dalen L, De Meester L, Ekblom R, Fawcett KD, Fior S, Hajibabaei M, Hill JA, Hoezel AR, Hoglund J, Jensen EL, Krause J, Kristensen TN, Krutzen M, McKay JK, Norman AJ, Ogden R, Ouml;sterling EM, Ouborg NJ, Piccolo J, Popovic D, Primmer CR, Reed FA, Roumet M, Salmona J, Schenekar T, Schwartz MK, Segelbacher G, Senn H, Thaulow J, Valtonen M, Veale A, Vergeer P, Vijay N, Vila C, Weissensteiner M, Wennerstrom L, Wheat CW, Zielinski P (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30:78–87
Shaffer ML, Stein MA (2000) Safeguarding our precious heritage. In: Stein BA, Kutner LS, Adams JS (eds) Precious heritage: the status of biodiversity in the United States. Oxford University Press, New York, pp 301–321
Sillanpää MJ (2011) On statistical methods for estimating heritability in wild populations. Mol Ecol 20:1324–1332
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Smith DR, Allan NL, McGowan CP, Szymanski JA, Oetker SR, Bell HM (2018) Development of a Species Status Assessment process for decisions under the U.S. Endangered Species Act. J Fish Wildl Manag 9:302–320
Sork VL (2018) Genomic studies of local adaptation in natural plant populations. J Hered 109:3–15
Stanton-Geddes J, Yoder JB, Briskine R, Young ND, Tiffin P (2013) Estimating heritability using genomic data. Methods Ecol Evol 4:1151–1158
Steane DA, Potts BM, McLean E et al (2014) Genome-wide scans detect adaptation to aridity in a widespread forest tree species. Mol Ecol 23:2500–2513. https://doi.org/10.1111/mec.12751
Steffen W, Broadgate W, Deutsch L, Gaffney O, Ludwig C (2015) The trajectory of the Anthropocene: the Great Acceleration. Anthropocene Rev 2:81–98
Szymanski J, Smith T, Horton A, Parkin M, Ragan L, Masson G, Olson E, Gifford K, Hill L (2016a) Rusty patched bumble bee (Bombus affinis) species status assessment. U.S. Fish and Wildlife Service, Final Report, Version 1
Szymanski J, Pollack C, Ragan L, Redmer M, Clemency L, Voorhies K, JaKa J (2016b) Species status assessment for the eastern massasauga rattlesnake (Sistrurus catenatus). U.S. Fish and Wildlife Service, SSA Report Version 2
Taylor MFJ, Suckling KF, Rachlinski JJ (2005) The effectiveness of the Endangered Species Act: a quantitative analysis. Bioscience 55:360–367
Theimer TC, Smith AD, Mahoney SM, Ironside KE (2016) Available data support protection of the Southwestern willow flycatcher under the Endangered Species Act. Condor 118:289–299
Thomas L, Palumbi SR (2017) The genomics of recovery from coral bleaching. Proc R Soc B https://doi.org/10.1098/rspb.2017.1790
Thorpe RS, Reardon JT, Malhotra A (2005) Common garden and natural selection experiments support ecotypic differentiation in the Dominican anole (Anolis oculatus). Am Nat 165:495–504
Todd EV, Black MA, Gemmell NJ (2016) The power and promise of RNA-seq in ecology and evolution. Mol Ecol 25:1224–1241. https://doi.org/10.1111/mec.13526
U.S. Fish and Wildlife Service (1995) Endangered and threatened wildlife and plants; final rule determining endangered status for the Southwestern Willow Flycatcher. Fed Reg 60:10694–10715
[USFWS and NMFS] U.S. Fish and Wildlife Service and National Marine Fisheries Service (1996) Policy regarding the recognition of distinct vertebrate population segments under the Endangered Species Act. Fed Reg 61:4722–4725
U.S. Fish and Wildlife Service (2016) Endangered and threatened wildlife and plants; threatened species status for rusty patched bumble bee. Fed Reg 82:3186–3208
U.S. Fish and Wildlife Service (2017) Endangered and threatened wildlife and plants; endangered species status for the eastern massasauga rattlesnake. Fed Reg 81:667214–671936
Verhoeven KJF, Vonholdt BM, Sork VL (2016) Epigenetics in ecology and evolution: what we know and what we need to know. Mol Ecol 25:1631–1638
Visscher PM, Goddard ME (2015) A general unified framework to assess the sampling variance of heritability estimates using pedigree or marker-based relationships. Genetics 199:223–232
Visscher PM, Hill WG, Wray NR (2008) Heritability in the genomics era—concepts and misconceptions. Nat Rev Genet 9:255–266
Vucetich JA, Nelson MP, Phillips MK (2006) The normative dimension and legal meaning of endangered and recovery in the US Endangered Species Act. Conserv Biol 20:1383–1390
Walters C (1986) Adaptive management of renewable resources. MacMillan, New York
Waples RS (1991) Pacific salmon, Oncorhynchus spp., and the definition of ‘species’ under the Endangered Species Act. Act Mar Fish Rev 53:11–22
Waples RS, Nammack M, Cochrane JF, Hutchings JA (2013) A tale of two acts: endangered species listing practices in Canada and the United States. Bioscience 63:723–734
Waples RK, Larson WA, Waples RS (2016) Estimating contemporary effective population size in non-model species using linkage disequilibrium across thousands of loci. Heredity 117:233–240
Weeks AR, Sgro CM, Young AG et al (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4:709–725. https://doi.org/10.1111/j.1752-4571.2011.00192.x
White GC, Burnham KP (1999) Program MARK: survival estimation from populations of marked animals. Bird Study Suppl 46:120–138
Williams BK, Szaro RC, Shapiro CD (2007) Adaptive management: the U.S. Department of the Interior Technical Guide. Adaptive Management Working Group, U.S. Department of the Interior, Washington DC
Williams BK, Eaton MJ, Breininger DR (2011) Adaptive resource management and the value of information. Ecol Modell 222:3429–3436
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wright S (1938) Size of population and breeding structure in relation to evolution. Science 87:430–431
Wright S (1943) Isolation by distance. Genetics 28:114–138
Zink RM (2015) Genetics, morphology, and ecological niche modeling do not support the subspecies status of the endangered willow flycatcher (Empidonax traillii extimus). Condor 117:78–86
Acknowledgements
We first thank A. Rus Hoelzel for inviting WCF to the Discussion Meeting at Durham University in September 2017 on “Conserving Adaptive Potential and Functional Diversity.” Without Rus’ initiative and perseverance in organizing this meeting, we would not have been spurred to assemble the diverse team of coauthors needed to bring together the ideas outlined here. We also thank Robin Waples and Gordon Luikart for excellent suggestions for improving the manuscript. We acknowledge funding from Colorado State University and National Science Foundation Ecology of Infectious Diseases Grant (DEB 1413925). The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the US Fish and Wildlife Service.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Funk, W.C., Forester, B.R., Converse, S.J. et al. Improving conservation policy with genomics: a guide to integrating adaptive potential into U.S. Endangered Species Act decisions for conservation practitioners and geneticists. Conserv Genet 20, 115–134 (2019). https://doi.org/10.1007/s10592-018-1096-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-018-1096-1