
Abstract
This chapter describes a transient protoplast co-transfection method that can be used to quantitatively study in vivo the activity and function of promoters and promoter elements (reporters), and their induction or repression by transcription factors (effectors), stresses, hormones, or metabolites. A detailed protocol for carrying out transient co-transfection assays with Arabidopsis At7 protoplasts and calculating the promoter activity is provided.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
1 Introduction
For high-level constitutive expression, or for precise control of transgene activity in response to a specific stimulus, promoters are the key for successful genetic engineering strategies. Therefore, detailed knowledge about the concerted action of both cis- and trans-acting elements is necessary. Defining cis-acting elements and the characterization of transcription factors which bind and/or regulate a promoter of choice is a standard experimental approach. Since in vitro DNA-binding assays might not reflect the situation in a living cell, in vivo assays are favored. The yeast one-hybrid assay [1] is the method of choice for rapid detection of protein–DNA interactions. However, this experimental approach has some limitations for the analysis of plant transcription factors and promoters. Among these are different conditions inside the yeast nucleus compared to the situation in plants, additional transcriptional start sites in promoters larger than approximately 300 bps in length, and false positives due to regulatory proteins with high affinity to unspecific DNA regions [2]. For the analysis of (synthetic) plant promoters , the use of plant cell systems avoids most of the disadvantages of yeast systems. Furthermore, plant systems provide necessary perception and signaling systems for the study of signal-induced processes.
Transient protoplast co-transfection systems are used for the study of unbiased activity and function of promoter elements, as well as for promoter activation or repression by a single or multiple transcription factors or several stresses or hormone treatments. In this respect, the transfection assay is focused on the analysis of factors that control the activity of a given promoter, and the role of proximal 5′-upstream cis-regulatory elements or sequences. These elements are important and central components for accurate and refined synthetic promoter design. Transient protoplast co-transfection assays allow fast access to results, especially when compared with stable transformation . In addition, they are unaffected by position effects caused by features of the site of transgene integration and by the copy number of inserted transgenes. The drawback is that cell type specificity or developmental control of promoter activity can usually not be studied in protoplasts from cultured cells.
The protoplast assay system employs purified plasmid DNA introduced into the cells via PEG-mediated DNA uptake. Various plasmids can be introduced at the same time (co-transfection). The protoplast assay system relies on assessing the level of gene expression for an engineered promoter construction that drives the expression of a reporter gene. The level of expression of the reporter, in the case described here, β-glucuronidase ( uidA , GUS) is taken as a measure for promoter activity. Thus, transient protoplast co-transfection assays have provided a wealth of information about cis elements required for promoter function, transcription factors, and signaling proteins that regulate expression of genes and signals regulating inducible gene expression [3–8].
2 Materials
2.1 Cells, Buffers, and Solutions
-
1.
At7 cell culture : hypocotyl-derived A. thaliana Columbia cell culture [9], maintained at 26 °C in darkness on a rotary shaker, weekly subcultured.
-
2.
dam − E. coli: Methylation-deficient dam and dcm E. coli strain K12 ER2925 (NEB).
-
3.
1000× 2,4-Dichlorophenoxyacetic acid (2,4-D): 1 mg/mL.
-
4.
MS medium: 4.3 g Murashige and Skoog Basal Salt Mixture (MS, Sigma-Aldrich), 1 mL 1000× 2,4-D, 10 mL 1000× Gamborg’s Vitamin Solution (Sigma-Aldrich), 30 g sucrose. Adjust to pH 5.7 with 1 M KOH and bring to 1 L with deionized H2O. Autoclave.
-
5.
Cellulase-mazerozyme solution: 1.16 % (w/v) cellulase Onozuka R-10 (Serva), 0.27 % mazerozyme R-10 (Serva). Solve in 240 mM CaCl2, stir cautiously until enzymes are dissolved (1–1.5 h). Pass enzyme solution through a folded filter paper, then filter sterilize.
-
6.
B-5 floating medium (B5 solution): 3.1 g Gamborg’s B-5 Basal Salt Mixture (Sigma-Aldrich), 1 mL 1000× 2,4-D, 136 g sucrose. Adjust to pH 5.7 with 1 M NaOH and bring to 1 L with deionized H2O. Filter sterilize.
-
7.
240 mM CaCl2. Autoclave.
-
8.
PEG solution: 125 g PEG 6000, 11.8 g Ca(NO3)2 × 4H2O, 41 g mannitol. Adjust to pH 9 with 1 M KOH and bring to 0.5 L with deionized H2O. Filter sterilize and store in 5 mL aliquots at −20 °C.
-
9.
275 mM Ca(NO3)2: Adjust to pH 6.0 with 1 M KOH. Autoclave.
-
10.
0.1 M K2HPO4 and 0.1 M KH2PO4. Autoclave.
-
11.
0.1 M Potassium phosphate: Mix the appropriate volumes of 0.1 M K2HPO4 and 0.1 M KH2PO4 for a desired pH of 7.0. Store at 4 °C up to 1 month.
-
12.
Protein extraction buffer: 100 mM potassium phosphate, 1 mM DTT (Sigma-Aldrich). Filter sterilize and store at 4 °C.
-
13.
2× Luciferase assay stock solution: 40 mM tricine, 2.14 mM Mg(CO3)4Mg(OH)2 × 5H2O, 5.34 mM MgSO4 × 7H2O, 0.2 mM EDTA. Store at 4 °C.
-
14.
Luciferase substrate solution: 1× luciferase assay stock solution, 33.3 mM DTT (Sigma-Aldrich), 270 μM CoA trilithium salt (Sigma-Aldrich), 470 μM luciferin (Roche), 570 μM ATP (Sigma-Aldrich). CoA trilithium salt and luciferin are light-sensitive, keep in the dark. Check pH and adjust to pH 7.5 if necessary. Filter sterilize and store in 5 mL aliquots at −80 °C in light-tight tubes.
-
15.
0.5 M Na2HPO4 and 0.5 M NaH2PO4. Autoclave.
-
16.
0.5 M Sodium phosphate buffer: Mix the appropriate volumes of 0.5 M Na2HPO4 and 0.5 M NaH2PO4 for a desired pH of 7.0. Store at 4 °C up to 1 month.
-
17.
GUS buffer: 50 mM sodium phosphate buffer, 1 mM EDTA pH 8.0, 0.1 % (v/v) Triton X-100, 10 mM β-mercaptoethanol.
-
18.
4-MUG substrate solution: 20 mM 4-methylumbelliferyl-β-D-glucopyranosiduronic acid (4-MUG, Sigma-Aldrich). Solve in GUS buffer. Filter sterilize and store in 15 mL aliquots at −20 °C.
-
19.
MU stock solution: 10 mM 4-methylumbelliferone (MU, Sigma-Aldrich). Solve in ethanol. Store at 4 °C.
-
20.
MU dilution series: Dilute the MU stock solution with GUS buffer to 0, 2.5, 5.0, 12.5, 25, 50, 100, 150, 200, and 250 μM.
-
21.
BSA dilution series: Dilute a 10 mg/mL BSA stock solution with protein extraction buffer to 0, 2, 4, 8, and 16 μg per 10 μL buffer.
-
22.
Protein assay dye reagent: Dilute the protein assay dye reagent concentrate (Bio-Rad) 1:5 with deionized H2O.
2.2 Equipment
-
1.
Laminar flow cabinet.
-
2.
50 mL Falcon tubes.
-
3.
13 mL centrifuge tubes.
-
4.
1.5 mL reaction tubes.
-
5.
Cell-Saver tips (Biozyme Scientific).
-
6.
Petri dishes (145 mm).
-
7.
Folded filter paper.
-
8.
Sterile filter units with 0.22 μm pore size.
-
9.
Incubator.
-
10.
Rotary shaker.
-
11.
Centrifuge with swing-out rotor; programmable settings should include the specification of acceleration/deceleration rates (see Note 8 ).
-
12.
Benchtop centrifuge.
-
13.
Plasmid purification kit with prepacked gravity-flow anion-exchange columns in maxi (500 μg) or mega (2.5 mg) scale; e.g. Plasmid Mega Kit (Qiagen), JETstar Plasmid Purification MIDI Kit (Genomed).
-
14.
Fluid aspiration system.
-
15.
Hemocytometer.
-
16.
Vortexer.
-
17.
FLUOstar Optima microplate reader (BMG Labtech) equipped with an on-board syringe injector (see Note 1 ).
-
18.
96-well microplates white LUMITRAC 200 (Greiner) (see Note 1 ).
-
19.
96-well microplates black FLUOTRAC 200 (Greiner) (see Note 1 ).
-
20.
Nunc™ MicroWell™ 96-well microplates (Nunc) (see Note 1 ).
-
21.
Equipment for maintenance of a cell suspension culture.
-
22.
Nylon net filter with 70 μm pore size (optional, see Note 10 ).
2.3 Plasmids
-
1.
Reporter constructs
Reporter constructs are based on the vector pBT10GUS [5] or the Gateway-compatible derivative pDISCO [10]. In both cases the promoter (fragment) to analyze is inserted or recombined upstream of the uidA ORF:nos terminator cassette (see Notes 2 and 3 ).
In case of testing the activity of a transcriptional repressor, the construction of a weakly active synthetic promoter is needed (see Note 4 ).
-
2.
Effector expression constructs
Effectors were expressed under the control of the strong constitutive CaMV 35S promoter (positions −417 to +8), inserted into classical pBT vectors [3] or the Gateway-compatible derivative pBTdest [6].
-
3.
LUC standardization plasmid
The LUC standardization plasmid used in the co-transfection assays contains a Photinus pyralis luciferase (LUC) encoding open reading frame [11] under the control of the constitutive Petroselinum crispum UBI4-2 promoter [12] in pBT (see Notes 5 and 6 ).
-
4.
Filling plasmid
The promoter-deleted standardization plasmid pBT10-Δ-LUC, which in protoplasts leads to no detectable luciferase activity, is added to keep the total amount of the transfected plasmid DNA constant (25 μg).
3 Methods
This protocol uses A. thaliana At7 cells from a cell suspension culture which is maintained at 26 °C in the dark on a rotary shaker at 105 rpm. Cells are subcultured once a week by transferring approximately 2.8 g of cells to 40 mL fresh MS-medium. For protoplast isolation , additional Erlenmeyer flasks with 40 mL MS-medium were inoculated 5 days prior to the harvesting of the cells.
3.1 Protoplast Preparation
-
1.
For each subcultivated Erlenmeyer flask of At7 cells with 40 mL cell culture prepare 60 mL of cellulase-mazerozyme solution (see Note 7 ).
-
2.
Transfer each 5-day-old At7 cell suspension subculture to a 50 mL Falcon tube.
-
3.
Centrifuge 5 min at 130 × g with moderate acceleration (5/9) and deceleration (3/9) in a swing-out rotor (see Note 8 ).
-
4.
Discard the supernatant carefully.
-
5.
Detach cell pellet with caution by soft tapping.
-
6.
Add 50 mL 240 mM CaCl2 and resuspend the cells by gentle inversion of the tube.
-
7.
Centrifuge 5 min at 130 × g with moderate acceleration (5/9) and deceleration (3/9) in a swing-out rotor.
-
8.
Discard the supernatant carefully.
-
9.
For each harvested At7 cell suspension flask, prepare two 145 mm Petri dishes each with 10 mL cellulase-mazerozyme solution (see Note 7 ).
-
10.
Little by little, add the residual 40 mL of the enzyme solution to the cell pellet and gently resuspend by inversion. Avoid cell clumping.
-
11.
Add half of the cell suspension (about 20 mL) to each of the prepared Petri dishes.
-
12.
Incubate overnight at 26 °C in the dark, shake at 20 rpm.
-
13.
Intensify the shaking to 40 rpm for no longer than 20 min.
-
14.
From each Petri dish, carefully transfer the protoplasts into a 50 mL Falcon tube (see Note 9 ).
-
15.
Centrifuge 5 min at 90 × g with moderate acceleration (5/9) and deceleration (3/9) in a swing-out rotor.
-
16.
Discard the supernatant carefully.
-
17.
Detach the pellet with caution by soft tapping.
-
18.
Wash the protoplasts by little and little adding 25 mL 240 mM CaCl2 and resuspend the protoplasts by gentle inversion of the tube (see Note 10 ).
-
19.
Centrifuge 5 min at 90 × g with moderate acceleration (5/9) and deceleration (3/9) in a swing-out rotor.
-
20.
Discard the supernatant carefully.
-
21.
Detach the pellet with caution by soft tapping.
-
22.
Little by little add 20 mL B-5 floating medium to each pellet and combine two resuspended pellets from 50 mL Falcon tubes in one tube (see Note 11 )
-
23.
Centrifuge 5 min at 130 × g with maximal acceleration (9/9) and minimal deceleration (1/9) in a swing-out rotor.
-
24.
Transfer the floating protoplasts (2–5 mL) with a Cell-Saver tip into a new 50 mL Falcon tube (see Note 12 ).
-
25.
Cautiously fill the Falcon tube with B-5 floating medium.
-
26.
Centrifuge 5 min at 130 × g with maximal acceleration (9/9) and minimal deceleration (1/9) in a swing-out rotor.
-
27.
Pool all floating protoplasts in a 13 mL centrifuge tube using a Cell-saver tip.
-
28.
Assess the quality of the protoplast suspension (see Note 13 ).
-
29.
Use the protoplasts immediately for transfection. 200 μL of protoplasts (containing 1–2 × 106 At7 protoplasts) are needed per co-transfection (see Note 14 ).
3.2 Preparing DNA for Co-transfection
-
1.
Plasmids to be used in protoplast co-transfection experiments are retransformed into the dam and dcm methylation-deficient E. coli strain K12 ER2925 (NEB) (see Note 15 ).
-
2.
Plasmid DNA is prepared from methylation-deficient E. coli strain using a plasmid purification kit with prepacked gravity-flow anion-exchange columns in maxi (500 μg) or mega (2.5 mg) scale according to manufacturer’s instructions.
-
3.
The high concentrated dam − plasmid DNA should be stored at 4 °C (see Note 16 ).
-
4.
For co-transfection experiments, plasmid dilutions have to be made: reporter constructs, standardization constructs and “filling” constructs with 1 μg/μL, effector constructs with 0.1 μg/μL (Fig. 1).
Fig. 1 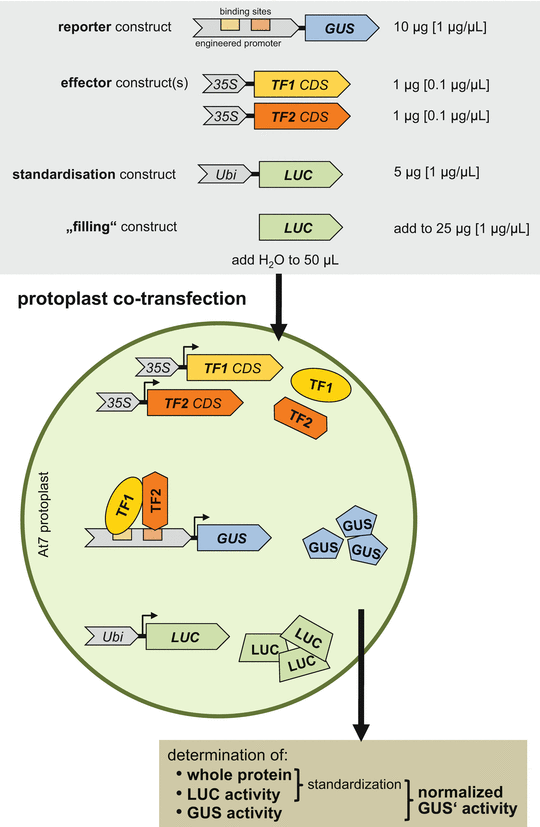
Schematic depiction of transient protoplast expression assay with co-transfected At7 protoplasts. The gray box at the top gives the plasmids (constructs) used in co-transfection experiments with the amounts and concentrations specified. The middle part depicts a transfected At7 protoplast with plasmids (italics) and the produced proteins (normal letters) and their interactions. The “filling” plasmid is not considered. The brown box at the bottom summarizes the calculation of normalized specific GUS′ activity, as a measure for promoter activity. Abbreviations: 35S cauliflower mosaic virus 35S promoter, CDS coding sequence, GUS β-glucuronidase , LUC luciferase , TF transcription factor, Ubi ubiquitin4-2 promoter
-
5.
Combined plasmid DNA solutions for co-transfection should be prepared in advance to enable a smooth execution of the protocol. All combined DNA solutions should contain an equal amount of plasmid DNA (25 μg) in a volume of 50 μL (Fig. 1). The use of a positive control (35S::GUS reporter construct) and a negative control (TATA::GUS reporter construct, containing only the truncated −46 minimal promoter (TATA)) is recommended in the experimental setup (see Note 17 ).

3.3 Protoplast Co-transfection
-
1.
Thaw PEG solution at room temperature (see Note 18 ).
-
2.
For each co-transfection transfer 200 μL of the protoplast suspension with a Cell-Saver tip into a 13 mL centrifuge tube.
-
3.
Pipet the prepared DNA solutions (25 μg in 50 μL) onto the protoplasts (see Note 19 ).
-
4.
Add 200 μL PEG solution and mix thoroughly but gently by soft shaking and tapping of the tube rack (see Note 20 ).
-
5.
Incubate the protoplast–DNA–PEG mixture at room temperature for 15 min (see Note 21 ).
-
6.
Stop the transfection reaction by stepwise adding 5 mL 275 mM Ca(NO3)2 (see Note 22 ).
-
7.
Centrifuge 5 min at 90 × g with maximal acceleration (9/9) and moderate deceleration (5/9) in a swing-out rotor.
-
8.
Discard the supernatant carefully.
-
9.
Stepwise add 7 mL B5 solution (see Note 23 ).
-
10.
Incubate at 26 °C for approximately 20 h in the dark, keeping the tubes in an almost horizontal position (see Note 24 ).
3.4 Harvesting of Transfected Protoplasts
-
1.
Prepare 50 mL Falcon tubes with 20 mL of cold 240 mM CaCl2 (4 °C) for each co-transfection.
-
2.
Add the protoplasts within the B5 solution to the tubes by decanting.
-
3.
Centrifuge for 10 min at 300 × g and 4 °C in a swing-out rotor.
-
4.
Remove the supernatant with a fluid aspiration system down to ca. 1 mL.
-
5.
Resuspend the pellet in the remaining supernatant and transfer the protoplast suspension into a 1.5 mL reaction tubes using Cell-Saver tips.
-
6.
Centrifuge 30 s at 10,000 × g and 4 °C.
-
7.
Remove the supernatant using the fluid aspiration system and instantly freeze the protoplast pellet in liquid nitrogen.
-
8.
Store the frozen protoplasts at −80 °C until use or thaw on ice for following protein extraction.
3.5 Extract Proteins from Protoplasts
-
1.
Thaw the protoplasts on ice.
-
2.
Add 750 μL of protein extraction buffer.
-
3.
Resuspend the pellet by vortexing rigorously for a minimum of 30 s.
-
4.
Centrifuge for 10 min at full speed on 4 °C in a benchtop centrifuge.
-
5.
Keep the reaction tubes on ice; the supernatant is used for the protein and reporter gene assays (see Note 25 ).
3.6 Reporter Gene Assays
3.6.1 Luciferase Assay
In this assay the luciferase activity is quantified as a measure for transfection efficiency (see Note 25 ).
-
1.
Pipet 10 μL of the protoplast protein extracts to the wells of a white LUMITRAC 96-well microplate.
-
2.
Adjust the instrument settings to luminescence detection.
-
3.
Fill the syringe injector of the FLUOstar Optima microplate reader with 100 μL luciferase substrate solution for each sample and start the luminescence measurement (see Note 26 ).
-
4.
Measure the produced light (relative light unit, RLU) during 10 s using the FLUOstar Optima microplate reader.
-
5.
Calculate the specific luciferase activity LUC i [RLU/s/μg] for each co-transfection by dividing the measured light [RLU/s/μL] by the protein concentration [μg/μL].
$$ {\mathrm{LUC}}_i=\frac{\frac{\mathrm{RLU}}{\mathrm{s}\times \mu \mathrm{L}}}{\frac{\mu \mathrm{g}\kern0.28em \mathrm{protein}}{\mu \mathrm{L}}} $$
3.6.2 GUS Assay
Fluorometric analysis allows quantification of GUS activity. In the presence of GUS, MUG is hydrolyzed to the fluorescent product 4-methylumbelliferone (MU). After the reaction, total fluorescence is measured and product concentration is calculated based on a MU standardization curve.
-
1.
Pipet 100 μL of the protein extracts into the wells of a black FLUOTRAC 200 96-well microplate.
-
2.
Add 100 μL of 4-MUG substrate solution to each protein extract.
-
3.
Pipet the MU dilution series to the microplate as well. This series is used to generate a MU standardization curve.
-
4.
Set excitation to 365 nm and read the sample emission at 455 nm after 20, 40, and 60 min at 37 °C in the FLUOstar Optima microplate reader.
-
5.
Determine the average change in measured MU fluorescence (ΔE455) from 20 to 40 min, and from 40 to 60 min (see Note 27 ).
-
6.
Calculate the line of best fit for the MU fluorescence in the MU dilution series (MU standardization curve). Determine the slope (m) of the MU standardization curve.
-
7.
The specific β-glucuronidase activity GUS i [pmol/min/mg] is determined by the formula:
$$ {\mathrm{GUS}}_i=\frac{\varDelta {\mathrm{E}}_{455}\times 1000\frac{\mu \mathrm{g}}{\mathrm{mg}}\times \frac{200\mu \mathrm{L}}{20\mu \mathrm{L}}}{20 \min \times \frac{m}{\mathrm{pmol}}\times \mu \mathrm{g}\kern0.28em \mathrm{protein}} $$The GUS activity measurement uses 20 μL of a 200 μL sample (consisting of 100 μL protein extract and 100 μL 4-MUG substrate solution).
The amount of protein [μg] in 100 μL protein extract is given.
3.7 Protein Concentration Measurement
To determine the protein concentration in the protein extract samples, a Bradford assay [13] is performed.
-
1.
Pipet 10 μL of the protein extracts to the wells of a Nunc™ MicroWell™ 96-well microplate.
-
2.
Pipet 10 μL of the BSA dilution series to the microplate as well. This series is used to generate a protein standardization curve.
-
3.
Mix with 200 μL protein assay dye reagent.
-
4.
Incubate for 5 min at 37 °C.
-
5.
Measure OD595 in the FLUOstar Optima microplate reader.
-
6.
Calculate the protein concentrations in the extracts with help of the BSA dilution series.
-
7.
Determine the amount of protein [μg] in 10 μL protein extract and calculate the protein concentration [μg/μL].
3.8 Calculation of the Normalized Specific GUS′ Activity (See Note 28 )
-
1.
We normally repeat each co-transfection experiment (with the same combination of plasmids) six times, with six independent (i) co-transfections with three different protoplast preparations, giving an “experimental block”. A “whole experiment”, including controls and all related experiments to answer a biological question, consists of several experimental blocks.
-
2.
Calculate the average of all specific LUC i values (LUC M ) from a whole experiment.
$$ {\mathrm{LUC}}_M=\frac{1}{n}\times {\displaystyle \sum }{\mathrm{LUC}}_i $$n: sum of all co-transfections in a whole experiment
-
3.
For standardization, a specific correction factor F i for each individual co-transfection experiment is determined by dividing LUC M by the specific LUC i value.
$$ {F}_i=\frac{{\mathrm{LUC}}_M}{{\mathrm{LUC}}_i} $$ -
4.
The standardized, corrected GUS activity (GUS ki ) is obtained by multiplying the specific correction factor F i with the specific GUS activity GUS i .
$$ {\mathrm{GUS}}_{ki}={F}_i\times {\mathrm{GUS}}_i $$ -
5.
The average of specific GUS ki values of an experimental block of six co-transfections is calculated as standardized GUS activity (GUS′).
$$ {\mathrm{GUS}}^{\prime }=\frac{1}{6}\times {\displaystyle \sum }{\mathrm{GUS}}_{ki} $$ -
6.
The standard deviation of GUS′ (SD(GUS′)) is determined by the formula:
$$ \mathrm{S}\mathrm{D}\left({\mathrm{GUS}}^{\prime}\right)=\frac{1}{\sqrt{6\left(6-1\right)}}\times \sqrt{{\displaystyle \sum }{\left({\mathrm{GUS}}_{ki}-{\mathrm{GUS}}^{\prime}\right)}^2} $$
4 Notes
-
1.
It is also possible to use alternative reaction containers and instruments for the protein, LUC , and GUS measurements.
-
2.
The transfection rate can be variable and depends on the plasmids used [5]. Generally, small vector sizes achieve higher transformation rates. The pBT vectors have a size-minimized backbone of 1989 bp containing colE1-ori and ampr [3].
-
3.
cis-acting elements often contain palindromic sequences. This has been interpreted as a reflection of the fact that DNA-binding proteins are in many cases active as dimers or tetramers, binding symmetrically to DNA matching the symmetry of the binding site [14]. Otherwise, several asymmetric cis-elements are known which are often recognized by heterodimeric trans-acting factors. Aiming to test the interaction of synthetic cis-elements and DNA binding proteins, we found it helpful to insert such binding elements in both orientations single copy, as a dimer, and as a tetramer fused to the GUS coding sequence in the pBT10GUS vector.
-
4.
A promoter with appreciable activity in At7 protoplasts in the absence of added effectors has been reported. This reporter construct contains the region between −90 to +8 of the CaMV 35S promoter (−90 CaMV 35S promoter), including an activation sequence factor 1 binding site (positions −83 to −63) [15], fused to uidA encoding GUS. Binding domains ( cis-elements ) to analyze can be cloned immediately upstream of the −90 CaMV 35S promoter in pBT10GUS [4].
-
5.
Addition of the LUC standardization plasmid in the co-transfection is used to determine specific LUC activity in a given sample to estimate the transfection rate (efficiency of transfection).
-
6.
The Photinus pyralis luciferase (LUC) encoding ORF contains three silent point mutations which remove XbaI, EcoRI, and ClaI sites [16].
-
7.
We observed strong differences between different lots of cellulase in terms of success in protoplasting. Each lot has to be tested and the amount of cellulase in the cellulase-mazerozyme solution has to be adjusted for successful protoplasting. Mazerozym is a multi-component enzyme mixture containing activities of pectinase, α amylase and hemicellulase; cellulase proteins hydrolyze 1,4-β-D-glucosidic bonds.
-
8.
We are using a Multifuge 1s (Heraeus) centrifuge with a TTH400 swing-out rotor (Heraeus) or a Multifuge 3s-r (Heraeus) centrifuge with a TTH750 swing-out rotor (Heraeus). These centrifuges have the option to specify acceleration and deceleration in ramps from 1 to 9. These parameters have been optimized to protect the delicate living samples.
-
9.
When harvesting the protoplasts from the Petri dishes, transfer the cell suspension into the Falcon tube by placing the brim of the Petri dish centrally over the tube and then decant carefully. Work above the inverted lid of the Petri dish to be able to recover eventually spilled protoplast suspension.
-
10.
When adding solutions to the protoplasts, never just pour the liquid on top of the protoplasts, rather hold the tube at a flat angle and let the liquid slowly run along the side of the tube. Slow rotation of the tube helps dissolving solid pellets.
-
11.
By washing with B5 floating medium, the living protoplasts are separated from the debris of broken cells. The high sugar concentration in the B5 floating medium causes floating of intact protoplasts.
-
12.
When pipetting living protoplasts, always use pipetting equipment which reduces the shearing force, such as Cell-Saver tips or pipettes with a wide tip orifice.
-
13.
When the floating protoplast suspension contains a high amount of cell clusters (caused by inefficient enzyme treatment for cell wall removal), it is possible to filter the protoplasts through a nylon net filter with 70 μm pore size. Although this filtering step drastically reduces the number of cells, the obtained protoplast suspension contains only the desired protoplasts.
-
14.
Protoplasts number is determined using a hemocytometer.
-
15.
The use of dam − plasmid DNA results in significantly reduced background activity, as shown for parsley protoplasts [17].
-
16.
In our hands, high-concentrated (>1 μg/μL) dam − plasmid DNA in TE (10 mM Tris–HCl pH 8, 1 mM EDTA) stored at 4 °C is stable for more than 10 years, and can be used in co-transfection experiments without known limitations.
-
17.
We recommend to generate and use a detailed pipetting plan.
-
18.
Thawing of PEG solution at room temperature could take a few hours.
-
19.
In order to ensure a simultaneous start of co-transfections for all experiments, do not mix the DNA and protoplasts at this point. If you are performing a small-scale experiment this is not as critical as if you are handling 40 co-transfection reactions at the same time.
-
20.
When using a dispenser to add the PEG solution, it is recommended to hold the tubes or the rack with the tubes at a flat angel and pipet to the side of the tube.
-
21.
Avoid to move the tubes during the incubation time.
-
22.
We found that splitting the pipetting of Ca(NO3)2 into two steps worked well to achieve a stopping of all reactions at around the same time. First pipet 1 mL to all the tubes and then add the remaining 4 mL.
-
23.
Split the pipetting of the 7 mL in at least two steps and first pipet 1 mL to all tubes. If you add the whole volume of B5 solution at once, it might result in clumping of the protoplasts.
-
24.
Almost horizontal incubation avoids contact of the liquid to the cap of the centrifugation tube, which might result in protoplasts stuck to the cap.
-
25.
The luciferase assay should be performed immediately after protein extraction as the luciferase is degraded in the extract. The recommended order in which the following measurements have to be performed is (1) luciferase assay, (2) GUS assay, and (3) protein concentration measurement.
-
26.
The luminescence measurement should immediately start after the addition of luciferase substrate solution. We implemented a program on the FLUOstar Optima microplate reader that adds 100 μL of luciferase substrate solution to the samples immediately before the luminescence is measured.
-
27.
Only assays with linear increase of the E455 values are taken into account.
-
28.
We recommend the generation and use of an Excel sheet for the calculations.
References
Fields S, Song O (1989) A novel genetic system to detect protein-protein interactions. Nature 340:245–247
Dobi KC, Winston F (2007) Analysis of transcriptional activation at a distance in Saccharomyces cerevisiae. Mol Cell Biol 27:5575–5586
Weisshaar B, Armstrong GA, Block A, da Costa e Silva O, Hahlbrock K (1991) Light-inducible and constitutively expressed DNA-binding proteins recognizing a plant promoter element with functional relevance in light responsiveness. EMBO J 10:1777–1786
Jin H, Cominelli E, Bailey P, Parr A, Mehrtens F, Jones J, Tonelli C, Weisshaar B, Martin C (2000) Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis. EMBO J 19:6150–6161
Sprenger-Haussels M, Weisshaar B (2000) Transactivation properties of parsley proline rich bZIP transcription factors. Plant J 22(1):1–8
Baudry A, Heim MA, Dubreucq B, Caboche M, Weisshaar B, Lepiniec L (2004) TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J 39:366–380
Zimmermann IM, Heim MA, Weisshaar B, Uhrig JF (2004) Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. Plant J 40:22–34
Stracke R, Favory J-J, Gubler H, Bartelniewöhner L, Bartels S, Binkert M, Funk M, Weisshaar B, Ulm R (2010) The Arabidopsis bZIP transcription factor HY5 regulates expression of the PFG1/MYB12 gene in response to light and ultraviolet-B radiation. Plant Cell Environ 33:88–103
Trezzini GF, Horrichs A, Somssich IE (1993) Isolation of putative defense-related genes from Arabidopsis thaliana and expression in fungal elicitor-treated cells. Plant Mol Biol 21:385–389
Stracke R, Jahns O, Keck M, Tohge T, Niehaus K, Fernie AR, Weisshaar B (2010) Analysis of production of flavonol glycosides-dependent flavonol glycoside accumulation in Arabidopsis thaliana plants reveals MYB11-, MYB12- and MYB111-independent flavonol glycoside accumulation. New Phytol 188:985–1000
Luehrsen KR, de Wet JR, Walbot V (1992) Transient expression analysis in plants using firefly luciferase reporter gene. Methods Enzymol 216:397–414
Kawalleck P, Somssich IE, Feldbrügge M, Hahlbrock K, Weisshaar B (1993) Polyubiquitin gene expression and structural properties of the ubi4-2 gene in Petroselinum crispum. Plant Mol Biol 21:673–684
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Takeda Y, Ohlendorf DH, Anderson WF, Matthews BW (1983) DNA-binding proteins. Science 221:1020–1026
Katagiri F, Lam E, Chua N-H (1989) Two tobacco DNA-binding proteins with homology to the nuclear factor CREB. Nature 340:727–730
Hartmann U, Valentine WJ, Christie JM, Hays J, Jenkins GI, Weisshaar B (1998) Identification of UV/blue light-response elements in the Arabidopsis thaliana chalcone synthase promoter using a homologous protoplast transient expression system. Plant Mol Biol 36:741–754
Tovar Torres J, Block A, Hahlbrock K, Somssich IE (1993) Influence of bacterial strain genotype on transient expression of plasmid DNA in plant protoplasts. Plant J 4:587–592
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Stracke, R., Thiedig, K., Kuhlmann, M., Weisshaar, B. (2016). Analyzing Synthetic Promoters Using Arabidopsis Protoplasts. In: Hehl, R. (eds) Plant Synthetic Promoters. Methods in Molecular Biology, vol 1482. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-6396-6_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6396-6_5
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6394-2
Online ISBN: 978-1-4939-6396-6
eBook Packages: Springer Protocols