
Abstract
Multiple Cdks (Cdk4, Cdk6, and Cdk2) and a mitotic Cdk (Cdk1) are involved in cell cycle progression in mammals. Cyclins, Cdk inhibitors, and phosphorylations (both activating and inhibitory) at different cellular levels tightly modulate the activities of these kinases. Based on the results of biochemical studies, it was long believed that different Cdks functioned at specific stages during cell cycle progression. However, deletion of all three interphase Cdks in mice affected cell cycle entry and progression only in certain specialized cells such as hematopoietic cells, beta cells of the pancreas, pituitary lactotrophs, and cardiomyocytes. These genetic experiments challenged the prevailing biochemical model and established that Cdks function in a cell-specific, but not a stage-specific, manner during cell cycle entry and the progression of mitosis. Recent in vivo studies have further established that Cdk1 is the only Cdk that is both essential and sufficient for driving the resumption of meiosis during mouse oocyte maturation. These genetic studies suggest a minimal-essential cell cycle model in which Cdk1 is the central regulator of cell cycle progression. Cdk1 can compensate for the loss of the interphase Cdks by forming active complexes with A-, B-, E-, and D-type Cyclins in a stepwise manner. Thus, Cdk1 plays an essential role in both mitosis and meiosis in mammals, whereas interphase Cdks are dispensable.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
The cell cycle is the biological process through which cells duplicate themselves. The universal cell cycle events are S phase (DNA replication) and M phase (mitosis), and these are under the control of Cyclin-dependent kinases (Cdks) [1]. Cdk activity during these cell cycle phases is regulated by the binding of regulatory molecules known as Cyclins. The temporal regulation of Cdks is a unique feature of the cell cycle machinery in eukaryotes and depends on the oscillations of Cyclin levels and the phosphorylation state of the Cdks [2]. Mammalian cells contain at least 20 different Cdks, but only a few subsets of Cdk–Cyclin complexes are directly involved in cell cycle progression. These include three interphase kinases (Cdk4, Cdk6, and Cdk2), a mitotic Cdk (Cdk1, also called M-Cdk) and ten Cyclins of four different classes (the A-, B-, D-, and E-type Cyclins) [2, 3]. Experiments in yeast have contributed to an in-depth understanding of cell cycle regulation, and these processes have been found to be highly conserved in mammalian cells.
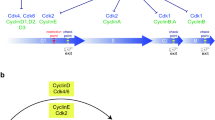
According to the long prevailing model of the cell cycle based on biochemical evidence (see Fig. 1a), stage-specific Cdk–Cyclin complexes orchestrate the various events during interphase in a sequential manner. In response to mitogen cues, cells initiate DNA synthesis and this is marked by specific binding of D-type Cyclins (D1, D2, and D3) to Cdk4 and Cdk6 resulting in their activation during G1 phase [4, 5]. This ‘preparatory’ phosphorylation leads to partial inactivation of the retinoblastoma protein (RB) and its homologues (p107 and p130), and facilitates the transcription of E2F-dependent genes such as Cyclin E [6]. Subsequently, E-type Cyclins (E1 and E2) bind to and activate Cdk2 [7]. Further phosphorylation of RB is carried out by the Cdk2–Cyclin E complex leading its complete inactivation. Additionally, several studies have indicated that the Cdk2–Cyclin E complex is essential for driving the G1–S transition [8]. During late S phase, Cyclin-A2 (Cyclin-A1 in germ cells) activates Cdk2 and the cell enters the G2 phase. At the end of G2 phase, A-type Cyclins activate Cdk1 to facilitate the onset of mitosis (the M phase). Furthermore, the phosphorylation of CDH1 (a component of the anaphase-promoting complex (APC)) by the Cdk2–Cyclin A complex causes its abrupt dissociation from the APC and leads to an increased level of Cyclin B that is required for cell cycle progression through the G2–M transition [9]. Nuclear envelope breakdown and A-type Cyclins degradation precedes the formation of the Cdk1–Cyclin B complex, which is responsible for driving M phase [10]. Cyclin binding alone is not sufficient to make Cdks fully functional and complete activation of Cdks requires their phosphorylation at a threonine residue (Thr161 in Cdk1, Thr160 in Cdk2) located adjacent to the kinase active site by Cdk-activating kinase (CAK). In mammalian cells, CAK phosphorylates the Cdks only after binding of Cyclins [11]. The regulation of the timing and coordination of crucial cell cycle events, therefore, depends on the finely tuned interactions of Cyclins (positive regulators), Cdk inhibitors (negative regulators), and both activating and inhibitory phosphorylations.

Cell cycle models. (a) Diagram showing the classical cell cycle model (based on biochemical analysis) that shows stage-specific functions of Cdks during cell cycle progression. (b) The essential cell cycle model (based on genetic evidence) that shows Cdk1 is the only essential Cdk needed to drive all the cell cycle events by binding with different Cyclins in the absence of interphase Cdks
1.1 Regulation of Cdks by Cyclins
In eukaryotic cells, Cyclins can be classified into G1-, G1/S-, S-, and M-Cyclins based on their specific functions and temporal expressions [12]. These specialized proteins bind to and activate Cdks in various phases of the cell cycle, and the Cdk–Cyclin complexes phosphorylate specific targets to ensure that cell cycle events occur in a timely manner. During the cell cycle, the levels of Cdks are held constant while the levels of Cyclins undergo cyclical fluctuations. The Cyclins that directly control cell cycle events are G1/S-, S-, and M-Cyclins, whereas the G1-Cyclins regulate cell cycle entry in response to extracellular cues. The D-type Cyclins typify the G1-Cyclins in mammals and their major catalytic partners are Cdk4 and Cdk6. Complexes of D-type Cyclins with Cdk4/6 are essential for exiting G1 and the initiation of S phase during the cell cycle [6]. Moreover, Cdk4/6-Cyclin D also regulates the expression of the E-type Cyclins. Levels of E-type Cyclins begin to rise in late G1 and decline in late S phase, which eventually initiates progression through the restriction point leading to DNA replication [2]. The commencement of S phase results in autophosphorylation of Cyclin E by the Cdk2–Cyclin E complex. The phosphorylated Cyclin E is then recognized by F-box protein FBW7 and is destined for proteasomal degradation [13]. This event allows A-type Cyclins (also known as S-Cyclins) to form complexes with Cdk2 to maintain DNA replication, and high levels of Cdk2 activity are maintained in S phase, G2 and early mitosis [14]. As the cell approaches mitosis, M-Cyclin (Cyclin B) comes into play and its activity peaks during metaphase. The major binding partner of Cyclin B is Cdk1 and the Cdk1–Cyclin B complex leads to mitotic spindle assembly and the alignment of sister chromatid on the metaphase plate [1, 15].
1.2 Regulation of Cdks by Cdk Inhibitors
The activity of the interphase and M-Cdks is regulated by the binding of Cdk inhibitors (CKIs). These polypeptide inhibitors of Cdks are categorized into two groups based on their specificities: the INK4 (INhibitors of Cdk4) family and the CIP/KIP (Cdk-Interacting Protein/Kinase Inhibiting Protein) family [11, 16]. The INK4 family includes four members (p16INK4a, p15INK4b, p18INK4c, and p19INK4d) that exclusively target Cdk4 and Cdk6. These highly conserved 15–19 kDa polypeptides are roughly 40 % homologous with each other, and it has been shown that mouse INK4 proteins share 90 % identity with those of humans [17]. INK4 proteins bind to Cdk4/6 and prevent phosphorylation of RB and thus promote cellular differentiation and inhibit inappropriate reentry into the cell cycle [18]. On the other hand, CIP/KIP family proteins (p21CIP1/WAF1, p27KIP1, and p57KIP2) prefer to bind Cdk–Cyclin complexes involved in G1 and G1/S control, and their promiscuous nature allows them to interact with G1 Cdks [5, 17].
1.3 Regulation of Cdks by Phosphorylation
The activities of the major Cdks involved in cell cycle progression are modulated by phosphorylation at positive (activating phosphorylation) and negative (inhibitory phosphorylation) regulatory sites [19–22]. The activating phosphorylation of Cdk1, Cdk2, Cdk4, and Cdk6 is catalyzed by CAK, a complex of Cdk7, Cyclin H, and Mat1 (ménage à trois 1) [23, 24]. Cyclin H activates Cdk7 and Mat1 regulates the substrate specificity of the complex [25]. Studies of Cdk2 showed that threonine 160 (Thr161 in Cdk1) located in the activation segment (T-loop) prevents binding of Cdk2 to its substrates. Phosphorylation of this segment enhances substrate binding by an additional 80- to 300-fold and leads to cell cycle progression [11, 12]. Conversely, further phosphorylation at threonine (Thr14) and tyrosine (Tyr15) of the Cdk2 and Cdk1 subunits inhibits the kinase activity of the Thr160/161-phosphorylated Cdk–Cyclin complexes [12, 19, 21, 22]. Crystallographic studies of Cdk2 showed that the inhibitory phosphorylation is Cyclin dependent, and that the phosphorylation alters the orientation of the catalytic site residues and the phosphates of ATP [12, 21]. The temporal regulation of mitosis entry by inhibitory phosphorylation is a unique feature of the cell cycle machinery [12, 26]. For example, inhibitory phosphorylation on Thr14 and Tyr15 of Cdk1 suppresses the activity of the Cdk1–Cyclin B complex during interphase. In mammalian cells, Wee1 and Myt1 kinases are responsible for the conserved Thr14 and Tyr15 phosphorylation activity. At the end of G2 phase, phosphatases abruptly dephosphorylate these residues leading to the activation of Cdk1 and subsequent entry of the cell into mitosis [27–29]. The tight coupling between activating and inhibitory phosphorylation of Cdk1 acts as a mitotic timer to prevent catastrophic mitotic failure [30].
2 Genetic Models for In Vivo Functional Studies of Cdks
The classical model of the cell cycle emphasizes the requirement of specific Cdks for each phase of cell cycle (see Fig. 1a). However, genetic experiments in cell cycle control in mice have challenged this concept and suggested that the functions of Cdks are cell-specific. Systematic loss-of-function studies in mice have given us a wealth of information regarding cell-specific roles of Cdks and their regulation at different cellular levels (Table 1).
2.1 Cdk4 and Cdk6 Knockout Mice
Two initially independent studies revealed the in vivo function(s) of Cdk4 in somatic cells [31, 32]. Cdk4 −/− mice display normal viability with defects in some organs, a similar phenotype to that seen in Cyclin D −/− mice. The prominent phenotype of Cdk4 −/− mice is both a small body size due to reduced cellularity compared to controls, and partial sterility. Despite a normal proliferation rate, Cdk4 −/− Mouse Embryonic Fibroblasts (MEFs) show delayed entry into S phase from their quiescence state. This is due to redistribution of p27, which attenuates Cdk2 activity. The double knockout of both Cdk4 and p27 demonstrated the importance of Cdk2 in normal S phase entry [32]. Additionally, pituitary hypoplasia and lactotroph (prolactin-producing cells) dysfunction occurs in Cdk4 −/− mice [33]. In these types of cells, Cdk6 does not compensate for the loss of Cdk4 and might not even be expressed in these cells. In adult Cdk4 −/− mice, degeneration of pancreatic beta cells resulted in a reduced cellularity of the pancreas leading to the development of diabetes mellitus in 80 % of the mutant mice. Besides exhibiting endocrine dysfunctions, Cdk4 −/− mice also display compromised fertility in both sexes. The male mutant suffer from testicular atrophy contributing to impaired spermatogenesis, whereas in females corpus luteum formation and ovulation are compromised rendering them infertile [31, 32]. The infertility in these mice is caused by perturbation of the neuro-endocrine axis in the absence of Cdk4 as opposed to developmental abnormalities of their gonads per se [31, 33]. The cell cycle inhibitor p16INK4a inhibits Cdk4 activity and prevents the phosphorylation of RB proteins [34]. Furthermore, the missense mutation of Cdk4 that replaces arginine 24 with a cysteine (R24C) abrogates inhibition of Cdk4 by p16INK4a without hindering binding of Cdk4 to D-type Cyclins. Generation of Cdk4 R24C/R24C mice corroborated the results from Cdk4 −/− mice and highlighted the highly specific role of Cdk4 in pancreatic beta cell proliferation and the maintenance of glucose homeostasis [31].
The results with Cdk4 knockout mice raised the question of whether or not Cdk2 could compensate for the loss of Cdk4 in the mammalian cell cycle. This was based on the fact that both of these genes are involved in the G1–S transition and there is the general notion that the lack of individual Cdks is compensated for by other Cdks [3]. Experiments with the Cdk4/Cdk2 double mutant showed the intricate roles these kinases play in mammalian cell cycle regulation. The predominant phenotype of these mutants is cardiac insufficiency due to their limited number of cardiomyocytes, which is caused by incomplete inactivation of the RB protein [36]. The diminished phosphorylation of RB resulted in suppression of E2F-dependent genes such as Cdk1 and Cyclin A2. The subsequently reduced Cdk1 expression level and failure of Cdk6 to compensate for the lack of Cdk4 and Cdk2 resulted in defective cell proliferation in the Cdk4/Cdk2 double mutant. This shows that Cdk4 and Cdk2 are crucial for embryonic cardiomyocyte proliferation whereas these kinases appear to be dispensable in other cell types and these two kinases cooperate to drive G1–S transition with mitosis [35, 36].
Cdk6 is widely distributed in mammalian tissues but is most prominent in lymphoid tissues [37]. Genetic studies in mice have revealed its specific function in the expansion of differentiated hematopoietic cells of erythroid lineage. Deletion of Cdk6 resulted in delayed G1 progression in lymphocytes, lower cellularity in the thymus and spleen, and mild anemia [38]. Nevertheless, the combined deletion of Cdk2 and Cdk6 had no effect on cell proliferation and revealed no synergism between Cdk6 and Cdk2 in driving cells through the G1–S transition. The phenotype of these mutants were similar to that of the Cdk6 and Cdk2 single mutants with limited hematopoietic defects and male and female sterility, respectively [38].
Whether or not any compensatory mechanism exists between Cdk4 and Cdk6 in the cell cycle has been highlighted by the study of the Cdk4/Cdk6 double knockout. Cdk4 and Cdk6 are functionally similar kinases that bind D-type Cyclins but govern different stages of erythroid maturation [39]. The Cdk4/Cdk6 double mutant exhibits late embryonic and postnatal lethality but normal cell proliferation kinetics. The cause of this phenotype is a lack of maturation of erythroid precursor cells that results in severe anemia. Based on the evidence from these genetic experiments, it can be assumed that these Cdks are dispensable in embryogenesis and are not involved in exiting cells from their quiescence state during cell cycle progression. In agreement with these observations, MEFs lacking both Cdk4 and Cdk6 showed normal proliferation and cell cycle profiles, which is similar to the phenotype of mice lacking the three D-type Cyclins [40]. In conclusion, Cdk4 and Cdk6 only play roles in cell cycle progression in specialized cells, such as hematopoietic cells [38, 41] and are dispensable in all other cell types.
2.2 Cdk2 Knockout Mice
Several lines of evidence indicate that Cdk2 governs the G1–S transition (in association with E-type Cyclins), S phase progression (in association with A-type Cyclins), progression to G2 phase, and, perhaps, mitotic entry [12, 42]. It has also been suggested that Cdk2 has specific roles in centrosome duplication [43], histone synthesis, and chromatin assembly [44]. These observations all portrayed Cdk2 as an essential regulator of the cell cycle for the majority of, if not all, somatic cells. However, deletion of Cdk2 in mice has refuted this idea and demonstrated that this seemingly crucial cell cycle kinase is dispensable for cell proliferation and development [42, 45]. Surprisingly, Cdk2 −/− mice are viable without any developmental anomalies, but display delayed S phase entry and dramatic meiotic failure [42, 45]. The obvious phenotype of Cdk2-deficient mice is infertility in both sexes with 100 % penetrance, emphasizing the essential role of Cdk2 in germ cell development beyond meiotic prophase I.
The germ cells of Cdk2 −/− males are arrested at the pachytene stage of prophase I. This results in increased apoptosis of primary spermatocytes combined with defective synaptonemal complex formation [45]. On the other hand, the oocytes at embryonic day 14.5–18.5 (E14.5–E18.5) had normal meiotic profiles with dictyate stage progression, normal synapsis, and apoptosis. Cdk2 −/− females displayed postnatal meiotic defects with improper distribution of synaptonemal complex protein 3 (a marker for the axial elements). Additionally, the ovarian morphology of prepubertal and adult mutant mice showed a depletion of functional oocytes along with an increase in apoptotic cells that resulted in ovarian atrophy [45]. Even when Cdk1 was expressed from the Cdk2 locus, male and female sterility could not be rescued in these genetically modified mice [46]. These studies indicate that Cdk2 is essential for cell cycle progression in germ cells in mice and that this function cannot be complemented by Cdk1. Deletion of Cdk2 from growing oocytes in mice shows the dispensability of Cdk2 in meiotic maturation and metaphase II arrest [47], but any roles of Cdk2 in early germ cell development in mice still remain to be tested.
2.3 Cdk1 Knockout Mice
The idea of a minimally essential Cdk in mammalian cells, such as is found in yeast, emerged from the results showing the dispensability of all interphase Cdks in mice [3, 48]. The triple knockout Cdk4 −/−; Cdk6 −/−; Cdk2 −/− MEFs show complete cell cycle progression, albeit with some delay in entry into different cell cycle stages. Moreover, the deletion of Cdk4, Cdk6, and Cdk2 did not affect the expression levels of Cdk1, Cdk7, Cdk9 or Cyclins (D1, D2, E1, A2, and B1), and the phosphorylation of RB at all four serine residues that are targets for the interphase Cdks was normal [48]. It was found that Cdk1 binds to all Cyclins, phosphorylates RB in the absence of these interphase Cdks, and causes the cells to leave their quiescent state [42]. Thus, Cdk1 is the only essential Cdk for cell division as Cdk1-null embryos fail to undergo even the early stages of development and die before E3.5, and Cdk1-null MEFs undergo premature senescence [48, 49].
A recent study used liver as an in vivo model to establish the function of Cdk1 in hepatocytes, which are terminally differentiated cell types that do not divide in adults [49]. The absence of Cdk1 in hepatocytes renders them unable to enter into mitosis, indicating that regeneration of liver can occur after partial hepatectomy without cell division [49]. To understand the underlying mechanism behind this, BrdU incorporation assays showed that Cdk1-null hepatic cells undergo some form of DNA replication. Moreover, loss of Cdk1 in these cells does not affect DNA replication in a wild-type Cdk2 background, but these cells are arrested at G2 phase and fail to enter into mitosis [49]. Approximately 30 % of Cdk1-null cells undergo DNA re-replication. This is due to Cdk2 activity because knocking down of Cdk2 in Cdk1-null cells significantly reduces the re-replication phenotype [49]. Furthermore, these studies also revealed that Cdk1 regulates Cdk2 activity by sequestering Cyclin A2 and preventing re-replication during late S phase. Concomitant deletion of Cdk1 with partial hepatectomy resulted in polyploidy, perhaps because of re-replication. Thus, this study suggests another layer of regulatory mechanisms for Cdk1 during cell cycle progression [49].
The above-mentioned studies have provided a wealth of information regarding functions of different Cdks in dividing cells such as hematopoietic cells, pancreatic beta cells, and pituitary lactotrophs. However, such details regarding the specific requirements of Cdks in case of non-dividing cells like oocytes had been lacking. In mammals, growing oocytes are arrested at prophase I of meiosis. The resumption of meiosis takes place with the preovulatory surge of luteinizing hormones, which is sequentially marked by germinal vesicle (GV) breakdown (GVBD), chromosome condensation, spindle formation, and the completion of meiosis I [47, 50]. Moreover, spontaneous meiosis resumption takes place when oocytes are liberated into suitable culture medium where they undergo meiosis II arrest until fertilization [50, 51]. Oocytes are mitotically inactive cells, but oocytes arrested at the GV stage are considered analogous to somatic cells at the G2–M transition phase of mitosis [52]. Furthermore, Cdk1 orchestrates the G2–M transition in dividing cells as demonstrated by several studies [46, 48, 49]. Nonetheless, such studies have not been extended to non-dividing cells like oocytes. Recently, this issue has been addressed by generating an oocyte-specific knockout of Cdk1 in mice [47]. This study revealed that Cdk1 is a central regulator for driving the resumption of meiosis in mouse oocytes as the deletion of Cdk1 from oocytes causes permanent arrest at the GV stage and renders females infertile. In mouse oocytes, Cdk1 phosphorylates PP1 (protein phosphatase 1) and suppresses its phosphatase activity. This maintains the phosphorylation of lamin A/C and eventually leads to GVBD and the resumption of meiosis [47]. Thus, the evidence provided by these recent studies suggests that Cdk1 is the only essential Cdk that is sufficient to drive both mitosis and meiosis [53].
3 Concluding Remarks
The Cdk family of serine/threonine kinases is made up of 20 proteins, of which only a few subsets have been shown to directly participate in cell cycle progression. The biological functions of these kinases are regulated by Cyclin binding, CKIs, and phosphorylation. More than two decades ago, a cell cycle model was proposed that was based solely on the biochemical analysis of egg extracts from different animal species. This cell cycle dogma claimed that specific Cdks are responsible for governing each phases of the cell cycle. However, genetic studies of deletions of different Cdks in mice challenges this biochemical cell cycle model. The interphase Cdks (Cdk4, Cdk6, and Cdk2) are not essential for cell cycle progression, but are stringently necessary for some specialized cells such as germ cells, hematopoietic precursor cells, pituitary lactotrophs, and pancreatic beta cells. Additionally, a new role for Cdk1 has emerged from these studies as a master regulator of the basic cell cycle machinery that can compensate for the absence of all interphase-Cdks in mammalian cells (see Fig. 1b). Moreover, Cdk1 has been found to be the only essential Cdk even for the resumption of meiosis in oocytes. Thus, both the mitosis and meiosis are driven by Cdk1. Further research is clearly needed towards delineating the regulatory mechanisms that ensure the precise spatial and temporal activation of Cdks in the specific cell types.
References
Nurse P (1990) Universal control mechanism regulating onset of M-phase. Nature 344(6266):503–508
Malumbres M, Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9(3):153–166
Satyanarayana A, Kaldis P (2009) Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 28(33):2925–2939
Massague J (2004) G1 cell-cycle control and cancer. Nature 432(7015):298–306
Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13(12):1501–1512
Lundberg AS, Weinberg RA (1998) Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 18(2):753–761
Harbour JW, Luo RX, Santi AD, Postigo AA, Dean DC (1999) Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98(6):859–869
Hochegger H, Takeda S, Hunt T (2008) Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol 9(11):910–916
Sørensen CS, Lukas C, Kramer ER, Peters J-M, Bartek J, Lukas J (2001) A conserved cyclin-binding domain determines functional interplay between anaphase-promoting complex–Cdh1 and Cyclin A-Cdk2 during cell cycle progression. Mol Cell Biol 21(11):3692–3703
Malumbres M, Barbacid M (2005) Mammalian cyclin-dependent kinases. Trends Biochem Sci 30(11):630–641
Morgan DO (1995) Principles of CDK regulation. Nature 374(6518):131–134
Morgan DO (1997) Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13:261–291
Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ (2001) Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294(5540):173–177
Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G (1992) Cyclin A is required at two points in the human cell cycle. EMBO J 11(3):961–971
Sherr CJ, Roberts JM (2004) Living with or without cyclins and cyclin-dependent kinases. Genes Dev 18(22):2699–2711
Tanner FC, Boehm M, Akyürek LM, San H, Yang Z-Y, Tashiro J, Nabel GJ, Nabel EG (2000) Differential effects of the cyclin-dependent kinase inhibitors p27Kip1, p21Cip1, and p16Ink4 on vascular smooth muscle cell proliferation. Circulation 101(17):2022–2025
Roussel MF (1999) The INK4 family of cell cycle inhibitors in cancer. Oncogene 18(38):5311–5317
Buchold GM, Magyar PL, Arumugam R, Lee MM, O’Brien DA (2007) p19Ink4d and p18Ink4c cyclin-dependent kinase inhibitors in the male reproductive axis. Mol Reprod Dev 74(8):997–1007
Kaldis P, Sutton A, Solomon MJ (1996) The Cdk-activating kinase (CAK) from budding yeast. Cell 86(4):553–564
Gould KL, Moreno S, Owen DJ, Sazer S, Nurse P (1991) Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J 10(11):3297–3309
De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgant DO, Kim S-H (1993) Crystal structure of cyclin-dependent kinase 2. Nature 363(6430):595–602
Kaldis P (1999) The cdk-activating kinase (CAK): from yeast to mammals. Cell Mol Life Sci 55(2):284–296
Desai D, Wessling HC, Fisher RP, Morgan DO (1995) Effects of phosphorylation by CAK on cyclin binding by CDC2 and CDK2. Mol Cell Biol 15(1):345–350
Fisher RP (2005) Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 118(22):5171–5180
Yankulov KY, Bentley DL (1997) Regulation of CDK7 substrate specificity by MAT1 and TFIIH. EMBO J 16(7):1638–1646
Dunphy WG (1994) The decision to enter mitosis. Trends Cell Biol 4(6):202–207
Mueller PR, Coleman TR, Kumagai A, Dunphy WG (1995) Myt1: a membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science 270(5233):86–90
Fattaey A, Booher RN (1997) Myt1: a Wee1-type kinase that phosphorylates Cdc2 on residue Thr14. Prog Cell Cycle Res 3:233–240
Lew DJ, Kornbluth S (1996) Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr Opin Cell Biol 8(6):795–804
Coulonval K, Kooken H, Roger PP (2011) Coupling of T161 and T14 phosphorylations protects cyclin B–CDK1 from premature activation. Mol Biol Cell 22(21):3971–3985
Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M (1999) Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in [beta]-islet cell hyperplasia. Nat Genet 22(1):44–52
Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H (1999) Targeted disruption of CDK4 delays cell cycle entry with enhanced p27Kip1 activity. Mol Cell Biol 19(10):7011–7019
Moons DS, Jirawatnotai S, Parlow AF, Gibori G, Kineman RD, Kiyokawa H (2002) Pituitary hypoplasia and lactotroph dysfunction in mice deficient for cyclin-dependent kinase-4. Endocrinology 143(8):3001–3008
Sherr CJ (2001) The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2(10):731–737
Berthet C, Klarmann KD, Hilton MB, Suh HC, Keller JR, Kiyokawa H, Kaldis P (2006) Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev Cell 10(5):563–573
Berthet C, Kaldis P (2006) Cdk2 and Cdk4 cooperatively control the expression of Cdc2. Cell Div 1:10
Meyerson M, Harlow E (1994) Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol 14(3):2077–2086
Malumbres M, Ro S, Santamaría D, Galán J, Cerezo A, Ortega S, Dubus P, Barbacid M (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118(4):493–504
Matushansky I, Radparvar F, Skoultchi AI (2000) Reprogramming leukemic cells to terminal differentiation by inhibiting specific cyclin-dependent kinases in G1. Proc Natl Acad Sci 97(26):14317–14322
Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, Akashi K, Sicinski P (2004) Mouse development and cell proliferation in the absence of D-cyclins. Cell 118(4):477–491
Barrière C, Santamaría D, Cerqueira A, Galán J, Martín A, Ortega S, Malumbres M, Dubus P, Barbacid M (2007) Mice thrive without Cdk4 and Cdk2. Mol Oncol 1(1):72–83
Berthet C, Aleem E, Coppola V, Tessarollo L, Kaldis P (2003) Cdk2 knockout mice are viable. Curr Biol 13(20):1775–1785
Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA (1999) Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol 1(2):88–93
Ma T, Van Tine BA, Wei Y, Garrett MD, Nelson D, Adams PD, Wang J, Qin J, Chow LT, Harper JW (2000) Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev 14(18):2298–2313
Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M (2003) Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35(1):25–31
Satyanarayana A, Berthet C, Lopez-Molina J, Coppola V, Tessarollo L, Kaldis P (2008) Genetic substitution of Cdk1 by Cdk2 leads to embryonic lethality and loss of meiotic function of Cdk2. Development 135(20):3389–3400
Adhikari D, Zheng W, Shen Y, Gorre N, Halet G, Kaldis P, Liu K (2012) Cdk1, but not Cdk2, is the sole Cdk that is essential and sufficient to drive resumption of meiosis in mouse oocytes. Hum Mol Genet 21(11):2476–2484
Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448(7155):811–815
Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, Kaldis P (2012) Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci 109(10):3826–3831
Adhikari D, Liu K (2009) Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocr Rev 30(5):438–464
Mehlmann LM (2005) Stops and starts in mammalian oocytes: recent advances in understanding the regulation of meiotic arrest and oocyte maturation. Reproduction 130(6):791–799
Solc P, Schultz RM, Motlik J (2010) Prophase I arrest and progression to metaphase I in mouse oocytes: comparison of resumption of meiosis and recovery from G2-arrest in somatic cells. Mol Hum Reprod 16(9):654–664
Adhikari D, Liu K, Shen Y (2012) Cdk1 drives meiosis and mitosis through two different mechanisms. Cell Cycle 11(15)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Risal, S., Adhikari, D., Liu, K. (2016). Animal Models for Studying the In Vivo Functions of Cell Cycle CDKs. In: Orzáez, M., Sancho Medina, M., Pérez-Payá, E. (eds) Cyclin-Dependent Kinase (CDK) Inhibitors. Methods in Molecular Biology, vol 1336. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2926-9_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2926-9_13
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2925-2
Online ISBN: 978-1-4939-2926-9
eBook Packages: Springer Protocols