
Abstract
Cellular proliferation is controlled by the orchestrated action of many cell cycle regulators. Among them are cyclins and cyclin-dependent kinases (CDKs), the activity of which is necessary to drive each phase of the cell cycle. Mitogenic stimulation leads to the expression of D cyclins which bind and activate CDK4 and CDK6. This event triggers a chain of events ultimately leading to cell division. Dissection of the functions of D cyclins and CDK4 in the regulation of proliferation of mammalian cells was greatly facilitated by the generation of genetically modified mice in which either D cyclins, their CDK partners, or other cell cycle regulators were ablated or replaced. In general, variable impact of germline loss of these cell cycle regulators on different tissues underscores specific roles for D cyclins and their partner CDKs in differentiation and development. These mouse models have also proved crucial for studies analyzing tumor development and for the discovery and evaluation of anticancer therapies, often linking tissue-specific functions to antineoplastic effects of inhibition of cyclin-D-dependent processes. This chapter summarizes the history of mice lacking D cyclins and CDK4/CDK6 and presents a synopsis of key findings from those animal models.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
2.1 Introduction
In 1855 Rudolf Virchow formulated his famous Omnis cellula e cellula —all cells come from cells. He also explained that “we must reduce all tissues to a single simple element, the cell (...), and from it emanate all the activities of life both in health and in sickness” [216]. Thus, to understand how the organism originates, develops, and matures, we have to understand how its cells proliferate, differentiate, become quiescent, die, or transform to malfunction and cause disease, such as cancer. Importantly, unicellular organisms , as well as cells of multicellular organisms , exploit the same molecular machinery governing their proliferation. This machinery ensures that the newly formed cell becomes ready to replicate its genetic material and that any mistakes occurring during replication will be removed. Next, this process dictates that cell division will produce two daughter cells properly prepared for either the next cell cycle or another fate, such as differentiation. Due to the wide variety of dividing cells, some aspects of cell cycle progression may be modified; however, the core of this process is constant and relies on the function of cyclin-dependent kinases (CDKs) and their regulatory cofactors (cyclins) .
In this chapter we will present one of the crucial cell cycle regulators, D-type cyclins and their CDK partners. We will center on the “ab ovo” part of the characterization of these proteins, i.e., their role in the development, and also outline their involvement in carcinogenesis . The majority of the studies presented here would not have been possible without the groundbreaking discoveries and techniques developed by Martin Evans and Matthew Kaufman as well as Gail Martin, who derived the first lines of mouse embryonic stem cells [58, 146], and Mario Capecchi and Oliver Smithies who showed how to genetically modify these cells [141]. Important input also came from Andrzej K. Tarkowski who was the first to create chimeric mice [229, 230], which established the basis for an indispensable method to generate knock-out or knock-in mice . Our major goal is to summarize what has been learned using mice in which cyclins D, CDK4, CDK6, or other cell cycle regulators were ablated or replaced. We are aware, however, that presenting all of the currently available data is not possible. Thus, we do regret the omission of any relevant finding and view. We are sure, however, that other chapters presented in this book will expand upon our summary of progress made in understanding D-type cyclin function derived from genetically modified mice.
2.2 The Core
The first studies leading to the discovery of the universal mechanisms governing cell cycle progression focused on Rana pipiens oocytes undergoing meiotic division—the so-called meiotic maturation . Experiments demonstrating that cytoplasm from dividing frog oocyte induced meiosis in prophase oocytes led to the discovery of the activity described as MPF (maturation-promoting factor or M-phase-promoting factor [148, 149, 219]). In a short time, similar activity was confirmed in mouse oocytes [11] and also in dividing somatic cells [183, 221] proving that MPF triggers not only meiosis but also mitosis and is responsible for interphase/metaphase transition . Next, a drop in MPF activity was shown to be prerequisite for metaphase exit. The biochemical nature of MPF was soon revealed—it was characterized as a complex of a protein kinase, later termed cyclin-dependent kinase (CDK), and a regulatory component, cyclin. The first CDK, i.e., CDK1, was discovered in yeast by cloning cdc2 and CDC28 [81, 82, 125, 215]. CDK1 activators were identified during analyses of dividing sea urchin and clam embryos [59, 192, 224]. They were named cyclins due to their periodic expression pattern. Cyclins accumulated in interphase and were abruptly degraded in M-phase just before each cleavage division of an embryo. These milestone discoveries were soon followed by characterization of other cyclins, CDKs, and their positive and negative regulators, present not only in yeast and animal cells but also in plant cells. It was also shown that specific CDKs can be regulated only by particular cyclins, the synthesis of which leads to activation of these enzymes. Precisely orchestrated destruction of the cyclins results in a drop in CDK activity.
Next, G1- and S-phase-specific CDK-cyclin complexes were identified along with their cell cycle-specific substrates. Thus, cyclin D-CDK4 or CDK6 (CDK4/CDK6) complexes regulate G1 phase, CDK2 together with E- and A-type cyclins controls S phase, and CDK1 activated by A- and B-type cyclins coordinates M-phase progression (Fig. 2.1). Current evidence supports a simplified model showing that in order to be active, a CDK has to be postranslationally modified. One of the crucial modifications is introduced by CAK, i.e., CDK-activating kinase that phosphorylates the T-loop of monomeric CDK [66]. Interestingly, CAK is itself composed of a CDK, CDK7, which is activated by cyclin H and MAT1. Further, in addition to its involvement in cell cycle control, CDK7 is also a component of transcription factor TFIIH, which plays a role in the regulation of gene expression [34] as do other CDKs, such as CDK8 or CDK9 (for review see [142]).

Simplified summary of cell cycle regulation. Cell cycle progression is precisely controlled by periodically active cyclin-CDK complexes. Mitogen stimulation drives cell cycle reentry by induction of D-type cyclin synthesis. Once synthesized, D cyclins bind and activate CDK4 and CDK6 kinase subunits, which in turn phosphorylate and inactivate pRb allowing activation of E2F transcription factors and their cofactors. One of the first products of E2F-controlled transcription is cyclin E which binds and activates CDK2 allowing initiation of S-phase. Next, S-phase is controlled by cyclin A-CDK2 complexes. Cyclin A-CDK1 activation that is necessary for M-phase entry is followed by cyclin B-CDK1 activation. Completion of each cell cycle phase requires degradation of the specific cyclin and, as a result, CDK inactivation. Each of the CDKs is blocked by specific inhibitors—CDK4/CDK6 by INK4 family members, e.g., p16Ink4a, CDK2, and CDK1 by KIP/CIP inhibitors, i.e., p21cip1, p27kip1, and p57kip2 (Modified from Ciemerych et al. [42])
In addition to activation by posttranslational modification , CDKs are also subject to inhibitory phosphorylation, e.g., p-T14/Y15 in case of CDK1, which needs to be removed by CDK-specific phosphatases [208], and also to inhibition by members of the INK family (e.g., p16Ink4a—specific for CDK4 and CDK6) or CIP/KIP family (e.g., p21Cip1, p27Kip1, p57Kip2—specific for CDK2 and CDK1) of protein inhibitors. Interestingly, cyclin D-CDK4/CDK6 complexes bind and sequester CIP/KIP proteins , such as p27kip1, avoiding being inhibited by them and promoting activation of other CDKs [9, 23, 114]. Nevertheless, it is the binding of CDK and cyclin that is a sine qua non condition for CDK activation, with each of the other events described serving to add exquisite layers of regulatory control on these vital cell cycle control enzymes.
The core of the cell cycle regulatory machinery is operative in dividing cells. However, one has to be aware that many cell types utilize customized adjustments to fundamental cell cycle mechanisms. These sometimes subtle differences allow certain cells to adapt to specific developmental or environmental requirements. For example, proliferation of certain embryonic cells, such as embryonic stem cells (ESCs) , is not inhibited by p16Ink4a [201] raising the possibility that the cyclin D-CDK4/CDK6 pathway might be modified or not fully operative during early mammalian development [61, 238]. Thus, the regulation of cleavage divisions in developing embryos and proliferation of precursors of various cell types, or embryonic stem cells, can serve as illustration of such fine-tuning (for review see, e.g., [42, 76, 111, 161, 202]).
2.3 The Details
2.3.1 D-Type Cyclins: What Are They and What Do They Do?
Three D-type cyclins, i.e., cyclin D1, D2, and D3, are present in mammalian cells and tissues. They were first reported as products of genes responding to mitogen stimulation and involved in G1 phase regulation [128, 153, 165, 166, 241, 244, 245]. They are encoded by separate genes but share significant amino acid identity that reaches 50–60% throughout the entire coding sequence and 75–78% within the most conserved cyclin box domain [91, 245]. As was mentioned above, expression of D-type cyclins is largely controlled by the extracellular environment —they are upregulated during cell cycle entry as a result of signals coming from extracellular matrix or soluble mitogens reaching the cell [106]. For example, cyclin D1 levels can be increased by mitogenic stimulation activating the MAPK canonical pathway , i.e., Ras-Raf-MEK-ERK1/ERK2 [3, 120], PI3K, Wnt, or other signaling pathways [106, 167]. Conversely, D-type cyclin expression declines when anti-mitogens are added and for these reasons they might be described as sensors of environmental changes.
The first identified function of D-type cyclins was to control cell cycle reentry by activating CDK4/CDK6 which phosphorylates pRb family members, i.e., pRb, p107, and p130 [18, 154, 155, 158, 253]. In its active, i.e., hypophosphorylated, state, pRb binds and prevents activity of the E2F transcription factors . Phosphorylation of pRB leads to the release of E2Fs and results in the activation of E2F-controlled genes, among them those encoding E- and A-type cyclins, i.e., cyclins involved in the activation of CDK2-regulating initiation and progression of S-phase [45, 54, 209, 210]. Cyclin D-CDK4/CDK6 complexes impact cell cycle progression also by controlling other proteins, such as SMAD family members. Phosphorylation of SMAD3 , a factor playing a crucial role in the antiproliferative TGF-β pathway , leads to the inhibition of its antiproliferative function [133, 156] and promotion of cell cycle progression. Systemic screening for cyclin D1-CDK4/CDK6 and cyclin D3-CDK4/CDK6 substrates revealed, apart from pRB family members and SMAD3, another 68 potential targets for these kinases [4]. Among identified targets were such factors as Myc or forkhead box M1 (FOXM1) , proteins which when phosphorylated and stabilized activate the expression of G1/S phase genes [4]. Interestingly, the number of substrates uncovered as a result of these analyses seemed to depend on the cyclin D type. Cyclin D1-CDK4/CDK6 substrates were less abundant than those phosphorylated by cyclin D3-CDK4/CDK6. In recent years CDK-independent functions of D-type cyclins were also uncovered (see below).
2.3.2 D-Type Cyclins: Where Are They Expressed?
All three D-type cyclins, as well as their CDK partners, are detectable during oogenesis [107, 164], spermatogenesis [19, 103, 254], and also at each step of pre- and post-implantation mammalian development [40]. Interestingly, they are expressed with significant overlap (Fig. 2.2). For example, in the developing nervous system, cyclins D1 and D2 are detectable in distinct cellular compartments, and their synthesis dynamically changes along the course of development [2, 239]. In some tissues, such as stratified squamous epithelia , cyclin D1 synthesis is associated with proliferating cells, whereas cyclin D3 is present in cells more advanced in differentiation [15]. In proliferating skeletal myoblasts , cyclin D1 prevents exit from the cell cycle and terminal differentiation [184, 217, 218]. Thus, the formation of mature myotubes is associated with decrease in cyclins D1 and D2 and increase in cyclin D3 expression [31, 104, 145]. In embryonic and also in adult tissues, some cellular compartments express a combination of two or even three D-type cyclins (e.g., [15, 16, 41, 73, 175, 186, 226, 248]) (Fig. 2.2). Expression of CDK4 and CDK6 does not seem to be so finely assigned as is the case for D-type cyclins [41].

Cyclin D, CDK4, and CDK6 expression in E13.5 mouse embryos. Sagittal sections of mouse embryos were hybridized with riboprobes specific for cyclin D1, cyclin D2, cyclin D3, CDK4, and CDK6 (Technical details in Ciemerych et al. [41]). Black color represents hybridization signal. B brain, Ret retina, L liver, V vertebrae, H heart, Lu lungs
The precisely timed expression patterns of D-type cyclins suggested that each of them may play some non-redundant and/or CDK-unrelated functions. Experiments aiming at the verification of this hypothesis started with the generation of mutant mice lacking a single D-type cyclin [62, 211, 213, 214] and soon was followed by experiments analyzing results of ablation of two and finally all three D-type cyclins [43, 109]. Next, kinase-dependent functions were tested in CDK4- and CDK6-null mice [144, 157, 182, 231]. As a result it was uncovered that lack of D-type cyclins or CDK4/CDK6 had dramatic consequences for the proper development of certain cellular compartments.
2.3.3 D-Type Cyclins and Their Partners: How to Live Without Them?
2.3.3.1 Single Knock-Out Mice
In 1995, the phenotype of the first cyclin D knock-out mice was described. It was only a few years after D-cyclin-encoding genes were cloned, and as not much was known about their specific function, a lot was to be discovered. Although all three D cyclins showed very high sequence similarity, it was suspected that each of them could play unique functions. This notion was supported by observations showing that despite widespread expression of each D-type cyclin, phenotypes of knock-out mice were limited to a narrow subset of cellular compartments. Importantly, mice lacking CDK4 or CDK6 displayed abnormalities within tissues and organs similar to those affected by the lack of D-type cyclins suggesting that at least some phenotypes resulting from D-cyclin loss are CDK related (Table 2.1).
The first cyclin studied using a genetic mouse model was cyclin D1. Its expression was independently disrupted by Sicinski (Weinberg group) and Fantl (Dickson group) [62, 213]. D1-null mice display reduced body mass, a spastic leg-clasping reflex, and a partially penetrant premature mortality within the first weeks of life. The latter phenotype is explained by abnormalities in the development and function of the nervous system. Despite the neurological deficiencies, brain size and neural progenitor cell number is comparable to that observed in wild-type controls [36, 74]. The loss of cyclin D1 does not reduce the number of neural progenitor cells in the subgranular zone (SGZ) [108]. It impacts, however, Schwann and glial cell proliferation associated with postnatal injury [7, 171] but does not markedly prevent axonal regrowth during induced regeneration [105]. Two initial studies on D1−/− mice revealed abnormal development of retinas [62, 213] resulting from restricted proliferation of retinal cells and increased photoreceptor cell death [137]. Interestingly, functional redundancy among D-cyclin subtypes was documented by the analysis of knock-in mice expressing cyclin D2 in place of cyclin D1 in that development of retinas was nearly normal [30]. Cyclin D2 could also replace cyclin D1 function in estrogen-induced proliferation of other tissues, i.e., mouse uterine epithelium [33]. However, other studies suggested that neither cyclin D2 nor cyclin D3 could fully ameliorate the retinal phenotype [52]. Interestingly, the function of cyclin D1 was replaceable by a downstream cell cycle regulator—cyclin E [70]. A second dramatic phenotype of D1-null mice is associated with the failure of mammary glands to undergo normal lobuloalveolar development during pregnancy [61,62,64, 213]. As a result D1−/− females cannot feed their pups. This phenotype could also be rescued by cyclin D2 [30].
Dissection of specific functions of cyclin D1 led to the generation of two knock-in mouse strains. At first, knock-in mice carrying a version of cyclin D1 that lacks the ability to activate CDK4/CDK6 were analyzed [117]. Such animals manifest only slightly underdeveloped retinas. Also pregnancy-induced mammary gland epithelial expansion is not substantially affected in uniparous knock-in females [117]. Further studies showed, however, that abrogation of cyclin D1-associated kinase activity influences mammary gland progenitor cell self-renewal and impacts their differentiation and tissue regeneration [96], as well as leads to upregulation of autophagy [27]. Similar to cyclin D1 knock-out animals, “knock-ins” are also characterized by some growth deficiency and neurological phenotypes , i.e., leg clasping. In contrast, mutation in the LxCxE motif, which is required for binding of D-type cyclins with pRb [55] and is essential for cell cycle regulatory functions of these proteins, impacts neither retinal development nor mammary gland function [10, 118]. In vitro studies exposed, however, that the LxCxE motif is crucial for cyclin D2 function, documenting that these two cyclins might not play redundant roles in cell cycle control [10].
The phenotype of cyclin D2-deficient mice is also very narrow. Females are sterile as a result of the inability of ovarian granulosa cells to proliferate in response to follicle-stimulating hormone (FSH) . As a consequence, ovarian follicles do not form properly, and oocytes cannot be ovulated. D2−/− males are fertile but testes are hypoplastic [214]. Lack of cyclin D2 also impacts the proliferation of peripheral B-lymphocytes [115, 220] and pancreatic β-cells [71, 112, 113]. Next, several neurological phenotypes are characteristic for cyclin D2−/− mice. Among them are mild cerebellar abnormalities [75, 87], a decrease in intermediate progenitor cells in the embryonic cortex [74], as well as impaired adult neurogenesis [5, 108]. D3-deficient mice, in turn, are viable but display abnormalities in T and B cell [48, 150, 211] as well as erythrocyte development [199]. D3-null mice are also characterized by deficient maturation of granulocytes in bone marrow and a reduced number of granulocytes and neutrophils in the blood [212]. Interestingly, cyclin D3 was also shown to be involved in pancreatic β-cell function, since in the nonobese diabetic (NOD) type 1 diabetes -prone mouse, lack of this cyclin exacerbates diabetes and impairs glucose responsiveness [195].
D-type cyclins bind and activate CDK4 and CDK6. As with their cyclin partners, the expression of these catalytic subunits during mammalian development is generally overlapping [41] (Fig. 2.2). Both kinases were shown to share 71% amino acid identity and phosphorylate the same substrates, e.g., pRb family members; thus, it was initially widely accepted that they play a redundant function [158, 159]. However, some lines of evidence document that subtle functional differences between these two kinases might exist. For example, CDK4 was shown to preferentially phosphorylate pRb at the threonine 826 residue, while CDK6 phosphorylates threonine 821 [225]. Next, in T lymphocytes , CDK6 is activated before CDK4 [136], and their actions seem to be different in such distinct cells as thymocytes [85, 86], osteoblasts [56, 57], or astrocytes [173], strongly suggesting a tissue-specific role for CDK6 in cellular differentiation [77, 78].
Generation of CDK4- and CDK6-deficient mice showed that both mutants are viable and characterized by rather mild phenotypes, suggesting that either CDK4 and CDK6 could substitute for each other in many tissues, or they can be replaced by CDK2, which indeed is able to provide functional compensation by interacting with D-type cyclins [144]. Importantly, some defects of CDK4- and CDK6-deficient mice mimic those observed in single D-type cyclin mutants (Table 2.1). CDK4−/− mouse phenotypes essentially equate with those of both cyclin D1 and cyclin D2 knock-out mice, i.e., retarded growth (similar to cyclin D1−/−), ovarian and testicular defects, and also pancreatic hypoplasia (similar to cyclin D2−/−) [147, 157, 182, 231]. Female infertility , however, is not caused by a defect in granulosa cell proliferation, as was shown for cyclin D2−/− females. Rather, infertility in CDK4−/− mice results from a failure in the development of pituitary lactotroph cells that leads to a deficiency in prolactin production and defective formation of corpus luteum and as a consequence prevents embryo implantation [98, 162, 163, 182]. Other affected processes include adipogenesis [1] and T lymphocyte maturation [39]. Lack of CDK4 also causes some neurological deficiencies , e.g., compromised locomotion [182] and a decrease in the proliferation of Schwann cells, but only during early postnatal development [8]. CDK6 deficiency results in hematopoietic defects (similar to cyclin D3−/− mice) manifested by abnormal spleen and thymus development, decreased number of peripheral blood cells [86, 144], as well as a partial deficiency in hematopoietic stem cell function , i.e., impaired repopulation after competitive transplantation [203]. The fact that some cell types fail to properly develop and function when either a single D-type cyclin CDK or CDK6 is absent in tissues that express most or all of these subunits underscores their unique functions.
2.3.3.2 Double and Triple Knock-Out Mice
In 2002 we stated that “These single-knock-out experiments are illuminating, but their analyses are greatly confounded by the presence of two remaining, intact D-cyclins, which may compensate for the ablated protein. We decided to reduce this complexity by creating mouse strains expressing only a single D-type cyclin. In doing so, we hoped to be able to directly test which proliferative and developmental functions can be executed solely by cyclin D1, D2, or D3” [43]. The double knock-out mice, i.e., D1−/−D2−/− (expressing only cyclin D3), D1−/−D3−/− (expressing only cyclin D2), and D2−/−D3−/− (expressing only cyclin D1), so-called “single-cyclin” mice, displayed the additive defects characteristic for mice lacking a single D-type cyclin. Animals expressing only cyclin D3 were born alive but died within 3 weeks after birth, likely due to enhanced neurological abnormalities affecting locomotive ability and proper feeding. These mice were also characterized by abnormal, underdeveloped cerebella. The majority of mice expressing only cyclin D2 died immediately after birth, with a small number able to survive up to 2 months. Again, the cause of their death was likely related to neurological defects leading to the aspiration of meconium into their lungs. Finally, development relying on cyclin D1 was terminated before birth, i.e., at 17.5–18.5 days of pregnancy. Analysis of surviving embryos revealed that they suffered from severe megaloblastic anemia [43]. Searching for the mechanisms allowing nearly normal development of the majority of tissues and organs in single-cyclin mice, we discovered upregulation of the remaining cyclin. This suggested the existence of a negative feedback loop in which a D-type cyclin that plays a key role in given tissue might repress the expression of the remaining ones. Interestingly, in tissues that failed to develop, e.g., cerebellum of D1−/−D2−/− mice, the remaining cyclin (i.e., cyclin D3) was not upregulated. In the case of the cerebellum, this failure is caused by the inability of N-myc, which plays the crucial role in the proliferation of granule neuron precursors, to communicate with cell cycle machinery via cyclin D3. Thus, this result suggested the existence of transcription factor—cyclin D dependency [43]. The requirement of D1-associated kinase activity for cerebellar development was documented by the analysis of mice lacking cyclin D2 and expressing kinase-deficient cyclin D1. Such mice were characterized by severely retarded cerebellar development, leading to the conclusion that cyclin D-CDK4/CDK6 activity is necessary for morphogenesis of this organ [117]. Moreover, we also showed another feedback loop involving D-type cyclins, i.e., facilitation of cell cycle progression-mediated downregulation of p27kip1 levels [43].
Analysis of double knock-out mice suggested that either the presence of a single cyclin D allows nearly normal development of a majority of tissues and organs or proliferation of at least some cell types may occur in the absence of D-type cyclins. The latter scenario was proven by us by the generation of mice lacking all three D-type cyclins [109]. Cyclin D1−/−D2−/−D3−/− embryos developed until mid-gestation and died before 17.5 day of pregnancy. Detailed analyses of triple knock-out embryos revealed that indeed the majority of cell types can proliferate normally. Proliferative failure was limited to myocardial cells and hematopoietic stem cells making these lineages critically dependent on D-type cyclins [109]. Importantly, Cdk4 and Cdk6 double knock-out mouse embryos were also embryonic lethal, severely anemic, and displayed various defects in variety of hematopoietic lineages [144]. Interestingly, these embryos were able to survive a few days longer than cyclin D triple “knock-outs” most probably because D cyclins were able to interact and activate CDK2 [144]. Thus, cellular proliferation was shown to be possible without D-type cyclin-associated kinases, also suggested by the observation that in cyclin D2−/−D3−/− embryos, in the presence of cyclin D1 only, CDK4 activity was not detectable [41]. Therefore, proliferation of only selected cell lines depends on cyclin D-CDK4/CDK6. At that time, however, it was uncertain if these cells fail to proliferate only because of their strict cell cycle requirements or because they require specific cyclin D functions, independent of CDK.
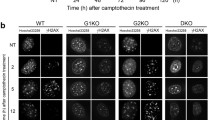
The fact that mouse embryos with acute ablation of all D-type cyclins failed to develop to term [109] made detailed studies of D cyclin function in peri- and postnatal development impossible. However, the development of the Cre-loxP system refining the conventional method of gene knock-out offered an excellent opportunity to avoid embryonic lethality and investigate consequences of acute ablation of a chosen gene in a tissue- and time-specific manner [129]. The technology of conditional gene knock-out is based on insertion of specific sequences (loxP or FRT) upstream and downstream of the target gene or gene fragment. Depending on the orientation of the sequences, the flanked region can be either irreversibly removed or inverted thanks to the activity or either Cre or FLP recombinase, respectively. By breeding mice carrying a floxed gene (f/f) with mice expressing Cre recombinase under the control of a tissue-specific promoter, tissue-/cell-specific deletion of a floxed gene is possible. Thus, using this method one can analyze the function of a selected gene in chosen tissue and at the chosen moment of embryonic or postnatal development, including adult organisms (reviewed in [193, 256]).
Based on the Cre-loxP system , conditional “knock-outs” of all three D-type cyclins were created that allowed a more precise test of the requirement for D-type cyclins in adult mice [38]. Using this technique conditional triple mouse mutants were generated by crossing “original” cyclin D2−/− mice with animals carrying conditionally modified cyclin D1- and D3-encoding genes. Next, these triple mutant mice were intercrossed with Mx1-Cre animals characterized by induced expression of Cre recombinase in hematopoietic cells [38]. A controlled shutdown of all D-type cyclins leads to abrupt disappearance of hematopoietic stem cells (HSCs) , while the number of mature bone marrow cells remains unaffected, demonstrating that HSCs depend on D-type cyclins for their survival. The pro-survival function of D-type cyclins involves regulation of the death receptor Fas and its ligand FasL which, upon deletion of D-type cyclins , are strongly upregulated leading to the initiation of caspase-8-dependent apoptosis [38]. Thus, analysis of conditional knock-out of all D-type cyclins unraveled unexpected, non-cell cycle-related, functions of D-type cyclins in quiescent HSCs. This pro-survival role of D-type cyclins in adult hematopoietic cells was not demonstrated in the initial analysis of the “conventional” triple , i.e., cyclin D1, D2, and D3, knock-out model [109].
Conditional knock-out mice also allowed analyses of mice lacking a single D-type cyclin which, due to the embryonic or early postnatal lethality of mice, were impossible using traditionally derived knock-out mouse strains (e.g., [43, 109, 200]). Conditional deletion of cyclin D1 in liver proved that lack of this particular cyclin does not hamper liver development but uncovered the role of this protein in glucose metabolism regulation [127]. Lack of cyclin D1 does not induce changes in gluconeogenic gene expression and glycemia in fasting mice; however, in the re-fed state, it significantly increases expression of gluconeogenic genes, glycemia, glucose, and insulin intolerance. Thus, this study also revealed a cell cycle-unrelated function of cyclin D1, i.e., involvement in the regulation of nutrient and insulin signaling to regulate glucose metabolism. Importantly, inhibition of CDK4 activity fails to enhance this phenotype, suggesting that cyclin D1 alone mediates metabolic effects in the liver [127]. The conditional knock-out approach was also used in a study focusing on the cyclin-dependent kinases. Deletion of Cdk4 and Cdk2 results in lethality manifested shortly after birth [12]. Such mice die due to the failure of cardiac development. A decreased number of proliferating cardiomyocytes indicate that CDK4 and CDK2 play compensatory roles during heart development. Conditional ablation of Cdk4 in Cdk2-null mice produces animals with no obvious abnormalities, proving that the function of adult tissues does not depend on CDK4 and CDK2 activity [12].
2.3.3.3 Quadruple and Quintuple Knock-Out Cells Enter the Stage
Generation of triple cyclin D knock-out mice and cell lines was followed by the derivation and analysis of the cells lacking either all G1 cyclins, i.e., cyclins D1, D2, D3, E1, and E2 [134], or those lacking cyclins E1, E2, A1, and A2 [102]. Surprisingly depletion of cyclins D and E did not block the proliferation of quintuple knock-out ES cells but completely prevent the proliferation of MEFs [134]. These ES cells, however, attenuated their pluripotent character and become prone to differentiate into trophectoderm. Further studies showed that G1 cyclin-dependent CDK activity is necessary to stabilize the pluripotency factors, such as Nanog, Sox2, and Oct4. Interestingly, ablation of G1/S cyclins, i.e., cyclins E and A , had no impact on MEFs [102].
2.3.3.4 What About Mice Deficient in CDK Inhibitors or pRb Family Members?
The goal of this chapter is to present the role of D cyclins and CDK4/CDK6 in cell cycle regulation during embryogenesis and cancer and also to describe some cell cycle-independent functions of these proteins. However, at least briefly, we would like to discuss the phenotypes of mice lacking some of the factors interacting with cyclin D-dependent kinases, i.e., CDK4/CDK6 inhibitors and pRb family members. The consequences of ablation of the expression of these proteins, resulting in the development of a variety of cancers, have been published in an enormous number of research articles, and it would be extremely difficult to present here a comprehensive summary. Thus, we will focus on studies describing the development and proper function of adult tissues.
The INK family of inhibitors includes p16Ink4a, p15Ink4b, p18Ink4c, and p19Ink4d (e.g., [32, 79, 80, 83, 204]). Expression of p16Ink4a and p15Ink4b is detectable only in adult tissues and increases with age [257]. p18Ink4c and p19Ink4d, on the other hand, are expressed in tissues of developing embryos as well as of adult animals [257, 259]. At the time of generation of p16 Ink4a knock-out mice , it was not known that the Ink4a locus (CDKN2A) encodes not only p16Ink4a but also p19Arf [180]. Ablation of these two genes, however, did not result in obvious developmental abnormalities but promoted lymphomas and sarcomas [205]. Subsequently, knock-out mice were produced lacking p16 Ink4a exclusively, and these animals developed almost normally. They were characterized by hyperplastic thymi, increased lymphocyte proliferation, and again high tumor incidence in keeping with a tumor suppressor function of p16Ink4a [110, 206, 207]. p15 Ink4b-null mice displayed hyperplastic lymph nodes and spleen, as well as extramedullary hematopoiesis, and also an increased proliferation rate of lymphocytes [119]. Lack of p18Ink4c alone also does not affect development. With age, however, p18 Ink4c-null mice become larger and reveal a hypoplastic pituitary gland and development of pituitary tumors, enlarged spleen, thymus, and other organs, as well as deregulated proliferation of epithelia , e.g., mammary gland epithelium [68, 119]. Deletion of genes encoding both p15 Ink4b and p18 Ink4c added some new phenotypes to those characteristic of the single knock-outs, i.e., double mutant mice suffer from enlarged testes and hyperplastic Langerhans islets [119]. Deficiency in p19Ink4d leads to male infertility due to testicular hyperplasia and hearing loss due to the malfunction of the auditory epithelium [35, 258, 260]. Therefore, the lack of inhibitors of cyclin D-CDK4/CDK6 complexes does not demonstrably impact embryonic development but in adult mice increases proliferation and leads to the development of hyperplasia of many organs, and eventually tumor development. On the other hand, ablation of cyclin D-CDK4/CDK6 substrates, i.e., pRb family members, results in much more severe phenotypes.
pRb, together with two other pRb-related proteins, namely, p107 and p130, is the first identified cyclin D-CDK4/CDK6 target [50]. Their phosphorylation and as a result inactivation are prerequisite for cell cycle progression since, as mentioned above, in the active state, they bind E2F transcription factors and prevent expression of crucial positive regulators of the cell cycle. During development, pRb is expressed starting from the peri-implantation stage of mouse embryo development (i.e., blastocyst) [93]. At later stages of development, all three pRb family proteins are specifically expressed in certain tissues [97]. The first studies focusing on Rb-null mice strongly suggested that this protein is indispensable for embryonic development. Knock-out mice died between 12 and 15 days of pregnancy due to severe anemia. They were also characterized by defects in lens development and massive cell death in the central (CNS) and peripheral nervous system (PNS) [44, 94, 124]. Generation of chimeric mice in which Rb-null cells were able to participate in the formation of many lineages, including the erythroid lineage, put in doubt a crucial role of this protein in hematopoiesis [138, 240]. Also, the neuronal apoptotic defects were not as obvious as described in the characterization of the phenotype of Rb −/− embryos. Generation of mice in which the Rb gene was conditionally deleted only in CNS, PNS, and lens revealed that CNS mutant tissues displayed ectopic S-phase entry but no apoptosis [65, 139].
Increased expression of hypoxia-inducible genes in Rb-null embryos suggested that observed apoptosis was induced by hypoxia [139]. This hypoxia, in turn, was thought to have resulted from placental malfunction. Experiments involving the “tetraploid complementation” technique allowing generation of mutant mice developing within wild-type placentas verified this notion [242]. Wu et al. proved that abnormal proliferation and differentiation of trophoblast cells prevented development of the labyrinth within the placenta which resulted in deficient nutrient and oxygen supply. These mice also died prematurely; however, they were able to develop to term, allowing observation of nearly normal development of the erythroid compartment and nervous system. Further studies showed that ablation of pRb in trophoblast stem cells resulted in abnormal trophoblast and placenta development [237].
The experiments described above, the creation of conditional mice and analyses of mice carrying Rb hypomorphic alleles, revealed the crucial role of pRb in embryonic myogenesis—muscle lacking pRb is characterized by hypoplastic myofibers [53, 242, 252]. Deletion of Rb in differentiating myoblasts resulted in apoptosis and failure to produce myotubes [88]. pRb’s myogenic connection was also revealed during analysis of p130 mutant mice. p130-null mice on a BALB/cJ background (characterized by reduced activity of p16Ink4a) died in utero between days 11 and 13 of pregnancy due to defects in neuro- and myogenesis, i.e., reduced number of myocytes in the differentiating myotome [121]. The phenotype of p107-null mice is also influenced by the genetic background, i.e., BALB/cJ mutants were characterized by growth retardation and myeloid hyperplasia, but p107-deficient mice on a 129Sv/C57BL6 background displayed no obvious abnormalities [46, 122, 126]. p107 −/− p130 −/− animals were characterized by defects in chondrocyte proliferation and abnormal endochondral bone development [46]. Next, ablation of pRb either with p107 or p130 proved that these factors can substitute for each other, as the phenotype of either genetic combination is very similar—embryonic lethality occurs between 11 and 13 days of pregnancy due to liver and CNS abnormalities [126]. Finally, the consequence of deletion of genes encoding all three pRb family members analyzed in embryonic stem cells revealed that these proteins were crucial for successful differentiation and proper control of cellular proliferation [51, 196]. Again, as was the case with CDK inhibitors, deregulation of pRb protein expression led to tumor development, proving a crucial role of these cyclin D-CDK4/CDK6 regulators and substrates in the cell cycle control.
2.3.4 Cell Cycle-Independent Functions of Cyclins and CDKs
Many lines of evidence document that cyclins are involved in balancing proliferation and differentiation by impacting various tissue-specific transcription factors. Functions of cyclin D1 in this regard are the best studied so far, and the control of processes other than CDK4/CDK6 regulation is very well documented. Thus, it was shown that upregulation of cyclin D1 in cancer cells stimulates cellular migration by p27kip1 stabilization and also by impacting Rho protein function [130]. On the contrary, ablation of cyclin D1 negatively impacts cellular motility [170]. Cyclin D1 is also linked to DNA repair by data demonstrating that it can recruit RAD51 [131] and antagonize BRCA1-dependent repression of estrogen receptor α activity [234]. Further, cyclin D1 forms a complex with BRCA2, RAD51, and the Sp1 transcription factor [174, 228], interacts with PCNA [152, 243] and replication factor C (RFC) [233], all of which are also involved in DNA repair. In addition to the aforementioned functions, cyclin D1 involvement in the regulation of transcription is unquestionable. For example, cyclin D1 was shown to compete with androgen receptor for p300/CBP-associated factor (P/CAF) binding [189] and to inhibit the function of peroxisome proliferator-activated receptor gamma (PPARgamma) [235], to interact with transcription factors such as myb-like binding protein (DMP1) [92], repress STAT3 [20, 21] and inhibit NeuroD function [135, 185]. Moreover, both D1 and D2 cyclins inhibit transcription activated via the v-Myb DNA-binding domain [69].
The identification of additional novel cyclin D1 roles was possible due to the generation of knock-in mice expressing proteins labeled with such tags as Flag or hemagglutinin (HA) . This approach was initially used by Bienvenu et al. who generated transgenic mice expressing Flag- and HA-tagged cyclin D1 [22]. By sequential immunoaffinity purification using anti-Flag and anti-HA antibodies, followed by repeated rounds of high-throughput mass spectrometry, novel cyclin D1-interacting proteins were identified. Among cyclin D1 interactors identified were known cell cycle partners, such as CDK4 and CDK6, and those less typical, such as CDK1, CDK2, CDK5, and CDK11. This study also confirmed involvement of cyclin D1 in the regulation of transcription —it was shown to bind to promoter regions of more than 900 genes [22]. Importantly, this approach revealed the mechanism leading to the retinal phenotype characteristic of D1-deficient mice. In retinas, cyclin D1 physically binds and recruits CBP histone acetyltransferase to the Notch1 upstream regulatory region [22]. In the absence of cyclin D1, acetylation of histones was decreased, resulting in transcriptional repression of the targeted gene, i.e., Notch1. Cyclin D1 transcriptional function in the development of other tissues and cancer formation will be the next major goal of many research efforts using this approach. In the meantime, protein interactome analyses of human cancers proved cyclin D1 interaction with DNA repair proteins, including RAD51 [99, 100]. Thus, the generation of knock-in mice carrying genes encoding tagged proteins provided a unique chance to uncover a whole new world of previously unappreciated protein functions.
2.4 Cyclin D- and CKD4/6-Deficient Mice Versus Cancer
Oncogenic roles of D-type cyclins and their CDK partners are widely documented by analyses showing that these proteins are overexpressed in a variety of tumors and by experiments involving either their overexpression or elimination [142, 143, 168]. Aberrant cyclin D1 expression is observed in a wide spectrum of human cancers, such as colorectal cancer, uterine cancer, malignant melanoma, squamous cell carcinoma of head and neck, astrocytoma, non-small-cell lung cancer, soft tissue sarcoma, and others [14, 17, 67, 123, 140, 151, 198]. Importantly, breast cancer is perhaps the best documented malignancy involving cyclin D1. Approximately 15–20% of mammary tumors contain amplification of the CCND1 gene whereas its overexpression is detected in over 50% [14, 72, 116, 262]. Interestingly, overexpression of cyclin D1 is more common than can be explained by gene alteration. Therefore, other mechanisms such as deregulation of mitogenic signaling pathways or aberrant proteolytic degradation must underlie cyclin D1 overexpression. Indeed, elevated levels of cyclin D1 protein were observed in the absence of increased mRNA reflecting a defect in its proteolysis [194]. This effect was confirmed in transgenic mice expressing phosphorylation-deficient cyclin D1 under the control of the tissue (i.e., mammary gland)-specific MMTV promoter. Disruption of cyclin D1 phosphorylation led to the accumulation of the protein in the nucleus, prevented its cytoplasmic proteolysis and accelerated mammary carcinogenesis [132].
MMTV-driven expression in transgenic mice has facilitated analysis of mammary gland-specific expression of various oncogenes, including cyclins, associated kinases, inhibitors, Ras, Myc, and others [227]. In 1994, MMTV-cyclin D1 mice were shown to develop mammary adenocarcinomas within 22 months of age [236]. The relatively late occurrence of these mammary tumors suggests involvement of other oncogenic pathways. Interestingly, intercrossing MMTV-cyclin D1 with p53 +/− mice did not result in mammary neoplasia [84]. In mice heterozygous for p53 deficiency and simultaneously carrying the MMTV-cyclin D1 transgene, only tumors typical for p53-deficient mice developed, and interestingly, their growth was significantly accelerated by cyclin D1 overexpression. Surprisingly, mammary tumors were not observed. More rapid development of non-mammary tumors in MMTV-cyclin D1/p53 +/−, as compared with p53 +/−, raise the possibility that p53 inactivation might complement or cooperate with cyclin D1 deregulation during the development of some types of non-mammary tumors.
The connection between cyclin D1, and also D2 and D3, and tumorigenesis was strengthened by the analyses of mice, or cells derived from them, that lacked single, two, or all three D-type cyclins or their CDK partners. Lack of cyclin D1 prevents not only physiological but also pathological proliferation of mammary gland epithelium. Yu et al. revealed that breast tumors arising in MMTV-ras and MMTV-neu mice expressed almost exclusively cyclin D1, very low levels of cyclin D3, and no cyclin D2 [249]. In contrast, several tumors arising in MMTV-Wnt-1 and MMTV-myc females expressed, in addition to cyclin D1, also high levels of D2. Importantly, all tumors arose from luminal epithelial cells , indicating that, in mammary epithelial cells, Ras and Neu oncogenes communicate with the cell cycle machinery through cyclin D1, whereas Wnt-1 and Myc can signal through other targets. Therefore, therapies involving cyclin D1 inhibition might be highly selective in shutting off the growth of human breast cancers, particularly those characterized by amplification and/or overexpression of c-Neu (ErbB-2, HER-2). This hypothesis was recently challenged in a genetic mouse model that allows controlled expression of cyclin D1 in progressing mammary tumors [255]. Zhang observed that cyclin D1 deficiency delayed the development of tumors; however, it did not protect against ErbB2-driven mammary carcinogenesis as previously reported [249]. Moreover, in the absence of cyclin D1, cyclin D3 was upregulated. Knockdown of cyclin D3 in tumor-derived cells lacking cyclin D1−/− resulted in significant tumor growth impairment in comparison to cells expressing cyclin D3. It is, therefore, possible that only the combined inhibition of cyclin D1 and D3 might serve as an effective strategy for breast cancer therapy. Further studies demonstrated that cyclin D1 absence suppressed Neu- and mutant Neu (activated c-neu)-driven mammary tumor formation confirming that cyclin D1 is required for the Neu-driven signal transduction pathway [25]. Interestingly, no significant changes in either cyclin D2 or cyclin D3 expression were detected in MMTV-c-neu/cyclin D1−/−-derived mammary tumors. However, increased levels of cyclin E and higher activity of cyclin E-CDK2 complexes were demonstrated. Thus, Bowe et al. suggested that neither cyclin D2 nor D3 compensate for the absence of cyclin D1 to promote the oncogenic potential of Neu [25]. The above discrepancies were addressed by Choi et al. who created conditional cyclin D1 and D3 knock-out mice allowing acute ablation of individual cyclins [37]. Contrary to what was presented by Zhang et al., induced ablation of cyclin D1 in the whole body, including ErbB2-driven mammary carcinomas, resulted in cessation of tumor progression [37].
Mice expressing a mutated form of cyclin D1 proved that cyclin-associated CDK activity is crucial for oncogene-induced breast cancer development [117, 247]. Knock-in mice expressing kinase-deficient cyclin D1-CDK4 complexes are resistant to mammary carcinomas triggered by ErbB-2 [117]. Also, analyses of CDK4-deficient mice confirmed the role of CDK4 in breast cancer [187, 188, 251]. Therefore, it was not surprising that administration of PD0332991, a specific and potent inhibitor of cyclin D-CDK4/CDK6 kinases, halted the progression of breast cancers [37]. Interestingly, cyclin E was shown to be able to replace the function of cyclin D1 in Wnt-induced tumors [70], however, CDK4 function seems to be unreplaceable by CDK6 [188]. Remarkably, cyclin D1-CDK2 complexes were present in mammary carcinoma cells; hence, they might be an additional factor contributing to the oncogenic effects of cyclin D1 overexpression [223]. Indeed, transgenic mice expressing a cyclin D1-CDK2 fusion protein under the control of the MMTV promoter developed breast tumors [49].
Cyclin D1 gene amplification was demonstrated in breast cancers in which CCND1 overexpression was linked to estrogen and progesterone receptor status (reviewed in [178]). This connection is attributed to cyclin D1 regulation by estrogen (ER) and interaction of cyclin D1 with ER coactivators to activate estrogen receptor binging elements (ERE) in a CDK4−/CDK6-independent manner [169, 197, 263]. Furthermore, cyclin D1 was shown to regulate progesterone receptor (PR) expression, through an estrogen- and cyclin D1-responsive enhancer localized on the 3’UTR [246]. Loss of cyclin D1 led to decreased PR mRNA levels in mammary glands. In addition, a higher risk of development of tumors that express estrogen receptor is associated with elevated prolactin (PRL) and PRL receptor (PRLR) levels—both critical for epithelial proliferation during development and pregnancy [222, 232]. Cyclin D1−/− mouse epithelial cells fail to proliferate in response to prolactin [26]. Although deletion of cyclin D1 in transgenic mice overexpressing PRL markedly decreased tumor incidence, cyclin D1−/− females overexpressing PRL developed significantly more preneoplastic lesions than D1−/− females [6]. Interestingly, tumors that formed in this background exhibited elevated levels of cyclin D3 and a squamous histotype similar to those that developed in MMTV-cyclin D3 mice [179].
Cyclin D1-deficient mice were also shown to be “resistant” to other cancers. For example, they do not develop Ras-triggered skin papillomas [190] or intestinal polyps in the Apcmin background [89]. Extending results with cyclin D1 overexpressing or null mice, the involvement of other D cyclins in carcinogenesis was also documented. Despite the fact that cyclin D2 was shown to be a direct target of Myc [24, 177], much less attention has been devoted to investigation of cyclin D2 involvement in breast cancer. Clinical data demonstrate, however, that cyclin D2 is absent in breast cancer cell lines and tumors [28, 60]. Mice lacking cyclin D2 are characterized by reduced susceptibility to gonadal tumors [29] and insensitivity to BCR/ABL-driven transformation [95] and, similarly to D1-deficient animals, to Apcmin-induced formation of intestinal polyps [47]. Aberrant accumulation of cyclin D3 was also documented in a subset of breast carcinomas [13, 194]. As mentioned above, ablation of cyclin D3 in cyclin D1−/− mice further reduces mammary tumor development. Lack of cyclin D3 was shown to result in delayed development of thymomas caused by p56lck and resistance to Notch-driven leukemias (acute lymphoblastic leukemia, T-ALL) [211], and acute ablation of cyclin D3 in abnormal CD4+CD8+ cells blocked the development of Notch1-driven T-ALL in vivo [37].
Although all D-type cyclins are highly related and are expressed in a largely overlapping fashion, it is clear that there are differences in their specificity to transmit specific oncogenic signals to the cell cycle machinery. Thus, requirement for D-type cyclins in oncogenic transformation was also tested using mouse embryonic fibroblasts lacking two or all three D-type cyclins [109, 250]. Each of the D-type cyclins is certainly sufficient to mediate the action of such oncogenes as Ras and c-Myc [250]. However, triple knock-out fibroblasts are resistant to the action of Ras, c-Myc, or Ras combined with c-Myc, dnp53, or E1A [109]. Also CDK4-deficient cells are unaffected by Ras and dnp53 [181, 261]. Further, CDK4 deficiency in mice resulted in decreased incidence of skin tumors [191] as well as Myc-induced tumors in the oral mucosa [160]. Similarly, CDK6-deficient mice were shown to be resistant to Akt-driven lymphoma [86] as well as BCR-ABLp2101-driven leukemia [203].
Current development of high-throughput platforms allows to study interactomes of various factors involved in oncogenesis, such as cyclin D1 or CDK4 [100, 101, 172, 176]. Results of such analyses document cell cycle-dependent functions of cyclins and CDKs as well as reveal their non-canonical properties (for the summary, see [90]). Such studies are of the vital importance for the development of future therapeutic approaches.
2.5 Concluding Remarks
Deletion of the genes encoding D-type cyclins and their partners provided valuable clues about their role in embryonic development and in cell cycle progression of different cell types. Consequently, observing the characteristics of mice lacking these genes has provided copious information that can be used to better understand D cyclin contributions to cancer formation. Taking advantage of conditional knock-out mice lacking one or more D cyclins, it has been possible to determine if a particular D cyclin is required at different developmental stages or for tumor initiation and maintenance. Since aberrant expression of cell cycle regulators is very frequent in tumorigenesis, it is of outmost importance to test if these proteins could be potentially targeted in various therapeutic approaches. Another burning problem that can be addressed using conditional mouse models is whether therapeutic targeting of those proteins will have negative consequences in tumor-free organs. Furthermore, dissection of the mammalian cell cycle machinery, including uncovering novel cyclin D1 roles, is possible based on the generation of variety of knock-in mice, including animals expressing tagged proteins. The exploration of such new tools has already brought surprising results although the process has just begun. Thus, the discovery of novel D cyclin roles in mechanisms regulating normal and tumor cell cycles is ongoing, and these studies will be invaluable in extending the understanding and application of current therapies targeting D-cyclin-dependent kinases.
References
Abella A, Dubus P, Malumbres M, Rane SG, Kiyokawa H, Sicard A, Vignon F, Langin D, Barbacid M, Fajas L. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metab. 2005;2:239–49.
Aguzzi A, Kiess M, Rued D, Hamel PA. Cyclins D1, D2 and D3 are expressed in distinct tissues during mouse embryogenesis. Transgenics. 1996;2:29–39.
Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–97.
Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai HL, Vidal M, Gygi SP, Braun P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–34.
Ansorg A, Witte OW, Urbach A. Age-dependent kinetics of dentate gyrus neurogenesis in the absence of cyclin D2. BMC Neurosci. 2012;13:46.
Asher JM, O'Leary KA, Rugowski DE, Arendt LM, Schuler LA. Prolactin promotes mammary pathogenesis independently from cyclin D1. Am J Pathol. 2012;181:294–302.
Atanasoski S, Shumas S, Dickson C, Scherer SS, Suter U. Differential cyclin D1 requirements of proliferating Schwann cells during development and after injury. Mol Cell Neurosci. 2001;18:581–92.
Atanasoski S, Boentert M, De Ventura L, Pohl H, Baranek C, Beier K, Young P, Barbacid M, Suter U. Postnatal Schwann cell proliferation but not myelination is strictly and uniquely dependent on cyclin-dependent kinase 4 (cdk4). Mol Cell Neurosci. 2008;37:519–27.
Bagui TK, Jackson RJ, Agrawal D, Pledger WJ. Analysis of cyclin D3-cdk4 complexes in fibroblasts expressing and lacking p27(kip1) and p21(cip1). Mol Cell Biol. 2000;20:8748–57.
Baker GL, Landis MW, Hinds PW. Multiple functions of D-type cyclins can antagonize pRb-mediated suppression of proliferation. Cell Cycle. 2005;4:330–8.
Balakier H, Czolowska R. Cytoplasmic control of nuclear maturation in mouse oocytes. Exp Cell Res. 1977;110:466–9.
Barriere C, Santamaria D, Cerqueira A, Galan J, Martin A, Ortega S, Malumbres M, Dubus P, Barbacid M. Mice thrive without Cdk4 and Cdk2. Mol Oncol. 2007;1:72–83.
Bartkova J, Zemanova M, Bartek J. Abundance and subcellular localisation of cyclin D3 in human tumours. Int J Cancer. 1996;65:323–7.
Bartkova J, Lukas J, Strauss M, Bartek J. Cyclin D1 oncoprotein aberrantly accumulates in malignancies of diverse histogenesis. Oncogene. 1995;10:775–8.
Bartkova J, Lukas J, Strauss M, Bartek J. Cyclin D3: requirement for G1/S transition and high abundance in quiescent tissues suggest a dual role in proliferation and differentiation. Oncogene. 1998;17:1027–37.
Bartkova J, Rajpert-De Meyts E, Skakkebaek NE, Bartek J. D-type cyclins in adult human testis and testicular cancer: relation to cell type, proliferation, differentiation, and malignancy. J Pathol. 1999;187:573–81.
Bartkova J, Lukas J, Muller H, Strauss M, Gusterson B, Bartek J. Abnormal patterns of D-type cyclin expression and G1 regulation in human head and neck cancer. Cancer Res. 1995;55:949–56.
Bates S, Bonetta L, MacAllan D, Parry D, Holder A, Dickson C, Peters G. CDK6 (PLSTIRE) and CDK4 (PSK-J3) are a distinct subset of the cyclin-dependent kinases that associate with cyclin D1. Oncogene. 1994;9:71–9.
Beumer TL, Roepers-Gajadien HL, Gademan IS, Kal HB, de Rooij DG. Involvement of the D-type cyclins in germ cell proliferation and differentiation in the mouse. Biol Reprod. 2000;63:1893–8.
Bienvenu F, Gascan H, Coqueret O. Cyclin D1 represses STAT3 activation through a Cdk4-independent mechanism. J Biol Chem. 2001;276:16840–7.
Bienvenu F, Barre B, Giraud S, Avril S, Coqueret O. Transcriptional regulation by a DNA-associated form of cyclin D1. Mol Biol Cell. 2005;16:1850–8.
Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature. 2010;463:374–8.
Blain SW, Montalvo E, Massague J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem. 1997;272:25863–72.
Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18:5321–33.
Bowe DB, Kenney NJ, Adereth Y, Maroulakou IG. Suppression of Neu-induced mammary tumor growth in cyclin D1 deficient mice is compensated for by cyclin E. Oncogene. 2002;21:291–8.
Brisken C, Ayyannan A, Nguyen C, Heineman A, Reinhardt F, Tan J, Dey SK, Dotto GP, Weinberg RA. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Dev Cell. 2002;3:877–87.
Brown NE, Jeselsohn R, Bihani T, Hu MG, Foltopoulou P, Kuperwasser C, Hinds PW. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012;72:6477–89.
Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, de Fazio A, Watts CK, Musgrove EA, Sutherland RL. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–33.
Burns KH, Agno JE, Sicinski P, Matzuk MM. Cyclin D2 and p27 are tissue-specific regulators of tumorigenesis in inhibin alpha knockout mice. Mol Endocrinol. 2003;17:2053–69.
Carthon BC, Neumann CA, Das M, Pawlyk B, Li T, Geng Y, Sicinski P. Genetic replacement of cyclin D1 function in mouse development by cyclin D2. Mol Cell Biol. 2005;25:1081–8.
Cenciarelli C, De Santa F, Puri PL, Mattei E, Ricci L, Bucci F, Felsani A, Caruso M. Critical role played by cyclin D3 in the MyoD-mediated arrest of cell cycle during myoblast differentiation. Mol Cell Biol. 1999;19:5203–17.
Chan FK, Zhang J, Cheng L, Shapiro DN, Winoto A. Identification of human and mouse p19, a novel CDK4 and CDK6 inhibitor with homology to p16ink4. Mol Cell Biol. 1995;15:2682–8.
Chen B, Pollard JW. Cyclin D2 compensates for the loss of cyclin D1 in estrogen-induced mouse uterine epithelial cell proliferation. Mol Endocrinol. 2003;17:1368–81.
Chen J, Larochelle S, Li X, Suter B. Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature. 2003;424:228–32.
Chen P, Zindy F, Abdala C, Liu F, Li X, Roussel MF, Segil N. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat Cell Biol. 2003;5:422–6.
Chen Z, Duan RS, Zhu Y, Folkesson R, Albanese C, Winblad B, Zhu J. Increased cyclin E expression may obviate the role of cyclin D1 during brain development in cyclin D1 knockout mice. J Neurochem. 2005;92:1281–4.
Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22:438–51.
Choi YJ, Saez B, Anders L, Hydbring P, Stefano J, Bacon NA, Cook C, Kalaszczynska I, Signoretti S, Young RA, et al. D-cyclins repress apoptosis in hematopoietic cells by controlling death receptor Fas and its ligand FasL. Dev Cell. 2014;30:255–67.
Chow YH, Zhu XD, Liu L, Schwartz BR, Huang XZ, Harlan JM, Schnapp LM. Role of Cdk4 in lymphocyte function and allergen response. Cell Cycle. 2010;9:4922–30.
Ciemerych MA, Sicinski P. Cell cycle in mouse development. Oncogene. 2005;24:2877–98.
Ciemerych MA, Yu Q, Szczepanska K, Sicinski P. CDK4 activity in mouse embryos expressing a single D-type cyclin. Int J Dev Biol. 2008;52:299–305.
Ciemerych MA, Archacka K, Grabowska I, Przewozniak M. Cell cycle regulation during proliferation and differentiation of mammalian muscle precursor cells. Results Probl Cell Differ. 2011;53:473–527.
Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H, Sicinski P. Development of mice expressing a single D-type cyclin. Genes Dev. 2002;16:3277–89.
Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–30.
Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–809.
Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA, Jacks T. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996;10:1633–44.
Cole AM, Myant K, Reed KR, Ridgway RA, Athineos D, Van den Brink GR, Muncan V, Clevers H, Clarke AR, Sicinski P, et al. Cyclin D2-cyclin-dependent kinase 4/6 is required for efficient proliferation and tumorigenesis following Apc loss. Cancer Res. 2010;70:8149–58.
Cooper AB, Sawai CM, Sicinska E, Powers SE, Sicinski P, Clark MR, Aifantis I. A unique function for cyclin D3 in early B cell development. Nat Immunol. 2006;7:489–97.
Corsino P, Davis B, Law M, Chytil A, Forrester E, Norgaard A, Teoh N, Law B. Tumors initiated by Constitutive Cdk2 activation exhibit transforming growth factor A resistance and acquire paracrine mitogenic stimulation during progression. Cancer Research. 2007;67(7):3135–44.
Dannenberg JH, te Riele HP. The retinoblastoma gene family in cell cycle regulation and suppression of tumorigenesis. Results Probl Cell Differ. 2006;42:183–225.
Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 2000;14:3051–64.
Das G, Clark AM, Levine EM. Cyclin D1 inactivation extends proliferation and alters histogenesis in the postnatal mouse retina. Dev Dyn. 2012;241:941–52.
de Bruin A, Wu L, Saavedra HI, Wilson P, Yang Y, Rosol TJ, Weinstein M, Robinson ML, Leone G. Rb function in extraembryonic lineages suppresses apoptosis in the CNS of Rb-deficient mice. Proc Natl Acad Sci U S A. 2003;100:6546–51.
Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–26.
Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, Weinberg RA. Physical interaction of the retinoblastoma protein with human D cyclins. Cell. 1993;73:499–511.
Ericson KK, Krull D, Slomiany P, Grossel MJ. Expression of cyclin-dependent kinase 6, but not cyclin-dependent kinase 4, alters morphology of cultured mouse astrocytes. Mol Cancer Res. 2003;1:654–64.
Ericson KK, Gocheva V, Slomiany P, Udoeyop I, Grossel MJ. Differences in Cdk4 and Cdk6 function in primary mouse astrocytes. Mol Biol Cell. 2002;13:438a.
Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–6.
Evans T, Rosenthal ET, Youngblom J, Distel D, Hunt T. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell. 1983;33:389–96.
Evron E, Umbricht CB, Korz D, Raman V, Loeb DM, Niranjan B, Buluwela L, Weitzman SA, Marks J, Sukumar S. Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res. 2001;61:2782–7.
Faast R, White J, Cartwright P, Crocker L, Sarcevic B, Dalton S. Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a). Oncogene. 2004;23:491–502.
Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 1995;9:2364–72.
Fantl V, Edwards PA, Steel JH, Vonderhaar BK, Dickson C. Impaired mammary gland development in Cyl-1(−/−) mice during pregnancy and lactation is epithelial cell autonomous. Dev Biol. 1999;212:1–11.
Fantl V, Creer A, Dillon C, Bresnick J, Jackson D, Edwards P, Rosewell I, Dickson C. Fibroblast growth factor signalling and cyclin D1 function are necessary for normal mammary gland development during pregnancy. A transgenic mouse approach. Adv Exp Med Biol. 2000;480:1–7.
Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, et al. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 2002;21:3337–46.
Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–80.
Florenes VA, Faye RS, Maelandsmo GM, Nesland JM, Holm R. Levels of cyclin D1 and D3 in malignant melanoma: deregulated cyclin D3 expression is associated with poor clinical outcome in superficial melanoma. Clin Cancer Res. 2000;6:3614–20.
Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899–911.
Ganter B, Fu SL, Lipsick JS. D-type cyclins repress transcriptional activation by the v-Myb but not the c-Myb DNA-binding domain. EMBO J. 1998;17:255–68.
Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–77.
Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–8.
Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, Barnes D, Peters G. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 1994;54:1812–7.
Glickstein SB, Alexander S, Ross ME. Differences in cyclin D2 and D1 protein expression distinguish forebrain progenitor subsets. Cereb Cortex. 2007;17:632–42.
Glickstein SB, Monaghan JA, Koeller HB, Jones TK, Ross ME. Cyclin D2 is critical for intermediate progenitor cell proliferation in the embryonic cortex. J Neurosci. 2009;29:9614–24.
Glickstein SB, Moore H, Slowinska B, Racchumi J, Suh M, Chuhma N, Ross ME. Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development. 2007;134:4083–93.
Gopinathan L, Ratnacaram CK, Kaldis P. Established and novel Cdk/cyclin complexes regulating the cell cycle and development. Results Probl Cell Differ. 2011;53:365–89.
Grossel MJ, Hinds PW. From cell cycle to differentiation – an expanding role for Cdk6. Cell Cycle. 2006;5:266–70.
Grossel MJ, Hinds PW. Beyond the cell cycle: a new role for cdk6 in differentiation. J Cell Biochem. 2006;97:485–93.
Guan KL, Jenkins CW, Li Y, O'Keefe CL, Noh S, Wu X, Zariwala M, Matera AG, Xiong Y. Isolation and characterization of p19INK4d, a p16-related inhibitor specific to CDK6 and CDK4. Mol Biol Cell. 1996;7:57–70.
Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–61.
Hartwell LH, Culotti J, Pringle JR, Reid BJ. Genetic control of the cell division cycle in yeast. Science. 1974;183:46–51.
Hindley J, Phear GA. Sequence of the cell division gene CDC2 from Schizosaccharomyces pombe; patterns of splicing and homology to protein kinases. Gene. 1984;31:129–34.
Hirai H, Roussel MF, Kato JY, Ashmun RA, Sherr CJ. Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol Cell Biol. 1995;15:2672–81.
Hosokawa Y, Papanikolaou A, Cardiff RD, Yoshimoto K, Bernstein M, Wang TC, Schmidt EV, Arnold A. In vivo analysis of mammary and non-mammary tumorigenesis in MMTV-cyclin D1 transgenic mice deficient in p53. Transgenic Res. 2001;10:471–8.
Hu MG, Deshpande A, Schlichting N, Hinds EA, Mao C, Dose M, Hu GF, Van Etten RA, Gounari F, Hinds PW. CDK6 kinase activity is required for thymocyte development. Blood. 2011;117:6120–31.
Hu MG, Deshpande A, Enos M, Mao DQ, Hinds EA, Hu GF, Chang R, Guo ZY, Dose M, Mao CC, et al. A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009;69:810–8.
Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME. Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development. 1999;126:1927–35.
Huh MS, Parker MH, Scime A, Parks R, Rudnicki MA. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J Cell Biol. 2004;166:865–76.
Hulit J, Wang C, Li Z, Albanese C, Rao M, Di Vizio D, Shah S, Byers SW, Mahmood R, Augenlicht LH, et al. Cyclin D1 genetic heterozygosity regulates colonic epithelial cell differentiation and tumor number in ApcMin mice. Mol Cell Biol. 2004;24:7598–611.
Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol. 2016;17:280–92.
Inaba T, Matsushime H, Valentine M, Roussel MF, Sherr CJ, Look AT. Genomic organization, chromosomal localization, and independent expression of human cyclin D genes. Genomics. 1992;13:565–74.
Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol. 1998;18:1590–600.
Iwamori N, Naito K, Sugiura K, Tojo H. Preimplantation-embryo-specific cell cycle regulation is attributed to the low expression level of retinoblastoma protein. FEBS Lett. 2002;526:119–23.
Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300.
Jena N, Deng M, Sicinska E, Sicinski P, Daley GQ. Critical role for cyclin D2 in BCR/ABL-induced proliferation of hematopoietic cells. Cancer Res. 2002;62:535–41.
Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, Hinds PW. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 2010;17:65–76.
Jiang Z, Zacksenhaus E, Gallie BL, Phillips RA. The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene. 1997;14:1789–97.
Jirawatnotai S, Aziyu A, Osmundson EC, Moons DS, Zou XH, Kineman RD, Kiyokawa H. Cdk4 is indispensable for postnatal proliferation of the anterior pituitary. J Biol Chem. 2004;279:51100–6.
Jirawatnotai S, Hu YD, Livingston DM, Sicinski P. Proteomic identification of a direct role for cyclin D1 in DNA damage repair. Cancer Res. 2012;72:4289–93.
Jirawatnotai S, Hu YD, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YYE, Clark AB, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011;474:230–4.
Jirawatnotai S, Sharma S, Michowski W, Suktitipat B, Geng Y, Quackenbush J, Elias JE, Gygi SP, Wang YYE, Sicinski P. The cyclin D1-CDK4 oncogenic interactome enables identification of potential novel oncogenes and clinical prognosis. Cell Cycle. 2014;13:2889–900.
Kalaszczynska I, Geng Y, Iino T, Mizuno SI, Choi Y, Kondratiuk I, Silver DP, Wolgemuth DJ, Akashi K, Sicinski P. Cyclin a is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell. 2009;138:352–65.
Kierszenbaum AL. Cell-cycle regulation and mammalian gametogenesis: a lesson from the unexpected. Mol Reprod Dev. 2006;73:939–42.
Kiess M, Gill RM, Hamel PA. Expression of the positive regulator of cell cycle progression, cyclin D3, is induced during differentiation of myoblasts into quiescent myotubes. Oncogene. 1995;10:159–66.
Kim HA, Pomeroy SL, Whoriskey W, Pawlitzky I, Benowitz LI, Sicinski P, Stiles CD, Roberts TM. A developmentally regulated switch directs regenerative growth of Schwann cells through cyclin D1. Neuron. 2000;26:405–16.
Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121:3853–7.
Kohoutek J, Dvorak P, Hampl A. Temporal distribution of CDK4, CDK6, D-type cyclins, and p27 in developing mouse oocytes. Biol Reprod. 2004;70:139–45.
Kowalczyk A, Filipkowski RK, Rylski M, Wilczynski GM, Konopacki FA, Jaworski J, Ciemerych MA, Sicinski P, Kaczmarek L. The critical role of cyclin D2 in adult neurogenesis. J Cell Biol. 2004;167:209–13.
Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–91.
Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–6.
Kubiak JZ, Ciemerych MA, Hupalowska A, Sikora-Polaczek M, Polanski Z. On the transition from the meiotic to mitotic cell cycle during early mouse development. Int J Dev Biol. 2008;52:201–17.
Kushner JA. Beta-cell growth – an unusual paradigm of organogenesis that is cyclin D2/Cdk4 dependent. Cell Cycle. 2006;5:234–7.
Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–62.
La Baer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–62.
Lam EW, Glassford J, Banerji L, Thomas NS, Sicinski P, Klaus GG. Cyclin D3 compensates for loss of cyclin D2 in mouse B-lymphocytes activated via the antigen receptor and CD40. J Biol Chem. 2000;275:3479–84.
Lammie GA, Fantl V, Smith R, Schuuring E, Brookes S, Michalides R, Dickson C, Arnold A, Peters G. D11S287, a putative oncogene on chromosome 11q13, is amplified and expressed in squamous cell and mammary carcinomas and linked to BCL-1. Oncogene. 1991;6:439–44.
Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22.
Landis MW, Brown NE, Baker GL, Shifrin A, Das M, Geng Y, Sicinski P, Hinds PW. The LxCxE pRb interaction domain of cyclin D1 is dispensable for murine development. Cancer Res. 2007;67:7613–20.
Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, Flores JM, Cordon-Cardo C, Barbacid M. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–506.
Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–16.
LeCouter JE, Kablar B, Whyte PF, Ying C, Rudnicki MA. Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development. 1998;125:4669–79.
LeCouter JE, Kablar B, Hardy WR, Ying C, Megeney LA, May LL, Rudnicki MA. Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol Cell Biol. 1998;18:7455–65.
Lee CC, Yamamoto S, Morimura K, Wanibuchi H, Nishisaka N, Ikemoto S, Nakatani T, Wada S, Kishimoto T, Fukushima S. Significance of cyclin D1 overexpression in transitional cell carcinomas of the urinary bladder and its correlation with histopathologic features. Cancer. 1997;79:780–9.
Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–94.
Lee MG, Nurse P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature. 1987;327:31–5.
Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E, Jacks T. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 1996;10:1621–32.
Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang X, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510:547–51.
Lew DJ, Dulic V, Reed SI. Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell. 1991;66:1197–206.
Lewandoski M. Conditional control of gene expression in the mouse. Nat Rev Genet. 2001;2:743–55.
Li Z, Wang C, Jiao X, Katiyar S, Casimiro MC, Prendergast GC, Powell MJ, Pestell RG. Alternate cyclin d1 mRNA splicing modulates p27(KIP1) binding and cell migration. J Biol Chem. 2008;283:7007–15.
Li ZP, Jiao XM, Wang CG, Shirley LA, Elsaleh H, Dahl O, Wang M, Soutoglou E, Knudsen ES, Pestell RG. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res. 2010;70:8802–11.
Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene. 2008;27:1231–42.
Liu F. Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 2006;17(1–2):9–17.
Liu L, Michowski W, Inuzuka H, Shimizu K, Nihira NT, Chick JM, Li N, Geng Y, Meng AY, Ordureau A, et al. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat Cell Biol. 2017;19:177–88.
Liu WD, Wang HW, Muguira M, Breslin MB, Lan MS. INSM1 functions as a transcriptional repressor of the neuroD/beta 2 gene through the recruitment of cyclin D1 and histone deacetylases. Biochem J. 2006;397:169–77.
Lucas JJ, Szepesi A, Modiano JF, Domenico J, Gelfand EW. Regulation of synthesis and activity of the plstire protein (cyclin-dependent kinase-6 (Cdk6)), a major cyclin-D-associated Cdk4 homolog in normal human T-lymphocytes. J Immunol. 1995;154:6275–84.
Ma CY, Papermaster D, Cepko CL. A unique pattern of photoreceptor degeneration in cyclin D1 mutant mice. Proc Natl Acad Sci U S A. 1998;95:9938–43.
Maandag EC, van der Valk M, Vlaar M, Feltkamp C, O'Brien J, van Roon M, van der Lugt N, Berns A, te Riele H. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–8.
MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol. 2003;23:1044–53.
Maeda K, Chung Y, Kang S, Ogawa M, Onoda N, Nishiguchi Y, Ikehara T, Nakata B, Okuno M, Sowa M. Cyclin D1 overexpression and prognosis in colorectal adenocarcinoma. Oncology. 1998;55:145–51.
Mak TW. Gene targeting in embryonic stem cells scores a knockout in Stockholm. Cell. 2007;131:1027–31.
Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15:122.
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–66.
Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type Cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504.
Mariappan I, Parnaik VK. Sequestration of pRb by cyclin D3 causes intranuclear reorganization of lamin A/C during muscle cell differentiation. Mol Biol Cell. 2005;16:1948–60.
Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78:7634–8.
Martin J, Hunt SL, Dubus P, Sotillo R, Nehme-Pelluard F, Magnuson MA, Parlow AF, Malumbres M, Ortega S, Barbacid M. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene. 2003;22:5261–9.
Masui Y. From oocyte maturation to the in vitro cell cycle: the history of discoveries of Maturation-Promoting Factor (MPF) and Cytostatic Factor (CSF). Differentiation. 2001;69:1–17.
Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool. 1971;177:129–45.
Mataraza JM, Tumang JR, Gumina MR, Gurdak SM, Rothstein TL, Chiles TC. Disruption of cyclin D3 blocks proliferation of normal B-1a cells, but loss of cyclin D3 is compensated by cyclin D2 in cyclin D3-deficient mice. J Immunol. 2006;177:787–95.
Mate JL, Ariza A, Aracil C, Lopez D, Isamat M, Perez-Piteira J, Navas-Palacios JJ. Cyclin D1 overexpression in non-small cell lung carcinoma: correlation with Ki67 labelling index and poor cytoplasmic differentiation. J Pathol. 1996;180:395–9.
Matsuoka S, Yamaguchi M, Matsukage A. D-type Cyclin-binding regions of proliferating cell nuclear antigen. J Biol Chem. 1994;269:11030–6.
Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–13.
Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–76.
Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, Sherr CJ. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–34.
Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–31.
Mettus RV, Rane SG. Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene. 2003;22:8413–21.
Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14:2077–86.
Meyerson M, Enders GH, Wu CL, Su LK, Gorka C, Nelson C, Harlow E, Tsai LH. A family of human cdc2-related protein kinases. EMBO J. 1992;11:2909–17.
Miliani De Marval PL, Macias E, Rounbehler R, Sicinski P, Kiyokawa H, Johnson DG, Conti CJ, Rodriguez-Puebla ML. Lack of cyclin-dependent kinase 4 inhibits c-myc tumorigenic activities in epithelial tissues. Mol Cell Biol. 2004;24:7538–47.
Momčilović O, Navara C, Schatten G. Cell cycle adaptations and maintenance of genomic integrity in embryonic stem cells and induced pluripotent stem cells. In: Kubiak JZ, editor. Cell cycle in development. Berlin/Heidelberg: Springer-Verlag GmbH; 2011.
Moons DS, Jirawatnotai S, Parlow AF, Gibori G, Kineman RD, Kiyokawa H. Pituitary hypoplasia and lactotroph dysfunction in mice deficient for cyclin-dependent kinase-4. Endocrinology. 2002;143:3001–8.
Moons DS, Jirawatnotai S, Tsutsui T, Franks R, Parlow AF, Hales DB, Gibori G, Fazleabas AT, Kiyokawa H. Intact follicular maturation and defective luteal function in mice deficient ford cyclin-dependent kinase-4. Endocrinology. 2002;143:647–54.
Moore GD, Ayabe T, Kopf GS, Schultz RM. Temporal patterns of gene expression of G1-S cyclins and cdks during the first and second mitotic cell cycles in mouse embryos. Mol Reprod Dev. 1996;45:264–75.
Motokura T, Keyomarsi K, Kronenberg HM, Arnold A. Cloning and characterization of human cyclin D3, a cDNA closely related in sequence to the PRAD1/cyclin D1 proto-oncogene. J Biol Chem. 1992;267:20412–5.
Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, Arnold A. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature. 1991;350:512–5.
Musgrove EA. Cyclins: roles in mitogenic signaling and oncogenic transformation. Growth Factors. 2006;24:13–9.
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72.
Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, Di Renzo J, Pestell RG, Hinds PW, Dowdy SF, Brown M, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol. 1997;17:5338–47.
Neumeister P, Pixley FJ, Xiong Y, Xie HF, Wu KM, Ashton A, Cammer M, Chan A, Symons M, Stanley ER, et al. Cyclin D1 governs adhesion and motility of macrophages. Mol Biol Cell. 2003;14:2005–15.
Nobs L, Nestel S, Kulik A, Nitsch C, Atanasoski S. Cyclin D1 is required for proliferation of olig2-expressing progenitor cells in the injured cerebral cortex. Glia. 2013;61:1443–55.
Odajima J, Saini S, Jung P, Ndassa-Colday Y, Ficaro S, Geng Y, Marco E, Michowski W, Wang YE, DeCaprio JA, et al. Proteomic landscape of tissue-specific cyclin E functions in vivo. PLoS Genet. 2016;12:e1006429.
Ogasawara T, Chikuda H, Ohba S, Chikazu D, Katagiri M, Yano F, Nakamura K, Chung U, Hoshi K, Takato T, et al. Functional switching of Runx2 by Cdk6 and Cdk4 in regulation of osteoblast differentiation. J Bone Miner Res. 2005;20:S5.
Opitz OG, Rustgi AK. Interaction between Sp1 and cell cycle regulatory proteins is important in transactivation of a differentiation-related gene. Cancer Res. 2000;60:2825–30.
Palmero I, Holder A, Sinclair AJ, Dickson C, Peters G. Cyclins D1 and D2 are differentially expressed in human B-lymphoid cell lines. Oncogene. 1993;8:1049–54.
Pauling JK, Christensen AG, Batra R, Alcaraz N, Barbosa E, Larsen MR, Beck HC, Leth-Larsen R, Azevedo V, Ditzel HJ, et al. Elucidation of epithelial mesenchymal transition-related pathways in a triple-negative breast cancer cell line model by multi-omics interactome analysis. Integr Biol-Uk. 2014;6:1058–68.
Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J. 1999;18:5310–20.
Peters G, Fantl V, Smith R, Brookes S, Dickson C. Chromosome 11q13 markers and D-type cyclins in breast cancer. Breast Cancer Res Treat. 1995;33:125–35.
Pirkmaier A, Dow R, Ganiatsas S, Waring P, Warren K, Thompson A, Hendley J, Germain D. Alternative mammary oncogenic pathways are induced by D-type cyclins; MMTV-cyclin D3 transgenic mice develop squamous cell carcinoma. Oncogene. 2003;22:4425–33.
Quelle DE, Ashmun RA, Hannon GJ, Rehberger PA, Trono D, Richter KH, Walker C, Beach D, Sherr CJ, Serrano M. Cloning and characterization of murine p16INK4a and p15INK4b genes. Oncogene. 1995;11:635–45.
Rane SG, Cosenza SC, Mettus RV, Reddy EP. Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol. 2002;22:644–56.
Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52.
Rao PN, Wilson B, Puck TT. Premature chromosome condensation and cell cycle analysis. J Cell Physiol. 1977;91:131–41.
Rao SS, Chu C, Kohtz DS. Ectopic expression of cyclin D1 prevents activation of gene transcription by myogenic basic helix-loop-helix regulators. Mol Cell Biol. 1994;14:5259–67.
Ratineau C, Petry MW, Mutoh H, Leiter AB. Cyclin D1 represses the basic helix-loop-helix transcription factor, BETA2/NeuroD. J Biol Chem. 2002;277:8847–53.
Ravnik SE, Rhee K, Wolgemuth DJ. Distinct patterns of expression of the D-type cyclins during testicular development in the mouse. Dev Genet. 1995;16:171–8.
Reddy HK, Grana X, Dhanasekaran DN, Litvin J, Reddy EP. Requirement of Cdk4 for v-Ha-ras-induced breast tumorigenesis and activation of the v-ras-induced senescence program by the R24C mutation. Genes Cancer. 2010;1:69–80.
Reddy HK, Mettus RV, Rane SG, Grana X, Litvin J, Reddy EP. Cyclin-dependent kinase 4 expression is essential for neu-induced breast tumorigenesis. Cancer Res. 2005;65:10174–8.
Reutens AT, Fu MF, Wang CG, Albanese C, McPhaul MJ, Sun ZJ, Balk SP, Janne OA, Palvimo JJ, Pestell RG. Cyclin D1 binds the androgen receptor and regulates hormone-dependent signaling in a p300/CBP-associated factor (P/CAF)-dependent manner. Mol Endocrinol. 2001;15:797–811.
Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L, Sicinski P, Tennant RW, Weinberg RA, Yuspa SH, Conti CJ. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–74.
Rodriguez-Puebla ML, Miliani de Marval PL, LaCava M, Moons DS, Kiyokawa H, Conti CJ. Cdk4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am J Pathol. 2002;161:405–11.
Rosenthal ET, Hunt T, Ruderman JV. Selective translation of messenger-Rna controls the pattern of protein-synthesis during early development of the surf clam, spisula-solidissima. Cell. 1980;20:487–94.
Rossant J, McMahon A. Creating mouse mutants-a meeting review on conditional mouse genetics. Genes Dev. 1999;13:142–5.
Russell A, Thompson MA, Hendley J, Trute L, Armes J, Germain D. Cyclin D1 and D3 associate with the SCF complex and are coordinately elevated in breast cancer. Oncogene. 1999;18:1983–91.
Saavedra-Avila NA, Sengupta U, Sanchez B, Sala E, Haba L, Stratmann T, Verdaguer J, Mauricio D, Mezquita B, Ropero AB, et al. Cyclin D3 promotes pancreatic beta-cell fitness and viability in a cell cycle-independent manner and is targeted in autoimmune diabetes. Proc Natl Acad Sci U S A. 2014;111:E3405–14.
Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–50.
Said TK, Conneely OM, Medina D, O'Malley BW, Lydon JP. Progesterone, in addition to estrogen, induces cyclin D1 expression in the murine mammary epithelial cell, in vivo. Endocrinology. 1997;138:3933–9.
Sallinen SL, Sallinen PK, Kononen JT, Syrjakoski KM, Nupponen NN, Rantala IS, Helen PT, Helin HJ, Haapasalo HK. Cyclin D1 expression in astrocytomas is associated with cell proliferation activity and patient prognosis. J Pathol. 1999;188:289–93.
Sankaran VG, Ludwig LS, Sicinska E, Xu J, Bauer DE, Eng JC, Patterson HC, Metcalf RA, Natkunam Y, Orkin SH, et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev. 2012;26:2075–87.
Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–5.
Savatier P, Lapillonne H, van Grunsven LA, Rudkin BB, Samarut J. Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene. 1996;12:309–22.
Savatier P, Lapillonne H, Jirmanova L, Vitelli L, Samarut J. Analysis of the cell cycle in mouse embryonic stem cells. Methods Mol Biol. 2002;185:27–33.
Scheicher R, Hoelbl-Kovacic A, Bellutti F, Tigan AS, Prchal-Murphy M, Heller G, Schneckenleithner C, Salazar-Roa M, Zochbauer-Muller S, Zuber J, et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 2015;125:90–101.
Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7.
Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, De Pinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37.
Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379–85.
Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91.
Shen T, Huang SL. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anti-Cancer Agent Me. 2012;12:631–9.
Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12.
Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–711.
Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 2003;4:451–61.
Sicinska E, Lee YM, Gits J, Shigematsu H, Yu Q, Rebel VI, Geng Y, Marshall CJ, Akashi K, Dorfman DM, et al. Essential role for cyclin D3 in granulocyte colony-stimulating factor-driven expansion of neutrophil granulocytes. Mol Cell Biol. 2006;26:8052–60.
Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell. 1995;82:621–30.
Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, et al. Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature. 1996;384:470–4.
Simanis V, Nurse P. The cell cycle control gene cdc2+ of fission yeast encodes a protein kinase potentially regulated by phosphorylation. Cell. 1986;45:261–8.
Simmons J. Virchow and the cell doctrine. In: Scientific 100 – a ranking of most influential scientists, past and present. Secaucus: Carol Publishing Group; 1996. p. 88–92.
Skapek SX, Rhee J, Spicer DB, Lassar AB. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–4.
Skapek SX, Rhee J, Kim PS, Novitch BG, Lassar AB. Cyclin-mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol Cell Biol. 1996;16:7043–53.
Smith LD, Ecker RE. The interaction of steroids with Rana pipiens oocytes in the induction of maturation. Dev Biol. 1971;25:232–47.
Solvason N, Wu WW, Parry D, Mahony D, Lam EW, Glassford J, Klaus GG, Sicinski P, Weinberg R, Liu YJ, et al. Cyclin D2 is essential for BCR-mediated proliferation and CD5 B cell development. Int Immunol. 2000;12:631–8.
Sunkara PS, Al-Bader AA, Riker MA, Rao PN. Induction of prematurely condensed chromosomes by mitoplasts. Cell Biol Int Rep. 1980;4:1025–9.
Swaminathan G, Varghese B, Fuchs SY. Regulation of prolactin receptor levels and activity in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:81–91.
Sweeney KJ, Swarbrick A, Sutherland RL, Musgrove EA. Lack of relationship between CDK activity and G1 cyclin expression in breast cancer cells. Oncogene. 1998;16:2865–78.
Swenson KI, Farrell KM, Ruderman JV. The clam embryo protein cyclin a induces entry into M phase and the resumption of meiosis in Xenopus oocytes. Cell. 1986;47:861–70.
Takaki T, Fukasawa K, Suzuki-Takahashi I, Semba K, Kitagawa M, Taya Y, Hirai H. Preferences for phosphorylation sites in the retinoblastoma protein of D-type cyclin-dependent kinases, Cdk4 and Cdk6, in vitro. J Biochem (Tokyo). 2005;137:381–6.
Tam SW, Theodoras AM, Shay JW, Draetta GF, Pagano M. Differential expression and regulation of Cyclin D1 protein in normal and tumor human cells: association with Cdk4 is required for Cyclin D1 function in G1 progression. Oncogene. 1994;9:2663–74.
Taneja P, Frazier DP, Kendig RD, Maglic D, Sugiyama T, Kai F, Taneja NK, Inoue K. MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert Rev Mol Diagn. 2009;9:423–40.
Tapias A, Ciudad CJ, Roninson IB, Noe V. Regulation of Sp1 by cell cycle related proteins. Cell Cycle. 2008;7:2856–67.
Tarkowski AK. Mouse chimaeras developed from fused eggs. Nature. 1961;190:857–60.
Tarkowski AK. Mouse chimaeras revisited: recollections and reflections. Int J Dev Biol. 1998;42:903–8.
Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–9.
Tworoger SS, Hankinson SE. Prolactin and breast cancer etiology: an epidemiologic perspective. J Mammary Gland Biol Neoplasia. 2008;13:41–53.
van der Kuip H, Carius B, Haque SJ, Williams BRG, Huber C, Fischer T. The DNA-binding subunit p140 of replication factor C is upregulated in cycling cells and associates with G(1) phase cell cycle regulatory proteins. J Mol Med-JMM. 1999;77:386–92.
Wang CG, Fan SJ, Li ZP, Fu MF, Rao M, Ma YX, Lisanti MP, Albanese C, Katzenellenbogen BS, Kushner PJ, et al. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer Res. 2005;65:6557–67.
Wang CG, Pattabiraman N, Zhou JN, Fu MF, Sakamaki T, Albanese C, Li ZP, Wu KM, Hulit J, Neumeister P, et al. Cyclin D1 repression of peroxisome proliferator-activated receptor gamma expression and transactivation. Mol Cell Biol. 2003;23:6159–73.
Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–71.
Wenzel PL, Wu L, de Bruin A, Chong JL, Chen WY, Dureska G, Sites E, Pan T, Sharma A, Huang K, et al. Rb is critical in a mammalian tissue stem cell population. Genes Dev. 2007;21:85–97.
White J, Stead E, Faast R, Conn S, Cartwright P, Dalton S. Developmental activation of the Rb-E2F pathway and establishment of cell cycle-regulated cyclin-dependent kinase activity during embryonic stem cell differentiation. Mol Biol Cell. 2005;16:2018–27.
Wianny F, Real FX, Mummery CL, Van Rooijen M, Lahti J, Samarut J, Savatier P. G1-phase regulators, cyclin D1, cyclin D2, and cyclin D3: up-regulation at gastrulation and dynamic expression during neurulation. Dev Dyn. 1998;212:49–62.
Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 1994;13:4251–9.
Won KA, Xiong Y, Beach D, Gilman MZ. Growth-regulated expression of D-type cyclin genes in human diploid fibroblasts. Proc Natl Acad Sci U S A. 1992;89:9910–4.
Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421:942–7.
Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71:505–14.
Xiong Y, Connolly T, Futcher B, Beach D. Human D-type cyclin. Cell. 1991;65:691–9.
Xiong Y, Menninger J, Beach D, Ward DC. Molecular cloning and chromosomal mapping of CCND genes encoding human D-type cyclins. Genomics. 1992;13:575–84.
Yang C, Chen L, Li C, Lynch MC, Brisken C, Schmidt EV. Cyclin D1 enhances the response to estrogen and progesterone by regulating progesterone receptor expression. Mol Cell Biol. 2010;30:3111–25.
Yang C, Ionescu-Tiba V, Burns K, Gadd M, Zukerberg L, Louis DN, Sgroi D, Schmidt EV. The role of the cyclin D1-dependent kinases in ErbB2-mediated breast cancer. Am J Pathol. 2004;164:1031–8.
Yang R, Bie W, Haegebarth A, Tyner AL. Differential regulation of D-type cyclins in the mouse intestine. Cell Cycle. 2006;5:180–3.
Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–21.
Yu Q, Ciemerych MA, Sicinski P. Ras and Myc can drive oncogenic cell proliferation through individual D-cyclins. Oncogene. 2005;24:7114–9.
Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32.
Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–64.
Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–46.
Zhang Q, Wang XY, Wolgemuth DJ. Developmentally regulated expression of cyclin D3 and its potential in vivo interacting proteins during murine gametogenesis. Endocrinology. 1999;140:2790–800.
Zhang Q, Sakamoto K, Liu C, Triplett AA, Lin WC, Rui H, Wagner KU. Cyclin D3 compensates for the loss of cyclin D1 during ErbB2-induced mammary tumor initiation and progression. Cancer Res. 2011;71:7513–24.
Zheng B, Sage M, Sheppeard EA, Jurecic V, Bradley A. Engineering mouse chromosomes with Cre-loxP: range, efficiency, and somatic applications. Mol Cell Biol. 2000;20:648–55.
Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–11.
Zindy F, van Deursen J, Grosveld G, Sherr CJ, Roussel MF. INK4d-deficient mice are fertile despite testicular atrophy. Mol Cell Biol. 2000;20:372–8.
Zindy F, Soares H, Herzog KH, Morgan J, Sherr CJ, Roussel MF. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 1997;8:1139–50.
Zindy F, den Besten W, Chen B, Rehg JE, Latres E, Barbacid M, Pollard JW, Sherr CJ, Cohen PE, Roussel MF. Control of spermatogenesis in mice by the cyclin D-dependent kinase inhibitors p18(Ink4c) and p19(Ink4d). Mol Cell Biol. 2001;21:3244–55.
Zou X, Ray D, Aziyu A, Christov K, Boiko AD, Gudkov AV, Kiyokawa H. Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to Arf/p53-independent senescence. Genes Dev. 2002;16:2923–34.
Zukerberg LR, Yang WI, Gadd M, Thor AD, Koerner FC, Schmidt EV, Arnold A. Cyclin D1 (PRAD1) protein expression in breast cancer: approximately one-third of infiltrating mammary carcinomas show overexpression of the cyclin D1 oncogene. Mod Pathol. 1995;8:560–7.
Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–15.
Acknowledgments
We want to thank Piotr Sicinski for being our mentor in the cyclin universe. We are also grateful to Katarzyna Koziak and Phil Hinds for their help at the very final stage of the manuscript preparation. During the preparation of this chapter, IK was supported by the Medical University of Warsaw statutory grant 1M15/N/2015 and the National Centre for Research and Development (NCBR) grant STRATEGMED3/307326/6/NCBR/2017 and MAC by the Faculty of Biology University of Warsaw funding 501/56/169600 and the National Science Centre Poland (NCN) grant 2012/05/N/NZ3/00314.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Kalaszczynska, I., Ciemerych, M.A. (2018). Mammalian Development and Cancer: A Brief History of Mice Lacking D-Type Cyclins or CDK4/CDK6. In: Hinds, P., Brown, N. (eds) D-type Cyclins and Cancer. Current Cancer Research. Springer, Cham. https://doi.org/10.1007/978-3-319-64451-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-64451-6_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64449-3
Online ISBN: 978-3-319-64451-6
eBook Packages: MedicineMedicine (R0)