
Abstract
Development and normal physiology of the nervous system require proliferation and differentiation of stem and progenitor cells in a strictly controlled manner. The number of cells generated depends on the type of cell division, the cell cycle length, and the fraction of cells that exit the cell cycle to become quiescent or differentiate. The underlying processes are tightly controlled and modulated by cyclin-dependent kinases (Cdks) and their interactions with cyclins and Cdk inhibitors (CKIs). Studies performed in the nervous system with mouse models lacking individual Cdks, cyclins, and CKIs, or combinations thereof, have shown that many of these molecules control proliferation rates in a cell-type specific and time-dependent manner. In this review, we will provide an update on the in vivo studies on cyclins, Cdks, and CKIs in neuronal and glial tissue. The goal is to highlight their impact on proliferation processes during the development of the peripheral and central nervous system, including and comparing normal and pathological conditions in the adult.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The development of the vertebrate nervous system involves a series of coordinated morphological events. The primordium of the entire central nervous system (CNS) is the neural tube, which originates from the neural plate. The latter is a specialized epithelial sheet of the ectoderm, which invaginates during neurulation along a rostro-caudal axis. The resulting neural tube differentiates into the spinal cord and the brain [1].
Peripheral Nervous System
Shortly after the neural groove closes completely, some neuroectodermal cells delaminate from the dorsal-most region of the neural tube. These neural crest progenitor cells are a highly proliferative and migratory cell population. They give rise to a wide variety of cell and tissue types including neurons and glia of the peripheral nervous system (PNS). The majority of Schwann cells, the myelinating and non-myelinating glial cells of the PNS, arise from neural crest-derived Schwann cell precursor cells that migrate along axons of developing nerves in the embryo [2]. The proliferation rate of Schwann cell precursors gradually decreases during the first mouse postnatal week as differentiation into myelinating and non-myelinating Schwann cells proceeds. Mature Schwann cells are growth-arrested; however, they remain capable of re-entering the cell cycle, as observed in de- and regeneration processes after nerve damage [3].
Developing Central Nervous System
The CNS originates from neuroepithelial cells of the ectoderm which expand by symmetric divisions and line the wall of the neural tube. They give rise to radial glia cells, a heterogenous population acting as neural stem and progenitor cells during the development of the CNS. The neural stem and progenitor cells within the ventricular zone (VZ) of the head region, including the developing cortex, the cerebellum, and the retina, are characterized by an extremely high proliferation rate [5, 94]. Since the development of the forebrain requires a vast expansion of tissue, an additional germinal layer, the subventricular zone (SVZ), has developed during evolution. It is colonized by basal progenitor cells, an amplifier population that emerges through asymmetric division of stem cells within the VZ [6]. This pool is less proliferative, but gives rise to the majority of the neurons in the cortex through neurogenic divisions [7]. Cell cycle progression plays an important role in controlling the transition from proliferative to neurogenic divisions of neural progenitor cells. This transition is associated with an increase in the length of the cell cycle due to a lengthening of the G1 phase [4, 8]. At later stages, the cortical stem cells switch their fate and start producing glial cells such as oligodendrocytes and astrocytes. This process is continued and completed after birth [5, 95]. Differently, in the spinal cord, all the neural and glial cells, with the exception of microglia, originate from the neuroepithelium [1]. First, distinct classes of neurons arise, and as they differentiate, they move laterally to the intermediate zone of the spinal cord before reaching their final position in the mantle zone [1]. Following the neurogenic phase, the oligodendrocytes and astrocytes arise from progenitor cells in spatially restricted domains of the VZ [9].
Adult Central Nervous System
In adult mammals, multipotent stem cells persist in specialized niches throughout the body including the brain. Unlike the embryonal neuronal stem cells that are highly proliferative and responsible for tissue growth, the adult neural stem cells leave the cell cycle and become quiescent [10]. In mice, adult neurogenesis continues in two main regions of the brain: the SVZ of the lateral ventricles that generates neurons of the olfactory bulb and the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) [10]. Importantly, brain injuries can induce neurogenesis by triggering and enhancing proliferation of quiescent stem and progenitor cells in the germinal niches of the SVZ and SGZ. How de novo neurogenesis could be promoted to reach a better recovery after CNS injuries without provoking adverse consequences such as seizures is under intense investigation [11]. In addition, oligodendrocyte progenitor cells (OPCs) persist and continue to divide slowly throughout adulthood in the brain parenchyma [12, 13]. Following an injury, OPCs are activated, increase their proliferation rate, and eventually move towards the lesion site [14]. Similarly, quiescent astrocytes become activated and restart dividing. Increased astrocyte proliferation (astrocytosis) is also commonly observed in acute and chronic brain diseases [12, 15].
Summing up, the development of the nervous system, its normal physiology, and the ability to respond to injuries require a sophisticated coordination between cell division, growth arrest, and differentiation of progenitor cells.
Cell Division
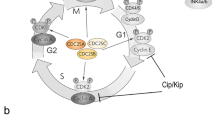
Cell division is a complex process that is regulated at multiple levels [5, 11, 96]. The cell cycle is divided in four phases organized as DNA synthesis (S), mitosis (M), and two gap phases, G1 and G2, preceding S and M phases, respectively (Fig. 1a). There are a number of checkpoints at which the cell examines external cues (like molecular signals) and internal cues (like DNA damage) and decides whether or not to move forward with division [102]. Once the cell passes the G1 checkpoint and enters S phase, it becomes irreversibly committed to division. The G2 checkpoint at the G2/M transition ensures that the replicated DNA is not damaged before the cell enters mitosis. If DNA has been damaged, the tumor suppressor gene p53 is activated. The cell cycle is blocked until the damage has been repaired or apoptosis is induced if the damage is unrepairable [102]. The mitotic checkpoint at the transition from metaphase to anaphase determines whether all the sister chromatids are correctly attached to the spindle microtubules. It is regulated by proteins centered around the activity of the anaphase-promoting complex (APC). To drive the cell cycle in one direction, mitotic proteins such as CyclinB are cyclically destroyed via ubiquitin (Ub)-mediated proteolysis. The rapid degradation is triggered by APC, a multi-subunit E3 ligase that facilitates conjugation of Ub to targeted substrates during mitosis [103].

Regulation of the cell cycle. a The cell cycle is divided into four phases: S-phase, in which DNA synthesis takes place, M-phase (mitosis) and two G (gap) phases, G1 and G2. The course of the cell cycle is regulated particularly at the transition between G1-S, G2-M, and M-phase, three check points that ensure the integrity of the genome. In G1, the cell responds to extrinsic signals. After the restriction point, the cell passes through the cell cycle without further mitogen stimulation. The Cdk complexes control the course of the cell cycle. Their catalytic activities are modulated by interactions with cyclins and Cdk inhibitors. Two classes of inhibitors regulate Cdk activities: the Ink4 family (p15, p16, p18, p19) and the Cip/Kip family (p27, p21, p27). b Unphosphorylated retinoblastoma (Rb) protein binds the transcription factor E2F and inhibits its activity. The CyclinD-Cdk4/6 and CyclinE-Cdk2 complexes phosphorylate Rb, which releases E2F. Free E2F binds to DP proteins and transcribes genes necessary for progression through the cell cycle
Cell cycle progression is initiated by mitogen stimulation during G1 phase, the only time period the cell cycle is responsive to extrinsic signals. In G1, at the restriction point, cells commit to a new round of cycle or exit from the cell cycle, either permanently (differentiation) or transiently (G0) [109]. Mitogen stimulation activates multiple signaling pathways that eventually induce the expression of D-type cyclins [104, 110]. These proteins, in association with their catalytic partners, Cdk4 and Cdk6, are fundamental to the regulation of the G1/S phase transition. CyclinD-Cdk4/6 and CyclinE-Cdk2 complexes mediate phosphorylation of the retinoblastoma protein pRb. This event disrupts the association of pRB with various family members of the E2F transcription factors that subsequently bind to their dimerization protein (DP) partners and activate the transcription of genes required for further progression through the cell cycle (Fig. 1b) [5]. The rate of cell cycle progression is tightly regulated by CKIs, which halt cell cycle progression under unfavorable conditions [5]. In mammals, CKIs are divided into two families: the Ink family includes p16, p15, p18, and p19, and the Cip/Kip family comprises p21, p27, and p57. Ink proteins specifically bind to and inhibit Cdk4/6, while Cip/Kip family members disrupt and inactivate cyclin/Cdk complexes, including CyclinE-Cdk2, Cyclin A-Cdk2, and CyclinB-Cdk1 (Fig. 1a).
In this review, we summarize in vivo studies of cyclins, Cdks, and CKIs in the nervous system, in which mouse models lacking individual members of these cell cycle molecules or combinations thereof were used. In doing so, we concentrate on those cyclins, Cdks, and CKIs that regulate the cell cycle of glial and neural progenitor cells [5, 11, 96].
Neurogenesis
There are numerous studies reporting on the expression of cell cycle activators and inhibitors in cells and tissues of the CNS and PNS. In Table 1, we have compiled the available information and provide an overview of the mRNA and protein expression patterns of cyclins, Cdks, and CKIs that are involved in proliferation processes during embryonic, postnatal, and adult stages in the mouse. Table 2 provides a list of references on in vivo studies in the mouse, in which the functional impact upon loss of cyclins, Cdks, and CKIs in neural and glial cells of the CNS and PNS was studied.
Neuronal Development
Most of the insight into the requirements for cell cycle proteins during mouse embryonic development has been gained from the analysis of neural progenitor cells in the cortex, the retina, and the cerebellum [5, 94]. In the cortex, it is well known that apical progenitor cells in the ventricular zone (VZ) have a high proliferative potential, whereas the basal progenitors in the subventricular zone (SVZ) divide only a few times [16]. All the more surprising is the fact that to date, only a few cell cycle proteins have been identified as regulators of cell cycle progression in distinct neural progenitor cells (NPCs) in the developing brain. For example, deletion of CyclinD1 affects the development of the retina by inducing a lengthening of the cell cycle in retinal progenitor cells [17, 18]. However, its loss in NPCs of the dorsal and ventral forebrain has no evident impact on cell division of these cells [19, 20]. A different outcome is observed upon loss of CyclinD2. In its absence, the basal progenitor pool in the dorsal and ventral SVZ of the developing cortex is diminished [19, 20]. Its lack also affects the progenitor pool in both the cerebellum and the retina during development [21,22,23]. These results demonstrate that a decrease in cell proliferation rates upon loss of distinct cell cycle proteins is cell-type specific. While CyclinD2 is indispensable for the proliferation of the progenitor cells in various developing brain tissues, CyclinD1 ablation appears to be compensated in most cases by an upregulation of other cyclin family members, such as CyclinD2 in the cortex [19, 20] or CyclinD3 in the retina [24].
To date, only a few publications report on the impact of Cdk deficiencies in the nervous system. Mice with single ablations of Cdk2, Cdk4, or Cdk6 survive, and their mutant apical and basal progenitors in the developing cortex show no major defects in cell cycle characteristics [25]. There are, however, contradictory results regarding Cdk6. Mi et al. reported that ablation of Cdk6 induced a reduction in the proliferation rate of cortical progenitors at embryonic day (E)12.5 [26]. Regional differences along the rostral-caudal axis might account for the discrepancy in the published studies and need to be further examined. Our group recently reported that concomitant deletion of Cdk4 and Cdk6 specifically affected basal but not apical progenitor cell populations in the ventral and dorsal cortex [25]. Similarly, Lim and Kaldis observed that Cdk2/Cdk4 double knockout embryos displayed a reduced thickness specifically of the SVZ and the cortical plate, but not of the VZ [27]. Notably, both studies came to the conclusion that proliferation rates of apical progenitors were not affected, which reveals substantial differences in the regulation of cell division in the SVZ vs. the VZ. Moreover, the finding that concomitant ablation of Cdk2 and Cdk6 had no effect on progenitor cell proliferation [25] demonstrates that only distinct combinations of Cdk molecules regulate the basal progenitor pool in the developing cortex.
Studies using specific shRNAs demonstrated that concomitant inhibition of Cdk4 and CyclinD1 in the developing cortex was required to induce a prolonged G1 phase in targeted cells accompanied by an increase in neurogenesis [28]. These findings substantiate the results obtained in the knockout mice, where single ablations of Cdk molecules were less effective in inducing a proliferation phenotype.
In addition to the role of cyclins and Cdks, CKIs have also been identified to be essential players in regulating proliferation of neural progenitor cells. In the retina, the absence of the inhibitors p19 and p27 caused an extended period of progenitor cell proliferation [29]. Similarly, p57-deficient mice displayed a hyperplastic anterior pituitary gland caused by increased proliferation of progenitors during development [30]. In accordance, forced overexpression of cell cycle inhibitors induced opposite effects: in the cortex, overexpression of p57 and p27 led to cell cycle exit of cortical progenitors [31,32,33], while in the retina, overexpression of p27 caused retinal progenitors to prematurely leave the cell cycle [34].
Adult Neurogenesis
In adult mice, cell division continues throughout life in two main regions of the CNS, the SVZ, and the SGZ (for adult neurogenesis, see [35]). The resulting continuous addition of new neurons into the circuitry is the focus of extensive research. Several studies have shown that dysregulation of the cell cycle is a primary cause of the alteration of the homeostasis in the adult neurogenic niches leading to neurological diseases [10]. One of the critical players in adult neurogenesis is CyclinD1. Its deletion inhibits the proliferation of neuronal progenitor cells in both the SVZ and SGZ [36], as well as the retina [37]. Similarly, mice lacking CyclinD2 display a reduction in the size of the hippocampus and the olfactory bulbs. This is in line with direct measurements of cell proliferation by in vivo labeling of newly synthesized DNA, where reduced neurogenesis in both the SVZ and SGZ was observed upon loss of CyclinD2 [38]. Interestingly, CyclinD2 only becomes indispensable 4 weeks after birth in the SGZ [39], and its loss is not compensated by overexpression of CyclinD1 in the adult hippocampus [40]. These data reveal remarkable functional differences between the two closely related family members during development and adulthood.
Proliferation of adult neural progenitor cells is also highly dependent on Cdk expression. Cdk6-deficient neural committed progenitors displayed reduced proliferation rates accompanied by a reduction of neurogenesis in the SVZ and SGZ [41]. Loss of Cdk2 began to affect progenitor proliferation in the SVZ only at 2 weeks after birth. Elevated levels of Cdk4 expression in the Cdk2-deficient mice point to potential compensatory mechanisms during the first days after birth [42]. Unlike Cdk2 and Cdk6, lack of Cdk4 did not significantly affect adult neurogenesis [41].
Studies on the requirement for CKIs in adult neural progenitor cells are scarce and have produced contradicting findings. Qiu et al. reported that the deletion of p21 had no effect on the proliferation rate in the SGZ [43], while Kippin et al. demonstrated the opposite [44]. During adulthood, the production of neural progenitors and neurons declines with age [45]. In the SVZ, the expression levels of the p16 inhibitor increase with age. Accordingly, deletion of p16 prevents the age-dependent decline in neural progenitor proliferation and neuron production specifically in the SVZ but not in the SGZ [46]. Similarly, the deletion of p57 promoted neurogenesis in both young and aged mice, but led to an exhaustion of the neural progenitor pool in the long term [47].
Gliogenesis
Gliogenesis in the CNS
During mouse brain development, the generation of glial cells starts shortly before birth. Hence, the prenatal time window is rather short and only few cell cycle proteins have been studied in this context. The loss of one of them, the p27 inhibitor, was shown to enhance the proliferation of glia progenitors in many areas, such as the spinal cord, the subcortical white matter, and the cerebellum. Interestingly, this effect was found to be transient, as the proliferation rate of OPCs equaled that of the wild-type counterpart in the spinal cord by E19 and in the cerebellum by postnatal day (P) 16 [48].
Gliogenesis mainly occurs postnatally, and it can be activated during the entire lifespan in response to stimulation such as traumatic injuries, as discussed later in the text. Cortices of mice lacking CyclinD1 exhibit a cell- and stage-specific requirement for CyclinD1 in the various glial lineages. While proliferation of fast dividing OPCs at early postnatal stages becomes gradually dependent on CyclinD1, this particular G1 regulator is strictly required for the slow divisions of OPCs in the adult cerebral cortex [49]. Indeed, in the adult mutant mice, the number of mature oligodendrocytes is reduced in both prefrontal cortex and corpus callosum with a consequent reduction in the number of myelinated axons. In contrast, the pool of microglia cells is diminished already a few days after birth, while the number of astrocytes is not affected [49]. Regarding the role of Cdks, it has been shown that the loss of Cdk2 has neither an effect on OPC proliferation nor on the rate of myelination in the corpus callosum [50]. Little more data is available regarding the cell cycle inhibitors: p27 has been identified as an important player in OPC proliferation in the postnatal cerebellum, since lack of p27 leads to an expansion of the proliferating progenitors. Moreover, glial cell numbers were increased in the optic nerve consistent with the function of p27 as an inhibitor of cell cycle progression [48, 51]. In contrast, p21-deficient OPCs exit from the cell cycle in a manner comparable to that in control wild-type cells [52].
Gliogenesis in the PNS
The involvement of cell cycle proteins during postnatal glial development has been studied more extensively in the PNS. Using CyclinD1 and CyclinD2-deficient mice, it was demonstrated that immature Schwann cells depend on neither CyclinD1 nor CyclinD2 for cell cycle progression [53, 54]. Immunohistochemical analyses revealed that immature Schwann cells express CyclinD1 in the cytoplasm but not in the nucleus (Fig. 2a), providing an explanation for why its absence does not affect proliferation at these early developmental stages [54]. Studies with mice lacking Cdk proteins showed that lack of Cdk4 affected Schwann cell proliferation during early postnatal stages, while neither Cdk2 nor Cdk6 appeared to be essential for Schwann cells proliferation [55]. Notably, proliferation of embryonic Schwann cells was not affected by the loss of Cdk4, suggesting that prenatal and postnatal proliferation are regulated by distinct molecular mechanisms. Regarding the cell cycle inhibitors, p21 and p16 were identified to be individually required for proper withdrawal of Schwann cells during postnatal development. Interestingly, p21 only appears in the cytoplasm at P7, when Schwann cells have mostly stopped dividing. Ablation of p21 after P7 induces a prolonged proliferative phase pointing to a novel function attributed to cytoplasmic p21 [56].

CyclinD1 localization in Schwann cells. a In uninjured proliferating Schwann cells during postnatal development, as well as in quiescent myelinating Schwann cells in the adult, CyclinD1 is localized to the perinuclear region, where it is not required for cell division. b In response to injury, CyclinD1 translocates to the nucleus and induces cell division in both developing and adult Schwann cells. CD1, CyclinD1
Injury
Cell cycle activation is a prominent feature in both acute and chronic neurodegenerative disorders. This has been extensively discussed in some recent reviews [57,58,59,60,61,62, 105]. Here, we will focus on the requirements for cell cycle proteins in response to neural injures in mice. An increase in the expression of cyclins and Cdks accompanied by decreased expression levels of CKIs is a typical feature observed in various types of injuries [63]. Depending on the cell type, these changes in expression levels of cyclins, Cdks, or CKIs lead to opposing effects in mice: in glial cells, they affect proliferation, while in neurons, they trigger apoptosis. In some cases, terminally differentiated neurons that replicate their DNA do not die, but remain alive with double the amount of DNA [105, 111]. There are various types of injuries that are applied in mouse models to study the influence of cell cycle proteins in degeneration and regeneration processes in the CNS and PNS. For example, mechanical insults such as spinal cord injury (SCI) or middle cerebral artery occlusion (MCAO) to mimic ischemic/hypoxic damage are typically used as injury models in the CNS [97, 98]. Systemic applications of brain-lesioning compounds such as excitotoxic kainic acid (KA) are also common approaches [99]. Further, ibotenic acid (IBO), a glutamate receptor agonist, induces injuries within well-defined borders [65], whereas lysolecithin (LPC) is used to induce focal de-myelination in the CNS [100]. In the PNS, nerve crush injuries are typically applied to study degeneration and regeneration, while nerve cut injuries serve as a model to investigate terminal degeneration.
Injury in the CNS
In mice, tissue damage induces mature neurons to re-enter the cell cycle, which in turn triggers apoptosis and leads to the loss of these cells (Fig. 3a). Simultaneously, glial proliferation is activated, which ultimately contributes to scar formation at the injury site (Fig. 3b). Altogether, these effects on neurons and glial cells make cell cycle proteins in this context a very interesting topic [64].

Activation of the cell cycle upon injury in the CNS. Damaging stress to the mouse nervous system induces CyclinD1 expression in both neurons and glial cells, but leads to opposite effects in vivo. a In neurons, re-entry into the cell cycle induces cell death, and ablation of CyclinD1 leads to improved neuronal survival. b In glial cells, expression of CyclinD1 promotes cell division, and lack of CyclinD1 reduces glial proliferation and limits reactive gliosis
In the literature, most of the in vivo studies in mice related to neuronal injuries focus on CyclinD1. Mice lacking CyclinD1 often display better responses to various types of insults (Fig. 3a, b). Typical examples are reduced lesion areas upon administration of IBO in the cortex [65], improved locomotor scores in response to SCI [66], or limited reactive gliosis and neuronal apoptosis induced by ischemic damage [15, 64, 67, 68]. Moreover, knockdown of CyclinD1 with antisense oligonucleotides is protective against KA-induced neuronal apoptosis [69]. In addition, ablation of CyclinD1 leads to reduced proliferation rates of various glial subpopulations in mutant compared to control cortices in IBO-induced injury models [65]. Similarly, in response to IBO, the proliferation rates of cortical Olig2-positive glial cells were reduced upon loss of Cdk4 compared to wild-type mice [65], while antisense oligonucleotides targeting Cdk4 were protective against KA-induced apoptosis of neurons in the cortex and the amygdala [69]. Loss of Cdk2 was studied in a different context: experiments performed with LPC to induce focal demyelination in the corpus callosum showed that in the absence of Cdk2, OPC proliferation was reduced and remyelination was promoted in the CNS [50]. The role of CKIs was studied in adult stem cells and neurons using ischemic models [64]. p21-null mice displayed a significant activation of the quiescent stem cell pool in the SGZ and in the SVZ after brain ischemia, promoting cell division and neurogenesis [43]. Similarly, neural progenitor proliferation was elevated upon loss of p27 in the ischemic SGZ [70].
Altogether, these results indicate that cell cycle proteins play an important role in various pathological conditions. It is under intensive debate whether their manipulation could be a valuable approach for supporting the survival of neurons or even promoting de novo neurogenesis after an insult.
Injury in the PNS
Injuries to peripheral nerves have profound effects on Schwann cell proliferation both during postnatal development and in the adult. Hence, several studies focused on the impact of cell cycle components during degeneration and regeneration processes in the PNS. Studies on CyclinD1 showed that this G1-cyclin only translocates into nuclei of immature and mature Schwann cell upon axonal damage (Fig. 2b). Consistent with this observation, proliferation of both developing and adult Schwann cells was significantly reduced following an injury in the absence of CyclinD1 compared to wild type [53]. CyclinD2, however, was dispensable for the proliferation response of Schwann cells after peripheral nerve damage [53, 54]. Interestingly, replacing CyclinD1 coding sequences with human CyclinE was not sufficient to rescue the phenotype observed following an insult [54]. These data support the notion that individual G1-cyclin members exert specific functions and cannot always compensate for each other under any circumstances. Similarly, members of the Cdk family were differentially required after nerve injury. While Schwann cells lacking Cdk4 were unable to re-enter the cell cycle, loss of Cdk2 or Cdk6 had only a minor effect on proliferation rates compared to controls [55]. Few data are available on the role of CKIs. Absence of p16 and p21 was found to be required for correct cell cycle control at the peak of Schwann cell proliferation in denervated nerves [56]. In summary, knowledge about the requirement for specific cell cycle components in this context is limited, but the available data suggest that a more detailed understanding of the proteins would be beneficial towards a better understanding of Schwann cell function in response to damage.
Differentiation and Cell Fate Determination
The manipulation of cell cycle proteins is not only elucidating their effects on proliferation processes, but is also providing increasing evidence that some members, in particular CKIs, can directly control the differentiation fate of progenitor cells. For example, p21 inhibits Sox2 expression in the SVZ by directly binding to a Sox2 enhancer element. Upon loss of p21, Sox2 accumulates at the protein level inducing a replicative stress that leads to growth arrest of progenitor cells in the SVZ [71]. p21 was also shown to modulate Bmp2 signaling, and loss of p21 induced premature differentiation of progenitor cells into the astrocytic fate [72]. In the developing cortex, lack of p27 leads to an increase in the generation of superficial layer neurons [33]. Thereby, p27 promotes neuronal differentiation through a mechanism that is independent of its function as cell cycle regulator: it stabilizes the transcription factor Neurogenin2 and consequently enhances the expression of pro-neural genes [73]. In addition, it increases neuronal migration by blocking RhoA signaling [73]. In the same context, the effects of p57 overexpression depended on the developmental stage. Early in development, forced expression of p57 enhanced neurogenesis, while at later stages, it induced the switch to gliogenesis. The effects on both neurogenesis and gliogenesis were mediated by the N-terminal cyclin/Cdk binding site [32]. In the retina, the simultaneous deletion of p19 and p27 increased the number of horizontal cells postnatally, although this type of neuron is normally born only in the embryonic period [29]. Conversely, the overexpression of p27 induced the generation of small clones, and only few contained bipolar cells or Müller glia (for retinal development, see [34]). In the SGZ, during the transition from early to late postnatal stages, neurogenesis becomes gradually dependent on CyclinD2 [39]. Lack of CyclinD2 not only diminished neuronal production in the adult tissue, but the few progenitor cells that were still generated preferentially differentiated towards the gliogenic lineage [38]. Moreover, in the CyclinD2 mutant cortex, a significant reduction of parvalbumin positive- (PV+) interneurons associated with electrophysiological defects was observed [74].
In conclusion, cell cycle activators and inhibitors not only regulate proliferation of progenitor cells, but can directly influence their fate determination through cell cycle-independent mechanisms.
Conclusions
Development and normal physiology of the nervous system require proliferation and differentiation events that have to be fine-tuned in a spatio-temporal manner. In this work, we reviewed studies that describe in vivo expression and functions of cyclins, Cdks, and CKIs in the nervous system during development and in the adulthood, under normal and pathological conditions.
We specifically focused on studies that apply knockout strategies in mice. In addition to this approach, further valuable insight into the molecular mechanisms that regulate proliferation and differentiation processes in the nervous system in vivo has been gained from overexpression experiments. For example, forced expression of CyclinD1/Cdk4 in the developing cortex using in utero electroporation prevented G1 lengthening, inhibited neurogenesis, and led to an increase of the basal progenitor population [28]. A separate study by Pilaz et al. showed that the sole overexpression of CyclinD1 or CyclinE was sufficient to shorten the G1 phase and delay neurogenesis [75]. In another tissue, the adult hippocampus, overexpression of CyclinD1/Cdk4 induced the expansion of the neuronal stem cell pool but did not affect the pool of committed progenitors. Similar to the results using knockout mice, also these findings point to differences in cell cycle dynamics between embryonic and adult progenitor cells, suggesting that the experimental approaches applied are faithfully revealing some fundamental physiological principles applied by neural and glial progenitor cells in vivo [101]. First, cell cycle proteins act in a cell-type-specific, context-, and time-dependent manner. Second, there are remarkable functional differences between closely related family members during development and adulthood. Third, there is growing evidence that components of the cell cycle machinery also play central roles during neuronal and glial differentiation.
In compiling the available in vivo data, it was striking to notice how few types of cyclins, Cdks, and CKIs have been studied in the nervous system to date, and how limited our knowledge on the in vivo functions of these cell cycle components is. In contrast, much effort was put into the investigation of Cdk5, an unconventional Cdk that is predominantly activated in post-mitotic neurons of the developing cerebral cortex and cerebellum, where it is involved in neurogenesis, neuronal migration, and axon and dendrite formation. Moreover, there is extensive data on the multiple molecular and cellular functions of Cdk5, which are based on the fact that Cdk5 phosphorylates many proteins involved in various cellular events [106]. Hyperactivation of Cdk5 due to binding to p35, p39, or p25 molecules is associated with a number of neurological disorders and neurodegenerative diseases such as Alzheimer’s, Huntington’s, or ischemia [107, 108]. Future studies are needed to expand current information, and they will possibly require double or triple knockout and knock-in strategies. In this context, it is worthwhile to consider that straight knockouts might exacerbate compensatory mechanisms. This could explain why many studies found that a significant number of cell cycle molecules seem to be dispensable for the survival of the organism. These effects appear to be stronger during embryonic development than in adulthood. Conditional knockout strategies are needed to capture the individual functions of cell cycle components in more detail in vivo and to expand our knowledge for the benefit of treating neurodegenerative diseases, injuries, and cancers.
References
Butler SJ, Bronner ME (2015) From classical to current: analyzing peripheral nervous system and spinal cord lineage and fate. Dev Biol 398(2):135–146
Newbern JM (2015) Molecular control of the neural crest and peripheral nervous system development. Curr Top Dev Biol 111:201–231
Jessen KR, Mirsky R (2016) The repair Schwann cell and its function in regenerating nerves. J Physiol 594(13):3521–3531
Gotz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6(10):777–788
Cunningham JJ, Roussel MF (2001) Cyclin-dependent kinase inhibitors in the development of the central nervous system. Cell Growth Differ 12(8):387–396
Lui JH, Hansen DV, Kriegstein AR (2011) Development and evolution of the human neocortex. Cell 146(1):18–36
Govindan S, Jabaudon D (2017) Coupling progenitor and neuronal diversity in the developing neocortex. FEBS Lett 591(24):3960–3977
Dehay C, Kennedy H (2007) Cell-cycle control and cortical development. Nat Rev Neurosci 8(6):438–450
Rowitch DH, Kriegstein AR (2010) Developmental genetics of vertebrate glial-cell specification. Nature 468(7321):214–222
Kriegstein A, Alvarez-Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32:149–184
Yu TS, Washington PM, Kernie SG (2016) Injury-induced neurogenesis: mechanisms and relevance. Neuroscientist 22(1):61–71
Domingues HS et al (2016) Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol 4:71
Dimou L, Gotz M (2014) Glial cells as progenitors and stem cells: New roles in the healthy and diseased brain. Physiol Rev 94(3):709–737
Scheller A, Bai X, Kirchhoff F (2017) The role of the oligodendrocyte lineage in acute brain trauma. Neurochem Res 42(9):2479–2489
Stoica BA, Byrnes KR, Faden AI (2009) Cell cycle activation and CNS injury. Neurotox Res 16(3):221–237
Lange C, Calegari F (2010) Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle 9(10):1893–1900
Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT et al (1995) Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82(4):621–630
Das G, Choi Y, Sicinski P, Levine EM (2009) Cyclin D1 fine-tunes the neurogenic output of embryonic retinal progenitor cells. Neural Dev 4:15
Glickstein SB, Monaghan JA, Koeller HB, Jones TK, Ross ME (2009) Cyclin D2 is critical for intermediate progenitor cell proliferation in the embryonic cortex. J Neurosci 29(30):9614–9624
Glickstein SB, Alexander S, Ross ME (2007) Differences in cyclin D2 and D1 protein expression distinguish forebrain progenitor subsets. Cereb Cortex 17(3):632–642
Huard JM et al (1999) Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development 126(9):1927–1935
Leto K, Bartolini A, di Gregorio A, Imperiale D, de Luca A, Parmigiani E, Filipkowski RK, Kaczmarek L et al (2011) Modulation of cell-cycle dynamics is required to regulate the number of cerebellar GABAergic interneurons and their rhythm of maturation. Development 138(16):3463–3472
Marcucci F, Murcia-Belmonte V, Wang Q, Coca Y, Ferreiro-Galve S, Kuwajima T, Khalid S, Ross ME et al (2016) The ciliary margin zone of the mammalian retina generates retinal ganglion cells. Cell Rep 17(12):3153–3164
Tong W, Pollard JW (2001) Genetic evidence for the interactions of cyclin D1 and p27(Kip1) in mice. Mol Cell Biol 21(4):1319–1328
Grison A, Gaiser C, Bieder A, Baranek C, Atanasoski S (2018) Ablation of cdk4 and cdk6 affects proliferation of basal progenitor cells in the developing dorsal and ventral forebrain. Dev Neurobiol 78(7):660–670
Mi D, Carr CB, Georgala PA, Huang YT, Manuel MN, Jeanes E, Niisato E, Sansom SN et al (2013) Pax6 exerts regional control of cortical progenitor proliferation via direct repression of Cdk6 and hypophosphorylation of pRb. Neuron 78(2):269–284
Lim S, Kaldis P (2012) Loss of Cdk2 and Cdk4 induces a switch from proliferation to differentiation in neural stem cells. Stem Cells 30(7):1509–1520
Lange C, Huttner WB, Calegari F (2009) Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5(3):320–331
Cunningham JJ, Levine EM, Zindy F, Goloubeva O, Roussel MF, Smeyne RJ (2002) The cyclin-dependent kinase inhibitors p19(Ink4d) and p27(Kip1) are coexpressed in select retinal cells and act cooperatively to control cell cycle exit. Mol Cell Neurosci 19(3):359–374
Bilodeau S, Roussel-Gervais A, Drouin J (2009) Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol Cell Biol 29(7):1895–1908
Mitsuhashi T, Aoki Y, Eksioglu YZ, Takahashi T, Bhide PG, Reeves SA, Caviness VS (2001) Overexpression of p27Kip1 lengthens the G1 phase in a mouse model that targets inducible gene expression to central nervous system progenitor cells. Proc Natl Acad Sci U S A 98(11):6435–6440
Tury A, Mairet-Coello G, DiCicco-Bloom E (2011) The cyclin-dependent kinase inhibitor p57Kip2 regulates cell cycle exit, differentiation, and migration of embryonic cerebral cortical precursors. Cereb Cortex 21(8):1840–1856
Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS (2005) Overexpression of p27 Kip 1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex 15(9):1343–1355
Dyer MA, Cepko CL (2001) Regulating proliferation during retinal development. Nat Rev Neurosci 2(5):333–342
Ming GL, Song H (2011) Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70(4):687–702
Ma J, Yu Z, Qu W, Tang Y, Zhan Y, Ding C, Wang W, Xie M (2010) Proliferation and differentiation of neural stem cells are selectively regulated by knockout of cyclin D1. J Mol Neurosci 42(1):35–43
Ma C, Papermaster D, Cepko CL (1998) A unique pattern of photoreceptor degeneration in cyclin D1 mutant mice. Proc Natl Acad Sci U S A 95(17):9938–9943
Kowalczyk A, Filipkowski RK, Rylski M, Wilczynski GM, Konopacki FA, Jaworski J, Ciemerych MA, Sicinski P et al (2004) The critical role of cyclin D2 in adult neurogenesis. J Cell Biol 167(2):209–213
Ansorg A, Witte OW, Urbach A (2012) Age-dependent kinetics of dentate gyrus neurogenesis in the absence of cyclin D2. BMC Neurosci 13:46
Urban N, Guillemot F (2014) Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front Cell Neurosci 8:396
Beukelaers P, Vandenbosch R, Caron N, Nguyen L, Belachew S, Moonen G, Kiyokawa H, Barbacid M et al (2011) Cdk6-dependent regulation of G(1) length controls adult neurogenesis. Stem Cells 29(4):713–724
Jablonska B, Aguirre A, Vandenbosch R, Belachew S, Berthet C, Kaldis P, Gallo V (2007) Cdk2 is critical for proliferation and self-renewal of neural progenitor cells in the adult subventricular zone. J Cell Biol 179(6):1231–1245
Qiu J, Takagi Y, Harada J, Rodrigues N, Moskowitz MA, Scadden DT, Cheng T (2004) Regenerative response in ischemic brain restricted by p21cip1/waf1. J Exp Med 199(7):937–945
Kippin TE, Martens DJ, van der Kooy D (2005) p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev 19(6):756–767
Lee SW, Clemenson GD, Gage FH (2012) New neurons in an aged brain. Behav Brain Res 227(2):497–507
Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443(7110):448–452
Furutachi S, Matsumoto A, Nakayama KI, Gotoh Y (2013) p57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J 32(7):970–981
Casaccia-Bonnefil P, Hardy RJ, Teng KK, Levine JM, Koff A, Chao MV (1999) Loss of p27Kip1 function results in increased proliferative capacity of oligodendrocyte progenitors but unaltered timing of differentiation. Development 126(18):4027–4037
Nobs L, Baranek C, Nestel S, Kulik A, Kapfhammer J, Nitsch C, Atanasoski S (2014) Stage-specific requirement for cyclin D1 in glial progenitor cells of the cerebral cortex. Glia 62(5):829–839
Caillava C, Vandenbosch R, Jablonska B, Deboux C, Spigoni G, Gallo V, Malgrange B, Baron-van Evercooren A (2011) Cdk2 loss accelerates precursor differentiation and remyelination in the adult central nervous system. J Cell Biol 193(2):397–407
Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich V, Chao MV, Koff A (1997) Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes Dev 11(18):2335–2346
Zezula J, Casaccia-Bonnefil P, Ezhevsky SA, Osterhout DJ, Levine JM, Dowdy SF, Chao MV, Koff A (2001) p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep 2(1):27–34
Atanasoski S, Shumas S, Dickson C, Scherer SS, Suter U (2001) Differential cyclin D1 requirements of proliferating Schwann cells during development and after injury. Mol Cell Neurosci 18(6):581–592
Kim HA, Pomeroy SL, Whoriskey W, Pawlitzky I, Benowitz LI, Sicinski P, Stiles CD, Roberts TM (2000) A developmentally regulated switch directs regenerative growth of Schwann cells through cyclin D1. Neuron 26(2):405–416
Atanasoski S, Boentert M, de Ventura L, Pohl H, Baranek C, Beier K, Young P, Barbacid M et al (2008) Postnatal Schwann cell proliferation but not myelination is strictly and uniquely dependent on cyclin-dependent kinase 4 (cdk4). Mol Cell Neurosci 37(3):519–527
Atanasoski S, Boller D, de Ventura L, Koegel H, Boentert M, Young P, Werner S, Suter U (2006) Cell cycle inhibitors p21 and p16 are required for the regulation of Schwann cell proliferation. Glia 53(2):147–157
Braun SM, Jessberger S (2014) Adult neurogenesis: mechanisms and functional significance. Development 141(10):1983–1986
Greene LA, Liu DX, Troy CM, Biswas SC (2007) Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta 1772(4):392–401
van Leeuwen LA, Hoozemans JJ (2015) Physiological and pathophysiological functions of cell cycle proteins in post-mitotic neurons: implications for Alzheimer's disease. Acta Neuropathol 129(4):511–525
Nguyen MD, Mushynski WE, Julien JP (2002) Cycling at the interface between neurodevelopment and neurodegeneration. Cell Death Differ 9(12):1294–1306
Patricio P et al (2013) Re-cycling paradigms: cell cycle regulation in adult hippocampal neurogenesis and implications for depression. Mol Neurobiol 48(1):84–96
Sharma R, Kumar D, Jha NK, Jha SK, Ambasta RK, Kumar P (2017) Re-expression of cell cycle markers in aged neurons and muscles: Whether cells should divide or die? Biochim Biophys Acta Mol basis Dis 1863(1):324–336
Alunni A, Bally-Cuif L (2016) A comparative view of regenerative neurogenesis in vertebrates. Development 143(5):741–753
Rashidian J, Iyirhiaro GO, Park DS (2007) Cell cycle machinery and stroke. Biochim Biophys Acta 1772(4):484–493
Nobs L, Nestel S, Kulik A, Nitsch C, Atanasoski S (2013) Cyclin D1 is required for proliferation of Olig2-expressing progenitor cells in the injured cerebral cortex. Glia 61(9):1443–1455
Byrnes KR, Stoica BA, Fricke S, di Giovanni S, Faden AI (2007) Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain 130(Pt 11):2977–2992
Zhu Z, Zhang Q, Yu Z, Zhang L, Tian D, Zhu S, Bu B, Xie M et al (2007) Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia 55(5):546–558
Rashidian J, Iyirhiaro G, Aleyasin H, Rios M, Vincent I, Callaghan S, Bland RJ, Slack RS et al (2005) Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc Natl Acad Sci U S A 102(39):14080–14085
Ino H, Chiba T (2001) Cyclin-dependent kinase 4 and cyclin D1 are required for excitotoxin-induced neuronal cell death in vivo. J Neurosci 21(16):6086–6094
Qiu J, Takagi Y, Harada J, Topalkara K, Wang Y, Sims JR, Zheng G, Huang P et al (2009) p27Kip1 constrains proliferation of neural progenitor cells in adult brain under homeostatic and ischemic conditions. Stem Cells 27(4):920–927
Marques-Torrejon MA et al (2013) Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell Stem Cell 12(1):88–100
Porlan E, Morante-Redolat JM, Marqués-Torrejón MÁ, Andreu-Agulló C, Carneiro C, Gómez-Ibarlucea E, Soto A, Vidal A et al (2013) Transcriptional repression of Bmp2 by p21(Waf1/Cip1) links quiescence to neural stem cell maintenance. Nat Neurosci 16(11):1567–1575
Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts JM et al (2006) p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev 20(11):1511–1524
Glickstein SB, Moore H, Slowinska B, Racchumi J, Suh M, Chuhma N, Ross ME (2007) Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development 134(22):4083–4093
Pilaz LJ, Patti D, Marcy G, Ollier E, Pfister S, Douglas RJ, Betizeau M, Gautier E et al (2009) Forced G1-phase reduction alters mode of division, neuron number, and laminar phenotype in the cerebral cortex. Proc Natl Acad Sci U S A 106(51):21924–21929
Schmetsdorf S, Gartner U, Arendt T (2005) Expression of cell cycle-related proteins in developing and adult mouse hippocampus. Int J Dev Neurosci 23(1):101–112
Geng Y, Yu Q, Whoriskey W, Dick F, Tsai KY, Ford HL, Biswas DK, Pardee AB et al (2001) Expression of cyclins E1 and E2 during mouse development and in neoplasia. Proc Natl Acad Sci U S A 98(23):13138–13143
Koeller HB, Ross ME, Glickstein SB (2008) Cyclin D1 in excitatory neurons of the adult brain enhances kainate-induced neurotoxicity. Neurobiol Dis 31(2):230–241
Lukaszewicz AI, Anderson DJ (2011) Cyclin D1 promotes neurogenesis in the developing spinal cord in a cell cycle-independent manner. Proc Natl Acad Sci U S A 108(28):11632–11637
Chen Z, Duan RS, Zhu Y, Folkesson R, Albanese C, Winblad B, Zhu J (2005) Increased cyclin E expression may obviate the role of cyclin D1 during brain development in cyclin D1 knockout mice. J Neurochem 92(5):1281–1284
Tsunekawa Y, Osumi N (2012) How to keep proliferative neural stem/progenitor cells: a critical role of asymmetric inheritance of cyclin D2. Cell Cycle 11(19):3550–3554
Dyer MA, Cepko CL (2000) Control of Muller glial cell proliferation and activation following retinal injury. Nat Neurosci 3(9):873–880
Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S et al (2011) Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell 21(4):655–668
Belachew S, Aguirre AA, Wang H, Vautier F, Yuan X, Anderson S, Kirby M, Gallo V (2002) Cyclin-dependent kinase-2 controls oligodendrocyte progenitor cell cycle progression and is downregulated in adult oligodendrocyte progenitors. J Neurosci 22(19):8553–8562
van Lookeren Campagne M, Gill R (1998) Cell cycle-related gene expression in the adult rat brain: selective induction of cyclin G1 and p21WAF1/CIP1 in neurons following focal cerebral ischemia. Neuroscience 84(4):1097–1112
Tikoo R, Zanazzi G, Shiffman D, Salzer J, Chao MV (2000) Cell cycle control of Schwann cell proliferation: role of cyclin-dependent kinase-2. J Neurosci 20(12):4627–4634
Li Y, Chopp M, Powers C, Jiang N (1997) Immunoreactivity of cyclin D1/cdk4 in neurons and oligodendrocytes after focal cerebral ischemia in rat. J Cereb Blood Flow Metab 17(8):846–856
Kaya SS et al (1999) Expression of cell cycle proteins (cyclin D1 and cdk4) after controlled cortical impact in rat brain. J Neurotrauma 16(12):1187–1196
Di Giovanni S et al (2005) Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A 102(23):8333–8338
Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R et al (2001) Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci 21(14):5045–5053
Zindy F et al (1997) Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ 8(11):1139–1150
Pechnick RN, Zonis S, Wawrowsky K, Pourmorady J, Chesnokova V (2008) p21Cip1 restricts neuronal proliferation in the subgranular zone of the dentate gyrus of the hippocampus. Proc Natl Acad Sci U S A 105(4):1358–1363
Pechnick RN, Zonis S, Wawrowsky K, Cosgayon R, Farrokhi C, Lacayo L, Chesnokova V (2011) Antidepressants stimulate hippocampal neurogenesis by inhibiting p21 expression in the subgranular zone of the hipppocampus. PLoS One 6(11):e27290
Donovan SL, Dyer MA (2005) Regulation of proliferation during central nervous system development. Semin Cell Dev Biol 16(3):407–421
Agirman G, Broix L, Nguyen L (2017) Cerebral cortex development: an outside-in perspective. FEBS Lett 591(24):3978–3992
Schafer KA (1998) The cell cycle: a review. Vet Pathol 35(6):461–478
Fluri F, Schuhmann MK, Kleinschnitz C (2015) Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther 9:3445–3454
Steward O, Willenberg R (2017) Rodent spinal cord injury models for studies of axon regeneration. Exp Neurol 287(Pt 3):374–383
Mohd Sairazi NS et al (2015) Kainic acid-induced excitotoxicity experimental model: protective merits of natural products and plant extracts. Evid Based Complement Alternat Med 2015:972623
Wang C, Kotter MR (2018) Experimental demyelination and remyelination of murine spinal cord by focal injection of lysolecithin. Methods Mol Biol 1791:233–241
Artegiani B, Lindemann D, Calegari F (2011) Overexpression of cdk4 and cyclinD1 triggers greater expansion of neural stem cells in the adult mouse brain. J Exp Med 208(5):937–948
Ovejero S, Bueno A, Sacristán MP (2020) Working on genomic stability: from the S-phase to mitosis. Genes 11(2):225
Jillian A et al (2008) Regulation of APC/C activators in mitosis and meiosis. Annu Rev Cell Dev Biol 24(1):475–499
Sherr CJ, Kato J, Quelle DE, Matsuoka M, Roussel MF (1994) D-type cyclins and their cyclin-dependent kinases: G1 phase integrators of the mitogenic response. Cold Spring Harb Symp Quant Biol 59:11–19
Frade JM, Ovejero-Benito MC (2015) Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle 14(5):712–720
Kawauchi T (2014) Cdk5 regulates multiple cellular events in neural development, function and disease. Develop Growth Differ 56(5):335–348
Su SC, Tsai LH (2011) Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol 27(1):465–491
Gupta KK, Singh SK (2019) Cdk5: a main culprit in neurodegeneration. Int J Neurosci 129(12):1192–1197
Foster DA, Yellen P, Xu L, Saqcena M (2010) Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1(11):1124–1131
Buchakjian M, Kornbluth S (2010) The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol 11:715–727
López-Sánchez N, Fontán-Lozano Á, Pallé A, González-Álvarez V, Rábano A, Trejo JL, Frade JM (2017) Neuronal tetraploidization in the cerebral cortex correlates with reduced cognition in mice and precedes and recapitulates Alzheimer's-associated neuropathology. Neurobiol Aging 56:50–66
Acknowledgments
We thank Dr. Ned Mantei for comments on the manuscript.
Funding
A. G. was supported by the University of Basel Research Fund for excellent early career researchers. S. A. was supported by the Swiss National Science Foundation and the Swiss Federal Government (SystemsX.ch The Swiss Initiative in Systems Biology).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Grison, A., Atanasoski, S. Cyclins, Cyclin-Dependent Kinases, and Cyclin-Dependent Kinase Inhibitors in the Mouse Nervous System. Mol Neurobiol 57, 3206–3218 (2020). https://doi.org/10.1007/s12035-020-01958-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01958-7