
Abstract
SK-MEL-5 is a human melanoma cell line that has been used in several researches to explore new therapies against melanoma. Based on this study we report on the development of quantitative structure–activity relationship (QSAR) model and molecular docking simulation able to predict the cytotoxic effect of diverse chemical compounds on this cancer cell line. The dataset of seventy-two (72) cytotoxic compounds were downloaded from the National Cancer Institute database. It contains the data of compounds for which cytotoxicity results expressed by pGI50 was recorded. The QSAR model was built using fifty (50) compounds and the best-generated model based on multiple linear regression showed, respectively good quality of fits [\(R^{2}\) (0.864), \(R_{adjusted}^{2}\) (0.846), Q2cv (0.841) and \(R_{pred}^{2}\) (0.885)]. The model’s predictive ability was determined by a test set of twenty-two (22) compounds and the applicability domain was assessed through leveraged approach. Compounds 41 and 69 were selected as templates for in silico design because they had high pGI50 activity and are within the model’s applicability domain. The obtained information from the model was explored to design novel compounds by introducing various substituents. Moreover, the designed compounds were docked into the active site of the protein (PDB CODE: 3OG7) which is responsible for melanoma cancer to elucidate their binding mode. Ia (− 12.4 kcal mol−1) and IIb (− 12.3 kcal mol−1) showed a better binding affinity for the target when compared with (vemurafenib, − 11.3 kcal mol−1) the known inhibitor of the target (V600E-BRAF). These results may be of great help in early anticancer drug discovery.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
SK-MEL-5 is a human melanoma cell line derived from a metastatic axillary node of a young female patient, and is characterized by a high level of expression of the V600E mutation of BRAF and the wild-type NRAS [1], as well as by relatively high levels of the ABCB1 transcript [2]. It has been used in various studies to explore new therapies against melanoma in various in vitro experiments [3, 4]. Several potent drugs are now available in clinical trials against melanoma including kinase inhibitors with different degree of success. Sorafenib one of the multi-kinase inhibitor, which inhibit EGFR tyrosine receptor kinase, BRAF serine and threonine kinase [5]. vemurafenib (Zelboraf 1), a more distinct BRAF inhibitor was approved in 2011 by the FDA for melanoma (metastatic) [6]. However, Treatment with the use of BRAF inhibitors can result in the development of inhibitor (drug) resistance which restrict their usage [7]. Melanoma is a dangerous form of cancer that has a very bad prognosis and is resistant to many standard anti-cancer therapies, this helps these cancer cells to evade the immune system [8]. For example, majority of patients that were administered vemurafenib (standard V600E-BRAF inhibitor) eventually develop resistance towards it. Therefore, there is a need to develop new potent drugs that targets melanoma and several techniques are being used from exploring better delivery system for an old compounds to assessing new targets [9, 10].
For new drugs to be available for commercialization, requires a long period of about twelve (12) years and the estimated cost for the marketed drug is also very high [11, 12]. This lengthy and expensive processes may cause delays and failure of the drugs development. Therefore, it is very vital to predict the failures before the clinical stage to reduce the costs of drug development [13]. To filter out the potential failures in the drug development stage, several methods are being used such as in silico, in vitro, etc. An example of an in silico approach is the modeling of a quantitative structure activity relationships (QSARs) that can be used to screen chemical libraries, understand drugs action and design novel compounds [14, 15]. Combinatorial approaches are an influential technique used in the selection to speed up drug discovery and with various mechanisms of action and it has being used to cure cancer [16, 17]. QSAR modeling has become very crucial in the molecular interpretation of biological and physicochemical properties [18, 19]. This method is the most essential tool adopted in ligand-based drug design and have been used broadly for the determination of assorted properties like stability, carcinogenicity, toxicity, ADME, retention time and other physicochemical properties apart from biological activity prediction [20,21,22,23,24].
It is, therefore, necessary to construct a QSAR model for the prediction of the activity of the designed leads before their synthesis. Because a successful QSAR model not only helps in understanding relationships between the structural features and biological activity of any class of compounds but also provides researchers a deep analysis of the lead compounds to be used in further studies [13]. Furthermore, understanding the mechanism of the ligand/receptor interactions is very significant in drug development, and the molecular docking simulation method is a proper tool for gaining such understanding. Molecular docking simulation is a computational technique used to predict the binding ability of the active site residues to specific groups on the receptor and to reveal the strength of interaction [25]. Molecular docking is a very useful and popular tool used in the drug discovery arena to evaluate the binding of small molecules (inhibitors) to the receptor (macromolecule) [26, 27]. This study was aimed to design new potent compounds on the SK-MEL-5 cell line through QSAR modeling and molecular docking simulations methods based on the compounds collected from the National Cancer Institute (NCI).
2 Computational methods
2.1 Data collection and structure preparation
Seventy two (72) set of compounds and their pGI50 activities on SK-MEL-5 melanoma cell line was retrieved from the drug discovery and development arm of the National Cancer Institute (NCI) database, which is openly available to the general public on the DTP Web site (https://wiki.nci.nih.gov/display/NCIDTPdata/NCI-60+Growth+Inhibition+Data). The anticancer activity, chemical name and NSC number of the studied compounds are presented in Table 1. The 2D structure of the studied compounds was transformed into the 3D structure using Spartan 14 on a Dell Intel(R)Core(TM)i7-5500U CPU), 16.00 GB RAM @ 2.400 GHz 2.400 GHz processor, 64-bit Operating system, a × 64-based processor on Windows 8.1 Pro). Geometry optimizations were conducted with Spartan 14 using Density Functional Theory (DFT) at the Becke88 three-parameter hybrid exchange potentials with Lee–Yang–Parr correlation potential (B3LYP) level of theory and 6-31G* basis set, the double zeta basis set with two polarized basis functions (d- and p- orbitals), where a d type orbital was added to all atoms except the hydrogen atoms, and a p type orbital was added to all hydrogen atoms.
2.2 Descriptors calculation and scaling
The fully optimized 3D structure in SD file were imported to paDEL descriptor tool kit [28, 29], in order to compute various molecular descriptors (1D, 2D and 3D descriptors) such as atom-type electrotopological state descriptors, 2D-Autocorrelations, WHIM, Petitjean shape index, count of chemical substructures and binary fingerprints of chemical substructures for each molecules and these descriptors were considered as possible input candidates to the model. The calculated descriptors and their corresponding activity values for each molecule were arranged in an n × m matrix format. This constituted the dataset, n is the number of molecule and m is the number of descriptors.
Descriptors are usually measured in different units and data modeling often favors data with higher absolute value. To minimize this bias and assign equal weight, importance and opportunity to each descriptor to be part of the final equation, descriptors values are usually scaled such that each descriptor column have equal variance [30]. Molecular descriptors in the study were scaled via auto-scaling approach:
where X′ is the standardized descriptor values, Xi is the value of each descriptor for a given molecule, \(\overline{X}\) and σ are the average and standard deviation value for each column of descriptors X respectively. For this purpose, all descriptors variables with constant or near constant values were removed and features containing missing values were also removed, because it is likely that for virtual screening purposes models built with such features will not be applicable for a part of the new compounds. Features highly correlated were also removed, using a threshold value of the coefficient correlation of 0.80 to reduce the redundancy in the data.
2.3 Data division and variable selection
The data set was splits into two subsets, the training set and test set using Kennard-Stone Algorithm [31, 32]. The training set is used in building the QSAR model which contains 70% of the data and the remaining 30% is for the test set that was used to describe the predictive ability of the model [33]. All the studied compounds was screened using the generated QSAR model for pGI50 activity prediction.
Material Studio Software 8.0 from BIOVIA-Accelrys, USA was adopted in performing GA and model building. The genetic algorithm (GA) was utilized in the selection of proper descriptors as this improves the model accuracy [34]. The Genetic algorithm method (GA) begins with the formation of a populace of randomly produced parameter sets. The probability of a given parameter from the active set is 0.5 in any of the initial population sets. The parameter set used for the GA incorporates the boundaries for mutation (0.1), hybrid (0.9), population (10,000), number of model generation (1000), R2 floor limit (50%), and target capacity (R2/N_par). The making of a successive generation includes crossovers between set substances and additionally changes. The calculation keeps running until the wanted number of generations is achieved. Equations were generated between the experimental biological activity and the descriptors. The best mathematical statement was taken in light of statistical parameters such as squared regression coefficient (R2) and leave-one-out cross validated regression coefficient (Q2cv).
2.4 QSAR model development and validation
Multiple Linear Regression (MLR) was used on the training set to determine the correlation between the dependent variable Y (pGI50) and independent variable, X (molecular descriptors). In this regression analysis, the contingent mean of the dependent variable (pGI50) Y relies on (descriptors) X. The best QSAR model was chosen based on the validation parameters such as the correlation coefficient (R2), Adjusted R2 (R2adj), Cross-validation coefficient (Q2CV) and correlation coefficient for an external prediction set (R2pred) all are represented in Eqs. (2–5):
where P is the number of descriptors in the model and, N is the sample size. Yexp; Ypred; Ymtraining is the activity (experimental), the activity (predicted) and the mean activity (experimental) of the compounds in the training set, respectively [33].
Further, in order to assess the robustness of the built model, the Y-randomization test was applied to the training data set as suggested by Tropsha et al. [33]. The dependent variable vector (activity data) was randomly shuffled and a new QSAR model was developed using the original independent variable matrix. For the built QSAR model to robust and reliable, the model is expected to have a low R2 and Q2 values for several trials. The coefficient of determination cR2p for Y-randomization is another parameter calculated which should be greater than 0.5 for passing this test as in Eq. (6):
cR2p is Coefficient of determination for Y-randomization, R is the coefficient of determination for Y-randomization and Rr is average ‘R’ of random models.
2.5 Ligand–protein preparation and docking studies
The selected ligands (compounds) were optimized and formatted to PDB files for docking utilizing Spartan 14. The X-ray structure of the V600E-BRAF kinase (receptor) in complex with PLX4032 (PDB CODE: 3OG7) [25, 35, 36] was retrieved from (www.rcsb.org). V600E-BRAF was imported into the Discovery studio and the PDB file was prepared by updating the hydrogen atoms and removing the excess water molecules present in the X-ray structure. This complex structure comprises of two homo-dimeric chains (A and B). Our goal was to target the mutated chain (chain A) of V600E-BRAF. Thus, chain B was removed from the structure of 3OG7 and the bound ligand also removed from chain A. All the compounds (ligands) were docked into the active kinase domain of V600E-BRAF using Autodock vina of Pyrx docking program software.
3 Results and discussion
3.1 QSAR model and validation
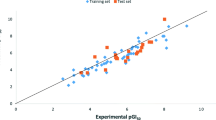
By using pGI50 values (activity) as dependent variables and calculated descriptors as independent variables, Kennard stone algorithm was applied in dividing the data set into two subsets, i.e., fifty compounds (50) as training set while twenty-two (22) compounds as the test set as presented in Table 1 and regression were executed for QSAR analysis. Robustness of generated QSAR model was depicted by the activity interactive graph which shows the predicted against experimental activity plot as in Fig. 1. The QSAR model is represented by Eq. (7):

The predicted pGI50 against the experimental values for the training and test sets
Further, the generated QSAR model has achieved high activity-descriptors relationship accuracy of 86.4% as shown by regression-coefficient (R2 = 0.864) and a high activity prediction accuracy of 84.1% as shown by the cross-validation regression-coefficient (Q2 = 0.841). Knowing the high predictive and descriptive ability, the generated model was considered to be highly robust for the prediction of anti-melanoma activity of these compounds against the SK-MEL-5 melanoma cancer cell line.
The developed model, which was built based on the training set compounds, was used to predict the activity of the test set of compounds. The predicted activities of the studied compounds for SK-MEL-5 cell line by the built QSAR model are shown in Table 1. Lower residual values found from both the training and test set as shown in Table 1 indicate that the model has a high potential to establish the correlation between the activity and structure. The correlation between the experimental and predicted activity based on the model was highly significant as indicated by statistical analysis. The closeness of regression-coefficient (R2) to 1.0 indicates that the developed model elaborated very high portion of the descriptor variation high enough for a good model. The 0.864 value demonstrates that 86.4% of the variation is found within the residual meaning that the model is very good.
The large adjusted regression-coefficient R2 (R2adj) value as presented in the generated model and its closeness to the value of regression-coefficient (R2) indicates that the model has perfect descriptive power to the descriptors in it and it also illustrates the real influence of used descriptors on the pGI50. Additionally, the high value and closeness of cross-validated regression-coefficient (Q2cv) to the regression-coefficient (R2) showed that the model was not over-fitted. The high R2pred as shown in the model explains that the generated model can provide good and valid predictions for the new compounds. A good and acceptable QSAR model must obey the following criteria: regression-coefficient (R2) and adjusted regression-coefficient (R2adj) values close to one. The Cross validated regression-coefficient (Q2cv) > 0.5, R2 − Q2cv ≤ 0.3, R2pred ≥ 0.6, and Ntest ≥ 5 [33, 37, 38]. The generated QSAR model satisfied the criteria and therefore acceptable statistically. Thus, we can conclude with confidence that the model will correctly predict the anti-melanoma pGI50 activity of a given compound.
Additionally, in order to assess the robustness of the model, the Y-randomization test was applied. The dependent variable vector (inhibitory activity) was randomly shuffled and a new QSAR model was developed using the original independent variable matrix. As was expected the new QSAR models (after several repetitions) have low R2 and Q2 values and also, the cR2p value was greater than 0.5 as presented in Table 2. This test affirms that the proposed model is powerful and not inferred by chance.
3.2 Contribution and interpretation of descriptors
Molecular descriptors are the physicochemical and structural information in the form of numerical values, each descriptor represents specific information that can be implored to improve the overall biological effect of a compound. By interpreting descriptors that appear in the model, it is possible to understand the factors which are related to the anti-melanoma activity. Therefore, brief descriptions of the selected descriptors are presented in Table 3 and an acceptable interpretation is provided. The contribution and significance of each descriptor in the built model were evaluated by the calculation of the mean effect (ME) value [39] of each descriptor by using Eq. (8) and the values for the ME are shown in Table 3:
where MEj is the mean effect (ME) of the descriptor j, βj represents the coefficient of the descriptor j, dij represents the value of the selected descriptors of each compound and m is the total number of the descriptors in the generated model.
The ME value indicates the significance of each descriptor when compare to the other descriptors. Descriptors found to have high ME values influences anti-melanoma activity (pGI50). The pGI50 changes with the ME values of a descriptor, as presented in Table 3. According to ME values, the selected descriptors were arranged in order about their contribution towards overall pGI50 of the compounds, in the following increasing order of pGI50 of compounds. Based on ME values, the associated descriptors are arranged in a sequence about their contributions towards the overall pGI50 of the studied compounds an increasing sequence of pGI50 of the compounds.
SM1_Dze is defined as Spectral moment of order 1 from Barysz matrix/weighted by Sanderson electronegativities. The negative ME value of the descriptor (Table 3) shows that the decrease in the value of this descriptor will increase the pGI50 of the compounds. SpMax4_Bhv is defined as the largest absolute eigenvalue of Burden modified matrix—n 4/weighted by relative van der Waals volumes. The SpMax4_Bhv ME has a negative sign. Negative contribution of SpMax4_Bhv indicates that inhibitory activity of the studied compound will increase with decrease of molecular descriptor.
SpMin5_Bhi a Burden modified eigenvalue descriptor is defined as the Smallest absolute eigenvalue of Burden modified matrix—n 5/weighted by relative first ionization potential. It is related to ionization potential of the compound. The ME of this descriptor was found to be -0.07565. Its negative sign suggest that decrease in the overall ionization potential of the compound increases the pGI50 values of the compounds. SpMin2_Bhs is the smallest absolute eigenvalue of Burden modified matrix—n 2/weighted by relative I-state. SpMin2_Bhs also has a negative mean effect value which suggests that the decrease of value for this descriptor will increase the anti-melanoma activity of a molecule. The SpMin5_Bhi has a negative mean effect (Table 3) and its decrease may improve the pGI50 activity.
Kier3 is defined as the Third kappa (κ) shape index and it has a positive ME value as shown in Table 3. This positive sign indicates the influence towards increasing the overall activity of the compounds when the value of this descriptor increases. vAdjMat (Vertex adjacency information (magnitude)) descriptor is the most significant descriptor in the model, with its ME 1.969472 as reported in Table 3. The mean effect of vAdjMat was found to positively influence the pGI50 activity of the compounds when increased. The descriptors selected for building the model in this research encoded electronic, topological and other geometrical aspects of the compounds. The presence of these descriptors in the QSAR model indicates the role of steric and electronic interactions in inducing anti-melanoma pGI50 activity on SK-MEL-5 cell line.
3.3 Applicability domain (AD) and in-silico screening
The applicability domain, AD of the QSAR model is the theoretical space in the chemical region comprising of both the descriptors of the model and modeled response. This domain permits prediction of uncertainty in the identification of a particular compound based on the data set of compounds used in the development of the model. The AD is also used to define the X-outliers in case of the training set and identify the molecules residing outside the defined AD in case of the test set utilizing the basic theory of standardization approach [38]. Several techniques had been used to define AD of QSAR models [40]. The commonly used one was demonstrated by Gramatica [41], which employed the leverages for each of the compound of the data set. The leveraged approach enables the evaluation of the position of a new compound in a QSAR model [41]. Therefore, Leverage method is utilized and is shown as hi in Eq. (9):
where x: represents the descriptor’s vector of the selected compound, X: refers to the descriptor matrix obtained from the training data set descriptor values and the h* (warning leverage) was calculated as in Eq. (10):
where N represents the number of the training set compounds and prefers to the number of the descriptors in the built model.
The defined AD was then visualized with a Williams plot. The plot of standardized residuals versus the leverage values (h). A molecule with hi > h* seriously influences the QSAR model performance and can be eliminated from the AD. Further, ± 3 value range of standardized residuals is always used as a threshold value for accepting predictions of a molecule, because points that lie within ± 3 of standardized residuals from the mean cover 99% of the normally distributed data [42]. In this regard, the leverage and the standardized residuals were combined for the determination and characterization of the AD. The Williams plot for the constructed QSAR model is presented in Fig. 2. The h* (warning leverage), was found to be 0.420 for the built QSAR model. According to the leverages (hi > 0.420), only two (2) training set compounds (45 and 47) and four (4) test set compounds (32, 54, 60 and 68) were found to be out of the defined AD (Fig. 2) of the constructed QSAR model, so, they were recognized as structurally influential compounds based on their large leverage values (hi > h*).

The Williams plot, the plot of the standardized residuals versus the leverage value
3.4 Molecular design and docking simulation studies
Compound 41 and 69 were selected as the templates due to their high activities (pGI50), low residual values as presented in Table 1 and were found to be within the model’s defined Applicability domain, this propose the possibility of improving the activity of the molecules by introducing some new substituents. The selected templates were docked and the best docking results were − 7.0 kcal mol−1 and − 7.3kcalmol−1 respectively. A library of substituent was imputed into the main structure of the selected compounds at appropriate positions and were docked within the binding site of the receptor so as to assess the chemical behavior the new structures.
Four novel compounds with similar structural profiles to the templates (Compound 41 and 69) were designed. The main strategies of the molecular modification in design of these new scaffolds were introduction of new substituent groups and adjustment the possibility of steric and electrostatic functionality and this changes can easily be done around the benzene ring moiety of the main structures. The structure of the templates used for modifications were presented in Figs. 3 and 4 respectively. The structures were altered in a careful way that can make the synthesis possible experimentally. A group of substituents were selected and added to the main structure of the templates (compound 41 and 69) at the benzene ring moiety as shown in Figs. 3 and 4 respectively. This group have some important features as hydrophobicity were increased.

Design of new scaffolds from the template (Compound 41) and Predicted activity (pGI50) with Leverage limit (h)

Design of new scaffolds from the template (Compound 69) and Predicted activity (pGI50) with Leverage limit (h)
In compound Ia we replaced the two “H” atoms of the benzene ring with “NH2” and “NHSO2CH3” groups as it supports the steric and hydrophobic prediction. In compound IIa we introduced “NH2” and “Cl” to replace “H” atoms from the benzene ring and “NH2” group at thiazole moiety to replace “H” atom which give better result. In compound Ib we replaced the two “H” atoms of the benzene ring with “Cl” and “NH2” groups. In compound IIb we introduced “Cl” and “NO2” to replace “H” from the benzene ring. All the designed compounds showed excellent predicted activity (Figs. 3 and 4) with respect to the templates used. Before the prediction of the pGI50 activity values of newly designed compounds by the developed model, their geometry has been optimized, descriptors computed and their leverages determined as explained for the training set. The warning leverage (limit) for the model (h∗ = 0.420) was used as the threshold value to screen the designed compounds. The positive leverage value of a molecule indicates that the compound was within the model AD and that the predicted pGI50 activity values by the developed model were reliable [43]. The pGI50 values of all the designed compounds were better than the lead compounds used for the design and out of which compound IIa has the best pGI50 activity (12.32). Therefore, it is observed that using a QSAR model, there is a chance to simultaneously identify and predict compounds with better activity and to evaluate which of the structural changes do not fall within the defined AD.
The designed anti-melanoma agents were evaluated for ADME properties and drug-likeness using SwissADME a free web tool used in evaluating ADME properties and drug-likeness of small molecules [44]. The Lipinski's rule of five is useful at pre-clinical stage of drug discovery which state that if any chemical violate more than 2 of these criteria (Molecular weight < 500, Number of hydrogen bond donors (nHBD) ≤ 5, Number of hydrogen bond acceptors (nHBA) ≤ 10, Calculated Log p ≤ 5 and Polar surface area (PSA) < 140 Å2), the chemical is said to be impermeable or badly absorbed [45].
From the results presented in Table 4 it can be seen that none of the molecules violate more than 2 of the criteria stated by Lipinski's rule of five, it means there is a high tendency all of these molecules might be pharmacologically active. It can also be observed from Table 5 that none of the molecules possess the BBB permeant. Thus, these molecules are said have good absorption, low toxicity level, orally bioavailable and permeable. The Bioavailability Radar gives an overview of the drug-likeness of a molecule (Fig. 5). The region painted pink indicates the range for each properties.

The Bioavailability Radar of (Ia), (IIa), (Ib) and (IIb)
Following the design of novel antimelanoma agents and filtering through the Lipinski’s rule of five for oral bioavailability and ADME compliance the designed compounds (Ia, IIa, Ib and IIb) were further evaluated for target binding affinity through molecular docking simulation studies. The best docking results for the studied compounds were presented in Table 6. The obtained docking poses from the Discovery Studio visualizer were shown in Figs. 6, 7, 8, 9 and 10 respectively. The designed compounds had negative free energy of binding higher in magnitude when compared with vemurafenib and this indicates a better binding affinity with the receptor. This shows that the designed compounds could be used as an anti-melanoma drug. The docking poses of the designed compounds showed that they interacted with the binding pocket of a protein target in a way similar to vemurafenib with additional number interactions.

a 3D and b 2D V600E-BRAF/Ia interactions

a 3D and b 2D V600E-BRAF/IIa interactions

a 3D and b 2D V600E-BRAF/Ib interactions

a 3D and b 2D V600E-BRAF/IIb interactions

a 3D and b 2D V600E-BRAF/Vemurafenib interactions
The free binding energy of Ia with a receptor is -12.4kcalmol−1, this interaction was achieved by nine (9) H-bonds with the ligand and Pi/sigma interaction that introduces stabilizing charges responsible for intercalating the drug within the protein (V600E-BRAF) as presented in Fig. 6. There were nine (9) conventional H-bonds present in the complex and seven (7) hydrophobic interactions with seven (7) amino acids (Table 6). The Pi/Cation interactions with PHE583, VAL471, and ALA481 which formed a Pi/donor H-bond with the amino acids in the binding segment of the receptor. Other identified interactions are the alkyl interaction with (ILE617 and ARG662), pi/alkyl interaction with PHE468 and Pi/Pi interaction with TRP531 similar to vemurafenib (Fig. 10).
The docked structure of IIa depicted in Fig. 7 shows negative free binding energy of (-11.8kcalmol−1) which indicates the possibility of stable interactions between the ligand and the protein target. There were seven (7) conventional H-bonds identified between this ligand and receptor; SER536, LYS578, ASN580, GLN530, CYS532, GLN530 and CYS532 and three (3) C-H with SER536, SER465 and ILE463. The complex stability may be attributed with an extra alkyl interaction with VAL471, Pi-sigma interaction with TYR538, Pi-alkyl interactions (PHE583, VAL471, ALA481, CYS532 and ILE463) and Pi-Pi interactions with TRP531 and PHE583 as reported in Table 6.
Ib docks with the V600E-BRAF domain with the free energy for binding of − 11.7 kcal mol−1 as presented in Table 6. Five (5) conventional H-bonds were present between the receptor and molecule (ligand) with GLN494, PHE595, ILE573 and HIS574. One (1) C-H bonds was found with PHE468. Besides, LEU485 form an alkyl interaction and ARG575 formed pi alkyl bonds with the ligand as depicted in Fig. 8.
The docked structure of IIb shown in Fig. 9 indicates a negative free binding energy of (− 12.3 kcal mol−1), suggesting that binding of is practicable, because most of the interactions (energies) are of H-bond type with these amino acids (CYS532, LYS578, LYS578, ASN580 and SER465), thus ensuing in the total negative value. Two (2) C-H bonds were also found with SER465 and CYS532 respectively. The complex stability result can be connected with an extra Pi/sigma interaction associated with ILE463, Pi/alkyl interactions associated with (PHE583 and VAL471) and Pi-Pi interactions (PHE583) as presented in Fig. 9.
It has been reported that H-bonding is the major force controlling the interaction between the docked ligands and the receptors and also the binding affinity of the ligand increases with the number of hydrogen bonding/distance [46, 47]. It can be seen that the number of amino acids involved in the conventional hydrogen bonding with the designed molecules was better than that of vemurafenib as presented in Table 6 and Figs. 6, 7, 8, 9 and 10 respectively. This might inform the better free binding affinity of the designed compounds for V600E-BRAF. Additionally, some of the designed compounds interacted more with the target amino acids through strong electrostatic forces especially compounds Ia and IIb.
4 Conclusion
In this research, we proposed the strategy to designed potent cytotoxic compounds on SK-MEL-5 human melanoma cell line by computational methods. A hybrid approach of QSAR with molecular docking simulation is applied for NCI data sets to investigate their proper binding mode.
The developed QSAR enable the design and prediction of activity (pGI50) of potent compounds on SK-MEL-5 cell line. The regression coefficients of the developed QSAR model showed an equally good model with sufficient statistical validation keys [\(R^{2}\) (0.864), \(R_{adjusted}^{2}\) (0.846), Q2cv (0.841) and \(R_{pred}^{2}\) (0.885)] for the internal and external data sets. This satisfies the criteria of acceptable QSAR models proposed by different groups. Compound 41 and 69 were selected because they had relatively high pGI50 activity; they are within the AD of the QSAR model and modification can be done easily around their benzene ring moiety. Based on results, we predicated the pGI50 activity of proposed compounds by the built QSAR model. Compound IIa was found to be the most active (pGI50 12.32) within all the designed compounds. Moreover, Molecular docking simulation was also applied to investigate the proper binding mode of the designed compounds on V600E-BRAF protein kinase. All the studied ligands were able to inhibit the receptor by totally occupying the active site in the target (receptor). The designed Ia, IIa, Ib and IIb with free binding energy (FBE) of − 12.4 kcal mol−1, − 11.8 kcal mol−1, − 11.7 kcal mol−1 and − 12.3 kcal mol−1 respectively were found to be more potent than the standard V600E-BRAF inhibitor (vemurafenib, FBE = −11.3 kcal mol−1) due to the introduction of the new substituents which has the ability of increasing the overall free binding energy by increasing the number of hydrogen bonds and hydrophobic interactions shown in their complexes. Therefore, in future studies, there is hope to include the synthesis, in vivo and in vitro evaluation of these ligands (inhibitors) which can establish them as potent V600E-BRAF inhibitors for the treatment melanoma cancer.
References
Fang Y, Chen X, Bai Q, Qin C, Mohamud AO, Zhu Z, Ball TW, Ruth CM, Newcomer DR, Herrick EJ (2015) IL-9 inhibits HTB-72 melanoma cell growth through upregulation of p21 and TRAIL. J Surg Oncol 111(8):969–974
Al-Qathama A, Gibbons S, Prieto JM (2017) Differential modulation of Bax/Bcl-2 ratio and onset of caspase-3/7 activation induced by derivatives of Justicidin B in human melanoma cells A375. Oncotarget 8(56):95999
Carbone C, Martins-Gomes C, Pepe V, Silva A, Musumeci T, Puglisi G, Furneri P, Souto E (2018) Repurposing itraconazole to the benefit of skin cancer treatment: a combined azole-DDAB nanoencapsulation strategy. Colloids Surf B 167:337–344
Al-Sanea MM, Ali Khan MS, Abdelazem AZ, Lee SH, Mok PL, Gamal M, Shaker ME, Afzal M, Youssif BG, Omar NN (2018) Synthesis and in vitro antiproliferative activity of new 1-phenyl-3-(4-(pyridin-3-yl) phenyl) urea scaffold-based compounds. Molecules 23(2):297
Shi J-H, Chen J, Wang J, Zhu Y-Y, Wang Q (2015) Binding interaction of sorafenib with bovine serum albumin: Spectroscopic methodologies and molecular docking. Spectrochim Acta Part A Mol Biomol Spectrosc 149:630–637
Wu C-P, Ambudkar SV (2014) The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm Sin B 4(2):105–111
Zubrilov I, Sagi-Assif O, Izraely S, Meshel T, Ben-Menahem S, Ginat R, Pasmanik-Chor M, Nahmias C, Couraud P-O, Hoon DS (2015) Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett 361(1):86–96
Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE, Piccart-Gebhart MJ (2013) Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev 39(8):935–946
Mioc M, Pavel IZ, Ghiulai R, Coricovac DE, Farcaş C, Mihali C-V, Oprean C, Serafim V, Popovici RA, Dehelean CA (2018) The cytotoxic effects of betulin-conjugated gold nanoparticles as stable formulations in normal and melanoma cells. Front Pharmacol 9:429
Theodosakis N, Micevic G, Langdon CG, Ventura A, Means R, Stern DF, Bosenberg MW (2017) p90RSK blockade inhibits dual BRAF and MEK inhibitor-resistant melanoma by targeting protein synthesis. J Invest Dermatol 137(10):2187–2196
Usta O, McCarty W, Bale S, Hegde M, Jindal R, Bhushan A, Golberg I, Yarmush M (2015) Microengineered cell and tissue systems for drug screening and toxicology applications: evolution of in-vitro liver technologies. Technology 3(01):1–26
Kraljevic S, Stambrook PJ, Pavelic K (2004) Accelerating drug discovery. EMBO Rep 5(9):837–842
Lionberger RA (2008) FDA critical path initiatives: opportunities for generic drug development. AAPS J 10(1):103–109
Yap C, Xue Y, Chen Y (2006) Application of support vector machines to in silico prediction of cytochrome p450 enzyme substrates and inhibitors. Curr Top Med Chem 6(15):1593–1607
Satbhaiya S, Chourasia O (2015) Scaffold and cell line based approaches for QSAR studies on anticancer agents. RSC Adv 5(103):84810–84820
Kamal A, Bharathi EV, Ramaiah MJ, Dastagiri D, Reddy JS, Viswanath A, Sultana F, Pushpavalli S, Pal-Bhadra M, Srivastava HK (2010) Quinazolinone linked pyrrolo [2, 1-c][1, 4] benzodiazepine (PBD) conjugates: design, synthesis and biological evaluation as potential anticancer agents. Bioorg Med Chem 18(2):526–542
Xie F, Zhao H, Zhao L, Lou L, Hu Y (2009) Synthesis and biological evaluation of novel 2, 4, 5-substituted pyrimidine derivatives for anticancer activity. Bioorg Med Chem Lett 19(1):275–278
Martin YC (1998) 3D QSAR: current state, scope, and limitations. 3D QSAR in Drug Design. Springer, New York, pp 3–23
Ashton PR, Fyfe MC, Hickingbottom SK, Stoddart JF, White AJ, Williams DJ (1998) Hammett correlations ‘beyond the molecule’1. J Chem Soc Perkin Trans 2(10):2117–2128
Reddy AS, Pati SP, Kumar PP, Pradeep H, Sastry GN (2007) Virtual screening in drug discovery-a computational perspective. Curr Protein Pept Sci 8(4):329–351
Srivani P, Sastry GN (2009) Potential choline kinase inhibitors: a molecular modeling study of bis-quinolinium compounds. J Mol Graph Model 27(6):676–688
Benigni R, Giuliani A (2003) Putting the predictive toxicology challenge into perspective: reflections on the results. Bioinformatics 19(10):1194–1200
Hansch C, Leo A, Mekapati SB, Kurup A (2004) Qsar and Adme. Bioorg Med Chem 12(12):3391–3400
Srivastava HK, Chourasia M, Kumar D, Sastry GN (2011) Comparison of computational methods to model DNA minor groove binders. J Chem Inf Model 51(3):558–571
Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467(7315):596
Abdulfatai U, Uzairu A, Uba S (2017) Quantitative structure-activity relationship and molecular docking studies of a series of quinazolinonyl analogues as inhibitors of gamma amino butyric acid aminotransferase. J Adv Res 8(1):33–43
Parthasarathy K, Praveen C, Saranraj K, Balachandran C, Kumar PS (2016) Synthesis, antimicrobial and cytotoxic evaluation of spirooxindole [pyrano-bis-2H-l-benzopyrans]. Med Chem Res 25(10):2155–2170
Amin SA, Gayen S (2016) Modelling the cytotoxic activity of pyrazolo-triazole hybrids using descriptors calculated from the open source tool “PaDEL-descriptor”. J Taibah Univ Sci 10(6):896–905
Yap CW (2011) PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32(7):1466–1474
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inf 29(6–7):476–488
Rajer-Kanduč K, Zupan J, Majcen N (2003) Separation of data on the training and test set for modelling: a case study for modelling of five colour properties of a white pigment. Chemom Intell Lab Syst 65(2):221–229
Kennard RW, Stone LA (1969) Computer aided design of experiments. Technometrics 11(1):137–148
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. Mol Inf 22(1):69–77
Leardi R (1996) Genetic algorithms in feature selection. Genetic algorithms in molecular modeling. Elsevier, Amsterdam, pp 67–86
Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A (2002) BRAF and RAS mutations in human lung cancer and melanoma. Can Res 62(23):6997–7000
Choi W-K, El-Gamal MI, Choi HS, Baek D, Oh C-H (2011) New diarylureas and diarylamides containing 1, 3, 4-triarylpyrazole scaffold: Synthesis, antiproliferative evaluation against melanoma cell lines, ERK kinase inhibition, and molecular docking studies. Eur J Med Chem 46(12):5754–5762
Wu W, Zhang C, Lin W, Chen Q, Guo X, Qian Y, Zhang L (2015) Quantitative structure-property relationship (QSPR) modeling of drug-loaded polymeric micelles via genetic function approximation. PloS One 10(3):e0119575
Umar BA, Uzairu A, Shallangwa GA, Sani U (2019) QSAR modeling for the prediction of pGI50 activity of compounds on LOX IMVI cell line and ligand-based design of potent compounds using in silico virtual screening. Netw Model Anal Health Inf Bioinform 8(1):22
Jalali-Heravi M, Konuze E (2002) Use of quantitative structure property relationships in predicting the Kraft point of anionic surfactants. Electron J Mol Des 1:410–417
Eriksson L, Jaworska J, Worth AP, Cronin MT, McDowell RM, Gramatica P (2003) Methods for reliability and uncertainty assessment and for applicability evaluations of classification-and regression-based QSARs. Environ Health Perspect 111(10):1361–1375
Gramatica P, Giani E, Papa E (2007) Statistical external validation and consensus modeling: a QSPR case study for Koc prediction. J Mol Graph Model 25(6):755–766
Jaworska J, Nikolova-Jeliazkova N, Aldenberg T (2005) QSAR applicability domain estimation by projection of the training set descriptor space: a review. Atla-Nottingham 33(5):445
Netzeva TI, Worth AP, Aldenberg T, Benigni R, Cronin MT, Gramatica P, Jaworska JS, Kahn S, Klopman G, Marchant CA (2005) Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships: the report and recommendations of ecvam workshop 52. Altern Lab Anim 33(2):155–173
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
Ismail S, Uzairu A, Sagagi B, Sabiu M (2018) In silico molecular docking and pharmacokinetic study of selected phytochemicals with estrogen and progesterone receptors as anticancer agent for breast cancer. J Turk Chem Soc Sect A Chem 5(3):1337–1350
Adedirin O, Uzairu A, Shallangwa GA, Abechi SE (2018) Optimization of the anticonvulsant activity of 2-acetamido-N-benzyl-2-(5-methylfuran-2-yl) acetamide using QSAR modeling and molecular docking techniques. Beni-Suef Univ J Basic Appl Sci 7(4):430–440
Umar BA, Uzairu A, Shallangwa GA, Uba S (2019) Rational drug design of potent V600E-Braf kinase inhibitors through molecular docking simulation. J Eng Exact Sci 5(5):0469–0481
Acknowledgements
The authors sincerely acknowledge the National Cancer Institute for providing the data set used in this study and Ahmadu Bello University, Zaria for providing the soft wares used.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared they have no conflict of interest.
Human and animal rights
This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Umar, A.B., Uzairu, A., Shallangwa, G.A. et al. Design of potential anti-melanoma agents against SK-MEL-5 cell line using QSAR modeling and molecular docking methods. SN Appl. Sci. 2, 815 (2020). https://doi.org/10.1007/s42452-020-2620-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2620-8