
Abstract
The anti-melanoma activity (pGI50) values of 71 compounds from the National Cancer Institute (NCI) data bank on LOX IMVI cell line were modeled to illustrate the Quantitative structure–activity relationship (QSAR) of the compounds. The genetic function algorithm (GFA) has been used to select the most relevant descriptors so as to improve the performance of the QSAR model. The statistical significance of the model was verified based on the values of validation parameters such as \( R_{{{\text{train}} }}^{2} \) (0.867), \( R_{{{\text{adj}} }}^{2} \) (0.848), \( Q_{{{\text{cv}} }}^{2} \) (0.809) and \( R_{{{\text{test}} }}^{2} \) (0.749) needed to evaluate the robustness and strength of the model. The result of the internal and external validation of the model indicates that the model is good and could be used to predict pGI50 of anti-melanoma compounds on LOX IMVI cell line for which no experimental data are available. Compound 41 was selected using in-silico screening method as a template due to its good pGI50 (9.793) and was utilized to design new potent compounds, thereby enhancing the activity of the parent structure. Ten (10) new potent compounds were deigned and predicted using the proposed model. The predicted pGI50 of the majority of the designed analogous were more than the lead compound 41 used for the design and among which compound N5 showed the best activity (pGI50=13.186). Thus, this study provides a valuable approach and new direction to novel drug discovery.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Melanoma is one of the tumors developed from melanocytes and among the most deadly cancers among young adults (Lee et al. 2015). It has a high capability of invasion and quick metastasis to other organs which are caused by abnormalities in the cells; this may be as a result of genes (inherited) or due to exposure of the body to radiation, chemicals, or even infectious agents (Liotta et al. 1991; Mignatti and Rifkin 1993). Patients with sophisticated melanoma have a median survival time of less than 1 year, and the guessed five-year survival rate is less than fifteen percent (15%) (Anderson et al. 1995; Barth et al. 1995). With the rapid increase of melanoma in the United States (US) and other developed countries, there is an urgent need to identify more effective drugs (Gray-Schopfer et al. 2007; Lee et al. 2015). Several novel drugs were approved by the US Food and Drug Administration (FDA) such as benzylideneoxindoles, ZM336372, sorafenib, isoquinolones, triarylimidazoles, PLX4032, and XL281 for treatment of melanoma (Wu and Ambudkar 2014). Unfortunately, treatment with the use of such drugs can result in the development of drug resistance and the metastases develop again increasing about 6 months the life expectancy of the patient (Saini et al. 2013; Zubrilov et al. 2015). Therefore, identification and prediction of anti-melanoma activity of novel drugs are of great importance for cancer (Melanoma) research (Roskoski 2012).
Optimal anti-cancer drugs would exterminate cancer cells without damaging normal tissues (Al-Suwaidan et al. 2016; Choi et al. 2011; Naik and Pardasani 2018). Regrettably, currently no available drugs meet this condition, and clinical use of drugs involves a weighing of benefits against toxicity in a search of favorable therapeutic index (Chabner 1990; Makrariya and Pardasani 2019). Thus, these limitations have made it necessary to search for novel anti-cancer drugs with diverse chemical structure as potential anti-cancer agents (Al-Suwaidan et al. 2016). Nevertheless, in the field of medicinal chemistry, activity prediction of new compounds is a primary goal for the drug design process (Vaidya et al. 2014). The chemical and molecular computing models are used in designing new drugs which helped in reducing the time and cost involved in designing more potent drugs. Among several computational methods used, quantitative structure–activity relationship (QSAR) has a remarkable role in designing a drug.
QSAR is an attempt to correlate structural descriptors of compounds quantitatively with biological activities. The molecular descriptors include parameters that account for conformational, constitutional, thermodynamic, steric effects and electronic properties of a molecule. Others like fragment constant, hydrophobicity, topology, hydrogen bond acceptor, and hydrogen bond-donor are also determined recently by computational methods (Arthur et al. 2016; Young 2004). QSAR models are mathematical equations which relate the chemical structure of compounds to their biological activity. Therefore, it is necessary to develop a model that could be used for the identification of new potent compounds and prediction of their anti-cancer activity before the synthesis. This will help to reduce the cost and time involved in drug discovery. This study was aimed to develop QSAR model based on the compounds collected from NCI data bank which can be used to predict the anti-melanoma activity of known and new potent compounds on LOX IMVI cell line. Additionally, an in silico screening technique is applied to the proposed QSAR model to predict the structure of new potent anti-melanoma compounds.
2 Materials and methods
2.1 Software and computer specifications
All the molecular modeling studies were carried out on a Dell Intel(R)Core(TM)i7-5500U CPU, 16.00 GB RAM @ 2.400 GHz 2.400 GHz processor, 64-bit Operating system, a 64× based processor on Windows 8.1 Pro. Spartan 14 (Hehre and Huang 1995) was employed to perform density functional theory calculations, Material studios 8.0 was used to develop the model and Microsoft office Excel 2013 was utilized for statistical analysis.
2.2 Data set
In this research, a data set of 71 anti-melanoma compounds and their pGI50 activities on LOX IMVI human melanoma cell line were collected from the drug discovery and development section of the National Cancer Institute (NCI) (https://wiki.nci.nih.gov/display/NCIDTPdata/NCI-60+Growth+Inhibition+Data). Their NSC number and anti-melanoma activity results as pGI50, which is the negative log (−LogGI50) of the concentration for 50% of cancer cell proliferation, are depicted in Table 2.
2.3 Computation of descriptors
The 2D structure of each of the compounds was converted into the 3D structure using Spartan 14. The structures were cleaned by minimizing and checking using a molecular mechanic force field (MM+) option on Spartan 14, so as to remove all strain from the structure of the molecule. Additionally, this will guarantee a well-defined and stable conformer relationship within the compounds in the study (Viswanadhan et al. 1989). Geometry optimization was set at the ground state utilizing the density functional theory (DFT) at the Becke88 three-parameter hybrid exchange potentials with Lee–Yang–Parr correlation potential (B3LYP) level of theory and for the basis set 6-311G (d) was selected. The fully optimized 3D structure in SD file was then imported into PaDEL descriptor software to compute both thermodynamic, topological, autocorrelation constitutional, electronic, and geometric descriptors (Amin and Gayen 2016) for further studies (Yap 2011).
2.4 Dataset division into modeling and prediction sets
The data set was divided into two sets, the modeling and prediction set. The modeling set is used in developing the model; it contains seventy percent (70%) of the entire data set. While the test set which constitutes the remaining thirty percent (30%) of the whole data set was not used in the construction of the model but to ascertain the predictive ability of the model (Tropsha et al. 2003). This partitioning ensures that a similar principle can be employed for the activity prediction of the test set. Kennard–Stone Algorithm was applied for dividing dataset into a modeling and test set (Kennard and Stone 1969; Rajer-Kanduč et al. 2003).
2.5 Model development
In QSAR studies, the identification and selection of descriptors which provide maximum information in activity variations and have minimum co-linearity are important. Therefore, a genetic function algorithm (GFA) (Leardi 1996) improves the model accuracy in the selection of proper descriptors. Multiple Linear Regression (MLR) was used on the modeling set to show the relationship between the dependent variable Y (pGI50) and independent variable X (molecular descriptors). In regression analysis, the contingent mean of the dependent variable (pGI50) Y relies on (descriptors) X.
2.6 QSAR model validation
In the validation of a QSAR model, the stability and predictive ability of the model is one of the key steps in QSAR modeling. Various statistical parameters have been utilized for the validation of the suitability of the built model for the prediction of the anti-cancer activity of the studied compounds (Asadollahi et al. 2011) this includes correlation coefficient (R2) which describes the fraction of the total variation attributed to the model. The closer the value of R2 is to 1.0, the better the regression and equation explain the Y variable. R2 is the most commonly used internal validation indicator and is expressed as in Eq. (1):
where Yexp, Ypred, and Ymtraining are the experimental property, the predicted property and the mean experimental activity of the compounds in the training set, respectively. The minimum recommended value for this parameter is shown in Table 1 (Wu et al. 2015).
Adjusted R2 (\( R_{{{\text{adj}} }}^{2} \)): R2 value varies directly with the increase in the number of descriptors; thus, R2 cannot be a useful measure for the goodness of model fit. Therefore, R2 is adjusted for the number of explanatory variables in the model. The adjusted R2 is defined as in Eq. (2):
where P = number of independent variables in the model and N = sample size (Abdulfatai et al. 2017). The minimum recommended value for this parameter is presented in Table 1.
Cross-validation coefficient parameter (\( Q_{{{\text{CV}} }}^{2} \)) is the most commonly used internal validation indicator and is expressed as in Eq. (3):
where Yexp is the experimental activity, Ypred is the predicted activity, and Ymntraining is the mean of the experimental activity of the validation set (Tropsha et al. 2003). However, it should be noted that a high \( Q_{{{\text{CV}} }}^{2} \) does not necessarily mean high predictability of the built model (Asadollahi et al. 2011). In other words, the high value of \( Q_{{{\text{CV}} }}^{2} \) is a necessary condition, but not sufficient for a developed model to have high predictability.
To assess the predictive ability and to check the statistical significance of the developed model, the proposed model was applied to predict the pGI50 values of an external (test) set compounds that were not used in building the model. The predictive powers of the proposed regression model on the training set were evaluated by predicted values of the prediction (test) set. Therefore, validation through an external prediction set (\( R_{{{\text{test}} }}^{2} \)) is a very important parameter that is used to test the external predictive ability of a QSAR model. The \( R_{{{\text{test}} }}^{2} \) value is calculated by Eq. (4):
where Yexp is the experimental activity, Ypred is the predicted activity, and Ymntraining is the mean of the experimental activity of the training set (Tropsha et al. 2003).
2.7 Y-randomization test
To assess the robustness of the built model, the Y-randomization test was applied to the training set data as suggested by Tropsha et al. (2003). The dependent variable vector (activity data) was randomly shuffled and a new QSAR model was developed using the original independent variable matrix. For the built QSAR model to be robust and reliable, the model is expected to have low R2 and Q2 values for several trials. The coefficient of determination \( {\text{cR}}_{{{\text{p}} }}^{2} \) for Y-randomization is another parameter calculated which should be greater than 0.5 for passing this test as in Eq. (5):
\( {\text{cR}}_{{{\text{p}} }}^{2} \) is coefficient of determination for Y-randomization, R is the coefficient of determination for Y-randomization and Rr is average ‘R’ of random models.
3 Results and discussion
On the basis of Kennard–Stones algorithm, 49 compounds out of 71 were selected as the modeling (training) set and the remaining 22 were selected as the prediction (test) set. GFA regression was used on the modeling data set to select the significant descriptors and it was found that among 1875 calculated descriptors, the SpMin-Bhv, SpMax4-Bhe, SpMin5-Bhi, SpMin3-Bhs, piPC1, and GGI4 build the best model and a new GFA-MLR QSAR regression equation was developed based on modeling set.
3.1 QSAR model for predicting pGI50 on LOX IMVI cell line
where N is the number of compounds in the training and test sets, \( R_{{{\text{train}} }}^{2} \) is the squared correlation coefficient, \( R_{{{\text{adjusted}} }}^{2} \) is the adjusted R-squared, \( Q_{{{\text{cv}} }}^{2} \) is the cross-validation coefficients of the training set and \( R_{{{\text{test}} }}^{2} \) is the squared correlation coefficient of the prediction (test) set.
3.2 QSAR model validation
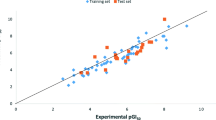
In a further study, the built QSAR model from the modeling data set was used to evaluate its predictive ability by predicting the pGI50 values in the prediction set (test set). The results are given in Table 2. The predicted pGI50 values for the training and test sets were plotted against the experimental pGI50 as shown in Fig. 1. The predicted pGI50 results obtained for both the modeling set and prediction set (Table 2) are in good agreement with the experimental pGI50 obtained from NCI. The residual values obtained between predicted and experimental pGI50 were very low.

The predicted pGI50 against the experimental values for the training and test sets
The result of the QSAR model is in conformity with the standard shown in Table 1 as seen from the built model. The closeness of coefficient of determination (R2) to its absolute value of 1.0 is an indication that the model explained a very high percentage of the response variable (descriptor) variation, high enough for a robust QSAR model. Its 0.867 value illustrates that 86.7% of the variation is residing in the residual meaning that the model is very good.
The high adjusted R2 (\( R_{{{\text{adj}} }}^{2} \)) value as seen in the model and its closeness in value to the value of R2 imply that the model has excellent explanatory power to the descriptors in it. It also demonstrates the real influence of applied descriptors on the pGI50. Also, the high and closeness of \( Q_{{{\text{cv}} }}^{2} \) to \( R_{{{\text{train}} }}^{2} \) revealed that the model was not over-fitted. The high \( R_{{{\text{test}} }}^{2} \) as seen in the model is an indication that the model is capable of providing valid predictions for new compounds.
Additionally, to assess the robustness of the model, the Y-randomization test was applied. The dependent variable vector (inhibitory activity) was randomly shuffled and a new QSAR model was developed using the original independent variable matrix. As was expected, the new QSAR models (after several repetitions) have low R2 and Q2 values and also, the \( {\text{cR}}_{{{\text{p}} }}^{2} \) value was greater than 0.5 as presented in Table 3. This test affirms that the proposed model is powerful and not inferred by chance.
3.3 Contribution and interpretation of descriptors in model
The six-variable QSAR model adequately represents the pGI50 data, based on direct statistics as well as validation methods. Each of the variables is a descriptor of an aspect of molecular structure and will be discussed to indicate the specific structural information encoded. By interpreting the descriptors contained in the QSAR model, it is possible to gain some insights into factors, which are related to the anti-cancer activity. For this reason, an acceptable interpretation of the selected descriptors is provided. The brief descriptions of the descriptors are shown in Table 4. The relative importance and contribution of each descriptor in the model were determined by the calculation of the value of the mean effect (MF) (Jalali-Heravi and Konuze 2002) for each descriptor using Eq. (6) and the MF values are presented in Table 4:
where MFj represents the mean effect for the descriptor j, βj is the coefficient of the descriptor j, did is the value of the interested descriptors for each molecule and m is the number of descriptors in the model.
The MF value shows the relative importance of each descriptor compared to the other descriptors. The MF of the descriptors SpMin-Bhv, SpMax4-Bhe, SpMin5-Bhi, SpMin3-Bhs, piPC1 and GGI4 are also shown in Table 4 and indicate that among the selected descriptors, the most important one is piPC1 (Conventional bond order ID number of order 1 (ln(1 + x)) as it has the highest mean effect value and has the largest effect on the pGI50 of the compound. On the basis of MF values, the associated descriptors are arranged in a sequence pertaining to their contribution towards overall pGI50 of the compounds, in the following increasing order of pGI50 of compounds.
The SpMin6_Bhv descriptors have been proposed as the chemical structure descriptors derived from a new representation of the molecular structure. SpMin6_Bhv is the smallest absolute eigenvalue of Burden modified matrix-n 6/weighted by relative van der Waals volumes. The SpMin6_Bhv mean effect has a positive sign as presented in Table 4. This sign suggests that the anti-melanoma activity is directly related to this descriptor.
SpMax4_Bhe is defined as the largest absolute eigenvalue of Burden modified matrix-n 4/weighted by relative Sanderson electro-negativities. The SpMax4_Bhe mean effect has a negative sign as shown in Table 4. This sign suggests that the decrease of value for this descriptor will increase the anti-cancer activity of a molecule and vice versa. SpMin5_Bhi is the smallest absolute eigenvalue of Burden modified matrix-n 5/weighted by relative first ionization potential. The negative sign of the mean effect (Table 4) of SpMin5_Bhi suggests that its decrease may increase the anti-cancer activity. SpMin3_Bhs is the smallest absolute eigenvalue of Burden modified matrix-n 3/weighted by relative I-state. SpMin3_Bhs also has a negative mean effect value which suggests that the decrease of value for this descriptor will increase the anti-melanoma activity of a molecule. The SpMin5_Bhi has a negative mean effect (Table 4) and its decrease may improve the anti-melanoma activity.
piPC1 is a 2D descriptor defined as the conventional bond order ID number of order 1 (ln(1 + x); it is also describe as the molecular multiple path counts of order 01; and the mean effect of piPC1 was found to positively influence the anti-melanoma activity of the compounds when increased as shown in Table 4. GGI4 is defined as topological charge index of order 4. The mean effect value for this descriptor has a negative sign (Table 4). This sign suggests that the anti-melanoma activity will increase with the decrease in its value. The descriptors used for building the QSAR model in this work encoded topological, electronic and geometrical aspects of molecules. Appearances of these descriptors in the model reveal the role of electronic and steric interactions in inducing anti-melanoma pGI50 activity on LOX IMVI cell line.
3.4 In silico screening
An in silico screening method is a very powerful tool used for identifying new biologically potent compounds with improved characteristics and predicting their activities before their actual synthesis (Muegge and Oloff 2006; Tropsha et al. 2003). Therefore, the in silico technique reduces the time and cost involved in identifying potent compounds. Virtual screening was performed by deletion, insertion, and substitution of different substitutes on the original template (molecule) (Melagraki et al. 2007, 2009) and the effects of the structural alterations on the biological activity were evaluated. Then, the applicability domain (AD) of the QSAR model was defined to use the model for predicting and screening new leads. Defining the domain of application of the QSAR model is essential in establishing the model capability to make predictions within the space (chemical) for which it was developed (Tropsha et al. 2003).
Various methods have been utilized to define the AD of the QSAR models (Eriksson et al. 2003). The most usual one was described by Gramatica et al. (2007) which used the leverage values for each compound. The leverage approach allows the determination of the position of new chemical in the QSAR model (Gramatica et al. 2007), In this regard, Leverage approach is used and is represented as hi in Eq. (7):
where x refers to the descriptor vector of the considered compound and X represents the descriptor matrix derived from the training set descriptor values. The warning leverage (h*) was determined as in Eq. (8):
where N is the number of training compounds and p is the number of descriptors in the model.
The defined applicability domain (AD) was then viewed via a Williams plot, the plot of the standardized residuals against the leverage values (h). A compound with hi > h* seriously influences the model performance and may be eliminated from the AD applicability, but it does not appear to be an outlier since its standardized residual could be small. Furthermore, a value range of ± 3 standardized residuals is often used as a cutoff value for accepting predictions of a molecule, because points which lie within ± 3 standardized residual from the mean cover ninety-nine percent (99%) of the normally distributed data (Jaworska et al. 2005). Thus, the leverage and the standardized residuals were used jointly for the characterization and determination of the applicability domain. The Williams plot for the built QSAR is shown in Fig. 2. The warning leverage (h*) was found to be 0.430 for the developed QSAR model. Based on the leverages (h i> 0.430), the two compounds among the training set (44 and 47) and five test set compounds (1, 51, 54, 55 and 67) were found to be outside of the defined AD (Fig. 2) of the QSAR model; so, they were identified as structurally influential chemical based on their large leverage values (hi> h*).

The Williams plot, the plot of the standardized residuals versus the leverage value
Furthermore, the in silico screening method was used for the design of new potent structures with pGI50 activity on LOX IMVI cell line according to the developed QSAR model. For this purpose, compound 41 (AC1L2OAS, NSC-376,128) listed in Table 2 with pGI50 of 9.793 was chosen as a template due to its high pGI50 activity, low residual value and was found to be within the defined AD (Fig. 2). The structure of compound 41 and the template used for modifications are shown in Fig. 3. The compound was altered in a way that will make its synthesis experimentally possible. Then, the in silico screening was applied by the insertion and substitution of different groups in the X, Y and Z positions as presented in Fig. 3; the results of this are presented in Table 5. The model endures various AC1L2OAS substituents since the majority of the newly designed analogous was within the applicability domain. The predicted pGI50 of the majority of the designed analogous were more than the lead compound (41) used for the design and among which compound N5 showed the best activity (pGI50 = 13.186). Thus, it is clear that using a simple QSAR model, there is a possibility to simultaneously predict and identify compounds with better activity and to determine which of the structural modifications do not fall within the AD. Lastly, the result in this research confirms the robustness and reliability of the developed QSAR model and it illustrates that with the modeling of the QSAR model and use of an in silico screening technique, it is possible to identify new potent synthetic targets for drug development.

a Structure of compound 41. b Structure of the template used for design
4 Conclusions
In this research, GFA-MLR modeling tool was used in the construction of a QSAR model for predicting pGI50 of anti-melanoma compounds on LOX IMVI cell line. The accuracy and predictability of the proposed model was illustrated by various criteria, the model is statistically fit both internally (\( R_{{{\text{train}} }}^{2} \) = 0.867, \( R_{{{\text{adj}} }}^{2} \) = 0.848 and \( Q_{{{\text{cv}} }}^{2} \) = 0.809), externally (\( R_{{{\text{test}} }}^{2} \) = 0.749), and Y-randomization. This satisfies the criteria of acceptable QSAR model proposed by different groups. Moreover, in silico screening method was applied to the developed QSAR model which enables the design and prediction of pGI50 of new potentially active compounds on LOX IMVI cell line. The predicted pGI50 of the majority of the designed analogous were more than the lead compound 41 used for the design. The proposed model was found to be useful for the prediction of pGI50 of anti-melanoma compounds for which no experimental data are available and it also helps in the reduction of time and cost involved in the synthesis and anti-melanoma activity prediction of compounds on LOX IMVI cell line.
References
Abdulfatai U, Uzairu A, Uba S (2017) Quantitative structure-activity relationship and molecular docking studies of a series of quinazolinonyl analogues as inhibitors of gamma amino butyric acid aminotransferase. J Adv Res 8(1):33–43
Al-Suwaidan IA, Abdel-Aziz AA-M, Shawer TZ, Ayyad RR, Alanazi AM, El-Morsy AM, Mohamed MA, Abdel-Aziz NI, El-Sayed MA-A, El-Azab AS (2016) Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4 (3H) quinazolinone analogues. J Enzyme Inhib Med Chem 31(1):78–89
Amin SA, Gayen S (2016) Modelling the cytotoxic activity of pyrazolo-triazole hybrids using descriptors calculated from the open source tool “PaDEL-descriptor”. J Taibah Univ Sci 10(6):896–905
Anderson CM, Buzaid AC, Legha SS (1995) Systemic treatments for advanced cutaneous melanoma. Oncology 9:4–5
Arthur DE, Uzairu A, Mamza P, Abechi S (2016) Quantitative structure–activity relationship study on potent anticancer compounds against MOLT-4 and P388 leukemia cell lines. J Adv Res 7(5):823–837
Asadollahi T, Dadfarnia S, Shabani AMH, Ghasemi JB, Sarkhosh M (2011) QSAR models for CXCR6 receptor antagonists based on the genetic algorithm for data preprocessing prior to application of the PLS linear regression method and design of the new compounds using in silico virtual screening. Molecules 16(3):1928–1955
Barth A, Wanek LA, Morton DL (1995) Prognostic factors in 1521 melanoma patients with distant metastases. J Am Coll Surg 181(3):193–201
Chabner BA (1990) Cancer chemotherapy: principles and practice. Lippincott Williams and Wilkins, United States, pp 341–355
Choi W-K, El-Gamal MI, Choi HS, Baek D, Oh C-H (2011) New diarylureas and diarylamides containing 1, 3, 4-triarylpyrazole scaffold: synthesis, antiproliferative evaluation against melanoma cell lines, ERK kinase inhibition, and molecular docking studies. Eur J Med Chem 46(12):5754–5762
Eriksson L, Jaworska J, Worth AP, Cronin MT, McDowell RM, Gramatica P (2003) Methods for reliability and uncertainty assessment and for applicability evaluations of classification-and regression-based QSARs. Environ Health Perspect 111(10):1361–1375
Gramatica P, Giani E, Papa E (2007) Statistical external validation and consensus modeling: a QSPR case study for Koc prediction. J Mol Graph Model 25(6):755–766
Gray-Schopfer V, Wellbrock C, Marais R (2007) Melanoma biology and new targeted therapy. Nature 445(7130):851
Hehre W, Huang W (1995) Chemistry with computation: an introduction to SPARTAN. Wavefunction. Inc, Irvine, CA
Jalali-Heravi M, Konuze E (2002) Use of quantitative structure property relationships in predicting the kraft point of anionic surfactants. Electron J Mol Des 1:410–417
Jaworska J, Nikolova-Jeliazkova N, Aldenberg T (2005) QSAR applicability domain estimation by projection of the training set descriptor space: a review. Atla-Nottingham 33:445
Kennard RW, Stone LA (1969) Computer aided design of experiments. Technometrics 11(1):137–148
Leardi R (1996) Genetic algorithms in molecular modeling. Elsevier, pp 67–86
Lee JA, Roh EJ, Oh C-H, Lee SH, Sim T, Kim JS, Yoo KH (2015) Synthesis of quinolinylaminopyrimidines and quinazolinylmethylaminopyrimidines with antiproliferative activity against melanoma cell line. J Enzyme Inhib Med Chem 30(4):607–614
Liotta LA, Steeg PS, Stetler-Stevenson WG (1991) Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell 64(2):327–336
Makrariya A, Pardasani K (2019) Numerical study of the effect of non-uniformly perfused tumor on heat transfer in women’s breast during menstrual cycle under cold environment. Netw Modeling Anal Health Inform Bioinform 8(1):9
Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O (2007) Optimization of biaryl piperidine and 4-amino-2-biarylurea MCH1 receptor antagonists using QSAR modeling, classification techniques and virtual screening. J Comput Aided Mol Des 21(5):251–267
Melagraki G, Afantitis A, Sarimveis H, Koutentis PA, Kollias G, Igglessi-Markopoulou O (2009) Predictive QSAR workflow for the in silico identification and screening of novel HDAC inhibitors. Mol Divers 13(3):301–311
Mignatti P, Rifkin DB (1993) Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 73(1):161–195
Muegge I, Oloff S (2006) Advances in virtual screening. Drug Discov Today Technol 3(4):405–411
Naik PA, Pardasani KR (2018) 2D finite-element analysis of calcium distribution in oocytes. Netw Model Anal Health Inform Bioinform 7(1):10
Rajer-Kanduč K, Zupan J, Majcen N (2003) Separation of data on the training and test set for modelling: a case study for modelling of five colour properties of a white pigment. Chemom Intell Lab Syst 65(2):221–229
Roskoski R (2012) MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem Biophys Res Commun 417(1):5–10
Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE, Piccart-Gebhart MJ (2013) Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev 39(8):935–946
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. Mol Inform 22(1):69–77
Vaidya A, Jain S, Jain S, Jain AK, Agrawal RK (2014) Quantitative structure-activity relationships: a novel approach of drug design and discovery. J Pharm Sci Pharmacol 1(3):219–232
Viswanadhan VN, Ghose AK, Revankar GR, Robins RK (1989) Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J Chem Inf Comput Sci 29(3):163–172
Wu C-P, Ambudkar SV (2014) The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm Sin B 4(2):105–111
Wu W, Zhang C, Lin W, Chen Q, Guo X, Qian Y, Zhang L (2015) Quantitative structure-property relationship (QSPR) modeling of drug-loaded polymeric micelles via genetic function approximation. PLoS One 10(3):e0119575
Yap CW (2011) PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32(7):1466–1474
Young D (2004) Computational chemistry: a practical guide for applying techniques to real world problems. Wiley, Hoboken, New Jersey, United States
Zubrilov I, Sagi-Assif O, Izraely S, Meshel T, Ben-Menahem S, Ginat R, Pasmanik-Chor M, Nahmias C, Couraud P-O, Hoon DS (2015) Vemurafenib resistance selects for highly malignant brain and lung-metastasizing melanoma cells. Cancer Lett 361(1):86–96
Acknowledgements
The authors sincerely acknowledge Ahmadu Bello University, Zaria for providing the softwares used and all the members of the group for their advice and encouragement in the cause of this research.
Funding
The authors received no direct funding for this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared they have no conflict of interest.
Human and animal rights statement
This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Umar, B.A., Uzairu, A., Shallangwa, G.A. et al. QSAR modeling for the prediction of pGI50 activity of compounds on LOX IMVI cell line and ligand-based design of potent compounds using in silico virtual screening. Netw Model Anal Health Inform Bioinforma 8, 22 (2019). https://doi.org/10.1007/s13721-019-0202-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-019-0202-8