
Abstract
Background
In the last decades, a higher incidence of congenital hypothyroidism (CH) has been recorded in Italy (1:1940) and worldwide, mainly due to the shift to lower screening TSH cutoffs. Although CH can also be caused by dysgenetic defects, most CH cases have recently been found to be more frequently associated with functional defects of an in situ thyroid gland. Although the clinical phenotype is milder with high prevalence of transient forms, some cases eventually prove to be permanent.
Results
Possible explanations of the raised incidence of CH are ethnic modifications of the screened population and the increasing incidence of preterm birth and multiple pregnancies. These findings are important in terms of public health and standardization of screening programmes for special at-risk categories such as preterms, acutely ill term neonates, low birth weight and very low birth weight infants, and newborns with specific drug exposure. Other environmental factors have contributed to the increased incidence of hypothyroidism, including thyroid disrupting chemicals, iodine supply (excess/deficiency), and drugs interfering with thyroid function. Finally, an increased prevalence of hypothyroidism has been documented in obese children and patients with syndromic forms (Williams, Down, Turner, pseudohypoparathyroidism). The clinical and molecular phenotype of patients with CH will be better defined thanks to novel genetic approach based on the systematic analysis of a panel of genes (TSHR, DUOX2, DUOXA, TPO, PDS, TG, NKX2.1, JAG1, GLIS3, FOXE1, PAX-8).
Conclusions
This review summarizes significant advances in the epidemiology and aetiology of non-autoimmune hypothyroidism, with a focus on thyroid dysfunction in preterm infants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Non-autoimmune hypothyroidism is defined as thyroid hormone deficiency not due to autoimmunity, and it is classified into congenital and acquired forms. Congenital hypothyroidism (CH) is an important congenital endocrine disorder and one of the most preventable causes of intellectual disability. CH can be classified according to site in primary (thyroidal) or secondary (central) hypothyroidism and according to severity in compensated [free-thyroxine (fT4) levels within the normal range for age] or decompensated (fT4 levels subnormal) hypothyroidism [1] (Table 1). Acquired hypothyroidism is a condition of impairment of thyroid function occurring after birth: it may consist in a failure of newborn screening procedures, it may be related to iodine or drug exposure, or due to infantile hemangiomas.
Central congenital hypothyroidism
Central congenital hypothyroidism (C–CH), also known as hypothalamic-pituitary CH, is a rare disease, in which thyroid hormone deficiency is caused by insufficient thyrotropin (TSH) stimulation of a normally located thyroid gland. The incidence of C–CH has been described worldwide with a variable rate: an incidence of 1:16,000 has been reported in the Netherlands [2], whereas in Japan it is about 1:30,000 and about 1:100,000 in the United States. This variability can be explained by diverse screening strategies and methods used in the different countries. Most patients with C–CH have low fT4 levels and inappropriately low or normal TSH levels [3]. C–CH leads to a presumed milder hypothyroid phenotype compared to primary CH. Only a few screening programmes measure total or free T4 and TSH (simultaneously or stepwise) enabling detection of C–CH [4]. Autosomal recessive TSH deficiency (OMIM 188540), due to mutations inactivating the TSH β-subunit, and thyrotropin-releasing hormone receptor (TRH) inactivating mutations (OMIM 188545) are known to be genetic causes of C–CH presenting in the absence of other syndromes. Mutations of transcription factors involved in pituitary development and differentiation, including POU1F, PROP1, HESX1, LHX3, and LHX4, lead to a multiple pituitary hormone deficiency [5]. Many of these cases are diagnosed when neonates present with clinical features such as cholestatic jaundice, hypoglycaemia (GH and/or ACTH/cortisol deficiency), micropenis and undescended testes (LH/FSH deficiencies), or with features of midline defects, such as decreased vision or nystagmus, as it is seen in the syndrome of septooptic dysplasia.
Primary congenital hypothyroidism
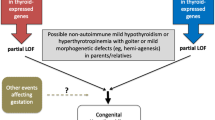
The aetiology of thyroidal CH (T-CH), the most common form of CH, includes thyroid dysgenesis (TD) (athyreosis, ectopy, hemiagenesis, hypoplasia), and inborn functional defects associated to an in situ thyroid gland (GIS) (thyroid dyshormonogenesis). TD describes a spectrum of defects of thyroid organogenesis. Isolated TD is generally a sporadic disease, but specific genetic forms of syndromic and non-syndromic TD and TSH resistance may be associated with five monogenetic forms due to mutations in TSHR, PAX8, NKX2-1, FOXE1, and NKX2-5. Thyroid dyshormonogenesis comprises defects at every step of thyroid hormone synthesis. Mutations in seven genes are well described causing iodine transport defect (NIS/SLC5A5), iodine organification defect (TPO, DUOX2, DUOXA2, SLC26A4/PDS), thyroglobulin (TG) synthesis or transport defect or iodotyrosine deiodinase (IYD/DEHAL1) deficiency [6]. Mutations in TSH receptor (TSHR) are associated with TSH resistance, a genetic defect characterized by a heterogeneous phenotype ranging from severe CH to non-autoimmune subclinical hypothyroidism (SH). This condition is usually characterized by elevated serum TSH concentrations with normal to very low levels of thyroid hormone, and a normal/reduced sized thyroid gland (absence of goitre) [7, 8]. The exact prevalence of SH due to TSHR mutations differs among populations: a prevalence between 11 and 29 % has been reported in children with non-autoimmune SH in two different Italian studies [9, 10]. The degree of insensitivity to TSH depends on the type and location of the TSHR mutations and whether the patient is homozygous or heterozygous. More severe loss-of-function TSHR mutations manifest as CH, whereas mild or heterozygous mutations present as SH in childhood or adulthood. It has been recently reported that SH in heterozygotes with TSHR mutations is a stable compensated condition that does not require replacement therapy. However, homozygous subjects with incompletely compensated SH show reduced fT4 levels over time and may require levothyroxine treatment [8].
Mutations in DUOX2 gene have been associated with an extreme phenotypic heterogeneity: monoallelic variations are usually associated with transient cases, whereas biallelic mutations with permanent ones [11]. The most significant features of patients carrying DUOX2 mutations are the presence of goitre and partial iodine organification defect, and/or the low fT4 and high TSH serum concentrations at the first postnatal serum sampling, despite borderline blood spot TSH [12].
In recent years, several observational studies reported an increasing incidence of CH confirmed at birth (permanent + transient), with an almost doubled number of cases (1:1500), compared to the usually reported incidence in the past decades (1:3000–1:4000) [13–15]. These studies were originally performed in the State of New York (1:1415) [16] and in Italy (1:1940 according to the INRICH, Lombardy 1:1446) [16, 17], but the same trend was observed worldwide [18, 19]. The increasing incidence was mostly explained by changes in screening programmes, with the introduction of progressively lower TSH cutoffs down to 10 mU/L, and special screening procedures for at-risk newborns [19, 20]. The epidemiological changes led to a detection of a higher number of mild forms of CH usually associated with a eutopic thyroid gland [17], and this led to a completely reversed aetiological classification. The incidence of CH confirmed at birth (permanent + transient) in the Italian population showed a clear-cut prevalence of functional defects with a eutopic thyroid (2/3) over dysgenetic ones (1/3) [17, 20, 21]. In a recent study, Olivieri et al. [16] investigated the incidence trend of permanent CH in Italy and demonstrated a dramatic increase of permanent CH with normal/hyperplastic thyroid over the years (increment 225 %), whereas the incidence of permanent CH with thyroid dysgenesis only showed a slight increase (increment 8 %).
A similar trend was reported in Quebec: the estimated incidence of CH was influenced by minimal changes in TSH screening cutoffs. Furthermore, the authors observed that the lowering of the TSH cutoff did not significantly increase the incidence of the most severe types of CH. Rather, the additional cases identified were predominantly milder functional disorders with a normal-sized GIS [19].
Ethnicity, preterm birth and multiple pregnancies
Other possible explanations for these findings are the changing demography with higher susceptibility of CH in Asian Indian and Hispanic children [22], and preterm birth, because the incidence and survival of premature infants is diffusely growing in the recent years [14, 23]. Premature birth is a condition with a 3 to 5-fold increased risk of CH with eutopic thyroid [17]. The study of Olivieri and collaborators reported a significant increment of 58 % of preterm babies with permanent CH in the period 1999–2008 compared with years 1987–1998. Moreover, in this period about 50 % of the increased incidence of both confirmed and permanent CH alone was attributable to preterm babies [16]. Preterm children were reported to be predisposed to transient hypothyroidism [24] as a likely consequence of multiple causes. The main factors that influence thyroid function in preterms are immaturity of hypothalamic-pituitary-thyroid axis [25], postnatal depletion of thyroid stores, neonatal comorbidities and non-thyroidal illness, and insufficient or excessive iodine intake [26]. Preterm infants typically have delayed TSH rise with normal TSH values at the first screening test, whereas later testing shows elevation of the TSH. Preterm babies represent the largest at-risk population, but the re-screened population also includes other special categories [1, 24, 27, 28]. In acutely ill term neonates admitted to Neonatal Intensive Care Unit (NICU) [29], babies with congenital anomalies and chromosomal alterations [30], same-sex twins [31], low birth weight and very low birth weight infants [17], and newborns with specific drug exposure (e.g. steroids, dopamine, iodine) [32], most newborn screening programmes now collect a second specimen.
The high number of CH infants born after multiple pregnancies has been underlined by the INRICH data [33]: the studies of Olivieri et al. [31] demonstrated an increased risk for both permanent and transient CH in multiple than in single pregnancies. These findings have important implications in term of public health given the high number of induced pregnancies in Italy as well in Western countries, because of the increasing use of assisted reproduction techniques [34, 35]. Most twins were discordant for thyroid disease: this idea supports the hypothesis of the presence of impairments in thyroidal genes caused by non-inheritable post-zygotic events that may include epigenetic determinants or intrauterine environmental modulators.
Iodine deficiency/excess
Iodine deficiency early in life impairs development and growth, and causes goitre. Iodine deficiency is an environmental factor which can explain different incidence of congenital/acquired hypothyroidism in iodine deficient or sufficient countries. Over the past decade, iodine nutritional status has been improved worldwide and the number of countries that are iodine deficient has fallen from 54 to 30, while the number iodine-sufficient countries has increased from 67 to 112 [36]. Based on the current estimates obtained from the International Council for the Control of Iodine Deficiency Disorders Global Network 2014 [37], the iodine intake of the Italian population is considered mildly insufficient (urinary iodine concentration 50–99 µg/L). A low iodine supply is possibly linked to different feeding habits and lifestyle of immigrant populations [38]. In addition, in our region Lombardy, in newborns whose TSH values whole blood are above 5 mU/L, a borderline-mild iodine deficiency is prevalent (5.4–6.3 %, according to WHO Iodine Deficiency Disorders indicators mild prevalence 3–19.9 %), despite a programme of iodine prophylaxis [17]. Therefore, more effective and safe iodine prophylaxis measures represent an urgent need for the Italian government. On the other hand, the increased iodine load may be observed in perinatal disinfection. However, iodine contamination is nowadays generally avoided during obstetric procedures and iodine has been removed from antiseptics used in most intensive care nurseries. An inadequate iodine supply might be especially dangerous in premature infants. An exposure to an iodine excess during the neonatal period, caused by iodine-containing antiseptics, is possible as preterms are able to escape the Wolff–Chaikoff effect. On the other hand, preterm infants have lower iodine stores and greater iodine requirements than term infants, and they are also at a risk for hypothyroxinemia due to iodine deficiency. The iodine intake of NICU hospitalized newborns is entirely dependent on the iodine concentration of enteral (breast milk and formulas) or parenteral nutrition. The minimum recommended dietary allowance is different depending on age groups. The iodine intake required is at least 15 µg/kg/day in full-term newborns and 30 μg/kg/day in preterms. However, the potential importance of the risk of iodine deficiency in preterm is highlighted by a study of iodine content in the nutrition provided to hospitalized preterm infants. The study of Belfort et al. demonstrated that a preterm infant would receive less than the recommended 30 µg/kg/day of iodine with nearly any standard nutritional regimen. The commercial preterm formulas tested contained only 16–26 µg/kg/day of iodine, even when concentrated to maximum caloric density. Samples of pooled donor breast milk contained surprisingly little iodine (median 9.0 µg/kg/day, range 5.0–17.6 µg/kg/day), and even fortification with human mild fortifier (HMF) failed to raise their iodine content to recommended levels. Notably, the parenteral nutrition solutions contain virtually no iodine (0.2–0.3 µg/kg/day) [39].
Thyroid disrupting chemicals (TCDs) and drugs
Another mechanism possibly contributing to the high incidence of functional defects includes thyroid disrupting chemicals (TCDs). Interestingly, TDC exposure may have influence on thyroid hormones for sensitive population like pregnant women and children [40]. In addition, it has been recently reported that dioxin contamination, particularly exposure before menarche, may have enduring impacts on women’s thyroid hormones [41]. Moreover, higher neonatal TSH values have been documented in babies born in Lombardy from mothers exposed 25 years before to dioxin during the ICMESA accident in Seveso during 1976 [42].
Another contributory factor to the high incidence of hypothyroidism includes drugs interfering with thyroid function (caffeine, antiepileptic agents, metformin, iodinated drugs). Aminophylline and caffeine are used as respiratory stimulants in preterm infants with recurrent apnoea. A single dose of intravenous aminophylline to asthmatics is followed by acute increase of TSH whereas the effect is absent after chronic administration to infants [43].
Obesity and syndromic forms
Childhood obesity has become a global epidemic, and hypothyroidism is usually associated with weight gain, decreased thermogenesis and metabolic rate. Although thyroid function is usually normal in obese subjects, it is known that TSH and body mass index (BMI) are positively correlated. In fact, many studies in children and adolescents have demonstrated that TSH and fT3 levels are slightly increased in obese subjects as compared to normal weight humans [44]: the elevation of thyroid hormone concentrations in obesity increases energy expenditure to avoid accumulation of energy as fat. These alterations in thyroid hormones are rather a consequence than a cause of obesity and these seem to be an adaptation process to reduce the availability of energy for conversion into fat. Furthermore, fasting and weight loss are associated with a decrease of thyroid hormone levels [45].
During the last decade, there has been a growing body of evidence indicating an increased prevalence of hypothyroidism in patients with syndromic forms and chromosomal alterations. Children affected by Williams syndrome (WS) have both structural (hypoplasia) [46, 47] and functional thyroid defects with transient TSH elevation and normal thyroid hormones (SH) [48]. Even in paediatric patients with Down syndrome, SH is a frequent finding either with normal or hypoplastic thyroid gland. In these children, the disorder is benign and usually remitting in the majority of cases (70 %) [49]. Additionally, an increased incidence of hypothyroidism has been largely documented in Turner syndrome [50]: these patients, especially those carrying X isochromosome, are more prone to develop thyroid autoimmunity [51]. Finally, children affected by pseudohypoparathyroidism show resistance towards parathyroid hormone (PTH) in the proximal renal tubules. In this rare disorder that is caused by GNAS methylation changes, resistance can occur towards other hormones, such as TSH, thus causing subclinical or overt hypothyroidism.
Consumptive hypothyroidism
Another form of non-autoimmune hypothyroidism is associated with hemangiomas: this rare form is described as consumptive hypothyroidism. This is a unique form of hypothyroidism resulting from increased production of type 3-iodothyronine deiodinase (D3), an enzyme responsible for the inactivation of thyroid hormones. As a consequence, the degradation of thyroid hormone exceeds the gland’s ability to synthesize it. Affected children usually present refractory hypothyroidism and require very high doses of l-thyroxine (l-T4) treatment to restore euthyroidism [52, 53].
Conclusions
A higher incidence of non-autoimmune hypothyroidism has been documented in children: this is mainly due to the detection of hypothyroid newborns with functional defects associated to GIS. However, many factors (preterm and twin birth, ethnic modifications, screening programmes, iodine supply, TCDs, drugs, obesity) have contributed to such increased incidence of hypothyroidism in most recent years.
In conclusion, it is likely that CH with GIS could not be a monogenic disease, and that epigenetic mechanisms and environmental factors could be involved in disease expression. The novel genetic approach based on the systematic analysis of a panel of genes will help in the molecular characterization of the phenotype of the “newly detected population” of hypothyroid children with GIS. This could lead to the definition of more appropriate and personalized therapeutic approach and follow-up protocols.
References
Léger J, Olivieri A, Donaldson M et al (2014) European society for paediatric endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. J Clin Endocrinol Metab 99(2):363–384
Lanting CI, van Tijn DA, Loeber JG et al (2005) Clinical effectiveness and cost-effectiveness of the use of the thyroxine/thyroxine-binding globulin ratio to detect congenital hypothyroidism of thyroidal and central origin in a neonatal screening program. Pediatrics 116(1):168–173
Tajima T, Nakamura A, Morikawa S et al (2014) Neonatal screening and a new cause of congenital central hypothyroidism. Ann Pediatr Endocrinol Metab 19:117–121
Zwaveling-Soonawala N, van Trotsenburg P, Verkerk H et al (2015) The severity of congenital hypothyroidism of central origin should not be underestimated. J Clin Endocrinol Metab 100(2):E297–E300
Tajima T, Ishizu K, Nakamura A (2013) Molecular and clinical findings in patients with LHX4 and OTX2 mutations. Clin Pediatr Endocrinol 22(2):15–23
Szinnai G (2014) Clinical genetics of congenital hypothyroidism. Endocr Dev 26:60–78
Persani L, Calebiro D, Cordella D et al (2010) Genetics and phenomics of hypothyroidism due to TSH resistance. Mol Cell Endocrinol 322(1–2):72–82
Tenenbaum-Rakover Y, Almashanu S, Hess O et al (2015) Long-term outcome of loss-of-function mutations in TSH receptor gene. Thyroid 25:292–299
Calebiro D, Gelmini G, Cordella D et al (2012) Frequent TSH receptor genetic alterations with variable signaling impairment in a large series of children with nonautoimmune isolated hyperthyrotropinemia. J Clin Endocrinol Metab 97:E156–E160
Nicoletti A, Bal M, De Marco G et al (2009) Thyrotropin-stimulating hormone gene analysis in pediatric patients with non-autoimmune subclinical hypothyroidism. J Clin Endocrinol Metab 94:4187–4194
Vigone MC, Fugazzola L, Zamproni I et al (2005) Persistent mild hypothyroidism associated with novel sequence variants of the DUOX2 gene in two siblings. Hum Mutat 26(4):395
Muzza M, Rabbiosi S, Vigone MC et al (2014) The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J Clin Endocrinol Metab 99(3):E544–E553
Dussault JH (1999) The anecdotal history of screening for congenital hypothyroidism. J Clin Endocrinol Metab 84(12):4332–4334
Rastogi MV, LaFranchi SH (2010) Congenital hypothyroidism. Orphanet J Rare Dis 5:17
Wassner AJ, Brown RS (2013) Hypothyroidism in the newborn period. Curr Opin Endocrinol Diabetes Obes 20(5):449–454
Olivieri A, Fazzini C, Medda E (2015) Multiple factors influencing the incidence of congenital hypothyroidism detected by neonatal screening. Horm Res Paediatr 83:86–93
Corbetta C, Weber G, Cortinovis F et al (2009) A 7-year experience with low blood TSH cutoff levels for neonatal screening reveals an unsuspected frequency of congenital hypothyroidism (CH). Clin Endocrinol (Oxf) 71(5):739–745
Mengreli C, Kanaka-Gantenbein C, Girginoudis P et al (2010) Screening for congenital hypothyroidism: the significance of threshold limit in false-negative results. J Clin Endocrinol Metab 95(9):4283–4290
Deladoëy J, Ruel J, Giguère Y, Van Vliet G (2011) Is the incidence of congenital hypothyroidism really increasing? A 20-year retrospective population-based study in Québec. J Clin Endocrinol Metab 96(8):2422–2429
Olivieri A, Corbetta C, Weber G et al (2013) Congenital hypothyroidism due to defects of thyroid development and mild increase of TSH at screening: data from the Italian National Registry of infants with congenital hypothyroidism. J Clin Endocrinol Metab 98(4):1403–1408
Rabbiosi S, Vigone MC, Cortinovis F et al (2013) Congenital hypothyroidism with eutopic thyroid gland: analysis of clinical and biochemical features at diagnosis and after re-evaluation. J Clin Endocrinol Metab 98(4):1395–1402
Hinton CF, Harris KB, Borgfeld L et al (2010) Trends in incidence rates of congenital hypothyroidism related to select demographic factors: data from the United States, California, Massachusetts, New York, and Texas. Pediatrics 125(Suppl 2):S37–S47
Beck S, Wojdyla D, Say L et al (2010) The worldwide incidence of preterm: a systematic review of maternal birth mortality and morbidity. Bull World Health Organ 88:31–38
Vigone MC, Caiulo S, Di Frenna M et al (2014) Evolution of thyroid function in preterm infants detected by screening for congenital hypothyroidism. J Pediatr 164(6):1296–1302
Radetti G, Fanolla A, Pappalardo L et al (2007) Prematurity may be a risk factor for thyroid dysfunction in childhood. J Clin Endocrinol Metab 92(1):155–159
Ares S, Quero J, de Escobar GM (2008) Iodine balance, iatrogenic excess, and thyroid dysfunction in premature newborns. Semin Perinatol 32(6):407–412
Cassio A, Corbetta C, Antonozzi I et al (2013) The Italian screening program for primary congenital hypothyroidism: actions to improve screening, diagnosis, follow-up, and surveillance. J Endocrinol Invest 36(3):195–203
LaFranchi SH (2010) Newborn screening strategies for congenital hypothyroidism: an update. J Inherit Metab Dis 33(Suppl 2):S225–S233
Goldsmit GS, Valdes M, Herzovich V et al (2011) Evaluation and clinical application of changes in thyroid hormone and TSH levels in critically ill full-term newborns. J Perinat Med 39(1):59–64
Olivieri A, Stazi MA, Mastroiacovo P et al (2002) A population-based study on the frequency of additional congenital malformations in infants with congenital hypothyroidism: data from the Italian Registry for Congenital Hypothyroidism (1991–1998). J Clin Endocrinol Metab 87(2):557–562
Olivieri A, Medda E, De Angelis S et al (2007) High risk of congenital hypothyroidism in multiple pregnancies. J Clin Endocrinol Metab 92(8):3141–3147
Filippi L, Pezzati M, Poggi C et al (2007) Dopamine versus dobutamine in very low birthweight infants: endocrine effects. Arch Dis Child Fetal Neonatal Ed 92(5):F367–F371
Olivieri A (2012) Epidemiology of congenital hypothyroidism: what can be deduced from the Italian registry of infants with congenital hypothyroidism. J Matern Fetal Neonatal Med 25(Suppl 5):7–9
Reynolds MA, Schieve LA, Martin JA et al (2003) Trends in multiple births conceived using assisted reproductive technology, United States, 1997–2000. Pediatrics 111(5 Pt 2):1159–1162
Sakka SD, Malamitsi-Puchner A, Loutradis D (2009) Euthyroid hyperthyrotropinemia in children born after in vitro fertilization. J Clin Endocrinol Metab 94(4):1338–1341
Zimmermann MB (2013) Iodine deficiency and excess in children: worldwide status in 2013. Endocr Pract 19(5):839–846
Global Iodine Nutrition Scorecard (2014) Available at: http//www.ign.org/cm_data/Scorecard_IGN_website_02_03_2015.pdf. Accessed 23 March 2015
Harris KB, Pass KA (2007) Increase in congenital hypothyroidism in New York State and in the United States. Mol Genet Metab 91(3):268–277
Belfort MB, Pearce EN, Braverman LE et al (2012) Low iodine content in the diets of hospitalized preterm infants. J Clin Endocrinol Metab 97(4):E632–E636
Andra SS, Makris K (2012) Thyroid disrupting chemicals in plastic additives and thyroid health. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 30(2):107–151
Chevrier J, Warner M, Gunier RB et al (2014) Serum dioxin concentrations and thyroid hormone levels in the Seveso Women’s Health Study. Am J Epidemiol 180(5):490–498
Baccarelli A, Giacomini SM, Corbetta C et al (2008) Neonatal thyroid function in Seveso 25 years after maternal exposure to dioxin. PLoS Med 5(7):e161
Williams FL, Ogston SA, van Toor H et al (2005) Serum thyroid hormones in preterm infants: associations with postnatal illnesses and drug usage. J Clin Endocrinol Metab 90(11):5954–5963
Knudsen N, Laurberg P, Rasmussen LB et al (2005) Small differences in thyroid function may be important for body mass index and the occurrence of obesity in the population. J Clin Endocrinol Metab 90(7):4019–4024
Reinehr T (2010) Obesity and thyroid function. Mol Cell Endocrinol 316(2):165–171
Selicorni A, Fratoni A, Pavesi MA et al (2006) Thyroid anomalies in Williams syndrome: investigation of 95 patients. Am J Med Genet A 140(10):1098–1101
Stagi S, Bindi G, Neri AS et al (2005) Thyroid function and morphology in patients affected by Williams syndrome. Clin Endocrinol (Oxf) 63(4):456–460
Cherniske EM, Carpenter TO, Klaiman C et al (2004) Multisystem study of 20 older adults with Williams syndrome. Am J Med Genet A 131(3):255–264
Claret C, Goday A, Benaiges D et al (2013) Subclinical hypothyroidism in the first years of life in patients with Down syndrome. Pediatr Res 73(5):674–678
Jørgensen KT, Rostgaard K, Bache I et al (2010) Autoimmune diseases in women with Turner’s syndrome. Arthritis Rheum 62(3):658–666
Grossi A, Crinò A, Luciano R et al (2013) Endocrine autoimmunity in Turner syndrome. Ital J Pediatr 39:79
Huang SA, Tu HM, Harney JW et al (2000) Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med 343(3):185–189
Vigone MC, Cortinovis F, Rabbiosi S et al (2012) Difficult treatment of consumptive hypothyroidism in a child with massive parotid hemangioma. J Pediatr Endocr Met 25(1–2):153–155
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, informed consent is not required.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vigone, M.C., Di Frenna, M. & Weber, G. Heterogeneous phenotype in children affected by non-autoimmune hypothyroidism: an update. J Endocrinol Invest 38, 835–840 (2015). https://doi.org/10.1007/s40618-015-0288-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-015-0288-5