
Abstract
Purpose
Congenital primary hypothyroidism (CH) is a state of inadequate thyroid hormone production detected at birth, caused either by absent, underdeveloped or ectopic thyroid gland (dysgenesis), or by defected thyroid hormone biosynthesis (dyshormonogenesis). A genetic component has been identified in many cases of CH. This review summarizes the clinical and biochemical features of the genetic causes of primary CH.
Methods
A literature review was conducted of gene defects causing congenital hypothyroidism.
Results
Mutations in five genes have predominantly been implicated in thyroid dysgenesis (TSHR, FOXE1, NKX2-1, PAX8, and NKX2-5), the primary cause of CH (85%), and mutations in seven genes in thyroid dyshormonogenesis (SLC5A5, TPO, DUOX2, DUOXA2, SLC6A4, Tg, and DEHAL1). These genes encode for proteins that regulate genes expressed during the differentiation of the thyroid, such as TPO and Tg genes, or genes that regulate iodide organification, thyroglobulin synthesis, iodide transport, and iodotyrosine deiodination. Besides thyroid dysgenesis and dyshormonogenesis, additional causes of congenital hypothyroidism, such as iodothyronine transporter defects and resistance to thyroid hormones, have also been associated with genetic mutations.
Conclusion
The identification of the underlying genetic defects of CH is important for genetic counseling of families with an affected member, for identifying additional clinical characteristics or the risk for thyroid neoplasia and for diagnostic and management purposes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Congenital hypothyroidism (CH), defined as thyroid hormone deficiency present at birth, is the most common neonatal metabolic disorder and one of the most preventable causes of mental retardation and neurological alterations in children [1]. It occurs in one of every 2 to 4 thousand newborns owing to complete or partial failure of thyroid gland development or function. Τhe incidence may vary by geographic location [2]. Females are more frequently affected than males (female/male ratio 2–4:1) [3]. The main causes of CH are shown in Table 1.
The clinical presentation is often subtle or not present at birth, possibly due to placental passage of maternal thyroid hormones or thyroid production of the embryo. Typical manifestations include myxedematous face, macroglossia, umbilical hernia, hypotonia, large fontanels, a distended abdomen, decreased activity and increased sleep, constipation, feeding difficulty, and prolonged jaundice. Timely diagnosis of CH is of the utmost importance because of the essential role of thyroid hormones in central nervous system maturation [4]. Intellectual disability can be prevented with early screening: this was introduced in 1974 by using a blood sample from the newborn via a heel prick collected on filter paper and has been established worldwide ever since. Ideally, screening should be done at 3–4 days of age. Levothyroxine is the treatment of choice and should ideally be started in the first trimester of life [5]. Thyroid hormone deficiency may become apparent clinically and biochemically at a later stage; hence, despite normal initial screening, CH should still be considered in the case of a suggestive clinical phenotype.
CH is classified as permanent and transient and can be caused by primary, secondary, or peripheral etiologies. Permanent CH refers to the state of persistent thyroid hormone deficiency and requires lifelong treatment. Transient forms usually occur in areas of endemic iodine deficiency and are characterized by recovery of thyroid hormone deficiency within a few months or years. Thyroid dysgenesis and dyshormonogenesis cause primary hypothyroidism. Secondary or central hypothyroidism at birth results from a deficiency of thyroid-stimulating hormone (TSH). Congenital TSH deficiency may be isolated or associated with congenital hypopituitarism, which is characterized by additional pituitary hormone deficiencies [2]. Peripheral etiologies include defects in thyroid hormone transport, metabolism, or action [2].
It is well known that a genetic component is frequently involved in CH. The known genes associated with primary congenital hypothyroidism are primarily divided into those causing thyroid dysgenesis and those causing dyshormonogenesis.
The exact cause of CH remains obscure in many instances; however, developmental abnormalities of the thyroid gland, i.e., thyroid dysgenesis, accounts for the majority of the known cases, followed by abnormal thyroid hormone synthesis. Of note, normal thyroid hormone synthesis requires a normally developed thyroid gland and adequate nutritional iodide intake.
In order to summarize the known genetic causes and highlight the importance of genetic abnormalities in the development of CH, additional clinical features and, in rare cases, neoplasia, this review focuses on the genetic defects of the genes involved in primary congenital hypothyroidism caused by thyroid dysgenesis, dyshormonogenesis, TSH resistance, defects in thyroid hormones transport, and defects in the bioactivation of the thyroid hormones.
Thyroid dysgenesis and congenital hypothyroidism
Thyroid dysgenesis represents the leading cause of CH, accounting for 85% of CH cases, and the most common cause of permanent CH. It involves defective thyroid gland development with different forms include thyroid aplasia or agenesis (35–40% of all cases), hypoplasia (5%), and ectopy (30–45%, more frequently located in a sublingual position), which is the most common form (48–61%) [6]. It is usually sporadic but 2% of cases are found to be familial [7].
Current evidence suggests that the development of the embryonic thyroid gland and its normal migration from the base of the tongue to the anterior neck is a multistage process of highly regulated biochemical steps that requires the participation of certain transcription factors, such as TTF-2 or FOXE1, TTF-1 or NKX2-1, PAX-8, NKX2-5, DUOX2, GL153, and transcription regulators, such as GLIS3. These proteins are known to regulate genes expressed during the differentiation of the thyroid, such as thyroid peroxidase (TPO) and Tg genes. Mutations in these factors can result in thyroid dysgenesis [8].
Genes associated with thyroid gland dysgenesis cause either nonsyndromic CH (TSH receptor) or syndromic CH (TITF-1, NKX2, PAX8, THOX2, CDCA8, TUBB1, JAG1, NTN1, and GL153) [9, 10].
Extrathyroid genes that regulate the migration of the median thyroid bud during embryogenesis have also been reported to cause faulty embryogenesis when mutated. These include adhesion molecules and vascular factors which promote the stabilization of the bilobed thyroid structure [9].
Thyroid dysgenesis and nonsyndromic CH
TSHR gene
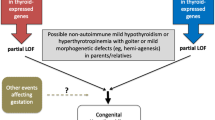
The TSH receptor belongs to a group of G protein-coupled receptors with seven transmembrane regions and mediates the effects of TSH, secreted by the anterior pituitary gland. It is divided into two subunits (α and β). Thyroid-stimulating hormone receptor (TSHR) activation results in intracellular signaling via Gsα protein, which leads to activation of cyclic adenosine monophosphate (AMP) cascade, and via Gq protein, that leads to activation of phospholipase C (PLC) cascade. cAMP binds to protein kinase A (PKA), which phosphorylates different components of intracellular signaling and activated PLC leads to the generation of inositol 1,4,5-triphosphate and diacylglycerol, which stimulate Ca2+ release into the cytoplasm and activate the protein kinase C (PKC) pathway. Increased concentrations of intracellular Ca2+ and PLC activity are important for H2O2 production, iodination of thyroglobulin, and iodide efflux, whereas adenylate cyclase and cAMP regulate the transcription of sodium-iodide symporter (NIS), Tg and TPO, as well as iodide uptake [11]. Homozygous or heterozygous mutations in the TSHR gene result in variable TSH resistance expressed as euthyroid hyperthyrotropinemia with a normal or a hypoplastic thyroid gland (fully compensated TSH resistance) [12,13,14,15,16] mild or borderline hypothyroidism with high TSH concentrations and a normal thyroid gland (partially compensated TSH resistance), or severe hypothyroidism with a hypoplastic thyroid gland or athyreosis (severe uncompensated TSH resistance) [25,26,27,28,29,30]. Heterozygous loss of function mutations in the TSHR gene have also been associated with mildly elevated TSH levels [12, 14, 16].
Thyroid dysgenesis and syndromic CH
Thyroid transcription factor-2
Thyroid transcription factor-2 (TTF-2), also known as forkhead box E1 (FOXE1), is a transcription factor involved in thyroid gland development. Although the role of TTF-2 in thyroid gland development has been well established, the underlying mechanisms remain obscure. It is a member of the forkhead/winged helix domain transcription factor family [17] and has been identified as a nuclear protein that recognizes and binds to a DNA sequence present in the promoters of Tg and TPO, regulating the expression of thyroid-specific genes [18, 19]. TTF-2 is encoded by the TITF2 gene, one of the thyroid dysgenesis-related genes, which is located on chromosome 9q22 and consists of a single exon [20]. Three homozygous loss-of-function missense mutations within the forkhead DNA-binding domain have been reported to result in clinical symptoms of CH including thyroid agenesis or an ectopic sublingual gland with added features of bilateral choanal atresia, spiky hair, hypoplastic bifid epiglottis and cleft palate (Bamforth syndrome) [21].
TTF-1
Thyroid transcription factor-1 (TTF-1), also known as thyroid-specific enhancer binding protein (T/EBP), is a homeodomain containing transcription factor which plays a critical role in the development of the thyroid gland by regulating the transcription of Tg, TPO, and TSH receptor genes in thyroid follicular cells [18, 22]. It is encoded by the NKX2 gene family, and more specifically by TITF1 gene, which is located on chromosome 14q13 and comprises 3 exons [23]. The TITF1 gene is expressed at the very beginning of thyroid differentiation and mutations can cause thyroid agenesis or ectopy due to pathological migration of the thyroid precursor cells. Reports show that TΙTF-1 is also expressed in the lungs and in the ventral forebrain [24]. In the lung, TTF-1 is present at the earliest stages of differentiation and is later confined to the branchial epithelium. It also regulates the transcription of surfactant protein B (SPB) in epithelial lung cells. Heterozygous TITF-1 gene mutations and an autosomal dominant TITF-1 gene mutation have been associated with compensated congenital hypothyroidism and unexplained respiratory distress due to lung hypoplasia in neonates with no pathological bronchial morphology [25]. In addition, de novo or dominant TITF-1 mutations have been associated with neurological symptoms including ataxia, benign chorea or choreathetosis, hypotonia, dysarthria, developmental delay, feeding difficulties, and microcephaly, suggesting a role of TITF-1 in brain development also [26, 27]. CH combined with unexplained respiratory distress is a strong indicator of the NKX2 mutation. The NKX2.5 gene has been found to be involved in cardiac development; hence, mutations lead to congenital heart defects in addition to CH.
PAX8
PAX8, a transcription factor, is one of the nine members of the mammalian paired homeodomain family. The PAX8 gene is mapped to chromosome 2q12-q14 and consists of 11 exons. It plays a fundamental role not only in the initiation of thyroid cell differentiation, but also in cell proliferation [28]. It activates the transcription of TPO, Tg, and NIS and acts in combination with TTF-1 to activate the promoter of the Tg gene. TPO is dependent on PAX8, and therefore, defective PAX8 function leads to reduced TPO expression and partial organification defect [28]. Heterozygous human PAX8 mutations have been described in patients with hypoplastic thyroid gland and others with ectopic thyroid gland. PAX8 is also expressed in the kidney, where it activates the Wilm’s tumor (WT1) gene promoter, as well as in the developing brain [29]. Heterozygous mutations have been associated with renal hemiagenesis, ipsilateral to thyroid hemiagenesis when present, and hypercalciuria. CH due to PAX8 mutations may be either syndromic or nonsyndromic.
NKX2-5
NKX2-5 is a homeodomain-containing transcription factor that has been shown to be involved in heart and thyroid development. However, data on the pathogenic role of NKX2-5 gene mutations on thyroid dysgenesis are inconclusive [30].
DUOX2
The gene DUOX2 or THOX2 encodes the human protein dual oxidase 2 (DUOX2), a member of the NADPH oxidase family. Hydrogen peroxide (H2O2) is essential for thyroperoxidase-mediated thyroid hormone synthesis in the follicular lumen of the thyroid gland. DUOX2 and its maturation factor and essential partner, DUOXA2, play a crucial role in H2O2 generation, necessary for the biological activation of TPO [31]. However, DUOX2 defects have been associated with cases of both thyroid dysgenesis and dyshormonogenesis [32].
GL153
GL153 is a transcription factor encoded by its gene located on chromosome 9q24. Its role in thyroid function is yet to be described. Mutation of the transcription factor is described with a rare syndrome of neonatal diabetes and congenital glaucoma along with CH [33].
GLIS3
The human GLIS3 gene, located on chromosome 9p24.2, encodes the transcription factor Gli-similar 3 (GLIS3), which belongs to the family of the Kruppel-like zinc finger transcription factor [34]. Heterozygous GLIS3 missense variants have been identified in Caucasian patients with primary congenital hypothyroidism, with half of the affected cases presenting with thyroid dysgenesis (athyreosis, thyroid hypoplasia, and thyroid ectopy) [35]. Neonatal diabetes, hepatitis, and congenital hypothyroidism in the form of thyroid aplasia with diminished colloid and temporary TSH resistance upon treatment with thyroxine have also been described [36].
TUBB1
Another gene associated with thyroid dysgenesis is TUBB1 (tubulin beta 1 class VI), a member of the β-tubulin protein family. β-Tubulins heterodimerize to form α/β-tubulin dimers, which are incorporated into one of the major cytoskeletal structures, microtubules [37]. An unsuspected role of tubulin isotype 1, TUBB1, in thyroid development has been reported and carriers of TUBB1 mutation present with thyroid dysgenesis in the form of ectopia, hypoplasia, hemithyroid or asymmetric thyroid gland, and abnormal platelet morphology [38]. Not all carriers of the mutation show thyroid phenotype, suggesting that a second hit, such as a somatic mutation in the thyroid gland or an epigenetic defect, may be necessary for the expression of hypothyroidism.
CDCA8 gene
The CDCA8 gene encodes borealin, a member of the chromosomal passenger complex, which plays a key role in cytokinesis and chromosome segregation [39]. It is expressed in human thyroid tissue during embryonic development and has been associated with congenital hypothyroidism due to defective migration and adhesion of human thyrocytes [40].
JAG1
Jagged canonical notch ligand 1, JAG1, is encoded by the JAG1 gene and loss-of-function mutations cause thyroid hypoplasia, liver, heart, skeleton, eye, and facial defects (Alagille syndrome) [41].
NTN1
Mutations in the NTN1 gene, encoding netrin 1, have been associated with congenital ventricular septum defect and thyroid ectopy in a single patient [42].
Several genes, including HHEX, HES1, HOXA3, and EYA1, have been associated with thyroid dysgenesis in mice.
HHEX
HHEX, hematopoietically expressed homeobox, refers to a transcription factor that is expressed in the thyroid gland in mice and contributes to thyroid gland differentiation [43, 44]. It has been reported that embryos with a mutation in the HHEX gene exhibit arrest of thyroid development at the budding stage (E9.5), whereas later in the developmental stages, the absence of HHEX expression is accompanied by a regression in the morphology of the developing gland [45, 46].
HES1
Helix-loop-helix protein HES1 (Hes family BHLH transcription factor 1), encoded by the HES1 gene, transactivates the NIS promoter and controls thyrocyte and C-cell precursors [47]. Mutations in the HES1 gene in mice have been associated with thyroid aplasia [48].
HOXA3
The HOXA3 gene encodes homeobox protein HOX-A3. Mutations in the HOXA3 gene in mice results in thymus and thyroid hypoplasia due to a reduced number of follicular and parafollicular cells [49].
EYA1
Thyroid hypoplasia, lack of isthmus, and reduction in calcitonin-producing cells have been reported in mice with inactivation of the eyes absent gene, EYA1, which is involved in the regulation of mature thyroid gland formation [50].
Dyshormonogenesis and congenital hypothyroidism
Dyshormonogenesis, also referred to as inborn errors of thyroid hormone biosynthesis, is a disorder in the enzymatic cascade of thyroid hormone synthesis and accounts for 10–15% of CH cases. Thyroid hormone biosynthesis, storage, and secretion compose a multistep process that occurs in the thyroid follicle (Fig. 1). After iodide uptake occurs in the basolateral membrane of NIS, iodide is transported to the apical membrane and then into the follicular lumen, partly by the protein pendrin (Fig. 1). Within the follicular lumen, iodine is oxidized by TPO in the presence of H2O2, the generation of which is catalyzed by dual oxidase DUOX2 and its maturation factor, DUOXA2. Subsequently, iodine is incorporated into tyrosyl residues of thyroglobulin (organification) to form monoiodotyrosine (MIT) and diiodotyrosine (DIT). Iodotyrosines are coupled by TPO to form the thyroid hormones, triidothyronine (T3), and thyroxine (T4), which remain bound to this protein until they are secreted into the bloodstream [51] (Fig. 1). TSH regulates all the stages of thyroid hormone synthesis. Defects in each of the aforementioned steps of thyroid hormone synthesis have been detected, leading to dyshormonogenesis and variable severity of hypothyroidism and goiter development, due to thyroid gland stimulation by TSH [52].

Schematic diagram of a follicular cell, illustrating the molecules involved in thyroid hormone synthesis
Specifically, gene mutations involved in dyshormonogenesis include enzymes that are required for iodide organification (TPO, DUOX2, DUOXA2, and SCL26A4), thyroglobulin synthesis (TG), iodide transport (SCL5A5), and iodotyrosine deiodination (DEHAL1) [53].
TPO
TPO is a heme-binding protein localized in the apical membrane of the thyroid follicular cell and consisting of 933 amino acids. TPO enzymatic activity is responsible for iodide oxidation, organification, and iodotyrosine coupling (52). The human TPO gene is mapped to chromosome 2p25 and includes 17 exons. Over 60 mutations in the TPO gene have been reported, accounting for the majority of cases of dyshormonogenesis [54]. The most common phenotype of this autosomal recessive disorder is a total iodide organification defect with severe symptoms of CH as a consequence.
NIS
NIS is responsible for the accumulation of iodide in the thyroid gland through active membrane transport [55]. It is a membrane protein of approximately 65 kDA that consists of 12 transmembrane domains, including both a carboxyl and amino termini location inside the cell [55]. NIS expression has been detected not only in normal and neoplastic thyroid tissue, but also in salivary glands, gastric mucosa, breast, colon, ovaries, placenta, skin, and the choroid plexus. The human gene SLC5A5 is located on chromosome 19p and contains 15 exons, encoding a protein of 643 amino acids. Several studies have shown that TSH up-regulates NIS gene expression, NIS protein abundance, and bioactivity. The first report of a mutation in the NIS gene was made in 1997, followed by the detection of several mutations, mostly inherited in an autosomal recessive manner [56]. The symptoms of CH may vary from fully compensated hypothyroidism to severe hypothyroidism. Goiter is not always present, and an absence of radioactive iodine intake is also described. People with heterozygous mutations are euthyroid. Patients with higher dietary iodine intake seem to present less severe symptoms of hypothyroidism; therefore, it seems that iodine supplementation is preferable to thyroxine treatment [57].
Pendred syndrome gene
Pendred syndrome (PDS) is an autosomal recessive disease caused by a homozygous or heterozygous mutation of the SLC26A4 (solute carrier family 26, member 4) or PDS gene. It is characterized by goiter and sensorineural deafness due to a Mondini cochlear defect, present at birth [58]. It represents the most common cause of syndromic deafness, accounting for 10% of cases [59]. The goiter becomes manifest in the second decade of life and may be multinodular or diffuse. Despite the presence of goiter, congenital hypothyroidism is rarely present and the subjects are usually euthyroid. However, TSH levels are often at the upper end of the normal range and, with the progression of time, hypothyroidism of variable severity may develop [59]. The relative gene is mapped to chromosome 7q: it contains 21 exons and is expressed in the thyroid gland as well as the cochlea. It encodes pendrin, a 760 amino acid chloride-iodide transporter with 11 transmembrane domains, localized in the apical membrane of the thyrocyte [60], but also in the inner ear, endometrium, and kidney [61]. Mutation in the gene leads to mild organification defect, due to disruption of iodide transportation across the apical membrane of the thyrocyte into the colloid space [62]. Both TPO defects and Pendred syndrome may present with hypothyroidism, goiter, partial iodide organification defects, and a positive perchlorate test [63]. A definite diagnosis is only possible with molecular testing.
SLC26A7
SLC26A7 has recently been described as being a novel iodide transporter in the human thyroid, with similar function to that of SLC26A4. It is predominantly expressed on the luminal side of thyroid follicular cells and homozygous nonsense mutations in the SLC26A7 gene result in defective thyroid hormonogenesis and congenital goitrous hypothyroidism [64].
NADPH oxidases (DUOX1, DUOX2)
The oxidation of the different substrates is dependent on the presence of hydrogen peroxide and the peroxide enzyme that catalyzes the process (TPO). Hydrogen peroxide is generated on the apical surface of the cell through the catalytic action of NADPH:O(2) oxidoreductase flavoproteins or NADPH oxidases, DUOX1 and DUOX2 [65]. NADPH oxidases are encoded by two genes, DUOX1 and DUOX2, mapped to chromosome 15, and are located at the apical membrane of thyrocytes where they play a fundamental role in H202 generation [66]. H2O2 is the substrate of TPO and essential for the biological activation of TPO so that iodide organification (iodination of tyrosine residues and incorporation of iodide into TG) and hormonogenesis are completed. Inactivating mutations of the DUOX2 gene have been associated with permanent or transient CH. Heterozygous mutations lead to mild transient CH and partial iodide organification defect. On the other hand, homozygosity leads to severe symptoms of CH and complete organification defect [67]. DUOXA2, a resident endoplasmic reticulum (ER) protein, is required for the maturation and plasma membrane localization of DUOX2 [68].
Tg
Thyroglonulin (Tg) is a homodimer with subunits of 330,000 Da, synthesized exclusively in the thyroid gland. The Tg gene is located on chromosome 8q24 and includes 42 exons [69]. The primary functions of Tg include iodide storage and thyroid hormonogenesis. Hormone synthesis from TG occurs via the iodination and coupling of pairs of tyrosine residues and is completed by Tg proteolysis. In the case of a mutated Tg gene, formation of T4 and T3 is ineffective due to a coupling defect. Gene mutations lead to mild to severe symptoms of CH with low concentrations of Tg and goiter [70].
DEHAL1
Iodotyrosine dehalogenase (DEHAL1) is a nitroreductase-related enzyme responsible for deiodination of iodotyrosines (monoiodotyrosine and diiodotyrosine) [71]. Homozygous mutations of the DEHAL1 gene have been reported to cause hypothyroidism from early infancy in the majority of cases, as well as intellectual deficits [72].
TSHR
The TSHR gene represents yet another gene that, when mutated, can cause nongoitrous CH [12]. As mentioned previously, TSHR is a G protein-coupled transmembrane receptor through which TSH mediates its effects towards thyroid hormone synthesis. Loss of function mutations in TSHR cause resistance to TSH, with a clinical presentation ranging from congenital nonautoimmune hyperthyrotropinaemia or compensated hypothyroidism (elevated TSH and normal thyroid hormone concentrations) to overt hypothyroidism with thyroid hypoplasia. Thus far, more than 60 biallelic inactivating TSHR mutations have been described [73]. In the case of complete resistance to TSH levothyroxine treatment is considered essential, but it remains controversial whether treatment is required in partial resistance to TSH [74].
GNAS1
Resistance to TSHR is also caused by mutations in the GNAS1 gene, which encodes for the α-stimulatory subunit of G protein (Gsα). Heterozygous mutations also cause hypocalcemia and hyperphosphatemia due to impaired parathyroid hormone (PTH) signaling. This syndrome is known as Albright hereditary osteodystrophy and is characterized by short stature, short metacarpals, round face, short neck, obesity, subcutaneous ossification, and mental retardation [75].
Concurrent loss-of-function TSHR mutations and DIO2 T92A polymorphism have also been described. TSH stimulates DIO2 activity; therefore this “double hit” results in a significant reduction in DIO2 activity, which converts inactive T4 to active T3. The identification of this form of abnormal thyroid hormone metabolism is important because combined L-T3 and L-T4 treatment is the optimal treatment [76]. Mutations in the TPO or Tg genes are the most frequent genetic defects in thyroid dyshormonogenesis.
Iodothyronine transporter defects and congenital hypothyroidism
The thyroid hormones, thyroxine (T4) and triiodothyronine (T3) are crucial for growth and brain development in infants and for metabolic activity in adults. In fact, they affect the function of virtually every organ system [77]. T4 is the principal hormone synthesized and secreted by the thyroid gland under the stimulation of TSH, which is secreted by the anterior pituitary gland. T4 is a prohormone and it is converted to the active form of thyroid hormone, T3, by 5′-monodeiodination in all tissues. Following its transmembrane passage into the target cells, T3 acts on nuclear receptors causing DNA structural changes and resulting in the transcription of different genes.
Several plasma membrane thyroid hormone transporters have been described, including the monocarboxylate transporter 8 (MCT8) and organic-anion transporting polypeptide 1C1 (OATP1C1) [78].
MCT8
MCT8, also referred to as SLC16A2 (solute carrier family 16, member 2), is a membrane thyroid hormone transporter, highly expressed in the brain, thyroid, pituitary, and placenta. The MCT8 gene is localized on the Xq13.2 chromosome and contains five exons [79]. Mutations in the MCT8 gene have been associated with Allan-Herndon-Dudley syndrome (AHDS), an X-linked condition of mental retardation. The syndrome is characterized by global developmental delay, central hypotonia, spastic quadriplegia, dystonia, poor head control, rotary nystagmus, absent speech, and impaired hearing [80]. Reported biochemical findings include elevated free T3 levels, low free T4 levels and TSH levels slightly elevated or varying from normal to high [80]. AHDS was first described in 1944 and only in 2005 was it clearly associated with MCT8 mutations [81]. This is the first example of X linked congenital hypothyroidism owing to a defect in target tissue and not in the pituitary-thyroid biosynthetic pathway.
OATP1C1
Organic-anion transporting polypeptide 1C1 (OATP1C1) is a high affinity T4 transporter expressed in cells of the blood-brain barrier, choroid plexus, human ciliary body epithelium, and testicular Leydig cells [82]. It is considered to play a major role in the transportation of T4 across the blood-brain barrier.
RTHs and congenital hypothyroidism
The pleiotropic actions of thyroid hormones are mediated through the thyroid hormone receptors (TRs), which represent a group of transcription factors also important for the development of mammalian nervous system [83].
The TRs, which bind T3 with high affinity, are members of the steroid/thyroid hormone receptor superfamily [84]. After binding to T3, they bind to hormone response elements (HREs), which are located in the promoter regions of target genes. Two major splice variants (TRα1 and TRα2) are encoded by the THRA gene and two isoforms (TRβ1 and TRβ2) are encoded by the THRB gene. TRα1 is mainly expressed in the heart, bone, and skeletal muscle. TRα2 is expressed throughout the body. TRβ1 is expressed predominantly in the brain, liver, and kidney and TRβ2 in the pituitary, retina, and cochlea. TRα1, TRβ1, and TRβ2 bind T3 with similar affinity [85].
Resistance to thyroid hormone β is a rare disorder (incidence 1/50,000 live births) and is characterized by elevated concentrations of free thyroid hormones, inappropriately normal or elevated TSH and decreased peripheral responses to iodothyronines [86]. More than 160 mutations in the THRB gene have been identified, and in 80% of the cases inheritance follows a dominant pattern [87].
The clinical phenotype of THRB mutations varies from the absence of clinical symptoms to severe manifestations, including goiter and hypothyroidism-related symptoms originating from tissues that express predominantly express TRβ (liver, kidney, and lung), growth impairment, mental retardation, attention deficit hyperactivity disorder (ADHD) in 50% of cases, deafness in 20% of cases, and symptoms of hyperthyroidism (tachycardia, growth acceleration, etc.) [84].
THRA mutations affect the hypothalamus-pituitary-thyroid axis less severely but can cause severe growth retardation, dysmorphic features, gastrointestinal disorders, such as severe constipation or megacolon, bradycardia, reduced muscle tone, and impaired fine and gross motor development due to resistance to T3 [87].
Genetic mutations in CH and malignancy
Albeit rare, malignant transformation in a dyshormonogenetic goiter is one of the most serious complications of CH. Both papillary or follicular thyroid cancer have been reported in the literature, with equal frequency [87]. One case of follicular thyroid cancer with anaplastic components has also been reported in a patient with Pendred syndrome [88], as well as a case of anaplastic thyroid cancer in a patient with thyroid dyshormonogenesis and TG gene mutation [89]. TPO and Tg mutations may predispose patients to thyroid cancer, with Tg representing the most common gene mutation implicated in dyshormonogenesis, CH and thyroid cancer [58, 59, 61]. Of note, elevated TSH concentrations and longstanding congenital goiter are present in all the reported cases, suggesting that TSH is a key player for the onset and progression of thyroid. TSH serves as a growth factor for thyroid epithelial cells and chronic TSH stimulation can promote the formation of thyroid nodules and cancer progression, hypothetically by activating genes involved in cancer promotion [90,91,92]. Moreover, long-term thyroid hormone deficiency, chronic TSH stimulation, and genetic defects may increase the incidence and aggressiveness of thyroid cancer in patients with thyroid dyshormonogenesis. Several case reports of thyroid dyshormonogenesis have been published in the literature revealing the presence of multiple thyroid nodules, large thyroid cysts, and thyroid cancer [93]. Approximately 25% of pediatric thyroid nodules are considered to place the children at risk for thyroid neoplasia, whereas in adults the corresponding percentage is 5% [94].
Since differentiating between neoplastic change and nuclear atypia in dyshormonogenic glands is of the utmost importance, particularly in the presence of thyroid nodules, careful follow-up of patients with thyroid dyshormonogenesis and timely genetic diagnosis is recommended. A lower threshold for thyroidectomy has also been proposed [95].
Although this review focuses on primary congenital hypothyroidism, a brief reference to central congenital hypothyroidism, an uncommon form of congenital hypothyroidism, follows. Central hypothyroidism is caused by insufficient or defective stimulation of the thyroid gland by TSH, resulting in inadequate thyroid hormone biosynthesis [96]. TSH deficiency may be isolated or may occur in the context of coexisting evolving pituitary hormone deficiencies [97]. In the first case, defects in genes that control the TSH biosynthetic pathway may present, including the thyrotropin-releasing hormone receptor (TRHR) gene, the β subunit of the thyroid stimulating hormone (TSHB), and the immunoglobulin superfamily member 1 (IGSF1) gene [98]. In the scenario of multiple pituitary hormone deficiencies, mutations in genes encoding signaling molecules and transcription factors that are involved in normal pituitary development, such as HESX1, LHX3, LHX4, SOX3, OTX2, PROP1, or POU1F1, may be identified [97, 99]. Despite recent advances in the current knowledge regarding the genetic component of congenital hypothyroidism, no identifiable defects in known genes are found in the majority of cases of central congenital hypothyroidism.
In conclusion, CH is the most common congenital endocrine disorder, diagnosed in early infancy in the majority of cases, after the initiation of neonatal screening programs. However, severe cognitive impairment can still be a major problem in the case of delayed treatment initiation or poor compliance, as can also be thyroid goiter and cancer in severe cases. The potential of genetic inheritability of several forms of CH has been recognized and the molecular and genetic mechanisms implicated in the pathogenesis of the disease have, to a significant degree, been elucidated. Gene mutations causing thyroid dysgenesis, organification defects, or other disorders involved in thyroid hormone biosynthesis have now been identified. Genetic characterization of patients with SC is important in order to enhance our understanding of this entity, to enable individualized treatment approaches, and to determine the risk for potential thyroid cancer in specific cases. Further studies are needed so that the genetic underlying causes of CH can be fully understood.
Data availability
Not applicable.
References
Toublanc J (1992) Comparison of epidemiological data on congenital hypothyroidism in Europe with those of other parts of the world. Horm Res (Basel) 38:230–235
Rastogi MV, LaFranchi SH (2010) Congenital hypothyroidism. Orphanet J Rare Dis 5:17
Lorey FW, Cunningham GC (1992) Birth prevalence of primary congenital hypothyroidism by sex and ethnicity. Hum Biol 64:531–538
Cao XY, Jiang XM, Dou ZH, Rakeman MA, Zhang ML, O’Donnell K, Ma T, Amette K, DeLong N, DeLong GR (1994) Timing of vulnerability of the brain to iodine deficiency in endemic cretinism. N Engl J Med 331:1739–1744
Rovet J, Ehrlich R (1995) Long-term effects of L-thyroxine treatment for congental hypothyroidism. J Pediatr 126:380–386
Grant DB, Smith I, Fuggle PW, Tokar S, Chapple J (1992) Congenital hypothyroidism detected by neonatal screening: relationship between biochemical severity and early clinical features. Arch Dis Child 67:87–90
Castanet M, Polak M, Bonaiti-Pellie C, Lyonnet S, Czernickow P, Leger J (2001) Nineteen years of national screening for congenital hypothyroidism: familial cases with thyroid dysgenesis suggest the involvement of genetic factors. J Clin Endocrinol Metab 86:2009–2014
Nilsson M, Fagman H (2017) Development of the thyroid gland. Development. 144:2123–2140
Deladoey J, Vassart G, Van Vliet G (2007) Possible non-Mendelian mechanisms of thyroid dysgenesis. Endocr Dev 10:29–42
Park SM, Chatterjee VK (2005) Genetics of congenital hypothyroidism. J Med Genet 42:379–389
Field JB, Ealey PA, Marshall NJ, Cockcroft S (1987) Thyroid-stimulating hormone stimulates increases in inositol phosphates as well as cyclic AMP in the FRTL-5 rat thyroid cell line. Biochem J 247:519–524
Sunthornthepvarakul T, Gootschalk ME, Hayashi Y, Refetoff S (1995) Resistance to thyrotropin caused by mutations in the thyrotropin-receptor gene. N Engl J Med 332:155–160
De Roux N, Misrahi M, Brauner R, Houang M, Carel JC, Granier M, Le Bouc Y, Ghinea N, Boumeddienne A, Toublanc JE, Milgrom E (1996) Four families with loss of function mutations of the thyrotropin receptor. J Clin Endocrinol Metab 81:4229–4235
Russo D, Betterle C, Arturi F, Chiefari E, Girelli ME, Filetti S (2000) A novel mutation in the thyrotropin (TSH) receptor gene causing loss of TSH binding but constitutive receptor activation in a family with resistance to TSH. J Clin Endocrinol Metab 85:4238–4242
Nagashima T, Murakami M, Onigata K, Morimura T, Nagashima K, Mori M, Morikawa A (2001) Novel inactivating missense mutations in the thyrotropin receptor gene in Japanese children with resistance to thyrotropin. Thyroid 11:551–559
Tonacchera M, Agretti P, De Marco G, Perri A, Pinchera A, Vitti P, Chiovato L (2001) Thyroid resistance to TSH complicated by autoimmune thyroiditis. J Clin Endocrinol Metab 86:4543–4546
Zannini M, Avantaggiato V, Biffali E, Arnone MI, Sato K, Pischetola M, Taylor BA, Phillips SJ, Simeone A, Di Lauro R (1997) TTF-2, a new forkhead protein, shows a temporal expression in the developing thyroid which is consistent with a role in controlling the onset of differentiation. EMBO J 16:3185–3197
Civitreale D, Lonigro R, Sinclair AJ, Di Lauro R (1989) A thyroid-specific nuclear protein essential for tissue-specific expression of the thyroglobulin promoter. EMBO J 8:2537–2542
Santisteban P, Acebron A, Polycarpou-Schwarz M, Di Lauro R (1992) Insulin and insulin- like growth factor I regulate a thyroid-specific nuclear protein that binds to the thyroglobulin promoter. Mol Endocrinol 6:1310–1317
Clifton-Bligh RJ, Wentworth JM, Heinz P, Crisp M, John R, Lazarus JH, Ludgate M, Chatterjee VKK (1998) Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet 19:399–401
Bamforth JS, Hughes IA, Lazarus JH, Weaver CM, Harper PS (1989) Congenital hypothyroidism, spiky hair, and cleft palate. J Med Genet 26:49–60
Francis-Lang H, Price M, Polycarpou-Schwartz M, Di Lauro R (1992) Cell-typespecific expression of the rat thyroperoxidase promoter indicates common mechanisms for thyroid-specific gene expression. Mol Cell Biol 12:576–588
Harvey RP (1996) NK-2 homeobox genes and heart development. Dev Biol 187:203–216
Lazzaro D, Price M, de Felice M, Di Lauro R (1991) The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development 113:1093–1104
Devriendt K, Vanhole C, Matthijs G, de Zegher F (1998) Deletion of the thyroid transcription factor-1 gene in an infant with neonatal thyroid dysfunction and respiratory failure. N Engl J Med 338:1317–1318
Doyle DA, Gonzalez I, Thomas B, Scavina M (2004) Autosomal dominant transmission of congenital hypothyroidism, neonatal respiratory distress, and ataxia caused by a mutation of NKX2–1. J Pediatr 145:190–193
Pohlenz J, Dumitrescu A, Zundel D, Martine U, Schonberger W, Koo E, Weiss RE, Cohen RN, Kimura S, Refetoff S (2002) Partial deficiency of thyroid transcription factor 1 produces predominantly neurological defects in humans and mice. J Clin Invest 109:469–473
di Magliano MP, Di Lauro R, Zannini M (2000) PAX8 has a key role in thyroid cell differentiation. Proc Natl Acad Sci U S A 97:13144–13149
Fraizer GC, Shimamura R, Zhang X, Saubders GF (1997) PAX8 regulates human WT1 transcription through a novel DNA binding site. J Biol Chem 272:30678–30687
van Engelen K, Mommersteeg MTM, Baars MJH, Lam J, Ilgun A, van Trotsenburg ASP, Smets AMJ, Christoffels VM, Mulder BJM, Postma AV (2012) The ambiguous role of NKX2-5 mutations in thyroid dysgenesis. PLoS One 7:e52685
Carvalho DP, Dupuy C (2013) Role of the NADPH oxidases DUOX and NOX4 in thyroid oxidative stress. Eur Thyroid J 2:160–167
Grasberger H (2010) Defects of thyroidal hydrogen peroxide generation in congenital hypothyroidism. Mol Cell Endocrinol 322(1–2):99–106
Desai MP (2012) Congenital hypothyroidism: screening dilemma. Indian J Endocrinol Metab 16:153–155
Lichti-Kaiser K, ZeRuth G, Jetten AM (2014) Transcription factor gli-similar 3 (Glis3): implications for the development of congenital hypothyroidism. J Endocrinol Diabetes Obes 2:1024
de Filippis T, Gelmini G, Paraboschi E et al (2017) A frequent oligogenic involvement in congenital hypothyroidism. Hum Mol Genet 26:2507–2514
Dimitri P (2017) The role of GLIS3 in thyroid disease as part of a multisystem disorder. Best Pract Res Clin Endocrinol Metab 31:175–182
Patel SR, Richardson JL, Schulze H, Kahle E, Galjart N, Drabek K, Shivdasani RA, Hartwig JH, Italiano JE Jr (2015) Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood 106:4076–4085
Stoupa A, Adam F, Kariyawasam D et al (2018) TUBB1 mutations cause thyroid dysgenesis associated with abnormal platelet physiology. EMBO Mol Med 10:e9569
Carré A, Stoupa A, Kariyawasam D et al (2017) Mutations in BOREALIN cause thyroid dysgenesis. Hum Mol Genet 26:599–610
Peters C, van Trotsenburg ASP, Schoenmakers N (2018) DIAGNOSIS OF ENDOCRINE DISEASE: congenital hypothyroidism: update and perspectives. Eur J Endocrinol 179:297–317
de Filippis T, Marelli F, Nebbia G et al (2016) JAG1 loss-of-function variations as a novel predisposing event in the pathogenesis of congenital thyroid defects. J Clin Endocrinol Metab 101:861–870
Opitz R, Hitz M-P, Vandernoot I et al (2015) Functional zebrafish studies based on human genotyping point to Netrin-1 as a link between aberrant cardiovascular development and thyroid dysgenesis. Endocrinology 156:377–388
Puppin C, Presta I, D'Elia AV et al (2004) Functional interaction among thyroid-specific transcription factors: Pax8 regulates the activity of hex promoter. Mol Cell Endocrinol 214:117–125
Puppin C, D'Elia AV, Pellizzari L et al (2003) Thyroid-specific transcription factors control hex promoter activity. Nucleic Acids Res 31:1845–1852
Barbera JPM, Clements M, Thomas P et al (2000) The homeobox gene hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development 127:2433–2445
Parlato R, Rosica A, Rodriguez-Mallon A et al (2004) An integrated regulatory network controlling survival and migration in thyroid organogenesis. Dev Biol 276:464–475
Ferretti E, Tosi E, Po A et al (2008) Notch signaling is involved in expression of Thyrocyte differentiation markers and is Down-regulated in thyroid tumors. J Clin Endocrinol Metab 93:4080–4087
Carre A, Rachdi L, Tron E et al (2011) Hes1 is required for appropriate morphogenesis and differentiation during mouse thyroid gland development. PLoS One 6:e16752
Manley NR, Capecchi MR (1995) The role of Hoxa-3 in mouse thymus and thyroid development. Development 121:1989–2003
Xu P-X, Zheng W, Laclef C et al (2002) Eya1 is required for the morphogen- esis of mammalian thymus, parathyroid and thyroid. Development 129:3033–3044
Kopp P (2013) Thyroid hormone synthesis. In: Braverman LE, Cooper D (eds) Werner and Ingbar’s the thyroid. A fundamental and clinical text, ed, vol 10. Lippincottt Williams & Wilkins, Philadelphia, pp 48–74
de Vijlder JJM, Vulsma T (1996) Hereditary metabolic disorders causing hypothyroidism. In: Braverman LE, Utiger RD (eds) Werner and Ingbar’s the thyroid, 7th edn. Lippincott-Raven p, Philadelphia, pp 749–755
Mangkalbruks A, Correa Billerbeck A-E, Wajchenberg B, Knobel M, Cox NJ, DeGroot LJ, Medeiros-Neto G (1991) Genetic linkage studies of thyroid peroxidase (TPO) gene in families with TPO deficiency. J Clin Endocrinol Metab 72:471–476
Fujiwara H, Tatsumi K, Miki K, Harada T, Miyai K, Takai SI, Amino N (1997) Congenital hypothyroidism caused by a mutation in the Na+/I2 symporter. Nat Genet 16:124–125
Kosugi S, Sato Y, Matsuda A, Ohyama Y, Fujieda K, Inomata H, Kameya T, Isozak O, Jhiang SM (1998) High prevalence of T354P sodium/iodide symporter gene mutation in Japanese patients with iodide transport defect who have heterogeneous clinical pictures. J Clin Endocrinol Metab 83:4123–4129
Reardon W, O’Mahoney CF, Trembath R, Jan H, Phelps PD (2000) Enlarged vestibular aqueduct: a radiological marker of Pendred syndrome, and mutation of the PDS gene. Q J Med 93:99–104
Reardon W, Trembath RC (1996) Pendred syndrome. J Med Genet 33:1037–1040
Batsakis JG, Nishiyama RH (1962) Deafness with sporadic goitre. Arch Otolaryngol 76:401–406
Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED (1997) Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17:411–422
Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED (2000) Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 14:839–845
Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED (2001) Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10:153–161
Scott DA, Wang R, Kreman TM, Andrews M, McDonald JM, Bishop JR, Smith RJH, Karniski LP, Sheffield VC (2000) Functional differences of the PDS gene products are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4). Hum Mol Genet 9:1709–1715
Pfarr N, Borck G, Turk A, Napiontek U, Keilmann A, Muller-Forell W, Kopp P, Pohlenz J (2006) Goitrous congenital hypothyroidism and hearing impairment associated with mutations in the TPO and SLC26A4/PDS genes. J Clin Endocrinol Metab 91:2678–2881
Ishii J, Suzuki A, Kimura T, Tateyama M, Tanaka T, Yazawa T et al (2019) Congenital goitrous hypothyroidism is caused by dysfunction of the iodide transporter SLC26A7. Commun Biol 2:270
Virion A, Michot JL, Deme D, Kaniewski J, Pommier J (1984) NADPH-dependent H2O2 generation and peroxidase activity in thyroid particular fraction. Mol Cell Endocrinol 36(1–2):95–105
Caillou B, Dupuy C, Lacroix L, Nocera M, Talbot M, Ohayon R, Deme D, Bidart JM, Schlumberger M, Virion A (2001) Expression of reduced nicotinamide adenine dinucleotide phosphate oxidase (ThoX, LNOX, Duox) genes and proteins in human thyroid tissues. J Clin Endocrinol Metab 86:3351–3358
Moreno JC, Bikker H, Kempers MJ, van Trotsenburg AS, Baas F, de Vijlder JJ, Vulsma T, Ris-Stalpers C (2002) Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med 347:95–102
Grasberger H, Refetoff S (2006) Identification of the maturation factor for dual oxidase: evolution of an eukaryotic operon equivalent. J Biol Chem 281:18269–18272
Berge-Lefranc JL, Cartonzon G, Mattei MG, Passage E, Malezet-Desmoulins C, Lissitzky S (1985) Localisation of the thyroglobulin gene by in situ hybridization to human chromosomes. Hum Genet 69:28–31
Hishinuma A, Takamatsu J, Ohyama Y, Yokozawa T, Kanno Y, Kuma K, Yoshida S, Matsuura N, Ieiro T (1999) Two novel cysteine substitutions (C1263R and C1995S) of thyroglobulin cause a defect in intracellular transport of thyroglobulin in patients with congenital goiter and the variant type of adenomatous goiter. J Clin Endocrinol Metab 84:1438–1444
Moreno JC (2003) Identification of novel genes involved in congenital hypothyroidism using serial analysis of gene expression. Horm Res 60:96–102
Moreno JC, Klootwijk W, van Toor H, Pinto G, D'Alessandro M, Leger A, Goudie D, Polak M, Gruters A, Visser TJ (2008) Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med 358:1811–1818
Nicoletti A, Bal M, De Marco G, Baldazzi L, Agretti P, Menabo S, Ballarini E, Cicognani A, Tonacchera M, Cassio A (2009) Thyrotropin-stimulating hormone receptor gene analysis in pediatric patients with non-autoimmune subclinical hypothyroidism. J Clin Endocrinol Metab 94:4187–4194
Cassio A, Nicoletti A, Rizzelo A, Zazzetta E, Bal M, Baldazzi L (2013) Current loss-of-function mutations in the thyrotropin receptor gene: when to investigate, clinical effects, and treatment. J Clin Res Pediatr Endocrinol 5:29–39
Weinstein LS, Yu S, Warner DR, Liu J (2001) Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 22(5):675–705
Park E, Jung J, Araki O, Tsunekawa K, Park SY, Kim J, Murakami M, Jeong SY, Lee S (2018) Concurrent TSHR mutations and DIO2 T92A polymorphism result in abnormal thyroid hormone metabolism. Sci Rep 8:10090
Van Herle AJ, Vassart G, Dumont JE (1979) Control of thyroglobulin synthesis and secretion. (first of two parts). N Engl J Med 301:239–249
Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ (2003) Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 278:40128–40135
Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S (2004) A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 74:168–175 Erratum in: Am J Hum Genet 2004;74:598
Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE (2005) Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77(1):41–53
Gao B, Huber RD, Wenzel A, Vavricka SR, Ismair MG, Remé C, Meier PJ (2005) Localization of organic anion transporting polypeptides in the rat and human ciliary body epithelium. Exp Eye Res 80(1):61–72
Rodriguez PA, Ibarrola N, Ifiguez MA, Mufioz A, Bernal J (1993) J Clin Invest 91:812–818
Sap J, Munoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennstrom B (1986) The c-erb-a protein is a high-affinity receptor for thyroid hormone. Nature 324:635–640
Singh BK, Yen PM (2017) A clinician’s guide to understanding resistance to thyroid hormone due to receptor mutations in the TRα and TRβ isoforms. Clin Diabetes Endocrinol 3:8
Ortiga-Carvalho TM, Sidhaye AR, Wondisford FE (2014) Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat Rev Endocrinol 10:582–591
Refetoff S, Weiss RE, Usala SJ (1993) The syndromes of resistance to thyroid hormone. Endocr Rev 14:348–399
Alzahrani AS, Baitei EY, Zou M, Shi Y (2006) Clinical case seminar: metastatic follicular thyroid carcinoma arising from congenital goiter as a result of a novel splice donor site mutation in the thyroglobulin gene. J Clin Endocrinol Metab 91:740–746
Camargo R, Limbert E, Gillam M, Henriques MM, Fernandes C, Catarino AL, Soares J, Alves VA, Kopp P, Medeiros-Neto G (2001) Aggressive metastatic follicular thyroid carcinoma with anaplastic transformation arising from a long-standing goiter in a patient with Pendred's syndrome. Thyroid 11:981–988
Yoon JH, Hong AR, Kim HK, Kang HC (2020) Anaplastic thyroid cancer arising from dyshormonogenetic goiter: c.3070T>C and novel c.7070T>C mutation in the thyroglobulin gene. Thyroid ahead of print https://doi.org/10.1089/thy.2020.0248
Hishinuma A, Fukata S, Kakudo K, Murata Y, Ieiri T (2005) High incidence of thyroid cancer in long-standing goiters with thyroglobulin mutations. Thyroid 15:1079–1084
Raef H, Al-Rijjal R, Al-Shehri S, Zou M, Al-Mana H, Baitei EY, Parhar RS, Al-Mohanna FA, Shi Y (2010) Biallelic p.R2223H mutation in the thyroglobulin gene causes thyroglobulin retention and severe hypothyroidism with subsequent development of thyroid carcinoma. J Clin Endocrinol Metab 95:1000–1006
Boelaert K (2009) The association between serum TSH concentration and thyroid cancer. Endocr Relat Cancer 16:1065–1072
Chertok Shacham E, Ishay A, Irit E, Pohlenz J, Tenenbaum-Rakover Y (2012) Minimally invasive follicular thyroid carcinoma developed in dyshormonogenetic multinodular goiter due to thyroid peroxidase gene mutation. Thyroid 22(5):542–546
Corrias A, Mussa A (2013) Thyroid nodules in pediatrics: which ones can be left alone, which ones must be investigated, when and how. J Clin Res Pediatr Endocrinol 5:57–69
Kaykhaei MA, Heidari Z, Mehrazin A (2014) Large thyroid cyst in a patient with congenital hypothyroidism. Arq Bras Endocrinol Metab 58:9
Persani L (2012) Clinical review: central hypothyroidism: pathogenic, diagnostic, and therapeutic challenges. J Clin Endocrinol Metab 97(9):3068–3078
Alatzoglou KS, Dattani MT (2009) Genetic forms of hypopituitarism and their manifestation in the neonatal period. Early Hum Dev 85(11):705–712
García M, Fernández A, Moreno JC (2014) Central hypothyroidism in children. In Paediatric Thyroidology. Endocr Dev 26:79–107 Ed G Szinnai Basel, Karger
Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT (2009) Genetic regulation of pituitary gland development in human and mouse. Endocr Rev 30(7):790–829
Author information
Authors and Affiliations
Contributions
Eirini Kostopoulou: conceptualization, literature review, original draft preparation, and writing. Konstantinos Miliordos: literature investigation and writing. Bessie Spiliotis: review, editing, and supervision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kostopoulou, E., Miliordos, K. & Spiliotis, B. Genetics of primary congenital hypothyroidism—a review. Hormones 20, 225–236 (2021). https://doi.org/10.1007/s42000-020-00267-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42000-020-00267-x