
Abstract
Purpose of Review
Fibroblast growth factor-23 (FGF23) is the key hormone produced in bone critical for phosphate homeostasis. Elevated serum phosphorus and 1,25-dihydroxyvitamin D stimulates FGF23 production to promote renal phosphate excretion and decrease 1,25-dihydroxyvitamin D synthesis, thus completing the feedback loop and suppressing FGF23. Unexpectedly, studies of common and rare heritable disorders of phosphate handling identified links between iron and FGF23 demonstrating novel regulation outside the phosphate pathway.
Recent Findings
Iron deficiency combined with an FGF23 cleavage mutation was found to induce the autosomal dominant hypophosphatemic rickets phenotype. Physiological responses to iron deficiency, such as erythropoietin production as well as hypoxia inducible factor activation, have been indicated in regulating FGF23. Additionally, specific iron formulations, used to treat iron deficiency, alter post-translational processing thereby shifting FGF23 protein secretion.
Summary
Molecular and clinical studies revealed that iron deficiency, through several mechanisms, alters FGF23 at the transcriptional and post-translational level. This review will focus upon the novel discoveries elucidated between iron, its regulators, and their influence on FGF23 bioactivity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fibroblast growth factor 23 (FGF23) is a crucial bone-derived hormone for regulation of serum phosphorus. Phosphorus concentrations are maintained within a narrow physiological range through balancing intestinal absorption from dietary intake, skeletal bone storage in the form of hydroxyapatite, and renal reabsorption. Studies of human phosphate handling disorders and genetic mouse models have demonstrated that FGF23 endocrine activity coordinates the communication in this multi-organ system for stable phosphorus levels [1,2,3,4,5]. The kidney is the primary target for FGF23 activity as it is the crucial site for short-term serum phosphate changes. Expression of the co-receptor α-Klotho (αKl) within the distal convoluted tubule is essential for a high affinity binding event between FGF23 and FGF receptor 1 (FGFR1) [6,7,8,9,10] to initiate the MAP kinase signaling cascade [11]. Subsequent to FGF23 signaling, the type II sodium phosphate co-transporters Npt2a and Npt2c proteins are internalized from the apical membrane, therefore promoting phosphaturia [12]. Additionally, FGF23 alters renal 1,25-dihydroxyvitamin D (1,25D) synthesis by downregulating the anabolic enzyme 1-alpha-hydroxylase (Cyp27b1) and enhancing expression of the catabolic enzyme 24-hydroxylase (Cyp24a1) [13]. Reduced circulating levels of 1,25D decreases intestinal phosphorus absorption [1]. As a secondary effect, serum calcium is also reduced with lower serum 1,25D and consequently promotes parathyroid hormone (PTH) release to counteract the FGF23 mediated changes in renal 1,25D synthesis [14]. The overall effect of negative phosphate balance completes the feedback loop to relieve FGF23 stimulation in osteoblasts/osteocytes [15].
Positional cloning performed from patients with the Mendelian disorder of phosphate wasting in autosomal dominant hypophosphatemic rickets (ADHR) identified gain-of-function mutations within FGF23 providing a secondary mechanism of regulation. FGF23 protein contains a subtilisin-like proprotein convertase (SPC) proteolytic cleavage site (176RHTR/S180AE) [16, 17]. Within this site, missense mutations seen in ADHR altered R176 or R179 residues to either glutamine (Q) or tryptophan (W). In vitro characterization of the R176 mutation demonstrated preferential secretion of the 32-kDa full-length intact form of FGF23 (iFGF23) which confers biological activity. However, wild-type protein is primarily secreted as the cleaved, inactive FGF23 (cFGF23). This finding of secondary FGF23 regulation correlates with the ADHR phenotype, whereby serum iFGF23 was significantly elevated despite prevailing hypophosphatemia.
The ability to regulate FGF23 processing at the post-transcriptional level was identified through studying rare heritable phosphate handling disorders that led to alterations in FGF23. Tumoral calcinosis (TC) arises from loss-of-function mutations in the gene encoding GalNac-transferase 3 (GALNT3) [18, 19]. Patients with TC and Galnt3-null mice show highly elevated cFGF23 and extremely low iFGF23, demonstrating an inability to secrete full-length, bioactive protein. While FGF23 protein contains multiple motifs for GALNT3 O-glycosylation, T178 within the SPC cleavage site has been shown to protect FGF23 from degradation with presence of this modification [20]. Alternatively, promoting cleavage and inactivation of FGF23 was determined with the identification of loss-of-function mutations within Family with sequence similarity 20, member C (FAM20C) that gives rise to autosomal recessive hypophosphatemic rickets-type 3 (ARHR-3) [21]. Novel mutations of this gene were described in surviving Raine syndrome patients that also exhibited bowing of the long bones [22,23,24]. As with other rachitic phenotypes, patients with R408W FAM20C mutations as well as Fam20c-null mice show highly elevated serum iFGF23 resulting in hypophosphatemia and profound osteomalacia [25,26,27]. Functional studies showed that FAM20C directly targets phosphorylation of FGF23, in addition to other secreted proteins at S-X-E recognition sequences [28]. Interestingly, FAM20C mediated phosphorylation at S180, immediately following the SPC motif-inhibited GALNT3 O-glycosylation of FGF23 leading to enhanced cleavage [21]. Thus, the interplay between GALNT3 and FAM20C activity within the endoplasmic reticulum and Golgi network dictates the secreted form of FGF23. Importantly, highly elevated serum FGF23 levels have been found to induce detrimental off-target effects in tissues outside of kidney [29, 30]. Recent studies haveidentified factors regulating FGF23 not involved in the phosphate and 1,25D feedback network [31, 32]. These may contribute to the pathogenic rises in FGF23 at both the transcriptional and post-translational level. Thus, knowledge of FGF23 regulation remains incomplete and understanding the effects of these new factors may elucidate novel therapeutic targets for rare and common forms of phosphate handling disorders.
FGF23 Regulation by Iron Deficiency
Iron Deficiency as a Driver of FGF23
Similar to phosphorus, iron is an important factor for many proteins in their enzymatic reactions and critical for incorporation into hemoglobin within red blood cells for oxygen transport [33]. Iron levels are tightly controlled at both the systemic and cellular level. Iron is taken up through intestinal enterocyte active transport [34] primarily regulated by the liver enzyme hepcidin [35]. Transportation through the circulation is facilitated by iron binding to transferrin and deposition into cells through the transferrin receptor where the iron is either incorporated into proteins or stored as ferritin [35]. Iron deficiency and iron deficiency anemia can occur in numerous situations and disorders such as pregnancy [36], poor diet [37,38,39], inflammation [40, 41], inability to absorb iron [42, 43], and renal failure [44, 45]. The observation that iron deficiency along with the classic ADHR mutation are both critical pieces of the gene-environment interaction necessary to produce the ADHR disease phenotype provided an interesting link between iron and FGF23 [6].
Low penetrance and the observation of ADHR patients to wax and wane in their disease manifestations were found to be unique features of the ADHR phenotype [16, 46, 47]. Women carrying the ADHR mutation more frequently portrayed later onset where phenotypes correlated with pubertal menses, a state often associated with iron deficiency [46, 47]. Interconnection between FGF23 and iron was identified, whereby low serum iron levels correlated with higher serum cFGF23 in both normal controls and ADHR patients. This negative correlation remained for low serum iron and iFGF23 in ADHR patients; however, no association with iFGF23 could be observed in normal controls [48]. Testing the hypothesis of iron levels modulating FGF23 was conducted in mice containing the ADHR R176Q-Fgf23 knock-in alleles (ADHR mice).These studies recapitulated the human data demonstrating that bone Fgf23 mRNA and serum cFGF23 was significantly induced in all mice during iron deficiency. ADHR mice exhibited elevated iFGF23 and hypophosphatemia during iron deficiency, whereas wild-type littermate controls maintained normal serum iFGF23 and phosphorus levels [31]. Importantly, low iron status in ADHR mice completely negated the normal feedback suppression of FGF23 from the reduced serum phosphorus. Thus, these data demonstrated that iron deficiency can enhance transcriptional activity of Fgf23, and that post-translational processing to cleave FGF23 is important for maintaining proper levels of serum iFGF23 and therefore phosphate balance.
In addition to diet-mediated iron deficiency, blood loss from trauma, surgery, and bowel disorders can also develop iron deficiency [49, 50]. Rabadi et al. found that acute bleeding of normal mice resulted in significantly increased cFGF23 beginning at 6 h post-bleed and persisted to 48 h [51]. In a prospective cohort study, patients admitted to the intensive care unit receiving transfusions were also examined as the need for red blood cells indicated blood loss. After assessing serum of 131 patients, a significant positive association was found between cFGF23 and the number of transfusions [51]. In both cases however, there were no measurable differences in serum iFGF23 demonstrating the ability to cleave the FGF23 protein and maintain normal phosphorus levels. These results are similar to controls in the ADHR study as well as premenopausal women with anemia [52]; in that, only cFGF23 is elevated with iron deficiency. In contrast, a recent study performed in a cohort of elderly men found a significant negative correlation between iFGF23 and both serum total iron levels as well as transferrin saturation, a measure of iron binding to transferrin. Hemoglobin levels were not significantly associated with iFGF23 as only a small percentage of the subjects demonstrated anemia along with the iron deficiency [53]. Thus, aging may affect the ability of cells to sense phosphate and properly cleave FGF23 in the setting of iron deficiency.
Hypoxia Inducible Factor Transcription Factor Activity
Iron responsive proteins (IRPs) regulate many of the iron homeostasis factors, including transferrin [54]. During iron deficiency, IRPs bind secondary structures within 3′ and 5′ untranslated regions generated by highly conserved iron responsive elements (IREs) to either enhance or block translation [55, 56]. Even though IRPs have been found to target factors not involved directly in iron handling [57], examination of FGF23 has not elucidated any putative IRE consensus sites. Thus, an alternative mechanism is utilized during iron deficiency to modulate FGF23 production. One important factor for low iron and low oxygen adaptation is hypoxia-inducible factor 1 alpha (HIF1α) [58, 59]. HIF1α is constantly expressed and undergoes regulation primarily at the post-translational level. Under normoxic conditions, prolyl hydroxylase domain isoforms (PHDs) catalyze hydroxylation at specific prolines (P402, P564) within the HIF1α protein [60, 61]. Upon recognition of this modification, von Hippel-Lindau (VHL) acts as an E3 ubiquitin-ligase resulting in rapid HIF1α degradation within the proteasome [26, 62].
Under hypoxic conditions, the PHD activity and subsequent VHL degradation is inhibited, therefore allowing for HIF1α stabilization and accumulation. HIF1α subsequently translocates to the nucleus and binds DNA at conserved hypoxia responsive elements (HREs) as a heterodimer with HIF1β to activate transcription of target genes [63]. Importantly, not only is HIF1α degradation dependent upon sufficient amounts of oxygen but PHDs require iron as a co-factor for the hydroxylation enzymatic reaction [64]. Thus, it is possible for HIF activation in low iron and oxygen replete conditions [65]. Indeed, in initial studies, osteoblastic cell lines treated with the iron chelator deferoxamine (DFO) under normoxic culturing conditions showed cellular HIF1α protein accumulation corresponding with a dramatic induction of Fgf23 mRNA expression [31]. In mice, functional iron deficiency can be induced with inflammation, a normal protective mechanism to sequester iron from pathogens [66]. Mice injected with IL-1β, to mimic a pro-inflammatory state, had significantly reduced iron levels with increases in both cFGF23 and iFGF23. Bone Fgf23 mRNA and serum cFGF23 levels were significantly reduced when the mice were treated with a HIF1α inhibitor prior to IL-1β injections, demonstrative of HIF1α targeting transcriptional activation of Fgf23 [67]. Interestingly, HIF1α inhibition combined with IL-1β injections increased iFGF23, suggesting that it is also involved in the secondary processing of the FGF23 protein. However, this study did not examine mRNA expression levels of the processing enzymes Fam20c, Galnt3, or Furin within the bone under these conditions.
HIF1α acts as a transcription factor by directly binding cis regulatory elements of target genes. In osteoblastic cells, HIF1α stabilization increased plasmid luciferase activity when cloned downstream of the mouse proximal Fgf23 promoter [68]. Additionally, chromatin immunoprecipitation showed direct binding of HIF1α within this segment and was ablated with the use of a HIF1α inhibitor [68]. However, the exact location of the consensus HRE was not identified. In a recent study by Onal et al., a novel enhancer region 16kb upstream of the Fgf23 transcriptional start site was identified that may mediate inflammation regulation [69••].
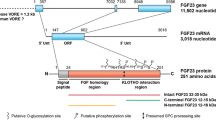
Interestingly, sequence annotation of the putative enhancer region identified an HRE displaying the highest matrix similarity score of all identified transcription factor binding sites. Deletion of the − 16-kb putative enhancer in mice significantly reduced bone Fgf23 mRNA levels without affecting serum FGF23 protein [69••]. As this enhancer was originally identified during inflammation, the enhancer knockout mice were subjected to inflammatory conditions including injections with IL-1β in a similar dose and time course to David et al. [67]. As in the previous study, IL-1β induced bone Fgf23 mRNA and serum protein in wild-type mice, whereas deletion of the − 16-kb enhancer attenuated IL-1β induction at both bone mRNA and serum protein levels. This study did not include measurement of serum iron levels so it is unclear whether iron deficiency occurred in these animals. Additionally, the putative enhancer also contained known downstream inflammatory transcription factor binding motifs including Stat3, Stat5, and NF-κB [70]. Thus, further study is needed to fully elucidate the contribution of the HRE within this enhancer to HIF1α-mediated induction of Fgf23 during iron deficiency (Fig. 1).

Emerging evidence has demonstrated multiple aspects of iron and iron-sensing pathways altering the regulation of FGF23. Iron deficiency activates hypoxia inducible factor 1α (HIF1α) within osteoblast/osteocytes (blue cell). This transcription factor then binds to DNA hypoxia responsive elements as a heterodimer with HIF1β to activate transcription of Fgf23 mRNA. In both normal mice and humans with iron deficiency, the intact bioactive FGF23 protein (iFGF23) can be cleaved (cFGF23) prior to secretion to maintain phosphorus levels. Iron deficiency, as well as prolyl-hydroxylase inhibitors (PHi), activates HIFs within the kidney to induce renal erythropoietin (EPO) production. Within the bone, EPO activates Fgf23 at the transcriptional level in both osteoblast/osteocytes as well as in hematopoietic lineage cells which increases serum cFGF23 levels. Pharmacological doses of EPO are also found to induce release of iFGF23. Iron polymaltose and ferric carboxymaltose intravenous iron preparations appear to block cleavage of the FGF23 protein, thereby promoting release of iFGF23
Iron Deficiency Therapeutics and Their Impact on FGF23
Erythropoietin
Erythropoietin (EPO) is a hematopoietic hormone produced primarily by the kidneys that plays a key role in the body’s physiologic response to iron deficiency. It is well established that EPO production is a key downstream target of HIF regulation, specifically through HIF2α (EPAS1) activation [71, 72]. This hormone is crucial for stimulation of red blood cell production by signaling through the EPO receptor (EPOR) on erythroblasts and activating the canonical JAK2-STAT5 pathway [73]. Additionally, EPO works to correct iron deficiency by indirectly reducing hepcidin to promote iron store release by activating the EPO responsive factor erythroferrone (ERFE) in erythroblasts [74, 75]. Indeed, mice fed a low iron diet demonstrate normal physiological responses with increased serum EPO compared to the control diet fed mice, which correlated with elevated cFGF23 [31]. In transgenic mice that overexpress EPO, both cFGF23 and iFGF23 are significantly increased compared to normal controls [76, 77], suggesting that EPO may have a direct effect on FGF23. A novel class of compounds have recently been developed that function to inhibit the PHDs (PHi) involved in HIF degradation [78]. The subsequent stabilization and activation of HIF complex proteins after PHi administration induces endogenous EPO production. Injection of the PHi in wild-type mice induced serum EPO and cFGF23 [79] as well as iFGF23 [76]. When the mice were also administered an EPO neutralizing antibody FGF23, induction from the PHi was abrogated [79], again suggesting a direct effect of EPO on FGF23 regulation independent of HIF. Anemia is a risk factor for patients with acute kidney injury (AKI) and FGF23 is often found significantly elevated with no relation to other known factors including phosphate, PTH, or 1,25D. A mouse model of AKI demonstrated coordinating increases in both serum EPO and cFGF23 after injury. AKI mice treated with the EPO antagonist, EMP9, resulted in a reduction of cFGF23, yet not to control levels [80]. Thus, multiple facets of iron sensing and homeostasis may be involved in regulation of FGF23.
However, these are conditions in which endogenous renal EPO production is exacerbated. It is unclear in these studies whether normal physiological levels of EPO are involved in regulating basal levels of FGF23. Additionally, while a direct effect of EPO on FGF23 production has been suggested, the factors involved in mediating transcriptional activation after EPO treatment have yet to be elucidated.
Iron deficiency and iron deficiency-induced anemia is a frequent co-morbidity arising in chronic kidney disease [81] with the incidence and prevalence of the diagnosis increasing as renal function declines [82]. Iron deficiency during renal failure is multifaceted, but is frequently due to reduced EPO synthesis [83]. Therefore, replacement therapy with recombinant EPO is a primary avenue for correcting iron deficiency and anemia in this patient population. However, the effect of these therapies on FGF23 was previously unknown. Acute injections of pharmacological doses of recombinant EPO into wild-type mice significantly increased not only cFGF23 [51] but also iFGF23 [76, 84••]. The mouse renal failure model of juvenile cystic kidney disease (Jck mice) was also treated with EPO in a similar manner. Serum levels of iFGF23 increased in all of the animals that received EPO. However, Jck mice treated with EPO further stimulated the induction of iFGF23 potentially due to effects of the underlying renal failure. To ensure that EPO did not cause a local iron deficit and subsequent HIF activation, EPO was co-administered with iron. Regardless of iron administration, EPO induced iFGF23 to a similar extent [84••]. In an adenine diet-induced model of chronic renal failure, EPO injections elicited a significant induction of cFGF23 but not iFGF23 [77]. These different observations may be due to the stage of renal failure in the mice at the time of injection and/or the EPO doses used in the two studies. In either case, EPO treatment was also found to induce bone Fgf23 mRNA. EPOR is well characterized within hematopoietic cells. However, whether EPOR is expressed on osteoblasts and osteocytes remains controversial [85]. Marrow ablation prior to EPO injections abolished the known EPO response of erythroferrone induction. Interestingly, cFGF23 was reduced following marrow ablation combined with EPO injections compared to the EPO injections alone. However, the levels were still above that of saline-injected control mice. Additionally, Fgf23 mRNA expression from the femur shaft was elevated with EPO injection and unaffected by marrow ablation [84••]. In contrast, other studies found EPO or epoetin alfa injections were unable to stimulate cortical bone production of Fgf23 mRNA [51, 76]. Taken together, these data suggested that EPO directly affected expression of Fgf23 mRNA and that marrow cells may contribute to circulating levels of FGF23. Immunostaining bone marrow after EPO injection highlighted co-localized expression of FGF23 with markers for erythroid, myeloid, and dendritic cells [76]. Flow cytometry of marrow cells demonstrated that the hematopoietic precursors designated as lineage- c-kit+Sca-1+ (LSK) cells significantly express Fgf23 mRNA after EPO administration [80, 84••]. Many of these studies have focused on the acute effects of EPO but CKD patients are maintained on EPO replacement therapy for prolonged durations to correct hematocrit and hemoglobin levels. Thus, future studies are needed to elucidate the chronic effects of EPO on FGF23 especially in the setting of renal failure.
Iron Supplementation
In light of the iron deficiency results from ADHR patients and ADHR mutant mice, iron repletion was tested as a therapeutic option to correct the elevated iFGF23 driving hypophosphatemia [48]. In ADHR mice, replacement of the low iron diet with an iron-repleted diet reversed the disease phenotype. Serum levels of iFGF23 of the ADHR mice returned to wild-type levels and serum phosphorus was restored to normal [86]. A patient with an R176Q FGF23 mutation exhibited late onset of the disease and when treated with intravenous iron II sulfate, rather than the standard phosphorus and 1,25D regimen, all disease-associated endocrine disturbances were rescued [87]. Serum phosphorus and 1,25D levels improved with normalization of serum FGF23. These data initiated a clinical trial for treating ADHR with iron supplementation beyond therapy using FGF23 stimulating factors. Whereas this iron preparation was able to reduce the pathogenic elevation of iFGF23, hypophosphatemia has been noted to occur with iron supplementation for other pathologies [88]. Two major categories exist for iron formulations including oral iron and intravenous iron. Oral iron has been a long-standing supplementation for the treatment of iron deficiency and anemia [89]. These preparations are extremely cost-effective, but due to the requirement of intestinal absorption, they are found to induce adverse side effects including nausea, vomiting, and diarrhea resulting in non-adherence. Intravenous (IV) iron preparations are chosen to circumvent gastrointestinal side effects and increase efficacy in correcting iron deficiency as measured by transferrin saturation or hemoglobin. This is especially important in late stage CKD patients as well as patients diagnosed with Crohn’s disease or inflammatory bowel disease when the intestinal absorption of iron is inhibited. Iron preparations for infusion are complexed with carbohydrate ligands to reduce unregulated release of labile iron known to cause oxidative stress [90]. Iron polymaltose was introduced in 1978 and is widely approved for the treatment of anemia. Hypophosphatemia and osteomalacia have been reported in multiple cases where patients harboring blood loss conditions were treated for their iron deficiency anemia with long-term iron polymaltose infusions [91]. While in both cases bone pain was associated with iron infusions, one patient sustained minimal trauma fractures due to persisting osteomalacia. Renal phosphate wasting was found secondary to increased iFGF23 and cessation of iron allowed for iFGF23 levels, and therefore serum phosphorus concentrations to return to normal. Ferric carboxymaltose (FCM) came on the market to replace iron polymaltose as it was well tolerated at higher doses and harbored less adverse events [92]. Hypophosphatemia was observed in a transient fashion when FCM was used to treat anemic premenopausal women [52]. In this initial study, cFGF23 was measured prior to treatment and was significantly elevated compared to controls owing to the effect of iron deficiency on FGF23 expression. Interestingly, immediately after FCM infusion cFGF23 levels decreased with a coordinating dramatic increase in serum iFGF23 leading to hypophosphatemia [52]. Iron dextran, also a high molecular weight-infused preparation, showed a similar reduction in cFGF23 in response to FCM with no change in iFGF23. Many other case reports have emerged in different patient populations receiving FCM that have exhibited hypophosphatemia due to significant elevations of iFGF23 [93,94,95,96]. These studies demonstrated that specific carboxyhydrate ligands reduce the ability to cleave iFGF23 into its inactivated fragments.
Treating functional iron deficiency and anemia in CKD is important as it is linked to increased morbidity and mortality in these patients [97,98,99]. This could be potentially mediated through FGF23 as iron deficiency and anemia can induce Fgf23 expression, and FGF23 has been linked to mortality through cardiovascular events in CKD [29, 100]. In a prospective randomized study, CKD patients with iron-deficiency anemia were treated with either oral iron (50 mg sodium ferrous citrate daily) or IV iron (40 mg of saccharated ferric oxide weekly) for 10 weeks. Serum cFGF23 levels were reduced in both groups, whereas only IV iron increased serum iFGF23 levels [101], suggesting oral preparations as preferential for iron supplementation to avoid further increases in iFGF23. This study was performed on CKD patients undergoing maintenance hemodialysis, and many of the patients received EPO or EPO-stimulating agents throughout the study. Thus, it is unclear whether EPO harbored effects on the ratio of iFGF23 to cFGF23. Besides iron deficiency, it is important to note that CKD is a multi-faceted disease state that has effects on additional factors known to induce FGF23. Among these changes is dysregulation of phosphate homeostasis that leads to elevated serum phosphorus due to a lack of excretion. To combat this loss of regulation, phosphate binders have been developed. The first iteration of calcium-containing phosphate binders was cost-effective. However, they increased the risk of hypercalcemia and vascular calcification. Sevelamer is a calcium-free phosphate binder, reducing the risk of hypercalcemia, yet its efficacy for reducing phosphorus is suboptimal and fails to significantly reduce FGF23 [102]. Iron-based phosphate binders have emerged as a new class in the form of sucroferric oxyhydroxide and ferric citrate. In comparison to sevelamer, sucroferric oxyhydroxide demonstrated an ability to lower serum phosphorus to a greater extent [103] and also modestly improved iron parameters [104] due to its low iron release. Importantly, CKD patients on hemodialysis treated with sucroferric oxyhydroxide also showed a significant reduction in serum FGF23. Ferric citrate acts similarly in that the efficacy in reducing serum phosphorus is superior to sevelamer [105] and also significantly improves iron parameters as measured by ferritin and transferrin saturation [106]. In hemodialysis CKD patients, ferric citrate improved serum iron and hemoglobin, which was also associated with a reduction in serum FGF23 [107]. In contrast, treatment with ferric citrate in non-dialysis CKD patients had a modest reduction in serum FGF23 that did not reach significance. However, the study utilized a small sample size, which likely did not have enough power to demonstrate significant effects [108]. It is therefore possible that simultaneously reduced serum phosphorus and iron deficiency correction provides the most beneficial reduction in iFGF23. Nevertheless, it is difficult with this compound to tease out the effects of each component independently on iFGF23 levels.
Conclusion
In summary, dovetailed mouse and human studies involving iron and iron-sensing pathways have elucidated novel aspects of FGF23 regulation. Iron deficiency, through HIF1α and EPO, independently activate Fgf23 mRNA expression as well as iFGF23 depending upon the context, including presence of ADHR mutation and status of renal function. These laboratory findings have therefore provided evidence for important considerations in the clinical application of iron supplementation. The evolutionary aspect of intertwined iron and phosphate homeostasis remains unclear. However, recent studies have established a strong foundation to interrogate these mechanistic questions further and eventually optimize therapeutic regimens.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–5.
Weber TJ, Liu S, Indridason OS, Quarles LD. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res. 2003;18(7):1227–34.
Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005;16(2):221–32.
Cho HY, Lee BH, Kang JH, Ha IS, Cheong HI, Choi Y. A clinical and molecular genetic study of hypophosphatemic rickets in children. Pediatr Res. 2005;58(2):329–33.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7.
Yu X, White KE. Fibroblast growth factor 23 and its receptors. Ther Apher Dial. 2005;9(4):308–12.
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444(7120):770–4.
Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;15(4):437–41.
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–3.
Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, et al. Alpha-klotho is a non- enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018;553(7689):461–6.
Farrow EG, Summers LJ, Schiavi SC, McCormick JA, Ellison DH, White KE. Altered renal FGF23-mediated activity involving MAPK and Wnt: effects of the Hyp mutation. J Endocrinol. 2010;207(1):67–75.
Yamazaki Y, Tamada T, Kasai N, Urakawa I, Aono Y, Hasegawa H, et al. Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J Bone Miner Res. 2008;23(9):1509–18.
Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, et al. Human fibroblast growth factor-23 mutants suppress Na+−dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278(4):2206–11.
Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. 2006;69(3):495–503.
Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17(5):1305–15.
Econs MJ, McEnery PT, Lennon F, Speer MC. Autosomal dominant hypophosphatemic rickets is linked to chromosome 12p13. J Clin Invest. 1997;100(11):2653–7.
ADHR-Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 2000;26(3):345–348.
Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36(6):579–81.
Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, et al. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab. 2006;91(10):4037–42.
Frishberg Y, Ito N, Rinat C, Yamazaki Y, Feinstein S, Urakawa I, et al. Hyperostosis- hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22(2):235–42.
Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014;111(15):5520–5.
Alem AM, Sherrard DJ, Gillen DL, Weiss NS, Beresford SA, Heckbert SR, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int. 2000;58(1):396–9.
Lefebvre P, Vekeman F, Sarokhan B, Enny C, Provenzano R, Cremieux PY. Relationship between hemoglobin level and quality of life in anemic patients with chronic kidney disease receiving epoetin alfa. Curr Med Res Opin. 2006;22(10):1929–37.
Jamal SA. Bone mass measurements in men and women with chronic kidney disease. Curr Opin Nephrol Hypertens. 2010;19(4):343–8.
Rolvien T, Kornak U, Schinke T, Amling M, Oheim R. A novel FAM20C mutation causing hypophosphatemic osteomalacia with osteosclerosis (mild Raine syndrome) in an elderly man with spontaneous osteonecrosis of the knee. Osteoporos Int. 2018.
Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr, Pavletich NP. Structure of an HIF- 1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296(5574):1886–9.
Hsu CY, McCulloch CE, Curhan GC. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the third National Health and nutrition examination survey. J Am Soc Nephrol. 2002;13(2):504–10.
Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108(12):3730–5.
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–408.
Grabner A, Mazzaferro S, Cianciolo G, Krick S, Capelli I, Rotondi S, et al. Fibroblast growth factor 23: mineral metabolism and beyond. Contrib Nephrol. 2017;190:83–95.
Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A. 2011;108(46):E1146–55.
Van Buren PN, Lewis JB, Dwyer JP, Greene T, Middleton J, Sika M, et al. The phosphate binder ferric citrate and mineral metabolism and inflammatory markers in maintenance Dialysis patients: results from Prespecified analyses of a randomized clinical trial. Am J Kidney Dis. 66(3):479–88.
Geissler C, Singh M. Iron, meat and health. Nutrients. 2011;3(3):283–316.
McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, et al. An iron- regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291(5509):1755–9.
Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85.
Brannon PM, Taylor CL. Iron Supplementation during Pregnancy and Infancy: uncertainties and implications for research and policy. Nutrients. 2017;9(12).
Muller O, Krawinkel M. Malnutrition and health in developing countries. CMAJ. 2005;173(3):279–86.
Skalicky A, Meyers AF, Adams WG, Yang Z, Cook JT, Frank DA. Child food insecurity and iron deficiency anemia in low-income infants and toddlers in the United States. Matern Child Health J. 2006;10(2):177–85.
Diaz-Castro J, Lopez-Frias MR, Campos MS, Lopez-Frias M, Alferez MJ, Nestares T, et al. Severe nutritional iron-deficiency anaemia has a negative effect on some bone turnover biomarkers in rats. Eur J Nutr. 2012;51(2):241–7.
Cartwright GE, Lauritsen MA, Humphreys S, Jones PJ, Merrill IM, Wintrobe MM. The Anemia associated with chronic infection. Science. 1946;103(2664):72–3.
Cartwright GE, Lauritsen MA, Jones PJ, Merrill IM, Wintrobe MM. The Anemia of infection. I. Hypoferremia, hypercupremia, and alterations in porphyrin metabolism in patients. J Clin Invest. 1946;25(1):65–80.
Qamar K, Saboor M, Qudsia F, Khosa SM. Moinuddin, Usman M. Malabsorption of iron as a cause of iron deficiency anemia in postmenopausal women. Pak J Med Sci. 2015;31(2):304–8.
Filmann N, Rey J, Schneeweiss S, Ardizzone S, Bager P, Bergamaschi G, et al. Prevalence of anemia in inflammatory bowel diseases in european countries: a systematic review and individual patient data meta-analysis. Inflamm Bowel Dis. 2014;20(5):936–45.
Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol. 2006;19(2):161–7.
Lankhorst CE, Wish JB. Anemia in renal disease: diagnosis and management. Blood Rev. 2010;24(1):39–47.
Econs MJ, McEnery PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82(2):674–81.
Imel EA, Hui SL, Econs MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res. 2007;22(4):520–6.
Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant Hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. 2011;96:3541–9.
Vieth JT, Lane DR. Anemia. Emerg Med Clin North Am. 2014;32(3):613–28.
Bryan LJ, Zakai NA. Why is my patient anemic? Hematol Oncol Clin North Am. 2012;26(2):205–30 vii.
Rabadi S, Udo I, Leaf DE, Waikar SS, Christov M. Acute blood loss stimulates fibroblast growth factor 23 production. Am J Physiol Renal Physiol. 2018;314(1):F132–F9.
Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res. 2013;28(8):1793–803.
Lewerin C, Ljunggren O, Nilsson-Ehle H, Karlsson MK, Herlitz H, Lorentzon M, et al. Low serum iron is associated with high serum intact FGF23 in elderly men: the Swedish MrOS study. Bone. 2017;98:1–8.
Erlitzki R, Long JC, Theil EC. Multiple, conserved iron-responsive elements in the 3′- untranslated region of transferrin receptor mRNA enhance binding of iron regulatory protein 2. J Biol Chem. 2002;277(45):42579–87.
Thomson AM, Rogers JT, Leedman PJ. Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int J Biochem Cell Biol. 1999;31(10):1139–52.
Piccinelli P, Samuelsson T. Evolution of the iron-responsive element. RNA. 2007;13(7):952–66.
Sanchez M, Galy B, Schwanhaeusser B, Blake J, Bahr-Ivacevic T, Benes V, et al. Iron regulatory protein-1 and -2: transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood. 2011;118(22):e168–79.
Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90(9):4304–8.
Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. 2007;117(7):1926–32.
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–72.
Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2002;99(21):13459–64.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL- mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–8.
Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High- resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117(23):e207–17.
Bailey PSJ, Nathan JA. Metabolic Regulation of Hypoxia-Inducible Transcription Factors: The Role of Small Molecule Metabolites and Iron. Biomedicines. 2018;6(2).
Bianchi L, Tacchini L, Cairo G. HIF-1-mediated activation of transferrin receptor gene transcription by iron chelation. Nucleic Acids Res. 1999;27(21):4223–7.
Weinberg ED. Iron withholding: a defense against infection and neoplasia. Physiol Rev. 1984;64(1):65–102.
David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89(1):135–46.
Zhang Q, Doucet M, Tomlinson RE, Han X, Quarles LD, Collins MT, et al. The hypoxia- inducible factor-1alpha activates ectopic production of fibroblast growth factor 23 in tumor- induced osteomalacia. Bone Res. 2016;4:16011.
•• Onal M, Carlson AH, Thostenson JD, Benkusky NA, Meyer MB, Lee SM, et al. A Novel Distal Enhancer Mediates Inflammation-, PTH-, and Early Onset Murine Kidney Disease- Induced Expression of the Mouse Fgf23 Gene. JBMR Plus. 2018;2(1):32–47 This study demonstrated the regulation of FGF23 by a distal upstream enhancer.
Bruning U, Fitzpatrick SF, Frank T, Birtwistle M, Taylor CT, Cheong A. NFkappaB and HIF display synergistic behaviour during hypoxic inflammation. Cell Mol Life Sci. 69(8):1319–29.
Jacobson LO, Goldwasser E, Fried W, Plzak L. Role of the kidney in erythropoiesis. Nature. 1957;179(4560):633–4.
Franke K, Gassmann M, Wielockx B. Erythrocytosis: the HIF pathway in control. Blood. 2013;122(7):1122–8.
Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. 2000;275(38):29338–47.
Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–84.
Arezes J, Foy N, McHugh K, Sawant A, Quinkert D, Terraube V, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. 2018;132:1473–7.
Daryadel A, Bettoni C, Haider T, Imenez Silva PH, Schnitzbauer U, Pastor-Arroyo EM, et al. Erythropoietin stimulates fibroblast growth factor 23 (FGF23) in mice and men. Pflugers Arch. 2018;470:1569–82.
Hanudel MR, Eisenga MF, Rappaport M, Chua K, Qiao B, Jung G, et al. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol Dial Transplant. 2018.
Gupta N, Wish JB. Hypoxia-inducible factor prolyl hydroxylase inhibitors: a potential new treatment for Anemia in patients with CKD. Am J Kidney Dis. 2017;69(6):815–26.
Flamme I, Ellinghaus P, Urrego D, Kruger T. FGF23 expression in rodents is directly induced via erythropoietin after inhibition of hypoxia inducible factor proline hydroxylase. PLoS One. 2017;12(10):e0186979.
Toro L, Barrientos V, Leon P, Rojas M, Gonzalez M, Gonzalez-Ibanez A, et al. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int. 2018;93(5):1131–41.
Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. 2012;23(10):1631–4.
Hinata A, Iijima M, Nakano Y, Sakamoto T, Tomita M. Chemical characterization of rabbit alpha 2-macroglobulin. Chem Pharm Bull (Tokyo). 1987;35(1):271–6.
Landau D, London L, Bandach I, Segev Y. The hypoxia inducible factor/erythropoietin (EPO)/EPO receptor pathway is disturbed in a rat model of chronic kidney disease related anemia. PLoS One. 2018;13(5):e0196684.
•• Clinkenbeard EL, Hanudel MR, Stayrook KR, Appaiah HN, Farrow EG, Cass TA, et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica. 2017;102(11):e427–e30 This study demonstrated EPO stimulation of FGF23 independent of HIF that occurs in both osteoblast/osteocytes as well as hematopoietic lineage cells.
Singbrant S, Russell MR, Jovic T, Liddicoat B, Izon DJ, Purton LE, et al. Erythropoietin couples erythropoiesis, B-lymphopoiesis, and bone homeostasis within the bone marrow microenvironment. Blood. 2011;117(21):5631–42.
Clinkenbeard EL, Farrow EG, Summers LJ, Cass TA, Roberts JL, Bayt CA, et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res. 2014;29(2):361–9.
Duval F, Mokrani MC, Monreal J, Weiss T, Fattah S, Hamel B, et al. Interaction between the serotonergic system and HPA and HPT axes in patients with major depression: implications for pathogenesis of suicidal behavior. Dialogues Clin Neurosci. 2002;4(4):417.
Okada M, Imamura K, Fuchigami T, Omae T, Iida M, Nanishi F, et al. 2 cases of nonspecific multiple ulcers of the small intestine associated with osteomalacia caused by long- term intravenous administration of saccharated ferric oxide. Nihon Naika Gakkai zasshi The Journal of the Japanese Society of Internal Medicine. 1982;71(11):1566–72.
Auerbach M, Macdougall IC. Oral Iron therapy: after three centuries, it is time for a change. Am J Kidney Dis. 2016;68(5):665–6.
Geisser P, Burckhardt S. The pharmacokinetics and pharmacodynamics of iron preparations. Pharmaceutics. 2011;3(1):12–33.
Bishay RH, Ganda K, Seibel MJ. Long-term iron polymaltose infusions associated with hypophosphataemic osteomalacia: a report of two cases and review of the literature. Ther Adv Endocrinol Metab. 2017;8(1–2):14–9.
Gilmartin CE, Hoang T, Cutts BA, Leung L. Retrospective cohort study comparing the adverse reactions and efficacy of intravenous iron polymaltose with ferric carboxymaltose for iron deficiency anemia. Int J Gynaecol Obstet. 2018;141(3):315–20.
Urbina T, Belkhir R, Rossi G, Carbonnel F, Pavy S, Collins M, et al. Iron supplementation-induced Phosphaturic Osteomalacia: FGF23 is the culprit. J Bone Miner Res. 2018;33(3):540–2.
Bartko J, Roschger P, Zandieh S, Brehm A, Zwerina J, Klaushofer K. Hypophosphatemia, severe bone pain, gait disturbance, and fatigue fractures after Iron substitution in inflammatory bowel disease: a case report. J Bone Miner Res. 2018;33(3):534–9.
Klein K, Asaad S, Econs M, Rubin JE. Severe FGF23-based hypophosphataemic osteomalacia due to ferric carboxymaltose administration. BMJ Case Rep. 2018;2018.
Tulewicz-Marti E, Moniuszko A, Rydzewska G. Management of anemia in inflammatory bowel disease: a challenge in everyday clinical practice. Przeglad gastroenterologiczny. 2017;12(4):239–43.
Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–92.
Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305(23):2432–9.
Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. 2011;22(5):956–66.
Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis. 2009;207(2):546–51.
Fukao W, Hasuike Y, Yamakawa T, Toyoda K, Aichi M, Masachika S, et al. Oral versus intravenous Iron supplementation for the treatment of Iron deficiency Anemia in patients on maintenance hemodialysis-effect on fibroblast growth Factor-23 metabolism. J Renal Nutr. 2018;28(4):270–7.
Liabeuf S, Ryckelynck JP, El Esper N, Urena P, Combe C, Dussol B, et al. Randomized clinical trial of Sevelamer carbonate on serum klotho and fibroblast growth factor 23 in CKD. Clin J Am Soc Nephrol. 2017;12(12):1930–40.
Koiwa F, Yokoyama K, Fukagawa M, Terao A, Akizawa T. Efficacy and safety of sucroferric oxyhydroxide compared with sevelamer hydrochloride in Japanese haemodialysis patients with hyperphosphataemia: a randomized, open-label, multicentre, 12-week phase III study. Nephrology (Carlton). 2017;22(4):293–300.
Covic AC, Floege J, Ketteler M, Sprague SM, Lisk L, Rakov V, et al. Iron-related parameters in dialysis patients treated with sucroferric oxyhydroxide. Nephrol Dial Transplant. 2017;32(8):1330–8.
Yang WC, Yang CS, Hou CC, Wu TH, Young EW, Hsu CH. An open-label, crossover study of a new phosphate-binding agent in haemodialysis patients: ferric citrate. Nephrol Dial Transplant. 2002;17(2):265–70.
Lee CT, Wu IW, Chiang SS, Peng YS, Shu KH, Wu MJ, et al. Effect of oral ferric citrate on serum phosphorus in hemodialysis patients: multicenter, randomized, double-blind, placebo- controlled study. J Nephrol. 2015;28(1):105–13.
Maruyama N, Otsuki T, Yoshida Y, Nagura C, Kitai M, Shibahara N, et al. Ferric citrate decreases fibroblast growth factor 23 and improves erythropoietin responsiveness in hemodialysis patients. Am J Nephrol. 2018;47(6):406–14.
Iguchi A, Yamamoto S, Yamazaki M, Tasaki K, Suzuki Y, Kazama JJ, et al. Effect of ferric citrate hydrate on FGF23 and PTH levels in patients with non-dialysis-dependent chronic kidney disease with normophosphatemia and iron deficiency. Clin Exp Nephrol. 2018;22(4):789–96.
Funding
The authors would like to acknowledge NIH grants F32-AR065389 (ELC); the Comprehensive Training Program in Musculoskeletal Research Grant T32-AR065971 (JAW) and a Center for Translational Sciences Institutional Biomedical Research Grant (ELC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jonathan A. Wheeler and Erica L. Clinkenbeard each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Molecular Control of Phosphorus Homeostasis
Rights and permissions
About this article

Cite this article
Wheeler, J.A., Clinkenbeard, E.L. Regulation of Fibroblast Growth Factor 23 by Iron, EPO, and HIF. Curr Mol Bio Rep 5, 8–17 (2019). https://doi.org/10.1007/s40610-019-0110-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-019-0110-9