
Abstract
Fibroblast growth factor 23 (FGF23) has emerged as an important regulator of phosphate and vitamin D homeostasis. It is important to understand how FGF23 interacts with vitamin D and parathyroid hormone (PTH) in a FGF23-Vitamin D-PTH axis to regulate mineral homeostasis. In this review, we discuss the genomic structure, and transcriptional, translational, and posttranslational regulation of FGF23. We describe its interaction with PTH and vitamin D, disorders of altered FGF23 states, and emerging therapies for diseases of FGF23 based upon these findings. This discussion helps redefine the role of PTH and vitamin D in relation to a complex bone-kidney-parathyroid loop, and points to areas within this complicated field in need of further clarification and research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
While parathyroid hormone (PTH) and 1,25 dihydroxy vitamin D3 (1,25-D) have long been known to be major regulators of blood calcium and phosphate, the discovery of fibroblast growth factor-23 (FGF23) revolutionized our understanding of phosphate and vitamin D homeostasis and established a PTH-vitamin D-FGF23 axis. PTH, 1,25-D, and FGF23 interact in a complex, multi-tissue feedback system that serve to maintain blood phosphate and calcium levels.
The existence of a “rachitogenic substance” was first postulated when a paraneoplastic disease characterized by phosphaturia and hypophosphatemia resolved following the removal of a small mesenchymal tumor, which presumably secreted such a factor [1]. Using a rodent model of X-linked hypophosphatemic rickets (XLH, the hyp mouse), Meyers et al. demonstrated that the renal phosphate wasting factor was transferrable to normal mice by parabiosis [2]. Thus, it was evident that it was a circulating hormone that was responsible for XLH. Positional cloning studies of a group of subjects with autosomal dominant hypophosphatemic rickets (ADHR) revealed missense mutations in the gene on chromosome 12 that coded for a protein with an FGF-like domain, and which was eventually labeled as FGF23 [3–7].
Genetic characterization of inherited disorders of phosphate wasting have helped define the role of FGF23 as a crucial regulator in the complex relationship between phosphate and vitamin D metabolism. Further confirmation came from the discovery that elevated levels of FGF23 were responsible for tumor-induced osteomalacia (TIO) [6, 7]. In rapid succession, FGF23 was implicated in other phosphate-wasting syndromes including autosomal recessive hypophosphatemic rickets [8–12], X-linked hypophosphatemic rickets [13], and fibrous dysplasia [14, 15]. There are currently at least 11 disorders of altered phosphate homeostasis associated with alternations in FGF23 physiology that have been well characterized (Table 1).
While we have made progress in describing the pathophysiology and genetic basis of FGF23 disorders, challenges persist. Reciprocal and homeostatic changes can often limit our ability to tease out a cause-and-effect mechanism. This is particularly evident in tightly controlled systems such as phosphate and vitamin D metabolism. Multilevel feedback and the kinetics of hormone action/reaction within the bone-kidney-parathyroid loop system can complicate the investigation of a hormone of interest. In addition, the shortcomings of animal and in vitro systems as models of human physiology can introduce confusion.
It is also becoming evident that FGF23 physiology as a primary disease of mineral metabolism is distinct from the secondary response in renal disease. Significant changes in FGF23 secretion and action in early through late stage renal disease may have broad health implications and is still being further elucidated. This review will focus on emerging areas of FGF23 molecular processing, physiologic regulation of FGF23 in non-renal disease versus renal disease, and human disorders of FGF23.
2 Genomic and structural organization and physiologic of FGF23
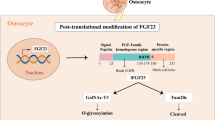
The mouse and human FGF23 orthologs were discovered based on structural similarity to other FGFs [16]. The human gene is located on chromosome 12p13, is 9386 nucleotides, and consists of 3 exons separated by 2 introns. It encodes a 32-kDa glycoprotein containing 251 amino acid residues (Fig. 1). The secretory hormone has a 24 amino acid hydrophobic signal sequence. The rat and mouse FGF23 amino acid sequences are 72 and 71 % homologous to human FGF23, respectively [17]. The N-terminal has a FGF homology domain and the C-terminal domain has a unique 72 amino acid sequence. FGF23 acts as an endocrine hormone by interaction of the C-terminus with its co-receptor α-klotho (αKl); the N-terminus contains the FGF receptor (FGFR) binding domain [18, 19]. In addition, while the 5′-upstream promoter region of FGF23 gene is largely conserved between mice and humans, an important difference is that while the mouse gene contains a consensus vitamin D receptor (VDR) the human gene does not (Fig. 1).

Human FGF23 structure including genomic organization, transcript profile and protein features. Information obtained from the National Center for Biotechnology Information (NCBI) databases regarding the nucleotide and amino acid sequences for the FGF23 gene (NC_000012), transcript (NM_020638) and protein (GenBank EAW88848). The three exons within the FGF23 gene are marked by boxes (blue), and the FGF23 coding region (open reading frame region; ORF) is also marked (blue box). FGF23 mRNA depicts the 5′- and 3′-untranslated regions (5′Unt and 3′Unt) as indicated. Putative O-glycosylation sites and phosphorylation sights are indicated. The subtilisin proprotein convertase (SPC) protease processing site where cleavage into N- and C-terminal fragments are indicated (protein and associated molecular weight depicted by yellow and green lines, respectively)
3 Phosphate and FGF23
Understanding the molecular mechanisms by which FGF23 participates in the PTH-vitamin D-FGF23 axis requires a familiarity with the mechanism of action of FGF23 to regulate serum phosphate. Phosphate is important in many metabolic processes, including intracellular signaling, enzymatic reactions, energy metabolism and bone structure. Therefore, the effort to maintain a normal serum phosphate level reflects a tightly regulated homeostasis between gastrointestinal absorption, bone storage (in the form of hydroxyapatite) and predominately renal phosphate excretion (one-third is lost in the feces) [20]. It is estimated that 80–85 % of filtered phosphate is reabsorbed in the proximal tubule via sodium-phosphate co-transporters (NaPi) 2a and 2c. Therefore, it is not surprising that when induced by various factors, FGF23 directly down-regulates the transcription, translation, and translocation of NaPi 2a and 2c transporters leading to reduced renal phosphate reabsorption. FGF23 also directly down-regulates renal 1α-hydroxylase which decreases production of active 1,25-D, thereby decreasing intestinal phosphate and calcium absorption. Finally, a compensatory increase in PTH occurs in response to relatively or frankly low 1,25-D to maintain eucalcemia [6, 21, 22].
In vitro experiments with proximal renal tubule cells have confirmed the FGF23-mediated regulation of transcription and translation of the NaPi2a and 1-α hydroxylase genes [23, 24]. FGF23 action at the proximal tubules would require local αKl expression. While some have suggested that αKl is not expressed in proximal tubule cells [25], others have demonstrated expression of αKl in these cells [26].
4 PTH and FGF23
The relationship between PTH and FGF23 is complex. One might predict that the parathyroid would be a target for FGF23, especially given parathyroid hormone has been considered the primary regulator of renal phosphate reabsorption until the discovery of FGF23. Animal and in vitro data suggests that FGF23 directly inhibits PTH synthesis and secretion in an αKl-dependent manner [27, 28]. Indeed, there is evidence that Klotho is expressed on both rodent and human parathyroid cells. However, clinical evidence that the parathyroid is a target of αKl-mediated FGF23 action is less clear. A single patient homozygous for a mis-sense mutation in αKl presented with hyperparathyroidism, thought to be due to a loss of an αKl-dependent inhibitory effect of FGF23 on PTH production [29]. Yet, in a patient with a translocation that resulted in increased alphaKL production and elevated FGF23, Brownstein et al. [30] also found increased PTH secretion, suggesting that PTH secretion is stimulated by FGF23 in a KL-dependent manner. In addition, FGF23 excess disorders such as TIO and XLH also do not demonstrate inhibition of PTH synthesis and secretion in the setting of markedly elevated FGF23 [31].
In the case of renal failure, where secondary hyperparathyroidism (sometimes leading to tertiary hyperparathyroidism) is a common finding, blood FGF23 levels can be exceptionally elevated in parallel with elevated PTH (Fig. 2). Therefore, in informative clinical states, evidence is lacking to support an inhibitory effect of FGF23 on PTH secretion.

Various levels of fibroblast growth factor 23 (FGF23) in healthy individuals and in different disease states, including chronic kidney disease (CKD; depicted by orange bars) and various primary hypophosphatemic disorders (depicted by blue bars). The dual y axis demonstrates FGF23 levels based on different commercially available assays, namely the intact FGF23 assay (iFGF23) and the C terminus assay (cFGF23) which detects both iFGF23 and cFGF23 [32]. The shaded gray area represents an incompletely defined normal range. ‘1° hypoP, FGF23’ refers to hypophosphatemic disorders caused by primary FGF23 excess, for example, X-linked hypophosphatemia [33] ‘1° hypoP, non-FGF23’ refers to hypophosphatemic disorders caused by mechanisms other than FGF23 excess, for example, hereditary hypophosphatemic rickets with hypercalciuria, in which FGF23 levels are secondarily suppressed [34]. ESRD, end-stage renal disease; Tx, transplantation. Reprinted by permission from Macmillan Publishers Ltd: Kidney International 82, 737–747, copyright (2012)
5 FGF23: modulators of transcription, translation, and post-translational processing
FGF23 transcriptional regulation by phosphate
Controversy exists regarding the regulation of FGF23 by dietary phosphate, as some studies have failed to show alternations in FGF23 levels based on dietary changes [35, 36]. However, in larger studies and those over a longer time period, dietary phosphate appears to modulate FGF23 levels [37–40]. A recent study [41] examined systemic phosphate loading (both intravenous and intestinal) and found almost identical early dose-dependent increases in plasma PTH followed by increases in FGF23. After a lag of about 36–48 h, 1,25-D decreased significantly. This suggests that serum phosphate levels are a direct secretagogue for FGF23, however, the temporal response to acute phosphate changes are slower than effects on PTH. It is unclear if the signal derives from the rise in PTH, as the phosphate-sensing mechanism in FGF23-secreting cells remains elusive.
There are also data to suggest that calcium may participate in a synergistic fashion with phosphate to regulate FGF23 levels [42]. In a particularly informative study that involved the PTH knockout mouse, the calcium-sensing receptor knockout mouse and/or the double knockout mouse, Quinn et al. showed that the greatest changes in serum FGF23 were accounted for by changes in the calcium x phosphate product, suggesting that both calcium and phosphorus may act in a synergistic fashion to regulate FGF23 levels.
FGF23 transcriptional regulation by PTH
PTH infusion studies in animals and human also yield conflicting results. Increases in murine osteocyte FGF23 mRNA expression in vitro have been demonstrated via activation of the PTHR1 receptor as well as increases in serum FGF23 via the PTHR1 ligand [43, 44]. Others have demonstrated decreased FGF23 with PTH treatment in animal studies [45–47]. In humans, Burnett-Bowie and colleagues [48] demonstrated that PTH infusion in healthy men increased FGF23 over 18 h, while Guiterrez [49] demonstrated acute lowering of FGF23 with PTH infusion within 6 h, suggesting either a role of 1,25-D as an acute regulator or an unknown alternative mechanism for PTH-FGF23 interaction.
One would predict that subjects with primary hyperparathyroidism might offer insight into an effect of PTH on FGF23, but the data from multiple studies are inconclusive [50–54]. Some report a direct effect of PTH on FGF23 levels, while others do not. The most common disease of elevated PTH is renal insufficiency/failure, which will be discussed in detail later. Other diseases of increased PTH signaling, such as Jansen’s metaphyseal chondrodysplasia (due to activating mutations of the PTH/PTHrP receptor) and fibrous dysplasia (FD) (due to activating mutations downstream of PTH at Gsα), demonstrate concomitant overexpression of FGF23 [15, 55] as well as increased processing into N- and C-terminal fragments. These inconsistencies, found across many studies in humans and animals, may be explained by several potentially confounding factors that are difficult to control for: the hypophosphatemic effect of PTH, the relatively elevated 1,25-D in hyperparathyroidism, the variable severity and possible independent effect of blood calcium levels.
FGF23 transcriptional regulation by 1,25-dihydroxyvitamin D3
The mechanism by which 1,25-D regulates FGF23 is presumed to be via the VDR. Our data [56] demonstrate that in patients with various forms of hypoparathyroidism, increases in serum 1,25-D was associated with an increase in serum FGF-23. Treatment with calcitriol in hypoparathyroid patients increased FGF23 and rapidly decreased serum phosphate, despite the presumed role of 1,25-D to increase phosphate reabsorption in the gut. Yet even with high FGF23 levels in these subjects, without a PTH effect, there was a diminished ability induce phosphaturia [56, 57].
Animal studies have supported a role for 1,25-D in the regulation of FGF23. VDR null mice have very low serum FGF23, but when given a rescue diet to normalize secondary hyperparathyroidism and low phosphate, FGF23 normalizes [58], but they do not respond to 1,25-D administration [59]. In both thyroparathyroidectomized rats and normal mice, administration of 1,25-D increases serum FGF23 [22, 59], much like the human experience. Ito et al. [60] also reported in vitro data in human chronic myelogenous leukemia K562 cells, demonstrating 1,25-D enhanced FGF23 promoter activity and mRNA expression. These differences may be a reflection of species differences (humans vs rodents) in terms of FGF23 responsivity owing in part the gene structure differences cited earlier, i.e., the lack of a strong consensus VDR in the human gene (Fig. 1).
It is also postulated that the regulation of FGF23 involves a proteolytic cleavage product, acidic serine aspartate-rich matrix extracellular phosphoglycoprotein (MEPE)-associated motif (ASARM) peptide. This peptide has been shown to cause mineralization defects in the mouse model of X-linked hypophosphatemic rickets, the hyp mouse, and “directly” inhibit renal phosphate uptake in vitro [61]. It is a complicated mechanism, the details of which go beyond the scope of this paper. Briefly, Rowe and colleagues [62, 63] have demonstrated that MEPE reversibly interacts with the X-lined phosphate regulating endopeptidase homolog, (PHEX). In normal individuals, this interaction of PHEX - MEPE protects MEPE from proteolysis and maintains mineralization. The ASARM motif plays a major role in the PHEX–MEPE interaction, and when PHEX is defective (hyp mouse model), the increased protease resistant ASARM peptides inhibit mineralization and disrupt renal phosphate handling, resulting in hypophosphatemia and rickets/osteomalacia.
The potential role of the ASARM peptide in regulating FGF23 calls into question the role of the gene products of several genes that have been shown to underlie diseases of FGF23 excess (Table 1).
FGF23 post-translational regulation
The regulation of FGF23 protein processing is an area that has made recent gains. We have known since 2001 that that the N- and C- terminal domains are separated by a consensus proteolytic cleavage site (RXXR, 176RHTR179), susceptible to proteolysis by a subtilisin-like proprotein convertase (SPC), such as furin [6]. SPC action and cleavage into N- and C-terminal fragments inactivates FGF23. This is a key step in the regulation of FGF23 activity because it appears only intact FGF23 (iFGF23) is biological active. This is supported by the fact that loss of function mutations in the GALNT3 cause the disease hyperphosphatemic familial tumoral calcinosis/hyperostosis syndrome (HFTC/HHS), a disorder of intact FGF23 deficiency [64]. In this disease, FGF23 is not glycosylated at 178T by GALNT3 and as a result is processed by a SPC into biologically inactive N- and C-terminal fragments. The SPC site in FGF23 is unique within the FGF family and conserved across all mammals; all of the original families with ADHR had mutations at this site [5].
Recent evidence suggests that phosphorylation may also play a role in the posttranslational regulation of FGF23 [65]. The current evidence suggests that the kinase family with sequence similarity 20, member C (FAM20C) phosphorylates FGF23 at Ser180. If phosphorylated, then glycosylation is prevented and FGF23 is degraded by an SPC to its biologically inactive N- and C-terminal. This is supported by findings in the Fam20c knockout mice, which are hypophosphatemic [66]. Confusing, however, is the fact that the human disease of FAM20C deficiency, Raine syndrome (OMIM 259775), is a lethal osteosclerotic disease without a mineral metabolism phenotype [67]. Therefore, additional work is needed to clarify the roles of GALNT3 and FAM20C in FGF23 biology and physiology.
FGF23 regulation by FGFR signaling
Perhaps the first evidence that FGF23 may be regulated in an autocrine/paracrine fashion via its own FGFR was the from the skeletal disease of FGF23 excess, osteglophonic dysplasia, which is a caused by activating mutations in FGFR1 [68]. Subsequently, FGFR kinase inhibitors were demonstrated to cause significant elevations in blood FGF23 levels [69, 70], as was the MAPK inhibitor, PD0325901, downstream of FGF signaling [71]. Further evidence for FGFR-mediated FGF23 regulation is demonstrated in mouse models in which various FGFRs have been deleted from osteocytes with the result that FGF23 levels were decreased, with the most compelling models pointing to FGFR1 as the mediator [72]. Recently, Han [73] and colleagues have used known ligands for FGFR1, including LMW-FGF-2 and HMW-FGF-2 (which are significantly increased in the Hyp and/or Dmp1 knock-out mouse models of FGF-23 excess [74, 75]) to demonstrate the various signaling pathways by which FGFR1 regulates FGF-23 promoter activity in osteoblasts. An autocrine/paracrine role for FGFR1 in regulating FGF23 was recently supported by the surprising finding that a significant number the of FGF23-secreting tumors that cause TIO may be driven by chromosomal translocations in which the fibronectin gene, which is highly-transcribed in extracellular matrix producing mesenchymal cells, is fused to a presumably active portion of FGFR1 [76]. All of this points to a potential therapeutic role for FGFR tyrosine kinase inhibitors in the treatment of FGF23 excess disorders.
6 Clinical consequences of FGF23 excess and deficiency and treatment
The disorders of FGF23 excess and deficiency are clinically challenging and can have severe consequences (Table 1). Most genetic FGF23 excess diseases typically manifest in childhood with short stature, bowing of the legs that starts with walking, and radiographic evidence of rickets. X-linked hypophosphatemic rickets (XLH) is the most frequent genetic cause of FGF23-related congenital hypophosphatemic rickets. Historically, limited treatment options required patients to take multiple daily doses of oral phosphate and activated vitamin D analogs in an attempt to reduce the skeletal morbidity. Despite aggressive supplementation, short stature persists into adulthood. Complications of treatment can include diarrhea, abdominal pain, and ectopic calcifications, including nephrocalcinosis and nephrolithiasis [13].
Several other gene mutations have been identified as the cause of disorders of FGF23 excess (Table 1). PHEX, ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP-1) and dentin matrix protein-1 (DMP-1) are bone cell-derived extracellular matrix proteins that appear to regulate FGF23 expression. The mechanisms by which mutations in these genes cause elevations in FGF23 and bone under-mineralization is not fully understood and may involve the ASARM peptide as cited above.
Osteoglophonic dysplasia (OGD), a rare genetic disorder characterized by a distinctive skeletal dysplasia, was identified in 2005 as being caused by germ line activating mutations in FGFR1 [68]. FGFR1 is known to be involved in the regulation of bone development and remodeling [77]. OGD is considered a “cross-over” disorder that has skeletal phenotypes of FGFR mutations 1 and 2 (most commonly known for their association with craniosynostosis) and FGFR3 mutations (associated with dwarfism). OGD presents with craniosynostosis, prominent supraorbital ridge, depressed nasal bridge, rhizomelic dwarfism and nonossifying bone lesions with failure to thrive. The term osteoglophonic refers to the ‘hollowed out’ appearance of the metaphyses in X-rays, which are the growth zones of long bones [78]. Hypophosphatemia in this disorder has been linked to elevations in FGF23, suggesting autocrine/paracrine regulation, as cited above.
In comparison to the inherited forms of FGF23 excess, tumor induced osteomalacia provides insight into a mechanism of neoplastic overexpression of FGF23. Lee and colleagues [76] recently describe the identification of fibronectin 1 (FN1) and FGFR1 fusion gene by high-throughput next-generation sequencing of the tumor transcriptome and fluorescence in situ hybridization (FISH). The identification of this fusion gene in 9 out of 15 phosphaturic mesenchymal tumors suggests an important role in phosphaturic mesenchymal tumors. The authors speculate that FN1 may induce its constitutively active promoter to overexpress the 3′ FGFR1 and possibly that the polymerization of fibronectin to superfibronectin may enable transphosphorylation and activation of multiple C′-FGFR1 molecules, leading to excess FGF23 expression.
7 New therapies
Several new therapies are on the horizon for treating disorders of FGF23 excess. Based upon the observation that the action of FGF23 appears to be at least in part PTH-dependent, the calcium-sensing receptor agonist, cinacalcet, which lowers blood PTH levels, has been successfully used to treat TIO [79] and XLH [80]. In addition, a monoclonal antibody to FGF23 has shown success in an animal model of XLH and in patients with XLH [18, 81, 82].
A potential application of the recent advances in FGFR1 signaling includes the use of the pan-specific FGFR tyrosine kinase inhibitor called NVP-BGJ398 in patients with FGF23-mediated hypophosphatemia. Pre-clinical animal data [69] in both Hyp and DMP-1 null mice (modeled for XLH and ARHR) demonstrates hopeful short term and long term efficacy [69]. Acute improvements in serum phosphate and calcium levels (via increased 1,25-D) were sustained over 8 weeks of testing, with structural changes including increased longitudinal bone growth, rescue of bone mass by increased mineralization, an re-organization of the growth plate. There was no evidence of hyperphosphatemia in these animal models, suggesting FGF23 deficiency in Hyp and DMP-1 null mouse didn’t occur (although effects on wild type mice were not reported).
8 Renal disease
It has been recently recognized by several cross-sectional studies [36, 83, 84] that not only are FGF23 levels elevated in CKD, but concentrations far exceed those found in any other disorder of mineral metabolism (Fig. 2). The significance of elevated FGF23 in renal disease is evolving as perhaps the most clinically relevant application of FGF23 research. Increased cardiovascular morbidity and mortality due to disordered mineral homeostasis in renal disease remains a diagnostic and therapeutic challenge. In a retrospective analysis of a cohort of subjects with CKD, Gutierrez and colleagues identified elevated FGF23 as an independent risk factor for morality [85]. In addition, in pre-dialysis CKD and healthy patients with normal serum phosphate, Gutierrez et al. found an independent association of elevated FGF23 with left ventricular hypertrophy (LVH) and mass [86]. LVH is known to progress over time to congestive heart failure and arrhythmia, both of which contribute to significant morbidity and mortality in CKD. Further support of this association comes from mice treated with FGF23 who develop heart failure through a Klotho-independent mechanism, suggesting that FGF23 acts directly on the myocardium [87]. Interestingly, while an association between hyperphosphatemia and vascular calcification has been suggested, a clear association between FGF23 and vascular calcification has not been established [88–91]. Thus, prospective data is needed to further define the effect of FGF23 on vascular calcification.
Evaluation of CKD by estimated glomerular filtration rate (eGFR) stratification has demonstrated that FGF23 levels increase progressively as eGFR declines, beginning in early CKD. In the prospective Chronic Renal Insufficiency Cohort (CRIC) study [92], where CKD stage 2–4 patients (n = 3879) were evaluated based on eGFR, elevated FGF23 proportionally surpassed elevations in PTH or serum phosphate in all cohorts [93]. This suggests that FGF23 excess may in fact be the first sign of disordered mineral metabolism, but prospective longitudinal data will confirm this [94].
Hasewaga and colleagues studied the effects of FGF23 neutralizing antibody on rats with progressive CKD [95]. Treated animals, as compared to non-treated animals, had lower fractional excretion of phosphate, thus increasing serum phosphate and normalizing 1,25-D (via increased 1α-hydroxylase activity and decreased 24-OHase activity). The net effect was increased serum calcium levels and decreased PTH, suggesting an important early role of FGF23 in normalizing serum phosphate and regulating 1,25-D. However, in a rat model of CKD treatment with a neutralizing FGF23 antibody, secondary hyperparathyroidism improved, yet resulted in increased mortality [96]. Therefore, the future of neutralizing antibody treatment of FGF23 excess in CKD remains unclear.
9 Conclusion: future directions
The discovery and progress that has been made in defining and understanding the PTH-Vitamin D-FGF23 axis truly highlights the importance of bone as a classic endocrine organ (Fig. 3). The interaction of the relatively newly described hormone FGF23 with the classic hormones PTH and vitamin D is an evolving field. The diseases of FGF23 excess and deficiency and the many related animal models have done much to help us understand how these hormones interact with each other and helped define the role of FGF23 in mediating the effects of PTH on phosphate and vitamin D homeostasis. Emerging therapies for the treatment of disorders of FGF23 excess hold great promise for the future, and offer new treatments for diseases for which there has not been new therapies for decades. A better understanding of the pathophysiologic role of FGF23 in CKD may lead to new treatments for this highly morbid condition.

The PTH-Vitamin D- FGF23 axis. FGF23 secretion from bone osteocytes acts on the kidney to induce phosphaturia via NaPi 2a/c transporters, similar to PTH, which acts via the PTH receptor. However, in contrast to the action of PTH to induce 1α hydroxylase, the enzyme that hydroxylates 25 vitamin D to 1,25-D, FGF23 acts to suppress 1α hydroxylase. 1,25-D is responsible for increased calcium and phosphate absorption from the gut and supports bone mineralization. The action of FGF23 on the parathyroid gland has been reported to suppress PTH secretion in vitro and in rodent models, but demonstration of a similar effect in humans is lacking
Abbreviations
- PTH:
-
Parathyroid hormone
- FGF23:
-
Fibroblast growth factor-23
- TIO:
-
Tumor induced osteomalacia
- 1,25-D:
-
1,25 dihydroxyvitamin D3
- XLH:
-
X-linked hypophosphatemic rickets
- ADHR:
-
Autosomal dominant hypophosphatemic rickets
- αKl:
-
α-klotho
- CKD:
-
Chronic kidney disease
- FGFR:
-
FGF receptor
- SPC:
-
Subtilisin-like proprotein convertase
- GALNT3:
-
O-glycosylation through ppGalNAc-T3
- NaPi:
-
Sodium-phosphate co-transporter
- P-ERK:
-
Phosphate epidermal growth factor
- EGR-1:
-
Early growth response gene 1
- G-CK:
-
Golgi casein kinase
- FD:
-
Fibrous dysplasia
- VDR:
-
Vitamin D receptor
- HYP:
-
X-linked hypophosphatemic rickets mice
- MEPE:
-
Matrix extracellular phosphoglycoprotein
- ASARM:
-
Acidic serine aspartate-rich matrix extracellular phosphoglycoprotein (MEPE)-associated motif
- PHEX:
-
Phosphate regulating endopeptidase homolog, X-linked
- FN1:
-
Fibronectin 1
- HFTC/HHS:
-
Hyperphosphatemic familial tumoral calcinosis/hyperostosis syndrome
- eGFR:
-
Estimated glomerular filtration rate
- LVH:
-
Left ventricular hypertrophy
- FAM20C:
-
Kinase family with sequence similarity 20, member C
- ENPP-1:
-
Ectonucleotide pyrophosphatase/phosphodiesterase 1
- DMP-1:
-
Dentin matrix protein-1
- OGD:
-
Osteoglophonic dysplasia
- FN1:
-
Fibronectin 1
- FISH:
-
Fluorescence in situ hybridization
References
Prader A et al. [Rickets following bone tumor]. Helv Paediatr Acta. 1959;14:554–65.
Meyer Jr RA, Meyer MH, Gray RW. Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res. 1989;4(4):493–500.
White KE et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60(6):2079–86.
White KE et al. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86(2):497–500.
ADHR C. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat Genet. 2000;26(3):345–8.
Shimada T et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–5.
Cai Q et al. Brief report: inhibition of renal phosphate transport by a tumor product in a patient with oncogenic osteomalacia. N Engl J Med. 1994;330(23):1645–9.
Feng JQ et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38(11):1310–5.
Lorenz-Depiereux B et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38(11):1248–50.
Turan S et al. Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia. Bone. 2010;46(2):402–9.
Levy-Litan V et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86(2):273–8.
Saito T et al. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone. 2011;49(4):913–6.
Carpenter TO et al. A clinician’s guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26(7):1381–8.
Bhattacharyya N et al. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res. 2012;27(5):1132–41.
Riminucci M et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112(5):683–92.
Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277(2):494–8.
Bhattacharyya N et al. Fibroblast growth factor 23: state of the field and future directions. Trends Endocrinol Metab. 2012;23(12):610–8.
Yamazaki Y et al. Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J Bone Miner Res. 2008;23(9):1509–18.
Goetz R et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27(9):3417–28.
Gattineni J, Baum M. Genetic disorders of phosphate regulation. Pediatr Nephrol. 2012;27(9):1477–87.
Shimada T et al. Targeted ablation of Ffg23 demonstrates an essential physiological role of FGF23 in pohsphate and vitamin D metabolism. J Clin Invest. 2004;113(4):561–8.
Shimada T et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–35.
Weinman EJ et al. Fibroblast growth factor-23-mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J Biol Chem. 2011;286(43):37216–21.
Baum M et al. Effect of fibroblast growth factor-23 on phosphate transport in proximal tubules. Kidney Int. 2005;68(3):1148–53.
Farrow EG et al. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009;20(5):955–60.
Andrukhova O et al. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone. 2012;51(3):621–8.
Ben-Dov IZ et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117(12):4003–8.
Krajisnik T et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195(1):125–31.
Ichikawa S et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117(9):2684–91.
Brownstein CA et al. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A. 2008;105(9):3455–60.
Chong WH et al. Tumor-induced osteomalacia. Endocr Relat Cancer. 2011;18(3):R53–77.
Smith ER et al. Biological variability of plasma intact and C-terminal FGF23 measurements. J Clin Endocrinol Metab. 2012;97(9):3357–65.
Jonsson KB et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348(17):1656–63.
Lorenz-Depiereux B et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78(2):193–201.
Nishida Y, et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int. 2006.
Larsson T et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64(6):2272–9.
Burnett SM et al. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21(8):1187–96.
Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90(3):1519–24.
Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91(8):3144–9.
Perwad F et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146(12):5358–64.
Scanni R et al. The human response to acute enteral and parenteral phosphate loads. J Am Soc Nephrol. 2014;25(12):2730–9.
Quinn SJ et al. Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Endocrinol Metab. 2013;304(3):E310–20.
Lavi-Moshayoff V et al. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010;299(4):F882–9.
Lopez I et al. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int. 2011;80(5):475–82.
Liu S et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17(5):1305–15.
Saji F et al. Regulation of fibroblast growth factor 23 production in bone in uremic rats. Nephron Physiol. 2009;111(4):59–66.
Samadfam R et al. Bone formation regulates circulating concentrations of fibroblast growth factor 23. Endocrinology. 2009;150(11):4835–45.
Burnett-Bowie SA et al. Effects of hPTH(1–34) infusion on circulating serum phosphate, 1,25-dihydroxyvitamin D, and FGF23 levels in healthy men. J Bone Miner Res. 2009;24(10):1681–5.
Gutierrez OM et al. (1–34) Parathyroid hormone infusion acutely lowers fibroblast growth factor 23 concentrations in adult volunteers. Clin J Am Soc Nephrol. 2012;7(1):139–45.
Singh RJ, Kumar R. Fibroblast growth factor 23 concentrations in humoral hypercalcemia of malignancy and hyperparathyroidism. Mayo Clin Proc. 2003;78(7):826–9.
Tebben PJ et al. Fibroblast growth factor 23, parathyroid hormone, and 1alpha,25-dihydroxyvitamin D in surgically treated primary hyperparathyroidism. Mayo Clin Proc. 2004;79(12):1508–13.
Kobayashi K et al. Regulation of plasma fibroblast growth factor 23 by calcium in primary hyperparathyroidism. Eur J Endocrinol. 2006;154(1):93–9.
Kawata T et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18(10):2683–8.
Mosekilde L. Primary hyperparathyroidism and the skeleton. Clin Endocrinol (Oxf). 2008;69(1):1–19.
Brown WW et al. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 2009;94(1):17–20.
Collins MT et al. Fibroblast growth factor-23 is regulated by 1alpha,25-dihydroxyvitamin D. J Bone Miner Res. 2005;20(11):1944–50.
Hill LF et al. Treatment of hypoparathyroidism with 1,25-dihydroxycholecalciferol. Clin Endocrinol (Oxf). 1976;5(Suppl):167S–73.
Yu X et al. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005;36(6):971–7.
Saito H et al. Circulating FGF-23 is regulated by 1alpha,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280(4):2543–9.
Ito M, et al. Vitamin D and phosphate regulate fibroblast growth factor-23 in K562 cells. Am J Physiol Endocrinol Metab. 2005.
Dubois SG et al. Role of abnormal neutral endopeptidase-like activities in Hyp mouse bone cells in renal phosphate transport. Am J Physiol Cell Physiol. 2002;283(5):C1414–21.
Rowe PS. Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit Rev Eukaryot Gene Expr. 2012;22(1):61–86.
Guo R et al. Inhibition of MEPE cleavage by Phex. Biochem Biophys Res Commun. 2002;297(1):38–45.
Benet-Pages A, et al. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2004.
Tagliabracci VS et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014;111(15):5520–5.
Wang X et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012;8(5), e1002708.
Simpson MA et al. Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet. 2007;81(5):906–12.
White KE et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76(2):361–7.
Wohrle S et al. Pharmacological inhibition of fibroblast growth factor (FGF) receptor signaling ameliorates FGF23-mediated hypophosphatemic rickets. J Bone Miner Res. 2013;28(4):899–911.
Wohrle S et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res. 2011;26(10):2486–97.
Zhang MY et al. Chronic inhibition of ERK1/2 signaling improves disordered bone and mineral metabolism in hypophosphatemic (Hyp) mice. Endocrinology. 2012;153(4):1806–16.
Xiao Z et al. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One. 2014;9(8), e104154.
Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J Biol Chem. 2015;290(16):10447–59.
Liu S et al. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol. 2009;23(9):1505–18.
Xiao L, Esliger A, Hurley MM. Nuclear fibroblast growth factor 2 (FGF2) isoforms inhibit bone marrow stromal cell mineralization through FGF23/FGFR/MAPK in vitro. J Bone Miner Res. 2013;28(1):35–45.
Lee JC et al. Identification of a novel FN1-FGFR1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. J Pathol. 2015;235(4):539–45.
Jacob AL et al. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol. 2006;296(2):315–28.
Beighton P, Cremin BJ, Kozlowski K. Osteoglophonic dwarfism. Pediatr Radiol. 1980;10(1):46–50.
Geller JL et al. Cinacalcet in the management of tumor-induced osteomalacia. J Bone Miner Res. 2007;22(6):931–7.
Alon US et al. Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clin J Am Soc Nephrol. 2008;3(3):658–64.
Aono Y et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24(11):1879–88.
Carpenter TO et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124(4):1587–97.
Shigematsu T et al. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis. 2004;44(2):250–6.
Gutierrez O et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16(7):2205–15.
Gutierrez OM et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359(6):584–92.
Gutierrez OM et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. 2009;119(19):2545–52.
Faul C et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121(11):4393–408.
Desjardins L et al. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos Int. 2012;23(7):2017–25.
Srivaths PR et al. Elevated FGF 23 and phosphorus are associated with coronary calcification in hemodialysis patients. Pediatr Nephrol. 2011;26(6):945–51.
Roos M et al. Relation between plasma fibroblast growth factor-23, serum fetuin-A levels and coronary artery calcification evaluated by multislice computed tomography in patients with normal kidney function. Clin Endocrinol (Oxf). 2008;68(4):660–5.
Inaba M et al. Role of fibroblast growth factor-23 in peripheral vascular calcification in non-diabetic and diabetic hemodialysis patients. Osteoporos Int. 2006;17(10):1506–13.
Feldman HI et al. The chronic renal insufficiency cohort (CRIC) study: design and methods. J Am Soc Nephrol. 2003;14(7 Suppl 2):S148–53.
Isakova T et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–8.
Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;82(7):737–47.
Hasegawa H et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010;78(10):975–80.
Shalhoub V et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest. 2012;122(7):2543–53.
Acknowledgments
This research was supported by the Division of Intramural Research, National Institute of Dental and Craniofacial Research, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services.
Conflict of Interest
JEB and MTC have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Blau, J.E., Collins, M.T. The PTH-Vitamin D-FGF23 axis. Rev Endocr Metab Disord 16, 165–174 (2015). https://doi.org/10.1007/s11154-015-9318-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-015-9318-z