
Abstract
Purpose of Review
Environmental pollutants contribute to the pathogenesis of numerous diseases including chronic cardiovascular, respiratory, and neurodegenerative diseases, among others. Emerging evidence suggests that extracellular vesicles (EVs) may mediate the association of environmental exposures with chronic diseases. The purpose of this review is to describe the impact of common environmental exposures on EVs and their role in linking environmental pollutants to the pathogenesis of chronic systemic diseases.
Recent Findings
Common environmental pollutants including particulate matter, tobacco smoke, and chemical pollutants trigger the release of EVs from multiple systems in the body. Existing research has focused primarily on air pollutants, which alter EV production and release in the lungs and systemic circulation. Air pollutants also impact the selective loading of EV cargo including microRNA and proteins, which modify the cellular function in recipient cells. As a result, pollutant-induced EVs often contribute to a pro-inflammatory and pro-thrombotic milieu, which increases the risk of pollutant-related diseases including obstructive lung diseases, cardiovascular disease, neurodegenerative diseases, and lung cancer.
Summary
Common environmental exposures are associated with multifaceted changes in EVs that lead to functional alterations in recipient cells and contribute to the pathogenesis of chronic systemic diseases. EVs may represent emerging targets for the prevention and treatment of diseases that stem from environmental exposures. However, novel research is required to expand our knowledge of the biological action of EV cargo, elucidate determinants of EV release, and fully understand the impact of environmental pollutants on human health.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Environmental exposures including air pollution, tobacco smoke, metals, and chemicals have wide systemic effects and contribute to the pathogenesis of chronic respiratory diseases, cardiovascular diseases, neurodegenerative diseases, and different types of cancer. Accumulating evidence suggests that associations of environmental exposures with numerous chronic diseases may be mediated in part by extracellular vesicles (EVs). EVs are nano-sized, membrane-bound particles that are released by human cells both under normal physiological conditions and in response to stressors including harmful environmental exposures [1]. EVs are heterogeneous in size and range from 30 to 1000 nm. EVs also differ by release mechanism: inward budding of the endosomal membrane generates smaller exosomes, while outward budding of the plasma membrane creates larger vesicles often referred to as microparticles or microvesicles [2].
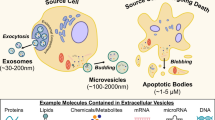
In addition to cellular waste management, EVs contribute to intercellular communication through the transmission of molecular cargo including proteins, lipids, and non-coding microRNAs (miRNAs) (Fig. 1) from the cell of origin to the cell of uptake. EVs are released by cells into their surroundings, including biofluids, and can be internalized by neighboring cells and by distant cells in the body, and in turn, EV cargo can be released into recipient cells. Studies of EV uptake have shown that EV cargo can be biologically active and potentially influence the phenotype of recipient cells [3].

Extracellular vesicle formation and cargo. Extracellular vesicles (EVs) range in size from 30 to 1000 nm and form through different mechanisms. Smaller exosomes are generated from inward budding of the endosomal membrane while larger microvesicles and microparticles form through outward budding of the plasma membrane. The outer EV membrane contains lipid particles and transmembrane proteins that are altered in response to environmental exposures. EVs also contain biologically active cargo including proteins, microRNAs, and mRNAs that mediate systemic effects after exposure to environmental pollutants. EVs are also known to contain metabolites, transfer RNAs (tRNAs), and mitochondrial DNA. This figure was created with BioRender.com
Prior research has shown that particle inhalation stimulates the release of EVs [4], which can travel across the pulmonary bed and a trigger a systemic pro-inflammatory response [5]. EVs have accordingly been implicated in the pathophysiology of chronic inflammatory lung diseases and various types of cancer and have been proposed as potential biomarkers between environmental exposures and chronic systemic diseases [6]. This review provides an overview of the impact of environmental exposures on EV biology and describes the role of EVs in linking environmental pollutants to the pathogenesis of chronic respiratory, cardiovascular, and neurodegenerative diseases.
Environmental Exposures and Extracellular Vesicles
Particulate Matter Air Pollution
Particulate matter (PM) is a mixture of aerosolized solid particles and liquid droplets that are generated by emissions from industrial facilities and motor vehicles [7]. Fine PM (diameter < 2.5 μm (PM2.5)) is small enough to deposit in the small airways of the lungs and generates a local inflammatory response [8, 9]. PM exposure also stimulates the release of EVs, which may contribute to local and systemic effects of PM exposure (Fig. 2) [10].

Conceptual model of particulate matter inhalation, EV signaling, and resulting systemic disease. Particulate matter inhalation triggers alveolar macrophages and alveolar epithelial cells in the lungs to release large quantities of pro-inflammatory EVs. Ultrafine particles also translocate through the lungs into the systemic circulation and trigger the release of EVs from endothelial cells, platelets, and circulating blood leukocytes, in addition to other organs including the brain. EVs amplify the production of inflammatory cytokines, generating lung damage, endothelial injury, and systemic diseases including obstructive lung disease, lung cancer, cardiovascular disease, and neurodegenerative diseases, among others. This figure was created with BioRender.com
Alveolar macrophages constitute the first line of defense against inhaled fine particles and release EVs in a dose-dependent manner [11]. A recent in vitro study demonstrated that macrophage-derived EVs stimulated respiratory epithelial cells to release inflammatory cytokines including interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) [12]. A separate study demonstrated that direct PM exposure triggered EV release from respiratory epithelial cells [13]. Lung-derived EVs may in turn contribute to the pathogenesis of chronic respiratory diseases after PM exposure. A recent in vitro study demonstrated that bronchial epithelial cells treated with PM2.5 secreted EVs that increased contractility of bronchial smooth muscle cells and contributed to airway hyperresponsiveness (Table 1) [14]. Accordingly, a follow-up study in mice demonstrated that PM-treated mice with PM-related EV release experienced exaggerated asthma symptoms. The same study also found that PM-related EV-miRNAs were overexpressed in the plasma of children with asthma [14]. Thus, EVs may mediate the relationship between PM exposure and reactive airways disease.
PM exposure also triggers the release of exosomes that may contribute to lung cancer development. A recent in vitro study demonstrated that PM2.5 exposure induced differential expression of exosomal miRNAs that contributed to tumorigenesis [15]. In subsequent animal models, chronic PM exposure triggered differential EV-miRNA expression that was tied to atypical bronchial epithelial cell hyperplasia, a precancerous lesion, in the airways of PM-exposed mice [15]. In a separate study, lung cancer cells treated with PM2.5 secreted exosomes that promoted tumor cell proliferation and tumor progression [16]. Thus, PM exposure may increase lung cancer risk in part through the release of tumorigenic EVs.
In addition to triggering the release of EVs that produce local effects in the lungs, PM exposure generates the release of blood-borne EVs that may exert systemic effects [10]. In a population-based study of adults, short-term PM exposure resulted in increased release of EVs into the bloodstream [4]. In a separate study of long-term ambient PM2.5 exposure, prolonged exposure was strongly associated with increased levels of circulating EVs in adults [17]. In a third study, PM2.5 exposure was associated with upregulation of EV-miRNAs involved in pathways related to cardiovascular disease pathogenesis including vascular inflammation and atherosclerosis [18]. A separate population-based study showed that long-term PM2.5 exposure increased circulating EV-miRNAs that amplified the magnitude of the association between PM2.5 exposure and elevated systolic blood pressure in adults [19]. Together, these studies suggest that PM exposure may trigger the release of circulating EV-miRNAs that modulate cardiovascular risk factors.
PM exposure may additionally modify cardiovascular disease risk by augmenting coagulation pathways. A prior in vitro study showed that PM exposure triggered microparticle release from blood and endothelial cells [20]. Endothelial-derived microparticles expressed functional tissue factor, and thereby exerted a pro-thrombotic effect through their ability to activate blood coagulation. In a separate study, PM stimulated EV release from platelets in vitro, which increased pro-inflammatory cytokines, induced vascular endothelial injury, and accelerated vascular thrombosis [21]. Animal models have recapitulated the impact of PM exposure on coagulation pathways. In a mouse model, PM exposure triggered the upregulation of platelet-derived microparticles, which contributed to a systemic pro-coagulant response [22]. A population-based study accordingly demonstrated that short-term PM exposure was associated with increased release of EV-miRNAs that increase blood fibrinogen levels, which is a marker of hypercoagulability [23]. A second study similarly showed that long-term PM exposure was associated with the expression of EV-miRNAs that alter pro-coagulant factors including endogenous thrombin [5]. In a separate longitudinal study of adults with diabetes, PM exposure increased pro-coagulant platelet-derived EVs with increased tissue factor expression [24]. PM exposure may therefore create a pro-thrombotic milieu through increases in the number and pro-coagulant effect of EVs. Hypercoagulability is linked in turn to an array of PM-associated diseases including cardiovascular disease and cerebrovascular disease [25].
In addition to increasing thrombotic risk, PM exposure triggers the release of EVs that may have direct neurotoxic effects. A recent in vitro study demonstrated that neuronal macrophages treated with PM2.5 released EVs with increased glutaminase protein levels [26]. EV glutaminase stimulated glutamate production in macrophages, which has a direct neurotoxic effect. Accordingly, a separate study demonstrated that PM2.5 triggered EV release from microglial cells in mice, which thereby increased glutaminase expression and glutamate generation in mouse neurons [27]. Increased glutamate is associated with neurodegeneration and may be attributed to glutaminase-containing EVs released in response to PM exposure.
Non-particulate Air Pollution
While the particulate matter has a significant impact on long-term health, other tropospheric pollutants also impact human health through the release of biologically active EVs. Ground-level ozone is created when nitrogen oxides and organic compounds from industrial emissions chemically react in the presence of UV light [28]. Animal models have shown that ozone inhalation increased the release of lung-derived EVs and changed the profile of lung EV-miRNAs, which may modulate ozone-induced lung inflammation and airway remodeling [29].
Industrial emissions also generate polycyclic aromatic hydrocarbons (PAHs), which are a group of organic compounds that contribute to air and soil pollution [30]. In animal models, PAH inhalation increased the release of the lung- and endothelial-derived microparticles, which were associated with endothelial dysfunction, platelet activation, thrombosis, and inflammation [31]. PAHs are metabolized in the liver after inhalation and have been shown to impact liver health. In prior murine models, PAH exposure triggered the release of hepatocyte-derived EVs, which were associated with hepatocyte toxicity and hepatic cell death [32]. Thus, PAH exposure may trigger the release of EVs that modulate both cardiovascular and liver disease risk.
Ionizing Radiation
Ionizing radiation consists of high-energy electromagnetic waves that stem from natural sources like the sun and man-made sources like nuclear reactors [33]. Ionizing radiation is ubiquitous in the environment and can generate DNA and tissue damage at high levels. In a prior study that exposed human blood samples to ionizing radiation, gamma rays altered the protein and miRNA cargo of plasma exosomes [34]. The radiation-responsive proteins and miRNAs were previously shown to impact cell cycle progression and proliferation and may play a role in tumorigenesis and invasion of cancer cells [35]. Thus, EVs may mediate the association of ionizing radiation with the development of human cancers.
Cigarette Smoke
Cigarette smoking aerosolizes thousands of toxic chemicals through the combustion of tobacco and chemical additives [36]. Tobacco smoke is an important risk factor for many chronic diseases including chronic obstructive pulmonary disease (COPD), asthma, cardiovascular disease, and lung cancer [37]. The impact of tobacco smoking on lung and heart health may be mediated in part by smoking-induced alterations in lung-derived and circulating EVs.
Cigarette smoking is the primary risk factor for lung cancer and emerging evidence suggests that smoking may induce lung carcinogenesis in part through alteration of EV-miRNA expression (Table 2). In vitro studies have shown that cigarette smoke-exposed human bronchial epithelial cells increased the production of exosomal miRNA miR-21, which stimulated angiogenesis and malignant transformation of bronchial epithelial cells [38]. Murine models confirmed that 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NKK), an abundant chemical in nicotine, altered the expression of two EV-miRNAs that were subsequently upregulated in the early stages of NNK-induced lung cancer [39]. Thus, smoking-induced EV-miRNA signaling may contribute or serve as a biomarker of lung carcinogenesis.
In addition to contributing to lung cancer development, smoking-induced EVs may play a role in COPD pathogenesis. In vitro studies have shown that cigarette smoke exposure stimulates microvesicle release from airway epithelial cells and alveolar macrophages [40]. Microvesicles released from human macrophages in response to tobacco smoke exposure had increased proteolytic activity that may contribute to emphysematous lung destruction in smokers [41]. Incubation of cigarette smoke-induced microparticles with lung epithelial cells also generated increased expression of pro-inflammatory cytokines, which damage lung tissue and induce airway remodeling [42].
Cigarette smoke exposure also alters EV-miRNA profiles in human bronchoalveolar lavage fluid [43, 44]. In particular, cigarette smoking upregulates EV-miRNA miR-21, which is responsible for myofibroblast differentiation [45]. Accordingly, EVs derived from cigarette smoke-exposed bronchial epithelial cells modify lung fibroblasts and stimulate differentiation into myofibroblasts [46]. Myofibroblast accumulation is a hallmark of COPD and contributes to airway remodeling and narrowing of small airways [47]. Smoking-induced exosomal miR-21 also influences the epithelial-mesenchymal transition in bronchial epithelial cells, which constitutes destabilization of airway epithelial cells and the emergence of mesenchymal-epithelial cells. Mesenchymal cells generate peribronchial fibrosis and airflow limitation, which are hallmark features of COPD [48]. Murine models have recapitulated that exosomes derived from the airways of cigarette smoke-exposed mice modulated the epithelial-mesenchymal transition, generated acute lung injury, and caused lung function impairment [49]. Human studies have additionally confirmed that cigarette smoke alters the composition of lipid membrane particles in lung-derived EVs, which may contribute to smoke-related airway inflammation [50].
While cigarette smoking triggers EV release in the lungs, smoking also triggers the release of EVs into the systemic circulation. Cigarette smoking causes endothelial damage and endothelial apoptosis in pulmonary capillaries and triggers the systemic release of endothelial-derived microparticles [51]. Endothelial-derived microparticles have been shown to correlate with lung function impairment in cigarette smoke-exposed rats [52]. In vitro studies have shown that endothelial-derived microparticles also impair macrophage clearance of apoptotic cells, which induces local inflammation and lung tissue destruction [53]. Population-based studies have also supported that upregulated endothelial-derived microparticles correlate with impaired diffusing capacity and emphysema in smokers [54]. Smoking-induced endothelial-derived EVs also have increased polyamine metabolites on their outer surface that generate smooth muscle cell constriction and proliferation, leading to pulmonary hypertension in smokers [55]. Smoking additionally alters the profile of plasma-derived EV-miRNAs, triggering differential expression of EV-miRNAs that regulate cytokine signaling, extracellular matrix deposition, and airway remodeling [6]. Animal and human models have also shown that smoking-induced pro-inflammatory cytokines in the lungs are encapsulated in EVs and transported to the bloodstream, thereby generating widespread systemic inflammation [56, 57].
While circulating smoking-related EVs are correlated with lung function decline, they may also contribute to hypercoagulability and cardiovascular disease risk. In population-based studies, cigarette smoking triggered an immediate increase in the number of circulating microparticles derived from endothelial cells, platelets, and circulating blood leukocytes [58]. Endothelial-derived microparticles correlate with impaired vasodilation and arterial stiffness and are independent predictors of cardiovascular disease [59]. Platelet-derived microparticles contribute to thrombosis and atherogenesis and are elevated in patients with coronary heart disease [60]. Compared to healthy volunteers, circulating exosomes in smokers also had increased surface cholesteryl ester transfer protein, which regulates lipid transport and is associated with cardiovascular disease in smokers [61]. Cigarette smoke also stimulates the production of pro-coagulant EVs with increased tissue factor expression and increased surface phosphatidylserine, a pro-coagulant phospholipid [40, 62,63,64]. In vitro studies have accordingly shown that cigarette smoke-induced EVs caused faster and more extensive thrombin generation in human plasma compared to control EVs [65]. Cigarette smoking additionally alters the EV-miRNA signature in adults and has been shown to upregulate EV-miRNAs associated with fibrosis and impaired infarct healing after ischemic injuries [66]. Thus, smoking-associated EVs may function as drivers of cardiovascular disease risk among individuals with cigarette smoke exposure.
Electronic Cigarette Smoke
Electronic cigarettes (e-cigarettes) vaporize a base liquid, which contains nicotine, flavorings, and a mixture of propylene glycol and vegetable glycerin [67]. Similar to conventional cigarette smoking, the vapor is then inhaled into the lungs. The chemicals contained in e-cigarettes have been shown to cause lung disease and cardiovascular disease [68]. Emerging evidence has demonstrated that the adverse health effects associated with e-cigarette use may be mediated in part by circulating EVs [69, 70]. In a randomized controlled trial, inhaling e-cigarette vapor was associated with increased levels of circulating endothelial- and platelet-derived EVs [71]. Platelet-derived EVs expressed platelet activation and inflammation markers, which are associated with increased incidence of acute coronary syndrome and strokes. Increased production of pro-coagulant EVs could therefore provide a mechanistic link between e-cigarette use and cardiovascular diseases.
Metals
Metals and metalloids are ubiquitous in the environment and derive from geologic and anthropogenic sources. Exposure to metals and metalloids is pervasive in global human populations, as common sources of exposure include food, drinking water, and air pollution. Several metals and metalloids including arsenic and manganese have well-characterized effects on human health and contribute to cardiovascular disease, cognitive impairment, and cancer incidence [72]. There is emerging evidence that certain metals and metalloids alter EV abundance and cargo, which can then propagate the effect of metals through the body to unexposed cells and generate harmful systemic effects.
Inorganic arsenic is a well-known carcinogen linked to multiple types of cancer including lung, prostate, and liver cancer [73]. Arsenic oxidation generates arsenite (AsIII), which is a soluble compound that can contaminate sources of groundwater [74]. In vitro studies demonstrated that AsIII-transformed prostate epithelial cells secreted more exosomes compared to control cells. In addition, AsIII exposure alters the packaging of exosomal miRNA cargo [75]. The exosomes from AsIII-transformed cells were notably enriched with pro-oncogenic factors including inflammation- and apoptosis-related gene transcripts and cancer-related miRNAs. Accordingly, the exosomes were shown to induce malignant transformation in recipient cells. In related studies, AsIII-transformed bronchial epithelial cells and hepatic epithelial cells similarly exhibited increased expression of pro-oncogenic and inflammation-related miRNAs [76, 77]. The exosomes derived from these transformed cells induced proliferation and pro-inflammatory activity in target recipient cells. Together, these studies show that AsIII induces exosomal changes that play key roles in intercellular communication and tumorigenesis. In addition, AsIII, but not methylated arsenic species, has been shown to trigger the release of pro-coagulant microparticles that alter platelet activity and stimulate thrombus formation in rats, providing a potential mechanism for the observed association between arsenic exposure and cardiovascular disease [78].
Chronic manganese (Mn) exposure causes neurotoxicity and has been linked to neurodegenerative disorders with symptoms similar to Parkinson’s disease [79]. The pathogenesis of many neurodegenerative disorders including Parkinson’s disease involves the aggregation of misfolded proteins, which are transferred from cell to cell via EVs [80]. Mn exposure induces the neuronal release of exosomes in vitro with increased expression of miRNAs that regulate inflammation and protein aggregation. In particular, exosomes from Mn-exposed cells have increased the loading of an adaptor protein that promotes neuroinflammation [81]. Manganese-exposed neuronal cells also released exosomes containing misfolded α-synuclein, a neurotoxic protein, and a key pathological feature of Parkinson’s disease [82]. Functionally, these exosomes also induced neuroinflammatory and neurodegenerative responses in mice akin to Parkinson’s disease [81, 82]. Thus, EVs may mediate the association of manganese exposure with neuroinflammation and neurodegenerative diseases.
Population-based studies have shown that metal exposure during pregnancy alters the expression of EV-miRNAs, which have been hypothesized to facilitate maternal–fetal communication [83]. Exposure to barium, cobalt, mercury, and thallium in early pregnancy was associated with altered EV-miRNA expression in the blood. Pathway analyses identified several putative biological pathways associated with these altered miRNAs, particularly the epidermal growth factor receptor (EGFR) pathway. EGFR is known to regulate placental development and endometrial function in early pregnancy and could underlie metal-related impacts on pregnancy health, though this hypothesis is not yet validated. Cadmium, molybdenum, nickel, antimony, and tin were not associated with any blood EV-miRNAs.
Plasticizers (Phenols and Phthalates)
Phthalates and phenols are two classes of synthetic organic chemicals commonly found in consumer products including food contact applications, medical devices, and flooring and wall coverings [84]. Specific phthalate and phenol compounds such as di(2-ethylhexyl) phthalate (DEHP) and bisphenol-A (BPA) have well-known endocrine-disrupting effects and have been associated with adverse reproductive and cardiometabolic effects. Emerging research suggests EVs may contribute to the endocrine-disrupting effects of plasticizer exposures.
In vitro studies have demonstrated that BPA exposure altered EV-miRNA expression in primary granulosa cells, which contributes to ovarian follicular development and oocyte maturation in women [85]. BPA exposure also altered protein expression in exosomes from placenta explants, and the implicated proteins have been shown to modulate pathways associated with inflammation and placental cellular injury [86]. In population-based studies of women undergoing in vitro fertilization (IVF), phthalate exposure was associated with altered EV-miRNA expression in the follicular fluid that surrounds ovarian follicles [87, 88]. These studies also reported several putative reproductive pathways related to these miRNAs, including gonadal cell proliferation, follicular development, oocyte development, and fertilization, further contributing to the overall evidence on mechanisms through which phenols and phthalates impact female reproductive health.
In addition to impacting female infertility, DEHP may trigger the release of EVs associated with cardiovascular disease risk. In a population-based study of young adults in Taiwan, DEHP metabolites were associated with increased serum endothelial- and platelet-derived microparticles [89]. Endothelial- and platelet-derived microparticles are established markers of vascular damage [89] and may contribute to cardiovascular disease risk among individuals with DEHP exposures. However, a second study of healthy adolescent children showed that increased phthalate metabolites were associated with decreased levels of plasma endothelial-derived microparticles [90]. The etiology of these disparate findings is unclear, though there were major differences in the methods, including the usage of plasma versus serum and different transmembrane protein markers to quantify endothelial and platelet microvesicles. These disparate methods may have resulted in the capture and quantifications of different EV sub-populations. Ultimately, there is some suggestion that phthalates are associated with systemic EV release and cargo, but further research is required to understand the functional consequences of these alterations in EV cargo.
Other Chemical Pollutants
In addition to phenols and phthalates, other chemical pollutants have been examined in relation to EV quantity and cargo. However, the evidence for these chemicals is limited. This is further complicated by the relative lack of evidence on their associations with health and disease. In light of these limitations, additional investigations are necessary to understand whether their impacts are local or systemic.
Perfluorinated chemicals (PFCs) are synthetic compounds used to make cookware, fabrics, clothes, and food packaging [91]. PFC exposure has been previously associated with endothelial damage and atherosclerosis. In the aforementioned Taiwanese study of young adults, serum endothelial- and platelet-derived microvesicle concentrations were higher across quartiles of four tested PFCs and were associated with markers of carotid atherosclerotic vascular disease [92].
Ethyl paraben is a chemical compound often used as a food additive. In a population-based study of women undergoing IVF, ethylparaben was associated with altered hsa-miR-375 expression. The EV-miRNA is thought to target pathways that regulate oocyte viability in women and suggests a potential association between ethylparaben exposure and oocyte viability and female fecundity, though this association is not yet definitively established [88].
Volatile organic compounds (VOCs) occur naturally in crude oil and enter the air through vehicle exhaust [93]. In vitro exposure to toluene alone [94] or a mixture of VOCs including toluene, xylene, and ethylbenzene [95] induced cellular and exosomal miRNA changes. There were incomplete overlaps between cellular and exosomal miRNA changes, which further supports the idea that environmental exposures affect not only cellular responses, but also the selective loading of EV cargo. These alterations were predicted to affect genes relevant for cancer and neurological diseases, which is consistent with the known impact of VOCs on cancer risk and neurological damage.
Rotenone is a chemical compound and environmental toxin that is often used as an insecticide [96]. In vitro studies have shown that rotenone-exposed neuronal cells demonstrated increased release of exosomes and increased loading of α-synuclein into exosomes and other EVs, which were not observed in control cells [97]. Thus, EVs may mediate the relationship of rotenone exposure and neurodegenerative diseases including Parkinson’s disease in a mechanism similar to Mn.
Current Challenges
Despite substantial advances in our knowledge of EV biology, several challenges remain. First, existing studies, both experimental and observational, have used different protocols that result in the isolation and characterization of different EV sub-populations. The nuances of these protocols are not universally discussed but likely have large impacts on the findings given the heterogeneity in EVs. Future studies should conform to the Minimal Information for Studies of Extracellular Vesicles (MISEV) guidelines [98], and researchers should clarify the details and intent of study protocols. Second, while future studies should continue to use sensitive untargeted methods to assess proteomic and RNA cargoes of EVs, advances in our understanding of miRNAs are necessary to interpret changes in EV-miRNA expression and speculate on the potential effects. Current strategies rely heavily on leveraging limited databases to identify putative mRNA targets and on using ontological and pathway enrichment analyses, but such analyses carry substantial uncertainty and need to be interpreted with caution. Third, human studies are often limited to studying the total EV population from easy to access biospecimen such as the blood or saliva. The resulting measures from this heterogenous EV population may reflect systemic changes but lack specificity to any specific target tissues and thus limits our inference. While system changes are intriguing and potentially useful, it is imperative that we go further and strive to understand the underlying source of change. To address this, human studies should adopt strategies that fractionate EVs based on the cell of origin or any other distinguishing features, which will provide opportunities to screen for early responses and changes from tissues impacted by noxious environmental exposures. Lastly, studies should expand beyond EV concentration as an indicator of injury or dysfunction. There is clear evidence that EVs released from cells in response to noxious environmental exposures are part of a larger cascade with significant downstream functional effects on recipient cells. Novel research is required to expand our knowledge of the biological action of EV cargo (e.g., miRNAs, proteins, lipids), elucidate determinants of EV release and uptake, and fully understand the impact of environmental pollutants on human health.
Conclusions
EVs function as biomarkers between harmful environmental exposures and chronic respiratory, cardiovascular, and neurologic diseases. Prior research has shown that air pollution, tobacco smoke, metals, and chemical pollutants trigger the systemic release of EVs that alter cell biology in every organ system in the body. Emerging research has identified specific biologically active EV-miRNAs that are upregulated in certain disease states and suggests that EV-miRNAs may represent novel targets for the prevention and treatment of diseases that stem from environmental exposures.
References
O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21(10):585–606.
Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol. 2016;36(3):301–12.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–9.
Bonzini M, Pergoli L, Cantone L, Hoxha M, Spinazze A, Del Buono L, et al. Short-term particulate matter exposure induces extracellular vesicle release in overweight subjects. Environ Res. 2017;155:228–34.
Pavanello S, Bonzini M, Angelici L, Motta V, Pergoli L, Hoxha M, et al. Extracellular vesicle-driven information mediates the long-term effects of particulate matter exposure on coagulation and inflammation pathways. Toxicol Lett. 2016;259:143–50.
Sundar IK, Li D, Rahman I. Small RNA-sequence analysis of plasma-derived extracellular vesicle miRNAs in smokers and patients with chronic obstructive pulmonary disease as circulating biomarkers. J Extracell Vesicles. 2019;8(1):1684816.
Nogueira JB. Air pollution and cardiovascular disease. Rev Port Cardiol. 2009;28(6):715–33.
Kodros JK, Volckens J, Jathar SH, Pierce JR. Ambient particulate matter size distributions drive regional and global variability in particle deposition in the respiratory tract. Geohealth. 2018;2(10):298–312.
Klein SG, Cambier S, Hennen J, Legay S, Serchi T, Nelissen I, et al. Endothelial responses of the alveolar barrier in vitro in a dose-controlled exposure to diesel exhaust particulate matter. Part Fibre Toxicol. 2017;14(1):7.
Benedikter BJ, Wouters EFM, Savelkoul PHM, Rohde GGU, Stassen FRM. Extracellular vesicles released in response to respiratory exposures: implications for chronic disease. J Toxicol Environ Health B Crit Rev. 2018;21(3):142–60.
Schneider DJ, Speth JM, Penke LR, Wettlaufer SH, Swanson JA, Peters-Golden M. Mechanisms and modulation of microvesicle uptake in a model of alveolar cell communication. J Biol Chem. 2017;292(51):20897–910.
Martin PJ, Heliot A, Tremolet G, Landkocz Y, Dewaele D, Cazier F, et al. Cellular response and extracellular vesicles characterization of human macrophages exposed to fine atmospheric particulate matter. Environ Pollut. 2019;254(Pt A):112933.
Bollati V, Angelici L, Rizzo G, Pergoli L, Rota F, Hoxha M, et al. Microvesicle-associated microRNA expression is altered upon particulate matter exposure in healthy workers and in A549 cells. J Appl Toxicol. 2015;35(1):59–67.
Zheng R, Du M, Tian M, Zhu Z, Wei C, Chu H, et al. Fine particulate matter induces childhood asthma attacks via extracellular vesicle-packaged Let-7i-5p-mediated modulation of the MAPK signaling pathway. Adv Sci (Weinh). 2022;9(3):e2102460.
Wang Y, Zhong Y, Sun K, Fan Y, Liao J, Wang G. Identification of exosome miRNAs in bronchial epithelial cells after PM2.5 chronic exposure. Ecotoxicol Environ Saf. 2021;215:112127.
Xu H, Jiao X, Wu Y, Li S, Cao L, Dong L. Exosomes derived from PM2.5-treated lung cancer cells promote the growth of lung cancer via the Wnt3a/β-catenin pathway. Oncol Rep. 2019;41(2):1180–8.
Rodosthenous RS, Coull BA, Lu Q, Vokonas PS, Schwartz JD, Baccarelli AA. Ambient particulate matter and microRNAs in extracellular vesicles: a pilot study of older individuals. Part Fibre Toxicol. 2016;13:13.
Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A. 2008;105(5):1516–21.
Rodosthenous RS, Kloog I, Colicino E, Zhong J, Herrera LA, Vokonas P, et al. Extracellular vesicle-enriched microRNAs interact in the association between long-term particulate matter and blood pressure in elderly men. Environ Res. 2018;167:640–9.
Neri T, Pergoli L, Petrini S, Gravendonk L, Balia C, Scalise V, et al. Particulate matter induces prothrombotic microparticle shedding by human mononuclear and endothelial cells. Toxicol In Vitro. 2016;32:333–8.
Kong L, Li K, Gao L, Yin A, Zhou L, Teng G, et al. Mediating effects of platelet-derived extracellular vesicles on PM2.5-induced vascular endothelial injury. Ecotoxicol Environ Saf. 2020;198:110652.
Wilson DW, Aung HH, Lame MW, Plummer L, Pinkerton KE, Ham W, et al. Exposure of mice to concentrated ambient particulate matter results in platelet and systemic cytokine activation. Inhal Toxicol. 2010;22(4):267–76.
Pergoli L, Cantone L, Favero C, Angelici L, Iodice S, Pinatel E, et al. Extracellular vesicle-packaged miRNA release after short-term exposure to particulate matter is associated with increased coagulation. Part Fibre Toxicol. 2017;14(1):32.
Emmerechts J, Jacobs L, Van Kerckhoven S, Loyen S, Mathieu C, Fierens F, et al. Air pollution-associated procoagulant changes: the role of circulating microvesicles. J Thromb Haemost. 2012;10(1):96–106.
Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, et al. Thrombosis: a major contributor to global disease burden. Arterioscler Thromb Vasc Biol. 2014;34(11):2363–71.
Liu F, Huang Y, Zhang F, Chen Q, Wu B, Rui W, et al. Macrophages treated with particulate matter PM2.5 induce selective neurotoxicity through glutaminase-mediated glutamate generation. J Neurochem. 2015;134(2):315–26.
Chen X, Guo J, Huang Y, Liu S, Huang Y, Zhang Z, et al. Urban airborne PM2.5-activated microglia mediate neurotoxicity through glutaminase-containing extracellular vesicles in olfactory bulb. Environ Pollut. 2020;264:114716.
Guenther A, Geron C, Pierce T, Lamb B, Harley P, Fall R. Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America. Atmos Environ. 2000;34(12–14):2205–30.
Smith GJ, Tovar A, Kanke M, Wang Y, Deshane JS, Sethupathy P, et al. Ozone-induced changes in the murine lung extracellular vesicle small RNA landscape. Physiol Rep. 2021;9(18):e15054.
Finlayson-Pitts BJ, Pitts JN Jr. Tropospheric air pollution: ozone, airborne toxics, polycyclic aromatic hydrocarbons, and particles. Science. 1997;276(5315):1045–52.
Malovichko MV, Abplanalp WT, McFall SA, Taylor BS, Wickramasinghe NS, Sithu ID, et al. Subclinical markers of cardiovascular toxicity of benzene inhalation in mice. Toxicol Appl Pharmacol. 2021;431:115742.
van Meteren N, Lagadic-Gossmann D, Chevanne M, Gallais I, Gobart D, Burel A, et al. Polycyclic aromatic hydrocarbons can trigger hepatocyte release of extracellular vesicles by various mechanisms of action depending on their affinity for the aryl hydrocarbon receptor. Toxicol Sci. 2019:kfz157.
McLean AR, Adlen EK, Cardis E, Elliott A, Goodhead DT, Harms-Ringdahl M, et al. A restatement of the natural science evidence base concerning the health effects of low-level ionizing radiation. Proc Biol Sci. 2017;284(1862):20171070.
Yentrapalli R, Merl-Pham J, Azimzadeh O, Mutschelknaus L, Peters C, Hauck SM, et al. Quantitative changes in the protein and miRNA cargo of plasma exosome-like vesicles after exposure to ionizing radiation. Int J Radiat Biol. 2017;93(6):569–80.
Imam JS, Plyler JR, Bansal H, Prajapati S, Bansal S, Rebeles J, et al. Genomic loss of tumor suppressor miRNA-204 promotes cancer cell migration and invasion by activating AKT/mTOR/Rac1 signaling and actin reorganization. PLoS One. 2012;7(12):e52397.
Kapp R. Tobacco Smoke. In: Wexler P, editor. Encyclopedia of toxicology (Second Edition). Lutherville, MD, USA: Elsevier; 2005.
Oberg M, Jaakkola MS, Woodward A, Peruga A, Pruss-Ustun A. Worldwide burden of disease from exposure to second-hand smoke: a retrospective analysis of data from 192 countries. Lancet. 2011;377(9760):139–46.
Liu Y, Luo F, Wang B, Li H, Xu Y, Liu X, et al. STAT3-regulated exosomal miR-21 promotes angiogenesis and is involved in neoplastic processes of transformed human bronchial epithelial cells. Cancer Lett. 2016;370(1):125–35.
Wu J, Yang T, Li X, Yang Q, Liu R, Huang J, et al. Alteration of serum miR-206 and miR-133b is associated with lung carcinogenesis induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Toxicol Appl Pharmacol. 2013;267(3):238–46.
Li M, Yu D, Williams KJ, Liu ML. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30(9):1818–24.
Li CJ, Liu Y, Chen Y, Yu D, Williams KJ, Liu ML. Novel proteolytic microvesicles released from human macrophages after exposure to tobacco smoke. Am J Pathol. 2013;182(5):1552–62.
Cordazzo C, Petrini S, Neri T, Lombardi S, Carmazzi Y, Pedrinelli R, et al. Rapid shedding of proinflammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on Ca2+ mobilization. Inflamm Res. 2014;63(7):539–47.
Heliot A, Landkocz Y, Roy Saint-Georges F, Gosset P, Billet S, Shirali P, et al. Smoker extracellular vesicles influence status of human bronchial epithelial cells. Int J Hyg Environ Health. 2017;220(2 Bt B):445–54.
Saxena A, Walters MS, Shieh JH, Shen LB, Gomi K, Downey RJ, et al. Extracellular vesicles from human airway basal cells respond to cigarette smoke extract and affect vascular endothelial cells. Sci Rep. 2021;11(1):6104.
Xu H, Ling M, Xue J, Dai X, Sun Q, Chen C, et al. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics. 2018;8(19):5419–33.
Fujita Y, Araya J, Ito S, Kobayashi K, Kosaka N, Yoshioka Y, et al. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J Extracell Vesicles. 2015;4:28388.
Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183–92.
Nowrin K, Sohal SS, Peterson G, Patel R, Walters EH. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer. Expert Rev Respir Med. 2014;8(5):547–59.
Wang L, Chen Q, Yu Q, Xiao J, Zhao H. Cigarette smoke extract-treated airway epithelial cells-derived exosomes promote M1 macrophage polarization in chronic obstructive pulmonary disease. Int Immunopharmacol. 2021;96:107700.
Hough KP, Wilson LS, Trevor JL, Strenkowski JG, Maina N, Kim YI, et al. Unique lipid signatures of extracellular vesicles from the airways of asthmatics. Sci Rep. 2018;8(1):10340.
Strulovici-Barel Y, Staudt MR, Krause A, Gordon C, Tilley AE, Harvey BG, et al. Persistence of circulating endothelial microparticles in COPD despite smoking cessation. Thorax. 2016;71(12):1137–44.
Liu H, Ding L, Zhang Y, Ni S. Circulating endothelial microparticles involved in lung function decline in a rat exposed in cigarette smoke maybe from apoptotic pulmonary capillary endothelial cells. J Thorac Dis. 2014;6(6):649–55.
Serban KA, Rezania S, Petrusca DN, Poirier C, Cao D, Justice MJ, et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep. 2016;6:31596.
Gordon C, Gudi K, Krause A, Sackrowitz R, Harvey BG, Strulovici-Barel Y, et al. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am J Respir Crit Care Med. 2011;184(2):224–32.
Zhu L, Xiao R, Zhang X, Lang Y, Liu F, Yu Z, et al. Spermine on endothelial extracellular vesicles mediates smoking-induced pulmonary hypertension partially through calcium-sensing receptor. Arterioscler Thromb Vasc Biol. 2019;39(3):482–95.
Feller D, Kun J, Ruzsics I, Rapp J, Sarosi V, Kvell K, et al. Cigarette smoke-induced pulmonary inflammation becomes systemic by circulating extracellular vesicles containing Wnt5a and inflammatory cytokines. Front Immunol. 2018;9:1724.
Chen Y, Li G, Liu Y, Werth VP, Williams KJ, Liu ML. Translocation of endogenous danger signal HMGB1 from nucleus to membrane microvesicles in macrophages. J Cell Physiol. 2016;231(11):2319–26.
Mobarrez F, Antoniewicz L, Bosson JA, Kuhl J, Pisetsky DS, Lundback M. The effects of smoking on levels of endothelial progenitor cells and microparticles in the blood of healthy volunteers. PLoS One. 2014;9(2):e90314.
Sinning JM, Losch J, Walenta K, Bohm M, Nickenig G, Werner N. Circulating CD31+/Annexin V+ microparticles correlate with cardiovascular outcomes. Eur Heart J. 2011;32(16):2034–41.
Viera AJ, Mooberry M, Key NS. Microparticles in cardiovascular disease pathophysiology and outcomes. J Am Soc Hypertens. 2012;6(4):243–52.
Kodidela S, Wang Y, Patters BJ, Gong Y, Sinha N, Ranjit S, et al. Proteomic profiling of exosomes derived from plasma of HIV-infected alcohol drinkers and cigarette smokers. J Neuroimmune Pharmacol. 2020;15(3):501–19.
Enjeti AK, Ariyarajah A, D’Crus A, Seldon M, Lincz LF. Circulating microvesicle number, function and small RNA content vary with age, gender, smoking status, lipid and hormone profiles. Thromb Res. 2017;156:65–72.
Amadio P, Baldassarre D, Tarantino E, Zacchi E, Gianellini S, Squellerio I, et al. Production of prostaglandin E2 induced by cigarette smoke modulates tissue factor expression and activity in endothelial cells. Faseb j. 2015;29(9):4001–10.
Stassen FRM, van Eijck PH, Savelkoul PHM, Wouters EFM, Rohde GGU, Briedé JJ, et al. Cell type- and exposure-specific modulation of CD63/CD81-positive and tissue factor-positive extracellular vesicle release in response to respiratory toxicants. Oxid Med Cell Longev. 2019;2019:5204218.
Benedikter BJ, Bouwman FG, Heinzmann ACA, Vajen T, Mariman EC, Wouters EFM, et al. Proteomic analysis reveals procoagulant properties of cigarette smoke-induced extracellular vesicles. J Extracell Vesicles. 2019;8(1):1585163.
Badrnya S, Baumgartner R, Assinger A. Smoking alters circulating plasma microvesicle pattern and microRNA signatures. Thromb Haemost. 2014;112(1):128–36.
Morris PB, Ference BA, Jahangir E, Feldman DN, Ryan JJ, Bahrami H, et al. Cardiovascular effects of exposure to cigarette smoke and electronic cigarettes: clinical perspectives from the prevention of Cardiovascular Disease Section Leadership Council and Early Career Councils of the American College of Cardiology. J Am Coll Cardiol. 2015;66(12):1378–91.
Sassano MF, Davis ES, Keating JE, Zorn BT, Kochar TK, Wolfgang MC, et al. Evaluation of e-liquid toxicity using an open-source high-throughput screening assay. PLoS Biol. 2018;16(3):e2003904.
Benedikter BJ, Koenen RR. Vaping, vapor, vesicles! Electronic cigarettes provoke vascular extracellular vesicle release in healthy volunteers. Atherosclerosis. 2020;301:79–81.
Kerr DMI, Brooksbank KJM, Taylor RG, Pinel K, Rios FJ, Touyz RM, et al. Acute effects of electronic and tobacco cigarettes on vascular and respiratory function in healthy volunteers: a cross-over study. J Hypertens. 2019;37(1):154–66.
Mobarrez F, Antoniewicz L, Hedman L, Bosson JA, Lundbäck M. Electronic cigarettes containing nicotine increase endothelial and platelet derived extracellular vesicles in healthy volunteers. Atherosclerosis. 2020;301:93–100.
Argos M, Kalra T, Rathouz PJ, Chen Y, Pierce B, Parvez F, et al. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): a prospective cohort study. Lancet. 2010;376(9737):252–8.
Arsenic and arsenic compounds. IARC Monogr Eval Carcinog Risk Chem Hum. 1980;23:39-141.
Dringen R, Spiller S, Neumann S, Koehler Y. Uptake, Metabolic effects and toxicity of arsenate and arsenite in astrocytes. Neurochem Res. 2016;41(3):465–75.
Ngalame NNO, Luz AL, Makia N, Tokar EJ. Arsenic alters exosome quantity and cargo to mediate stem cell recruitment into a cancer stem cell-like phenotype. Toxicol Sci. 2018;165(1):40–9.
Xu Y, Luo F, Liu Y, Shi L, Lu X, Xu W, et al. Exosomal miR-21 derived from arsenite-transformed human bronchial epithelial cells promotes cell proliferation associated with arsenite carcinogenesis. Arch Toxicol. 2015;89(7):1071–82.
Chen C, Luo F, Liu X, Lu L, Xu H, Yang Q, et al. NF-kB-regulated exosomal miR-155 promotes the inflammation associated with arsenite carcinogenesis. Cancer Lett. 2017;388:21–33.
Bae ON, Lim KM, Noh JY, Chung SM, Kim H, Lee CR, et al. Arsenite-enhanced procoagulant activity through phosphatidylserine exposure in platelets. Chem Res Toxicol. 2007;20(12):1760–8.
Bowman AB, Kwakye GF, Herrero Hernandez E, Aschner M. Role of manganese in neurodegenerative diseases. J Trace Elem Med Biol. 2011;25(4):191–203.
Harischandra DS, Ghaisas S, Rokad D, Zamanian M, Jin H, Anantharam V, et al. Environmental neurotoxicant manganese regulates exosome-mediated extracellular miRNAs in cell culture model of Parkinson’s disease: relevance to alpha-synuclein misfolding in metal neurotoxicity. Neurotoxicology. 2018;64:267–77.
Sarkar S, Rokad D, Malovic E, Luo J, Harischandra DS, Jin H, et al. Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Sci Signal. 2019;12(563):eaat9900.
Harischandra DS, Rokad D, Neal ML, Ghaisas S, Manne S, Sarkar S, et al. Manganese promotes the aggregation and prion-like cell-to-cell exosomal transmission of alpha-synuclein. Sci Signal. 2019;12(572):eaau4543.
Howe CG, Foley HB, Farzan SF, Chavez TA, Johnson M, Meeker JD, et al. Urinary metals and maternal circulating extracellular vesicle microRNA in the MADRES pregnancy cohort. Epigenetics. 2021:1–15.
Meeker JD, Sathyanarayana S, Swan SH. Phthalates and other additives in plastics: human exposure and associated health outcomes. Philos Trans R Soc Lond B Biol Sci. 2009;364(1526):2097–113.
Rodosthenous RS, Baccarelli AA, Mansour A, Adir M, Israel A, Racowsky C, et al. Supraphysiological concentrations of bisphenol A alter the expression of extracellular vesicle-enriched miRNAs from human primary granulosa cells. Toxicol Sci. 2019;169(1):5–13.
Sheller-Miller S, Radnaa E, Arita Y, Getahun D, Jones RJ, Peltier MR, et al. Environmental pollutant induced cellular injury is reflected in exosomes from placental explants. Placenta. 2020;89:42–9.
Barnett-Itzhaki Z, Knapp S, Avraham C, Racowsky C, Hauser R, Bollati V, et al. Association between follicular fluid phthalate concentrations and extracellular vesicle microRNAs expression. Hum Reprod. 2021;36(6):1590–9.
Martinez RM, Hauser R, Liang L, Mansur A, Adir M, Dioni L, et al. Urinary concentrations of phenols and phthalate metabolites reflect extracellular vesicle microRNA expression in follicular fluid. Environ Int. 2019;123:20–8.
Lin CY, Hsieh CJ, Lo SC, Chen PC, Torng PL, Hu A, et al. Positive association between concentration of phthalate metabolites in urine and microparticles in adolescents and young adults. Environ Int. 2016;92–93:157–64.
Kataria A, Levine D, Wertenteil S, Vento S, Xue J, Rajendiran K, et al. Exposure to bisphenols and phthalates and association with oxidant stress, insulin resistance, and endothelial dysfunction in children. Pediatr Res. 2017;81(6):857–64.
Kovarova J, Svobodova Z. Perfluorinated compounds: occurrence and risk profile. Neuro Endocrinol Lett. 2008;29(5):599–608.
Lin CY, Chen PC, Lo SC, Torng PL, Sung FC, Su TC. The association of carotid intima-media thickness with serum Level of perfluorinated chemicals and endothelium-platelet microparticles in adolescents and young adults. Environ Int. 2016;94:292–9.
Von Burg R. Toluene. J Appl Toxicol. 1993;13(6):441–6.
Lim JH, Song MK, Cho Y, Kim W, Han SO, Ryu JC. Comparative analysis of microRNA and mRNA expression profiles in cells and exosomes under toluene exposure. Toxicol In Vitro. 2017;41:92–101.
Lim JS, M; Cho, Y; Kim, W; Han, S; Ryu, J. Expression of exosomal and cellular microRNAs: as biomarkers for toluene, ethylbenzene, xylene (TEX) exposure. Mol Cell Toxicol. 2016;12:359–69.
Radad K, Al-Shraim M, Al-Emam A, Wang F, Kranner B, Rausch WD, et al. Rotenone: from modelling to implication in Parkinson’s disease. Folia Neuropathol. 2019;57(4):317–26.
Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan GA, Pal A, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898.
Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
Funding
Christina Eckhardt was supported by a clinical and translational science awards grant (TL1TR001875). Andrea Baccarelli is supported by R01ES025225, P30ES00908, R35ES031688, and R01ES027747 grants from the National Institute of Environmental Health Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Environmental Epigenetics
Rights and permissions
About this article

Cite this article
Eckhardt, C.M., Baccarelli, A.A. & Wu, H. Environmental Exposures and Extracellular Vesicles: Indicators of Systemic Effects and Human Disease. Curr Envir Health Rpt 9, 465–476 (2022). https://doi.org/10.1007/s40572-022-00357-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-022-00357-5