
Abstract
Objectives
Microparticles are membrane vesicles shed by cells upon activation and apoptosis. Agonists capable of inducing microparticle generation include cytokines, bacterial products, P-selectin, histamine. Cigarette smoke extract has also been recognized as an agonist involved in microparticle generation with an apoptosis-dependent mechanism. We investigated the possibility that cigarette smoke extract induces the rapid generation of proinflammatory microparticles by human mononuclear cells with a calcium-dependent mechanism.
Materials and methods
Human mononuclear cells were exposed to cigarette smoke extract. [Ca2+]i mobilization was assessed with the fluorescent probe Fluo-4 NW. Microparticles were quantified with a prothrombinase assay and by flow cytometry. Normal human bronchial epithelial cells and A549 alveolar cells were incubated with cigarette smoke extract-induced microparticles and the generation of ICAM-1, IL-8, and MCP-1 was assessed by ELISA.
Results
Exposure to cigarette smoke extract induced a rapid increase in [Ca2+]i mobilization. Microparticle generation was also increased. EGTA, verapamil and the calmodulin inhibitor, W-7, inhibited microparticle generation. Incubation of lung epithelial cells with cigarette smoke extract-induced microparticles increased the expression of proinflammatory mediators.
Conclusions
Exposure of mononuclear cells to cigarette smoke extract causes a rapid shedding of microparticles with a proinflammatory potential that might add to the mechanisms of disease from tobacco use.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microparticles (MP), also referred to as ectosomes, are submicron membrane vesicles shed by cells upon activation and/or apoptosis [1]. A growing body of evidence indicates that MP derived from leukocytes, platelets, and endothelial cells participate in numerous physiological processes such as blood coagulation and inflammation, including lung inflammation [2–5].
The mechanisms involved in MP formation have not been fully elucidated; however, two distinct, well-characterized pathways that can lead to MP formation have been identified, namely cell activation and apoptosis [1]. MP shedding caused by activation starts within seconds after addition of the relevant agonist [6] and is characterized by an increase in cytosolic calcium concentration [7, 8]; in contrast, in apoptosis-dependent MP formation, dynamic membrane blebbing follows cell contraction and DNA fragmentation, and its time-course is usually measured in hours [1, 9, 10].
Tobacco smoke is a worldwide epidemic currently considered the most important preventable cause of death in the Western countries [11]. Not surprisingly, considering that cigarette smoke comprises some 4,700 different compounds [12], the mechanisms whereby cigarette smoke causes disease are complex and not fully understood as yet. In vitro experiments have shown that cigarette smoke extract (CSE) exerts roles as diverse as, for example, upregulation of the synthesis of proinflammatory mediators by epithelial cells [13], induction of toll-like receptor expression in bone marrow mononuclear cells [14], pulmonary artery smooth muscle cell proliferation [15]. The role of CSE in the modulation of apoptosis has been extensively investigated. We [16] and others [17–19] have demonstrated that CSE induces apoptosis in human fibroblasts, nasal and bronchial epithelial cells and in mouse embryonic cells; a role for CSE in airway epithelial cell necrosis, rather than apoptosis, has also been demonstrated upon infection with the respiratory syncytial virus [20].
Li and coworkers [21] have demonstrated that CSE induces the generation of procoagulant MP from human mononuclear cells in a process requiring activation of the apoptotic process; this observation further contributes to our understanding of the pathogenetic role of CSE. The presence of tissue factor, the physiological initiator of the so-called extrinsic pathway of blood coagulation, on the surface of CSE-induced MP might play a role, for example, in the pathogenesis of smoking-related ischemic heart disease [22].
We have previously demonstrated that a number of agonists, including the calcium ionophore A23187, histamine, the peroxisome proliferator-activated receptor-γ agonist, rosiglitazone [5, 23], and angiotensin II [24] cause a rapid upregulation of MP shedding from leukocytes. The rapid time frame of these events rules out a role for apoptosis in the process, instead suggesting that other mechanisms, including calcium mobilization [24] are involved. It has been demonstrated that CSE causes an increase in [Ca2+]i in cardiac myocytes [25]. We, therefore, speculated that CSE, in addition to the apoptosis-mediated events described by Li and coworkers [21], might induce MP generation in a rapid, calcium-dependent, apoptosis-independent fashion. After testing this hypothesis, we also investigated the procoagulant and proinflammatory potential of MP generated through this mechanism.
Materials and methods
Reagents and kits
RPMI 1640 medium, high glucose-DMEM, penicillin, streptomycin, l-glutamine, fetal bovine serum (FBS), trypsin/EDTA, trypsin inhibitor, PBS, HEPES, sodium chloride, Ficoll–Hypaque, dextran, sodium citrate, calcium chloride, calcium ionophore A23187, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT), ethylene glycol tetraacetic acid (EGTA), Ham’s F12, DNAse, formaldehyde, o-phenylendiamine and peroxidase-conjugate anti-mouse IgG γ-chain specific were obtained from Sigma (Milan, Italy). Vitrogen was obtained from Tebubio (Milan, Italy). BEGM bullet kit was obtained from Cambrex (Caravaggio, BG, Italy). Thromboplastin standard was obtained from Beckman Coulter (Milan, Italy). Human anti-TF antibody was obtained from America Diagnostica (Instrumentation Laboratory, Milan, Italy). The Zymuphen MP-Activity kit was obtained from Hyphen BioMed (Neuville-sur-Oise, France). The Fluo-4 NW Calcium Assay kit was obtained from Molecular Probes (Invitrogen, Milan, Italy). Human IL-8 matched antibody pairs were obtained from Bender Medsystems GmbH (Vienna, Austria). Human CCL2 (MCP-1) ELISA Ready-SET-Go! was obtained from eBioscience (Hatfield, Ireland, UK).
The mouse anti-human-ICAM-1 monoclonal antibody (clone 15.2) and the mouse anti-human-IgG1 isotype control were obtained from Ancell (Bayport, MN, USA). Fluorescein isothiocyanate (FITC)-labeled Annexin V, phycoerythrin (PE)-labeled propidium iodide and annexin V binding buffer were purchased from Societá Italiana Chimici (Rome, Italy); allophycocyanin (APC)-labeled mouse anti human CD14 antibody was purchased from BD Pharmingen (San Jose, CA, USA). MegamixTM, a blend of monodisperse fluorescent beads of three diameters (0.5, 0.9 and 3 μm), was purchased from BioCytex (Marseille, France). All other chemicals were obtained from the hospital pharmacy and were of the best grade available.
Cell culture
Cells of the human alveolar epithelial line, A549 (American Type Culture Collection, CCL-195), were kindly provided by Dr. R. Danesi, University of Pisa, Italy. A549 cells were maintained in RPMI supplemented with 10 % (vol/vol) FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin in a humidified 95 % air–5 % CO2 atmosphere at 37 °C. Normal human bronchial epithelial (NHBE) cells were obtained from subjects undergoing diagnostic bronchoscopy according to Kelsen et al. [26] as previously described [27]. Briefly, after the subject’s informed consent to the procedure was received, the fiber-optic bronchoscope was positioned at the level of the carina and/or the level of second- or third-order bronchial branchings. The use of local anesthetics was kept as low as possible to minimize their effects on cell viability. Four to six brushings of grossly normal bronchial mucosa were obtained. The cells were then removed from the brush by vortexing in Ham’s F-12 medium–10 % FBS. The cells were brought to the laboratory on ice and incubated with DNase (50 μg/mL) to eliminate clumping. After a wash with ice-cold, serum-free Ham’s F-12 medium, the cells were resuspended in BEGM and plated on Vitrogen 100-coated culture flasks. Indirect immunofluorescence with anti-cytokeratin antibodies confirmed the epithelial origin of the cells [27]. Cells used in the specific experiments reported here were obtained from a patient with peripheral lung cancer undergoing diagnostic bronchoscopy and were harvested from the contralateral main bronchus. The cells were used at passages 3–4. Although not specifically enumerated, the number of cytokeratin positive cells represented an overwhelming majority. The procedure was approved by the local ethics committee.
Mononuclear cells isolation and culture
Mononuclear cells were isolated either from fresh buffy coats obtained from the local blood bank or from the peripheral blood of normal volunteers as described [5]. Briefly, a fresh buffy coat was diluted 1:1 with sodium citrate 0.38 % in normal saline solution, mixed gently with an equal volume of 2 % Dextran T500, and left for 30 min for erythrocyte sedimentation. The leukocyte-rich supernatant was recovered and centrifuged for 10 min at 200×g. The pellet was resuspended in 30 mL of sodium citrate solution, layered over 15 mL of Ficoll–Hypaque and centrifuged for 30 min at 350×g at 4 °C. The mononuclear cell-rich ring was recovered and washed twice in sodium citrate solution. Mononuclear cells were then resuspended in RPMI/10 % FBS and allowed to adhere for 18 h at 37 °C on 24-well plates (2 × 106 cells/well) or on 10-cm Petri dishes (40 × 106 cells/dish).
Cigarette smoke extract (CSE)
Camel cigarettes (R.J. Reynolds Tobacco Company), which contain 10 mg tar and 0.8 mg nicotine/cigarette, were used. CSE was prepared by a modification of the method of Carp and Janoff as previously described [16]. Briefly, a 5 cm cigarette without filter was combusted with a modified syringe-driven apparatus, the smoke was bubbled through 25 mL of serum-free RPMI and the resulting solution was considered 100 % CSE. The resulting suspension was filtered through a 0.22-μm pore filter to remove bacteria and large particles. To ensure the reproducibility among different CSE batches, the absorbance measured at 340 nm was used as a measure of the “strength” of the extract. Dilutions were made with RPMI to obtain the desired absorbance. Different concentrations of CSE (from 2.5 to 10 %) were applied to mononuclear cells cultures within 30 min of preparation.
MP generation and purification
Mononuclear cells were washed three times with pre-warmed serum-free RPMI. For MP generation, CSE or the calcium ionophore A23187 (12 μM) were added; after 15 min at 37 °C, the supernatants were recovered, cleared by centrifugation at 14,000×g for 5 min at room temperature to remove dead cells and big cell fragments that might have detached during the stimulation, and immediately used for further experiments. In selected experiments, MP (12 mL) were further purified by ultracentrifugation (100,000×g for 2 h, 4 °C); the pellet was resuspended in 250 μL of normal saline and used in a one-stage clotting assay to measure tissue factor (TF)-dependent coagulation.
Measurement of MP
Phosphatidlyserine (PS)-positive MP in each sample were detected using the Zymuphen MP-activity kit (Hyphen BioMed, Neuville-sur-Oise, France) according to the manufacturer’s instructions and expressed as PS equivalents (nM PS).
Measurement of [Ca2+]i
Molecular Probes Fluo-4 NW Calcium Assay kit was used to measure the changes in [Ca2+]i of mononuclear cells. Pre-washed mononuclear cells on 96-multiwell plate (0.5 × 106 cells/well) were loaded with 100 μL of the dye loading solution containing Fluo-4 NW dye and probenecid, according to the manufacturer’s instructions. The 96-well plate was incubated at 37 °C for 45–60 min in the dark and fresh CSE (10 %) and the calcium ionophore A23187 (12 μM) as a positive control were added to the cells. The changes in Fluo-4 NW fluorescence were measured by the Wallac 1420 Victor 2 (PerkinElmer, Milan, Italy) at λ ex 494 nm and λ em 516 nm. Calcium mobilization was observed over time (up to 80 s) and analyzed by the Wallac 1420 Software version 3 (PerkinElmer Life and Analytical Sciences, Wallac, Milan, Italy). The increase in [Ca2+]i fluorescence (expressed as relative fluorescence units, RFU) was calculated as the difference between the mean stimulation fluorescence value and the mean baseline fluorescence observed (ΔRFU).
ELISA for IL-8 and MCP-1 detection
MP generated by mononuclear cells were sedimented by ultracentrifugation (100,000×g for 2 h, 4 °C) and quantified by the Zymuphen MP-Activity kit. Resuspended MP were adjusted to 3.5 nM PS and incubated with A549 and NHBE cells grown to confluence in 96-well plates for 24 h at 37 °C. Following an 18-h incubation, the conditioned medium was harvested, cleared by centrifugation for 5 min at 12,000×g, and analyzed for IL-8 and MCP-1 content. IL-8 and MCP-1 in supernatants from A549 and NHBE cells were measured by a sandwich ELISA kit according to the manufacturer’s instructions.
Measurement of cell surface intercellular adhesion molecule-1 (ICAM-1) expression
ICAM-1 expression on HEBC and A549 cells was measured by direct cell ELISA as described with minor modifications [27]. Briefly, cells were seeded at 20 × 103 cells/well in 0.1 mL of medium in 96-well plates. When the cells reached subconfluence, they were washed three times with PBS and fixed with 3.7 % formaldehyde (w/v). Following additional extensive washing, nonspecific Ab binding sites were blocked by incubating the cells with PBS/3 % BSA (w/v) for 2 h. Cells were then incubated with mouse anti-human ICAM-1 Ab (5 mg/mL) in PBS 1 % BSA (w/v) for 2 h at 37 °C, then washed three times with wash buffer. Peroxidase-conjugated antimouse IgG Ab was then added at the concentration recommended by the manufacturer and incubated for 1 h at 37 °C. After extensive washing, bound enzyme was determined by adding o-phenylendiamine, 1 mg/mL in 50 mM sodium citrate (pH 4), containing 0.015 % H2O2. After blocking the reaction with 2 M H2SO4, the plate was read in a spectrophotometer (Titertek Multiskan MCC ELISA reader; Flow Laboratories, McLean, VA, USA) at 492 nm.
Flow cytometry detection of MP
Mononuclear cells were treated as described above and the supernatant submitted to flow cytometry using a FACScantoTM II flow cytometer (BD Biosciences, San Jose, CA, USA) as previously described [24]. Briefly, a mixture of 30 μL of supernatant, 3 μL of APC conjugated anti CD14 antibody, 3 μL of FITC labeled annexin V and 30 μL of annexin V 2 × binding buffer was incubated for 15 min at room temperature in the dark. Immediately prior to flow cytometric acquisition, 400 μL of PBS were added to each mixture. Events acquisition was obtained at high flow rate and was stopped after 210 s. The side scatter channel (SSC) and forward scatter channel (FSC) parameters were set at log scale. Specificity of CD14 labeling was confirmed with a non-immune, isotype-matched APC conjugated mouse IgG. Monocyte-derived MP were discriminated first by size, as events conforming to a light scatter distribution within the 0.5–0.9 μm bead range in a SSC vs. FSC window and further identified as CD14 and annexin V positive events in a APC vs. FITC window.
Cell viability assay
To assess mononuclear cells viability, cell counting was performed after MTT assays as previously described [28]. Briefly 2 × 106 cells/well were seeded into 24-well plates and incubated overnight. Mononuclear cells were then washed three times with pre-warmed serum-free RPMI and stimulated with RPMI in the presence or in the absence of CSE 10 % for 15 min. MTT (100 μL at 5 mg/mL) was added to each well and cells were incubated at 37 °C for a further 2 h. Formazan crystals formed by MTT metabolism were solubilized by addition of 100 μL of dimethyl sulphoxide to each well and absorbance was measured at a wavelength of 570 nm with background subtraction at 630–690 nm using a microplate reader (Titertek Multiskan MCC ELISA reader; Flow Laboratories, McLean, VA, USA).
Assessment of apoptosis
Apoptosis was measured using a commercially available kit as previously described [28]. In brief, mononuclear cells treated as described above were detached with a scraper after treatment and 5 × 105 cells were resuspended in 500 μL of binding buffer containing 5 μL of annexin V-FITC and 5 μL of propidium iodide–PE, and incubated in the dark at room temperature for 15 min. Cells were then analyzed in a FACScan flow cytometer (Beckton Dickinson, San Jose, CA, USA). Cisplatin (1 μM; 20 h) was used as a positive control.
Assessment of MP-bound TF activity
TF activity was measured in MP generated in vitro from human mononuclear cells by a one-stage clotting time assay as described [29], except that the normal human plasma was made MP-poor by ultracentrifugation (100,000×g for 2 h, 4 °C). The results were expressed in arbitrary units (AU) of procoagulant activity by comparison with a standard curve obtained using a human brain thromboplastin standard. This preparation was assigned a value of 1,000 UA for a clotting time of 30 s. An anti-human TF antibody (30 μg/mL) was used to assess the specificity of the test.
Results
CSE induces the mobilization of [Ca2+]i in mononuclear cells
Mononuclear cells were loaded with Fluo-4 NW probe and stimulated with fresh CSE. Figure 1 shows that CSE induces a rapid increase in [Ca2+]i. The figure also shows that verapamil, an L-type calcium channel current blocker, inhibits the CSE-induced increase in [Ca2+]i (see below).

[Ca2+]i mobilization in human mononuclear cells in baseline conditions (filled triangle) and upon incubation with 10 % CSE (filled circle), 10 % CSE after a 30 min preincubation with 1 μM verapamil (filled inverted triangle) and 12 μM A23187 (filled square), as assessed by Fluo-4 NW incorporation. Error bars represent SEM; (N = 3)
CSE induces a rapid generation of MP by mononuclear cells
To investigate whether CSE treatment induces a rapid release of MP following calcium mobilization, mononuclear cells were stimulated with fresh CSE at different dilutions (2.5, 5, 10 %) for 15 min. Figure 2 shows that mononuclear cells generate MP upon exposure to CSE for 15 min; the effect is concentration dependent (Fig. 2a) and reaches statistical significance at 10 % CSE (Fig. 2b). The upregulation of MP upon CSE treatment was confirmed by FACS analysis; furthermore, flow cytometry confirmed that the subpopulation of monocyte-derived, CD14 positive, MP is also increased (Fig. 2c).

Cigarette smoke extract induces the generation of MP by human mononuclear cells. (a) Concentration response curve; (b) Mean MP generation in baseline conditions and after incubation with 10 % CSE for 10 min; error bars represent SEM; (N = 9) (each experiment from an individual donor); *p < 0.05 by paired Student’s t test; c FACS analysis of CSE-induced MP (see text for details)
Rapid CSE-induced generation of MP by mononuclear cells is dependent on calcium mobilization
As shown in Fig. 3a, preincubation of cells with the calcium chelator EGTA (1 mM for 30 min) largely inhibited CSE-induced MP generation, confirming a role for calcium mobilization in the phenomenon.

Rapid CSE-induced generation of MP by mononuclear cells is dependent on calcium (a). Cells were exposed to CSE in the absence or in the presence of EGTA (1 mM) and MP generation was assessed. Error bars represent SEM; (N = 3) (each experiment from an individual donor); *p < 0.05 for CSE vs. baseline; **p < 0.05 for CSE + EGTA vs. CSE; repeated measures ANOVA. L-type channel calcium current blockage and calmodulin inhibition prevent rapid CSE-induced generation of MP by mononuclear cells (b). Verapamil (1 μM) and W-7 (1 μM) were preincubated for 30 min prior to CSE exposure. Error bars represent SEM (N = 4) (each experiment from an individual donor); p < 0.05 for both inhibitors vs. CSE alone; repeated measures ANOVA
To characterize further the role of calcium in the rapid induction of MP by CSE-stimulated mononuclear cells, we used two reagents involved in different steps of calcium signaling: verapamil, an L-type calcium channel current blocker and W-7, a calmodulin inhibitor. Figure 3b shows that both reagents inhibit CSE-induced MP generation. The inhibiting effect of verapamil on CSE-induced calcium mobilization is shown in Fig. 1.
Rapid CSE-induced generation of MP is independent of apoptosis
Apoptosis is a well known mechanism of MP generation. Specifically, Li et al. have demonstrated that CSE-induced, late (20 h) MP generation by human monocytes/macrophages is indeed dependent on apoptosis [21]. Figure 4 shows that, as expected, CSE did not induce apoptosis in the short time frame (minutes) of our experimental conditions. Cell viability was also assessed by an MTT assay and was confirmed to be unaffected (not shown).

CSE exposure of mononuclear cells does not induce apoptosis. Cells were exposed to vehicle (a) or 10 % CSE extract (b) for 15 min, and to vehicle (c) and 1 μM cisplatin (d) for 20 h prior to PI and annexin V staining. Data from one experiment representative of two. LL lower left quadrant; LR lower right quadrant; UL upper left quadrant; UR upper right quadrant
CSE-induced MP upregulate the synthesis of proinflammatory mediators by lung epithelial cells
Numerous studies have demonstrated the proinflammatory potential of leukocyte-derived MP [3, 5, 30–32]. In the present study, we investigated whether CSE-induced MP are able to upregulate the synthesis of proinflammatory mediators by lung epithelial cells.
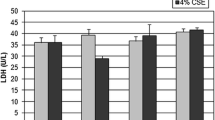
Lung epithelial cells were incubated with purified CSE-induced MP for 18 h, and IL-8 and MCP-1 were measured in the supernatants. Expression of membrane-bound ICAM-1 was assessed by c-ELISA. Figure 5 shows that CSE-induced MP upregulate the expression of proinflammatory mediators by A549 (panels a–c) and NHBE (panels d–e) cells. MCP-1 expression by NHBE cells was not apparently modulated by CSE-induced MP (not shown); these data were in agreement with previous results with A23187-induced MP and we did not further pursue this issue.

IL-8 (a), ICAM-1 (b) and MCP-1 (c) expression by A549 cells exposed to cigarette smoke extract-induced microparticles. Cells were incubated for 24 h with microparticles (3.5 nM phosphatidylserine) shed by human mononuclear cells upon incubation with 10 % cigarette smoke extract for 15 min. Error bars represent SEM (N = 5) for A549 (each experiment from an individual donor); *p < 0.05 by paired t test. IL-8 (d) and ICAM-1 (e) expression by NHBE cells exposed to cigarette smoke extract-induced microparticles. NHBE cells were incubated for 24 h with microparticles (3.5 nM phosphatidylserine) shed by human mononuclear cells upon incubation with 10 % cigarette smoke extract for 15 min. Error bars represent SEM (N = 7) (each experiment from an individual mononuclear cell donor); *p < 0.05 by paired t test
CSE-induced MP express TF on their surface
To evaluate whether MP induced upon short exposure of mononuclear cells to CSE express TF on their surface, we analyzed TF procoagulant activity of purified MP released by treated and untreated cells through a one-stage clotting test. As shown in Fig. 6, CSE treatment induces a 2.6-fold, significant increase in MP bound TF activity. A monoclonal antibody to TF (epitope specific for aa 1–25, American Diagnostica, Stamford, CT, USA; 30 μg/mL) inhibited most of the procoagulant activity (not shown), confirming the identity of this activity with TF.

Microparticle-bound tissue factor shed by human mononuclear cells upon incubation with cigarette smoke extract. Human mononuclear cells were incubated with cigarette smoke extract (10 %) for 15 min. Shed microparticles were purified by ultracentrifugation and analyzed for tissue factor activity with a one-stage clotting assay. Error bars represent SEM (N = 8) (each experiment from an individual mononuclear cell donor); *p < 0.05 by paired t test
Discussion
Originally considered laboratory artifacts or cell debris devoid of physiological significance, MP have more recently become the object of intense studies, since their role in human physiology and pathophysiology has been clearly recognized. In the field of respiratory diseases, Bastarache et al. [33] have recently investigated the potential role of MP in the pathogenesis of the acute respiratory distress syndrome, demonstrating a significant increase in MP concentration, likely of epithelial origin, in the edema fluid of patients with the syndrome compared with patients with hydrostatic edema. An increase in circulating MP of endothelial origin was observed in cigarette smokers with early lung destruction (normal spirometry and reduced diffusion capacity), compared to “healthy” smokers and normal controls [34]. Finally, it has been recently shown that MP of endothelial origin are increased during acute exacerbations of chronic obstructive lung disease [35].
Several pathophysiologically relevant agonists have been shown to induce the shedding of MP, including cytokines, such as TNF-α [36], bacterial products, such as lipopolysaccharides [37], histamine [5], P-selectin [38]. Of note, the composition and activity of MP differ not only based on the cell from which they originate, but also on the stimulus used for their induction [37]. The time course of MP stimulation with different agonists varies from minutes (e.g. A23187 [5]) to 1–2 h (e.g. histamine [5] and peroxisome proliferator activated receptor-γ agonists [23]) to several hours (e.g. LPS [37] and CSE [21]), likely reflecting the different pathways involved in MP generation. Based on the observation that CSE induces rapid calcium mobilization, we postulated that this agent might also induce rapid MP shedding. Indeed, we first demonstrated that calcium chelation with EGTA prevented MP shedding in our experimental conditions; we also showed that this phenomenon is inhibited with L-type calcium channel current blockage. This observation is consistent with the hypothesis that MP shedding in our conditions requires calcium entry via the L-type channels. According to this model, calmodulin inhibition would also be predicted to block MP generation, as indeed indicated by our results.
We then tested the hypothesis that MP induced by cigarette smoke, one of the most important causes of lung disease, are proinflammatory. The present data confirm that MP are generated by human mononuclear cells in a rapid, calcium-dependent fashion upon exposure to CSE and that they have a proinflammatory potential. The role of IL-8, MCP-1, ICAM-1 and, more broadly, of inflammation in lung diseases is well recognized [39–41]. Thus, CSE induced MP might contribute to mount the inflammatory response that characterizes these diseases.
Because they were detected through their annexin V binding properties, MP are procoagulant by definition, since binding to annexin V takes place via PS, an essential component of the prothrombinase and tenase complexes. However, we also investigated the presence of TF on the MP surface. TF is the initiator of the so-called extrinsic pathway of blood coagulation. The term extrinsic reflects the knowledge that TF is constitutively expressed on non-vascular cells—and is, therefore, extrinsic to blood—and comes in contact with circulating factor VIIa only upon disruption of the endothelial monolayer. However, it is now evident that TF can also be exposed on vascular cells, including endothelial cells and monocytes, upon activation. More recently, the expression of TF has also been demonstrated on circulating MP; this observation expands the procoagulant potential of these subcellular structures [2]. The current data extend Li’s observation of the presence of TF on MP generated via an apoptosis-dependent mechanism and confirm that MP originated by mononuclear cells upon exposure to CSE express fully functional TF and potentially contribute to the pathogenesis of cardiovascular diseases.
In conclusion, our data demonstrate that exposure of mononuclear cells to cigarette smoke causes rapid cell activation, via intracellular calcium mobilization, and shedding of MP with proinflammatory and procoagulant potential. Taken together with Li’s data, the results indicate that CSE has the potential to stimulate MP generation by two distinct mechanisms, one rapid, [Ca2+]i dependent, and one more slow, that requires the induction of apoptosis. Modulation of MP generation might represent in the future a viable approach to treat smoke-induced lung and vascular diseases.
References
VanWijk MJ, VanBavel E, Sturk A, et al. Microparticles in cardiovascular diseases. Cardiovasc Res. 2003;59:277–87.
Celi A, Lorenzet R, Furie BC, et al. Microparticles and a P-selectin-mediated pathway of blood coagulation. Dis Markers. 2004;20:347–52.
Ardoin SP, Shanahan JC, Pisetsky DS. The role of microparticles in inflammation and thrombosis. Scand J Immunol. 2007;66:159–65.
Chironi GN, Boulanger CM, Simon A, et al. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–51.
Cerri C, Chimenti D, Conti I, et al. Monocyte/macrophage-derived microparticles up-regulate inflammatory mediator synthesis by human airway epithelial cells. J Immunol. 2006;177:1975–80.
MacKenzie A, Wilson HL, Kiss-Toth E, et al. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. 2001;15:825–35.
Wiedmer T, Sims PJ. Participation of protein kinases in complement C5b-9-induced shedding of platelet plasma membrane vesicles. Blood. 1991;78:2880–6.
Gemmell CH, Sefton MV, Yeo EL. Platelet-derived microparticle formation involves glycoprotein IIb–IIIa. Inhibition by RGDS and a Glanzmann’s thrombasthenia defect. J Biol Chem. 1993;268:14586–9.
Aupeix K, Hugel B, Martin T, et al. The significance of shed membrane particles during programmed cell death in vitro, and in vivo, in HIV-1 infection. J Clin Invest. 1997;99:1546–54.
Zhang J, Driscoll TA, Hannun YA, et al. Regulation of membrane release in apoptosis. Biochem J. 1998;334:479–85.
Campbell SC, Moffatt RJ, Stamford BA. Smoking and smoking cessation—the relationship between cardiovascular disease and lipoprotein metabolism: a review. Atherosclerosis. 2008;201:225–35.
Yang SR, Wright J, Bauter M, et al. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–76.
Mio T, Romberger DJ, Thompson AB, et al. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med. 1997;155:1770–6.
Zhou J, Eksioglu EA, Fortenbery NR, et al. Bone marrow mononuclear cells up-regulate toll-like receptor expression and produce inflammatory mediators in response to cigarette smoke extract. PLoS ONE. 2011;6:e21173.
Zeng DX, Xu YJ, Liu XS, et al. Cigarette smoke extract induced rat pulmonary artery smooth muscle cells proliferation via PKCalpha-mediated cyclin D1 expression. J Cell Biochem. 2011;112:2082–8.
Carnevali S, Petruzzelli S, Longoni B, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284:L955–63.
Togo S, Sugiura H, Nelson A, et al. Hepatic growth factor (HGF) inhibits cigarette smoke extract induced apoptosis in human bronchial epithelial cells. Exp Cell Res. 2010;316:3501–11.
Chen ZL, Tao J, Yang J, et al. Vitamin E modulates cigarette smoke extract-induced cell apoptosis in mouse embryonic cells. Int J Biol Sci. 2011;7:927–36.
Lan MY, Ho CY, Lee TC, et al. Cigarette smoke extract induces cytotoxicity on human nasal epithelial cells. Am J Rhinol. 2007;21:218–23.
Groskreutz DJ, Monick MM, Babor EC, et al. Cigarette smoke alters respiratory syncytial virus-induced apoptosis and replication. Am J Respir Cell Mol Biol. 2009;41:189–98.
Li M, Yu D, Williams KJ, et al. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30:1818–24.
Cimmino G, D’Amico C, Vaccaro V, et al. The missing link between atherosclerosis, inflammation and thrombosis: is it tissue factor? Expert Rev Cardiovasc Ther. 2011;9:517–23.
Neri T, Cordazzo C, Carmazzi Y, et al. Effects of peroxisome proliferator activated receptors-gamma agonists on the generation of microparticles by monocytes/macrophages. Cardiovasc Res. 2012;11:243–7.
Cordazzo C, Neri T, Petrini S, et al. Angiotensin II induces the generation of procoagulant microparticles by human mononuclear cells via an angiotensin type 2 receptor-mediated pathway. Thromb Res. 2013;131:e168–74.
Yamada S, Zhang XQ, Kadono T, et al. Direct toxic effects of aqueous extract of cigarette smoke on cardiac myocytes at clinically relevant concentrations. Toxicol Appl Pharmacol. 2009;236:71–7.
Kelsen SG, Mardini IA, Zhou S, et al. A technique to harvest viable tracheobronchial epithelial cells from living human donors. Am J Respir Cell Mol Biol. 1992;7:66–72.
Celi A, Cianchetti S, Petruzzelli S, et al. ICAM-1-independent adhesion of neutrophils to phorbol ester-stimulated human airway epithelial cells. Am J Physiol. 1999;277:L465–71.
Carmazzi Y, Iorio M, Armani C, et al. The mechanisms of nadroparin-mediated inhibition of proliferation of two human lung cancer cell lines. Cell Prolif. 2012;45:545–56.
Celi A, Pellegrini G, Lorenzet R, et al. P-Selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci. 1994;91:8767–71.
Mesri M, Altieri DC. Endothelial cell activation by leukocyte microparticles. J Immunol. 1998;161:4382–7.
Berckmans RJ, Nieuwland R, Kraan MC, et al. Synovial microparticles from arthritic patients modulate chemokine and cytokine release by synoviocytes. Arthritis Res Ther. 2005;7:R536–44 Epub 2005 Mar 1.
Distler JH, Huber LC, Gay S, et al. Microparticles as mediators of cellular cross-talk in inflammatory disease. Autoimmunity. 2006;39:683–90.
Bastarache JA, Fremont RD, Kropski JA, et al. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1035–41.
Gordon C, Gudi K, Krause A, et al. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am J Respir Crit Care Med. 2011;184:224–32.
Takahashi T, Kobayashi S, Fujino N, et al. Increased circulating endothelial microparticles in COPD patients: a potential biomarker for COPD exacerbation susceptibility. Thorax. 2012;67:1067–74.
Combes V, Simon AC, Grau GE, et al. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J Clin Invest. 1999;104:93–102.
Bernimoulin M, Waters EK, Foy M, et al. Differential stimulation of monocytic cells results in distinct populations of microparticles. J Thromb Haemost. 2009;7:1019–28.
Hrachovinova I, Cambien B, Hafezi-Moghadam A, et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003;9:1020–5.
Pesci A, Balbi B, Majori M, et al. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J. 1998;12:380–6.
Traves SL, Culpitt SV, Russell RE, et al. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. 2002;57:590–5.
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445–54.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Bernhard Gibbs.
Rights and permissions
About this article
Cite this article
Cordazzo, C., Petrini, S., Neri, T. et al. Rapid shedding of proinflammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on Ca2+ mobilization. Inflamm. Res. 63, 539–547 (2014). https://doi.org/10.1007/s00011-014-0723-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-014-0723-7